

Abstract
Alcohol use disorders are a leading public health concern, engendering enormous costs in terms of both economic loss and human suffering. These disorders are characterized by compulsive and excessive alcohol use, as well as negative affect and alcohol craving during abstinence. Extensive research has implicated the dopamine system in both the acute pharmacological effects of alcohol and the symptomology of alcohol use disorders that develop after extended alcohol use. Preclinical research has shed light on many mechanisms by which chronic alcohol exposure dysregulates the dopamine system. However, many of the findings are inconsistent across experimental parameters such as alcohol exposure length, route of administration, and model organism. We propose that the dopaminergic alterations driving the core symptomology of alcohol use disorders are likely to be relatively stable across experimental settings. Recent work has been aimed at using multiple model organisms (mouse, rat, monkey) across various alcohol exposure procedures to search for commonalities. Here, we review recent advances in our understanding of the effects of chronic alcohol use on the dopamine system by highlighting findings that are consistent across experimental setting and species.
Keywords: Alcohol, Autoreceptors, Cross-species, Dopamine, Kappa Opioid receptors, Monkey, Mouse, Nonhuman primate, Rat, Uptake
1 Introduction
1.1 Alcohol Use Disorder
Alcohol use disorder (AUD) is a medical diagnosis describing a cluster of symptoms characterized by compulsive and excessive alcohol use despite negative health and social outcomes of drinking (Dziegielewski 2010; Hagman and Cohn 2011; O’Brien 2011). AUD is a leading public health concern, affecting roughly 14% of the adult population in the United States (Grant et al. 2015). The prognosis for AUD is often poor, and it is estimated that alcohol use leads to around 100,000 deaths per year in the United States alone, and 4% of deaths worldwide, making it the third leading preventable cause of death (Mokdad et al. 2004; Rehm et al. 2009). Beyond mental and physical deterioration, alcoholism and the larger spectrum of AUD also negatively affect civil and social responsibilities and interpersonal relationships. AUDs are associated with higher incidences of stress, anxiety, depression, and other mood disorders (Hasin et al. 2007), which may contribute to the maintenance of alcohol use as an anxiolytic (Blaine and Sinha 2017). In addition, AUD is a chronically relapsing disease (Dawson et al. 2007; Pickens et al. 1985). Given the severe negative outcomes of this prevalent brain disorder, the alcohol research field has focused its efforts on understanding the neurobiology of AUD in an effort to develop effective therapeutic strategies. Here we will review recent advances in understanding the mechanisms by which alcohol exposure affects dopamine signaling to produce aberrant behaviors seen in AUD.
1.2 The Role of Dopamine in Alcohol Use Disorder Symptomology
Many drugs of abuse exert their subjective effects (euphoria, or “high”), in part, via actions on the mesolimbic dopamine system (Di Chiara and Imperato 1988; Siciliano et al. 2015b; Volkow et al. 1997). Alcohol, like many abused substances, increases extracellular dopamine concentrations in the ventral striatum, an area known to be involved in reward and motivation (Humphries and Prescott 2010; Imperato and Di Chiara 1986). Acutely, alcohol increases dopamine signaling via directly targeting ion channels expressed on dopamine neurons, which alter the currents that shape cellular communication, and result in increased firing of dopamine neurons in the ventral tegmental area (VTA), leading to increased dopamine release downstream in the ventral striatum (Brodie 2002; Brodie et al. 1990, 1999; Nimitvilai et al. 2016). Alcohol enhances hyperpolarization-activated depolarizing cation currents (Ih), which increase intrinsic activity in dopamine neurons (Okamoto et al. 2006). In addition, alcohol modulates various potassium channel (K+)-mediated aspects of hyperpolarization (Appel et al. 2003). For example, alcohol has been shown to modulate large-conductance potassium (BK) channels (Chu et al. 1998), G-protein-coupled inwardly rectifying K+ channels (GIRK) (Aryal et al. 2009), and K+-channel mediated M currents (Koyama et al. 2007). Additionally, alcohol has been shown to alter L-type Ca2+ channels (Hendricson et al. 2003), which may alter dopamine cell firing. While it is important to note that alcohol acutely modulates dopaminergic activity, this review will focus on the adaptions to the dopamine system induced by chronic alcohol use. Further, this review will focus on receptor and circuit level analyses; for more in-depth discussions of the molecular mechanisms underlying these alterations we point the reader towards several other helpful reviews (Morikawa and Morrisett 2010; Abrahao et al. 2017; Morisot and Ron 2017; Ron and Barak 2016).
Changes in dopamine signaling are particularly important in the context of alcohol abuse, as the dopamine system plays a key role in mediating adaptive decision-making. Dopamine neurons in the VTA are required for adaptive encoding of reward-predictive cues, which allows organisms to successfully navigate complex environments and acquire rewards, such as food (Nicola 2010; Phillips et al. 2003; Schultz et al. 1997). Dynamic activity of dopamine neurons, and dopamine release downstream in areas such as the striatum, are critical to both initial learning of reinforcement contingencies and updating the value of these contingencies as they shift over time (Cools et al. 2009; Schultz 2013). Continued alcohol use can, in some individuals, induce a maladaptive shift in contingency valuation such that the motivational saliency of alcohol is increased, leading to behaviors aimed at acquiring alcohol at the expense of more adaptive rewards. Because alcoholics show a decreased ability to make adaptive decisions – and instead continue maladaptive behavioral strategies such as alcohol seeking – the dopamine system has been implicated as a probable locus of these behavioral aberrations.
In humans, it is known that chronic alcohol use dysregulates the dopamine system. For example, alcohol abusers show greatly reduced dopamine signaling in the ventral striatum (Diana et al. 1993; Martinez et al. 2005; Volkow et al. 1996, 2007). This reduced dopamine signaling is often referred to as a “hypodopaminergic state” and is also observed in other addictive disorders such as psychostimulant addiction (Melis et al. 2005). It is hypothesized that this low-dopamine state results in deficits in reward processing, which contributes to anhedonia during withdrawal from alcohol (Danjo et al. 2014; Schulteis et al. 1995). Because anhedonia, defined as a reduced ability to experience pleasure or reward, is thought to occur primarily in relation to non-alcohol stimuli, this may bias choices towards previously reinforced alcohol seeking behaviors over alternative options (Pierce et al. 1990; Rebec et al. 1997; Twining et al. 2015), resulting in continued and persistent alcohol use. Anhedonia, maladaptive decision-making, and alcohol seeking are sine qua non symptoms of AUD. The precise mechanisms by which alcohol disrupts dopamine system function are difficult to study in humans, and thus have been a major focus of preclinical alcohol abuse research.
1.3 Preclinical Models of Alcohol Abuse
A great deal of research into the neurobiological basis of AUD has been conducted in animal models, where hypotheses can be readily tested via direct measurements and manipulations of the receptors and circuits involved. Preclinical studies have overwhelmingly leveraged rodent (rat and mouse) and nonhuman primates as model organisms. Rodent and nonhuman primate models each offer specific advantages in exploring alcohol’s effects on the brain. For example, rodents can be procured and bred quickly, and are thus more practical for studies which require brain tissue to be harvested (e.g., for protein analysis, or ex vivo slice preparations) or where high-risk, invasive methods are needed (e.g., intracranial implants). Further, there are a wide array of tools that have been developed for use in rodents, including genetic modifications, in vivo microscopy, neurotransmitter sensors, and tools for manipulating neural circuit activity (Boyden et al. 2005; Flusberg et al. 2008; Koller and Smithies 1992; Wightman 1988; Wightman et al. 1976). Many of these techniques have been adapted for, and implemented in, nonhuman primates; however, they are generally less developed and often pose greater challenges (Ariansen et al. 2012; Eldridge et al. 2016; Stauffer et al. 2016).
Nonhuman primate models offer many advantages over rodents, although invasive approaches are often not feasible. First, they are genetically much closer to humans, as macaque monkeys share 95% gene homology with humans while mice share only 75% homology (Church et al. 2009). This genetic similarity manifests itself in, among other things, a high degree of correspondence in neuroanatomical structures between monkeys and humans (Seress 2007). High levels of homology increase the likelihood that discoveries will generalize from nonhuman primates to humans, compared to lower organisms. The advantages of nonhuman primates’ similarity to humans is even greater in studies of alcohol drinking, as the patterns of alcohol consumption in nonhuman primates are similar to humans (Grant et al. 2008; Majchrowicz and Mendelson 1970). While rodents metabolize alcohol at a much faster rate than humans, nonhuman primates have similar alcohol metabolism as humans. Further, AUD is a chronic disorder which can take years to develop, and lasts a lifetime (Dawson et al. 2008). Not only is AUD long lasting, but there are also interactions between drinking and age/developmental periods. For example, age at first drink is strongly predictive of problematic drinking behaviors later in life (Dawson et al. 2008). The large disparity between the human and rodent life span can make examining the effects of alcohol exposure over long periods of time or during specific developmental periods challenging (Silberberg and Silberberg 1954). Macaques, which have been used most frequently for alcohol studies in the nonhuman primate literature, have a lifespan of 25–35 years in captivity, and thus are often more appropriate for longitudinal experimental questions (Tigges et al. 1988).
1.4 Importance of Consistent Cross-Species Results
Rodent and nonhuman primate models have provided valuable insight into the neurobiological and pharmacological basis of AUD; here, we posit that the most important insights from these literatures are the consistencies that can be observed across species. Searching for these consistencies is particularly important because the effects of chronic alcohol exposure in preclinical models is extremely sensitive to experimental parameters such as alcohol concentration, exposure length, route of administration, withdrawal length, strain of rodent or species of nonhuman primate, and many other variables that can have large impacts on the observed effects (Bonthius and West 1990; Budygin et al. 2003; Hwa et al. 2011; Kashem et al. 2012; Rimondini et al. 2003; Siciliano et al. 2016b, 2017). However, the core behavioral symptomology of AUD (excessive alcohol consumption, craving/ seeking, and withdrawal behaviors) is relatively consistent across experimental parameters (Green and Grahame 2008; Le et al. 1998; Macey et al. 1996; Venniro et al. 2016), suggesting that neurochemical adaptations that are only observed under very specific experimental settings may not be driving the primary symptomology of AUD. Instead, it is likely that some of the alterations that are inconsistent across experimental setting may be “epiphenomenon” of the specific paradigm or model organism used. The large number of methodological differences across laboratories makes determining the source of any inconsistent effects difficult.
Comparing across species, strain, sex, and experimental parameters to search for consistent adaptations induced by chronic alcohol exposure is a powerful approach for filtering “noise” out of large sets of studies. Here, we will review commonalities and disparities in studies examining chronic alcohol-induced alterations to the dopamine system across multiple model organisms.
1.5 Alcohol Exposure Protocols Across Species
Each species utilized as a preclinical model of AUD described in this chapter provides unique assets to address and examine specific facets of alcoholism. The three model organisms discussed here (mouse, rat, and monkey) are distinct in terms of alcohol metabolism and intake pattern; thus, species-specific alcohol exposure protocols are often used. These protocols are described in the sections below and depicted in Fig. 1.
Fig. 1.

Example paradigms of chronic ethanol administration across species. Schematic outline of ethanol administration in (a) mice, (b) rats, (c) male cynomolgus monkeys, and (d) female rhesus monkeys (note that many parameters vary slightly across lab and study depending on the specific hypotheses being addressed). (a, b) In order to precisely regulate blood ethanol levels and achieve high levels of ethanol intake, vaporized ethanol administration paradigms are utilized for noncontingent chronic exposure in rodents. (a) Mice undergo multiple cycles, ranging from 1 to 5, of intermittent ethanol vapor exposure (inset: daily ethanol exposure during one cycle) separated by 3 days of abstinence. In many of the experiments discussed in this review, behavioral and physiological measurements were taken during acute withdrawal, immediately following removal from the vapor chamber, or during abstinence – typically 72 h after last exposure. (b) Rats undergo a similar vapor exposure paradigm; however, daily ethanol exposure is continuous and not divided into cycles by days of abstinence. (c, d) The pattern of ethanol consumption by nonhuman primates is similar to that of humans and volitional intake paradigms offer greater insight to consummatory behaviors and subsequent physiological alterations. In many of the experiments discussed in this review, following a 5-month schedule-induced polydipsia induction period, nonhuman primates were given free access to ethanol and water for 22 h/day for either 6 or 12 months, and neurophysiological experiments examining the dopamine system are performed during acute withdrawal
1.5.1 Chronic Intermittent Alcohol Exposure in Mice and Rats
Rodents (both mice and rats) will voluntarily consume alcohol under certain conditions; however, due to relatively low intakes and fast metabolism of alcohol, environmental or genetic manipulations are often required to produce high blood alcohol levels in these animals (Li et al. 1979; Penn et al. 1978; Rhodes et al. 2005, 2007). For example, selectively breeding animals with high alcohol intake has resulted in alcohol preferring strains of rats and mice (Penn et al. 1978). Environmental manipulations often include removing access to water or allowing alcohol access at specific times during the light cycle. Another approach to exposing animals to alcohol is a noncontingent method of alcohol administration in which rodents inhale tightly controlled levels of vaporized alcohol (for review see Gilpin et al. 2008). Because the levels are experimenter controlled (i.e., not dependent on the actions of the animal) this allows for titration of blood alcohol levels around a desired amount. Vapor alcohol exposure is often used to rapidly induced alcohol dependence by inducing very high blood alcohol levels (≥200 mg/dL) for extended periods of time (Anderson et al. 2016a; Diaz et al. 2011; Rose et al. 2016). This blood alcohol level is approximately three times the legal limit for motor vehicle operation in the United States; it is important to achieve such high blood alcohol concentrations intermittently in order to drive dependence in rodents (Griffin et al. 2009; Rose et al. 2016).
The primary utility of the vapor exposure model is that dependence-like symptoms can be induced in short periods of time, relative to models that require the animal to voluntarily drink. The exposure protocol used most often involves repeated exposures to vaporized alcohol separated by withdrawal periods. Typically, this is referred to as the chronic intermittent alcohol (CIE) vapor exposure model (Griffin et al. 2009). While the exact procedure varies between laboratories, the CIE exposure procedure often consists of exposure to vaporized alcohol for a large portion of the day (12–16 h) followed by withdrawal period (8–12 h) where room air is pumped into the chamber. This procedure is repeated daily (usually 4 consecutive days) before a longer withdrawal period (often 3 days) is imposed, for a total time of 1 week (referred to as one “cycle”). The CIE procedure in mice has typically been utilized to deliver one, two, three, four, or five cycles of alcohol vapor exposure, depending on the requirements of the study (Fig. 1a). Because mice metabolize alcohol at a high rate, to achieve desired blood alcohol levels it is necessary to inhibit the metabolic pathway of alcohol, and expose the animals to a “loading” dose of alcohol. This is typically achieved via systemic injection of the alcohol dehydrogenase inhibitor, pyrazole, mixed with alcohol. In this model, it is important to include a control group that is housed in a similar chamber and given injections of pyrazole, but not exposed to alcohol, to control for the effects of pyrazole as well as housing condition. Even though CIE exposure is a noncontingent exposure method (i.e., the animal has no choice but to be exposed to alcohol), it has been shown to drive augmented compulsive/anxiety-like behavior and an increase in alcohol drinking when animals are later given volitional access to alcohol, which suggests recapitulation of at least some aspects of alcohol dependence in humans (Anderson et al. 2016a, b; Rose et al. 2016).
Most protocols for exposing rats to alcohol vapor are very similar to mice. Rats are typically exposed to alcohol vapor for 12 h followed by 12 h of room air. This procedure is often repeated daily for 10–12 consecutive days (Gilpin et al. 2008) (Fig. 1b). Because rats do not metabolize alcohol as fast as mice, administration of pyrazole and a loading dose of alcohol is not required.
1.5.2 Chronic Alcohol Self-administration in Nonhuman Primates
Similar to rodents, there are many different procedures used to study nonhuman alcohol exposure. The most commonly used nonhuman primate model of alcohol consumption, and what we will focus on in this review, involves training animals to volitionally consume alcohol. Volitional consumption, as opposed to noncontingent exposure such as a vapor chamber or alcohol injection, is an important distinction. Indeed, humans consume alcohol volitionally, giving this approach strong face validity. Further, pattern of drug exposure and rate of onset/clearance are important factors in the pharmacological action of drugs, and can often affect the neurochemical adaptations induced by drug exposure (Allain et al. 2015; Calipari et al. 2013). Thus, allowing the animal to consume the drug in a self-imposed pattern is more likely to result in effects similar to those in humans.
The method of inducing alcohol self-administration often varies across laboratory and/or study. Generally, animals are trained to pull a lever or activate a finger-poke to receive access to a sipper containing alcohol (Grant et al. 2008; Vivian et al. 2001). In some cases, schedule-induced polydipsia is used to augment alcohol consumption during the initial exposure and training phase (Grant et al. 2008; Vivian et al. 2001). Schedule-induced polydipsia involves simply delivering small amounts of food at spaced intervals. Because most mammals tend to increase fluid consumption during times of feeding, the increase in number of feeding bouts produces a robust increase in fluid consumption (Falk 1966). Once trained to consume alcohol, animals are then allowed to drink, either freely with continuous access to alcohol in the home cage, or in daily sessions where alcohol becomes available. In this review we will primarily discuss studies in which nonhuman primates were given 22 h/day access to alcohol in the home cage, for a period of 6–18 months (Fig. 1c, d). Importantly, this model produces robust individual differences in alcohol intake between animals and between days within each animal. This allows for determining the effects of alcohol across a range of intake, as well as the factors that may predict individual differences in alcohol consumption (Cuzon Carlson et al. 2011; Grant and Bennett 2003; Grant et al. 2008; Nimitvilai et al. 2017; Pleil et al. 2015b; Vivian et al. 2001; Baker et al. 2014, 2017).
1.6 Tonic and Phasic Dopamine Signaling
Dopamine neurons originating from the VTA have two distinct types of firing patterns, tonic or phasic firing. Tonic firing is characterized by the periodic occurrence of single action potentials (2–5 Hz), while phasic firing is characterized by bursts of action potentials (10–25 Hz) occurring in close temporal proximity (Grace and Bunney 1983, 1984). These two types of signaling are critical in regulating reward processing and internal state, and can be controlled by both changes in VTA firing and local modulatory mechanisms directly at dopamine terminals in the nucleus accumbens (NAc; a subregion of the ventral striatum) (Exley and Cragg 2008). Below we will discuss the different methods for measuring tonic and phasic dopamine signaling.
Tonic dopamine levels, often referred to as extracellular levels, are comparatively low (usually 5–20 nM) and can be measured with relatively low temporal resolution over several minutes using techniques such as in vivo microdialysis. To conduct microdialysis, a concentric perforated probe is implanted into the area of interest. Artificial cerebrospinal fluid is perfused into the region of interest; neurotransmitters, such as dopamine, diffuse down their concentration gradient across this perforated membrane. This fluid is then collected over 5–30 min and analyzed using detection methods such as high performance liquid chromatography or mass spectrometry, which allow for quantification of analytes (e.g., neurotransmitters) within the sample. Changes in tonic dopamine levels have been shown to predictably alter thresholds for intracranial self-stimulation, which is used to monitor the function of brain reward systems and measure the motivational state of the animal, suggesting that tonic dopamine levels are involved in reward sensitivity and affective states (Carlezon and Chartoff 2007; Hernandez et al. 2012; Kokkinidis and McCarter 1990; Negus and Miller 2014; see Dobrossy et al. 2015 for review).
While microdialysis can give information about relative levels of synaptic neurotransmitters, it is important to understand how receptors and local regulation of dopamine neurons are influenced by alcohol exposure. Ex vivo fast-scan cyclic voltammetry (FSCV), typically performed in coronal brain slices, allows for measuring experimenter-stimulated dopamine release when the dopamine terminal is isolated from its endogenous inputs (due to severing these connections in the slicing process). FSCV detects electroactive analytes (including dopamine) by applying voltage to a microelectrode, which drives oxidation of dopamine to dopamine-o-quinone; the oxidation of dopamine results in the loss of electrons which are detected at the electrode as a change in current which is proportional to the concentration of dopamine molecules near the surface of the electrode. Thus, based on the electroactive properties of dopamine, FSCV is able to detect dopamine levels with high specificity, even within a heterogeneous environment of transmitter signaling. This detection can occur quickly (typically sampled at 10 Hz) allowing for information to be obtained about real-time dopamine release and clearance kinetics. With this technique, receptors expressed on dopamine terminals can be pharmacological targeted and their effects on the kinetics of dopamine signaling can be assessed (for review of the utility of ex vivo voltammetry see Ferris et al. 2013).
Phasic firing refers to bursts of activity that result in high concentrations (estimated to be around 100 μM) of dopamine within the synapse (Grace et al. 2007). These phasic dopamine signals are particularly important in the case of addiction where they act to encode information not just about rewards, but also about cues that predict their availability. For example, reward conditioning experiments have shown that phasic dopamine responses occur immediately following presentation of unexpected rewards; however, after multiple pairings of cue and reward, phasic dopamine responses shift to the cue predicting the reward instead of the reward itself (Phillips et al. 2003; Schultz 1998). Thus, understanding how this type of signaling is dysregulated by alcohol has implications not only for subsequent alcohol use, but also for decision-making and reward seeking outside of alcohol-related contexts.
Thus, understanding phasic and tonic dopamine signaling is crucially important as their interplay controls the execution of motivated behaviors, and the examination of these two aspects of dopaminergic signaling in tandem allows a greater understanding of the alcohol-induced maladaptive responses to external stimuli.
2 Dopamine Signaling Following Chronic Alcohol Exposure
2.1 Acute Effects of Alcohol on Dopamine Release
Acute alcohol administration has distinct, regionally specific effects on dopamine system activity. Systemic alcohol administration transiently increases extracellular tonic levels of dopamine in the NAc of rodents and monkeys as measured by microdialysis (Bradberry 2002; Karkhanis et al. 2016; Weiss et al. 1993; Yim et al. 1998). Similarly, an in vivo FSCV study in awake, behaving rats showed that alcohol administration resulted in an increase in phasic dopamine release (Shnitko and Robinson 2015). In contrast, ex vivo FSCV studies show that acute application of alcohol to brain slices results in a reduction of phasic dopamine release in the NAc of both rodents and monkeys (Siciliano et al. 2016b; Yorgason et al. 2014, 2015), which is dependent on alcohol concentration and frequency of stimulation. Reductions in dopamine release in these experiments were observed only at high concentrations of alcohol (80 mM and above) and during high frequency of stimulation (20 Hz and above) (Yorgason et al. 2015). While these findings may seem in opposition, the inconsistencies between in vivo (enhanced release) and ex vivo (reduced release) are likely driven by the fact that dopamine terminals are separated from the cell body in ex vivo slice preparations. In the ex vivo slice preparation, effects that are observed represent only the synaptic connections that are maintained within the slice, and do not assess the full spectrum of circuit connectivity between the region of interest and the rest of the brain. Without the contribution of alcohol-mediated excitation of VTA neurons to augment dopaminergic signaling, NAc terminals are inhibited by alcohol. However, the net effect of alcohol in the intact animal (i.e., inhibitory actions at the terminal and excitatory actions at the cell body) is increased extracellular dopamine levels in the striatum. These findings highlight both the complexity of this system and the need for multiple levels of exploration (from the slice to the whole animal) in order to fully appreciate the pharmacological actions of alcohol on the dopaminergic system. For more information on alcohol’s presynaptic actions, including non-dopaminergic systems, see Lovinger (2017) elsewhere in this volume.
2.2 Effects of Chronic Alcohol Exposure on Dopamine Release During Abstinence
While the section above outlines the acute effects of alcohol on dopamine release, chronic use leaves a lasting impact on dopamine release during alcohol-free periods, which may contribute to maladaptive decision making. In this section we will outline literature which has examined dopamine release in animals with a history of alcohol exposure, when there is no alcohol “on board” (i.e., during withdrawal).
Studies examining the role of chronic alcohol exposure on dopamine release have generally yielded mixed results, with species, sex, and experimental design appearing to have a strong influence on the findings. For example, stimulated dopamine release in ex vivo slices preparations was attenuated following three to five cycles of CIE exposure in mice (Karkhanis et al. 2015; Rose et al. 2016; but see Melchior and Jones 2017). However, in rats, shorter exposure to alcohol vapor over 5 or 10 days did not alter dopamine release as compared to control animals (Budygin et al. 2007). Data from nonhuman primates further “muddies the waters” in regard to interpreting the effects of alcohol exposure on stimulated dopamine release. In contrast to the decreased release observed in mice, male cynomolgus macaques were found to have increased stimulated dopamine release following 6 months of alcohol self-administration (Siciliano et al. 2015a). Complicating matters further, female rhesus macaques showed no change in dopamine release after 12 months of alcohol self-administration (Siciliano et al. 2016b). The driving factor underlying the inconsistency within the nonhuman primate studies is currently unclear, as sex, length of exposure (6 vs 12 months), and species (cynomolgus vs rhesus) were all divergent between the two studies.
Although these seemlying inconsistent results may suggest that alterations in dopamine release are not related to the primary pathology of AUD, it should also be noted again that ex vivo measurements of dopamine release can only give insight into certain aspects of the system. Indeed, many in vivo studies of dopamine release in response to stimuli have demonstrated that release can encode many different aspects of learned behaviors and drug associated cues, and is plastic throughtout the formation of these associations (Wanat et al. 2009). Because the afferent inputs that drive dopamine release in vivo are severed in an ex vivo slice preparation, ex vivo approaches provide insight to the size of the readily reasable pool of dopamine, but do not capture the complexity of dopaminergic encoding of these behaviors. It is also important to note that the tonic, extracelluar level of dopamine is depedent on many factors (discussed below); when tonic dopamine levels are measured via microdialysis, multiple studies have found them to be decreased following alcohol exposure in rats (Rossetti et al. 1992, 1999). Further, metabolic markers of dopaminergic activity are reduced in macaques after chronic alcohol use (Cervera-Juanes et al. 2016).
2.3 Dopamine Uptake
Extracellular dopamine levels are a complex interaction between dopamine release and its reuptake via the dopamine transporter (DAT) (Ferris et al. 2014). Not only is the DAT a major factor in determining tonic extracellular levels, but it is also thought to tightly regulate the “sphere of influence” and duration of dopamine effects at postsynaptic receptors as it flows out of release sites (Cragg and Rice 2004). Thus, DAT function and expression is an integral component in guiding dynamic dopamine neurotransmission.
Exposure to chronic alcohol modulates the DAT, both in terms of its function and expression. In mice, repeated cycles of CIE exposure augments dopamine reuptake, which is a primarily DAT mediated process, in the NAc (Karkhanis et al. 2015, 2016; Rose et al. 2016; Melchior and Jones 2017). In CIE models, uptake rate is enhanced immediately following cessation of the final alcohol exposure and are maintained for at least until 72 h into withdrawal, suggesting that this effect may be long lasting, though later time-points have not yet been tested (Karkhanis et al. 2015; Rose et al. 2016) (Fig. 2). Further, DAT density in the NAc is increased after CIE (Healey et al. 2008). Enhanced dopamine uptake rate likely contributes to reduced tonic dopamine levels by increasing the speed of dopamine removal from the extracellular space.
Fig. 2.
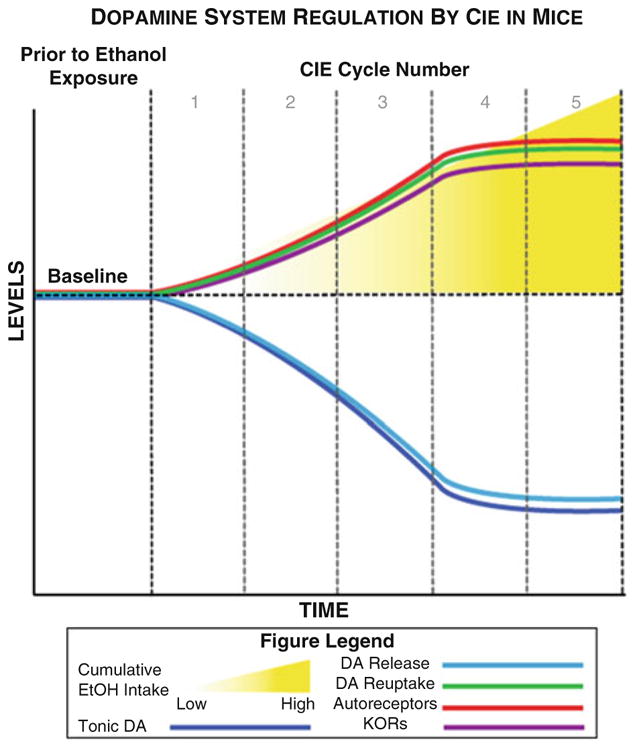
Alterations in NAc dopamine signaling in mice following successive cycles of CIE. This timeline represents schematized alterations in dopamine system regulation with increasing exposures to chronic intermittent ethanol cycles. With increasing cumulative ethanol exposure, tonic dopamine levels (measured via microdialysis) and stimulated dopamine release (measured via FSCV) are diminished, but dopamine uptake rate is enhanced, resulting in faster clearance of dopamine from the synaptic cleft. In addition, KOR and D2/D3 autoreceptor function is also augmented. Together these factors give rise to a systemic hypodopaminergic state in animals exposed to chronic intermittent ethanol. These effects appear to be dose-dependent on total cumulative ethanol exposure until they plateau after approximately three to five cycles. DA dopamine, EtOH ethanol, KORs kappa opioid receptors
Enhanced dopamine uptake rate following alcohol exposure is a phenomenon that is strongly conserved across species. Similar to mice, dopamine uptake rate is increased in rats exposed to CIE vapor (Budygin et al. 2007). In rats exposed to an alcohol-containing liquid diet for 1 year, DAT protein expression was increased in both the ventral and dorsal striatum (Rothblat et al. 2001), suggesting that increased functional uptake is a result of increased protein levels, although other mechanisms such as conformational alterations or changes in the affinity state of the DAT could also be at play. These findings have been extended to nonhuman primates, where dopamine uptake rate has been found to be increased in the NAc of male cynomolgus macaques and female rhesus macaques after 6 and 12 months of volitional access to alcohol, respectively (Siciliano et al. 2015a, 2016b). Together, these data demonstrate that, in the NAc, dopamine uptake is increased across species and experimental setting, suggesting that it may be an important factor in driving the core symptomology of AUD.
One exception is that in male cynomolgus macaques, uptake rate in the dorsolateral caudate (a subregion of the striatum typically thought to be involved in motor control and habit learning (Graybiel 1995, 2008; Porrino et al. 2004) were reduced after chronic access to alcohol (Siciliano et al. 2015a)). This decrease in dopamine uptake rate likely produces increases in extracellular dopamine levels. Interestingly, the ratio of uptake rates between the dorsolateral caudate and NAc was highly correlated with alcohol intake across animals. While the behavioral relevance of differential changes in dopamine uptake between these two regions remains to be determined, there is evidence that dysregulation of dopamine-mediated communication between these areas can lead to habitual and addiction-like behaviors (Belin and Everitt 2008; Everitt and Robbins 2013). Thus, enhancement of NAc dopamine reuptake and blunting of dorsolateral caudate dopamine reuptake together may contribute to maladaptive alcohol seeking and intake following chronic exposure.
Not only is dopamine uptake altered by chronic alcohol administration in pre-clinical models, there is also evidence that abnormal dopamine uptake may contribute to disease states in the human population. Genetic association studies point to alterations in the DAT in a subset of individuals with an AUD diagnosis, which may confer a heightened vulnerability towards development of the disease (Köhnke et al. 2005). The relationship between DATs and alcoholism in humans is not entirely clear, however. Alcoholism is associated with both increases and decreases in DAT availability as measured by positron emission tomography (PET) or single photon emission computed tomography (SPECT), depending on the study (Laine et al. 1999; Tiihonen et al. 1995; Tupala et al. 2006; Yen et al. 2015, 2016). These discrepancies may be due, at least in part, to different durations of withdrawal between studies and limitations of lower resolution, noninvasive approaches which are typically employed in human studies. However, given the abundant preclinical data showing alterations in uptake rate following chronic alcohol exposure discussed above, and implications for genetic DAT alterations in the clinical population, it is clear that the DAT plays a significant role in the etiology of AUD that should be further examined in future studies.
2.4 Autoreceptors
In the NAc, dopamine D2-type (D2, D3, D4) receptors are found on medium spiny projection neurons, local interneurons, and presynaptic terminals from afferent inputs (Alcantara et al. 2003; Ford 2014; Levey et al. 1993). D2-type receptors that are located on presynaptic dopamine terminals are autoreceptors, which function in a feedback-inhibitory manner, binding released dopamine and inhibiting future release. D2-type dopamine autoreceptors are G-protein coupled receptors expressed at both cell bodies and presynaptic terminals of dopamine neurons, where they inhibit action potential firing activity, release and synthesis of dopamine. Thus, in the NAc, when extracellular dopamine levels are high, autoreceptors inhibit dopamine release and synthesis, driving the system towards homeostasis. For this reason, D2-type autoreceptors are often conceptualized as the “brakes” on the dopamine system. Of the members of the D2-type receptor family, D2 receptors themselves have been found to mediate the majority of autoreceptor activity in the striatum, although D3 and D4 receptors are also present (Bello et al. 2011; Meador-Woodruff et al. 1994; Meller et al. 1993; Rubinstein et al. 1997).
Most studies examining D2-type autoreceptor sensitivity following chronic alcohol exposure have shown functional increases in activity/sensitivity, contributing to a reduction in dopamine signaling. Multiple cycles of CIE exposure result in greater autoregulation of release in the NAc in mice (Karkhanis et al. 2015), but shorter exposure times did not change the ability of autoreceptors to inhibit dopamine release in rats (Budygin et al. 2007) or their ability to inhibit dopamine synthesis in mice (Siciliano et al. 2017). Repeated CIE-induced increases in autoreceptor sensitivity appears to be relatively short-lived during abstinence, however, with sensitivity returning to control levels within a few days (Karkhanis et al. 2015). Typically, the sensitivity of these receptors has been assessed by examining the ability of D2-type dopamine receptor agonists to inhibit dopamine release ex vivo. When greater effects of D2-type specific agonists are observed, it is interpreted as an increase in the sensitivity of these receptors, which translates to increased inhibitory feedback when endogenous dopamine interacts with these receptors. Thus, increased sensitivity of D2-type autoreceptors likely contributes to a hypodopaminergic state via decreased probability of dopamine release from presynaptic terminals.
Similar findings have been observed in nonhuman primate models of AUD. In one study of male cynomolgus macaques it was found that, after 6 months of volitional alcohol drinking, there was no change in overall autoreceptor-mediated inhibition of dopamine release; however, there was a shift in the relative contribution of D2 vs D3 dopamine autoreceptors towards D2 receptors (Siciliano et al. 2016b). In other words, D2 sensitivity was increased, and D3 sensitivity was decreased such that the sum of autoreceptor inhibition remained the same while the contribution of the two receptor subtypes was shifted. In two studies of monkeys exposed to longer, 12- to 18-month periods of alcohol drinking, D2-type autoreceptor sensitivity was increased, but the relative contributions of receptor subtypes were not queried (Budygin et al. 2003; Siciliano et al. 2016a). Thus, it appears that, across species, overall changes in autoreceptor sensitivity occur after extended exposure to alcohol, but not after modest exposure lengths. These consistent cross-species findings of dopamine autoreceptor changes, with consistent relative time-courses and direction of change towards greater dopamine inhibition, provide confidence that autoreceptor changes may be functionally related to the core symptomology of AUD.
2.5 Kappa Opioid Receptors
2.5.1 Dopamine and Kappa Opioid Receptor Interactions
Like D2/D3 autoreceptors, kappa opioid receptors (KORs) are located on dopamine terminals (Ebner et al. 2010; Svingos et al. 2001; Werling et al. 1988) and act to reduce dopamine release. KOR activation results in reduced tonic dopamine levels as well as both decreased probability and magnitude of phasic release events (Steiner and Gerfen 1996). Given the role of dopamine in reward, it is not surprising that the activation of KORs, which inhibit dopamine release, is generally aversive (Land et al. 2009). Dynorphin, the endogenous ligand of KORs, is released during exposure to painful, noxious, or stressful stimuli (Bruchas and Chavkin 2010; Land et al. 2009; Nabeshima et al. 1992). KORs are believed to exert their behavioral and subjective effects in part through inhibition of dopaminergic signaling (Ebner et al. 2010; Svingos et al. 2001; Werling et al. 1988).
In the NAc, dopamine, dynorphin, and KORs have an intimate relationship of interconnectivity and feedback regulation. GABAergic medium spiny neurons (MSNs) account for more than 90% of all neurons in the striatum and are the major projection population. Striatal MSNs consist of two distinct populations, which are interspersed and equally distributed across the dorsal and ventral striatum, and differentiated based on the expression of dopamine receptors and opioid peptides. Approximately half of the MSN population expresses excitatory D1-type dopamine receptors and dynorphin peptides, and the other half of MSNs express inhibitory D2-type receptors and enkephalin peptides (ligand for mu and delta opioid receptors) (Gerfen et al. 1990). Dynorphin is generated in D1-MSNs in response to D1 receptor activation, and its release inhibits further dopamine release from the presynaptic terminal (Gerfen and Surmeier 2011; Steiner and Gerfen 1996). Dynorphin peptides are transported to recurrent collateral axons within the NAc and decrease dopamine release via presynaptic KORs (Steiner and Gerfen 1993). In this way, a feedback loop wherein dopamine release can increase the probability of dynorphin release, which in turn reduces dopamine release, is propagated.
2.5.2 Kappa Opioid Receptors and Chronic Alcohol Exposure
In humans, activation of KORs has been associated with feelings of dysphoria, and both KORs and dynorphin mRNA are upregulated in patients suffering from AUD (Bazov et al. 2013). In preclinical models, a single injection of alcohol in naïve rats results in a transient increase in dynorphin levels (Kuzmin et al. 2013; Lam et al. 2008; Marinelli et al. 2006). Following CIE exposure, inhibition of KORs has been shown to successfully mitigate negative affect-like behavioral alterations observed in mice and rats, without altering these behaviors in alcohol-naïve animals (Anderson et al. 2016b; Pleil et al. 2015a; Rose et al. 2016; Kissler et al. 2014). Interestingly, the effect of KOR activation on alcohol intake behaviors appears to be strongly influenced by the animal’s history of alcohol intake and state of dependency. Pharmacological blockade of KORs does not change alcohol consumption in non-alcohol-dependent animals; however, in animals that have been made dependent through CIE exposure, and show high dependence-induced volitional alcohol intake, KOR blockade reduces alcohol consumption to control levels (Rose et al. 2016; Walker et al. 2011; see Anderson and Becker 2017 for review). Conversely, in naïve animals, pretreatment with KOR agonists drives an increase in alcohol consumption comparable to animals that have been previously made dependent, again suggesting that alcohol-induced increased activity of these receptors is associated with excessive alcohol intake (Anderson et al. 2016b; Rose et al. 2016).
Behavioral studies, outlined above, have suggested that KORs are functionally altered by previous alcohol exposure, and subsequent studies directly measuring KOR regulation of dopamine signaling after chronic alcohol exposure support this hypothesis (Karkhanis et al. 2016; Rose et al. 2016; Siciliano et al. 2016a). Indeed, in mice and rats, the dopamine-decreasing effects of KOR activation, observed using ex vivo FSCV, are heightened dramatically following CIE exposure (Karkhanis et al. 2016; Rose et al. 2016). Thus, alcohol exposure augments the ability of KORs to reduce dopamine, contributing further to hypodopaminergia following chronic alcohol exposure. Importantly, dependence-induced increases in alcohol consumption can be reduced via microinfusion of a KOR antagonist directly into the NAc (Nealey et al. 2011). This suggests a causal role for increased KOR function in the NAc in aberrant alcohol consumption.
Augmentation of KOR sensitivity is conserved across species. Following 6 months of drinking in male cynomolgus macaques, KOR-mediated inhibitory regulation of dopamine signaling in the NAc is increased (Siciliano et al. 2016a). Further, KOR-mediated inhibition of dopamine release in the dorsolateral caudate is augmented after alcohol drinking (Siciliano et al. 2015a). Interestingly, while both regions appear to develop KOR hyper-function, KOR activity in the NAc, but not the dorsolateral caudate, is correlated with alcohol intake (Siciliano et al. 2015a), again suggesting that KOR signaling in the NAc is a critical node in driving excessive alcohol intake. Further, these effects are consistent across sex, as 1 year of alcohol self-administration also increased KOR regulation of dopamine release in the NAc of female rhesus macaques (Siciliano et al. 2016a). Together, these studies demonstrate that KOR regulation of dopamine release is increased across species as well as several other experimental parameters, including route of administration, exposure length, and withdrawal time, suggesting an integral role in the etiology of AUD. We hypothesize that alcohol dependence-induced increases in KOR function contribute to a hypodopaminergic state possibly leading to craving and excessive consumption of alcohol. These alterations in KOR changes at the dopamine terminal are illustrated in Fig. 3.
Fig. 3.

Putative synaptic alterations associated with chronic ethanol exposure across species. Synaptic changes in the NAc associated with withdrawal from chronic alcohol exposure in (a) male mice, (b) male rats, (c) male cynomolgus monkeys, and (d) female rhesus monkeys. (a) In mice, acute withdrawal from chronic intermittent ethanol exposure is associated with reduced dopamine release and augmented reuptake, D2R/D3R autoreceptor, and KOR sensitivity. With the exception of autoreceptor sensitivity, most of these effects are relatively long lasting, and remain altered 72 h following the final ethanol exposure. (b) In rats, dopamine release and autoreceptor function are unaffected during acute withdrawal; however, enhanced D1R and DAT sensitivity are observed. (c) Male cynomolgus monkeys show synaptic alterations remarkably similar to male mice during acute withdrawal from chronic ethanol intake, whereby DAT and KOR functions are augmented. However, dopamine release is increased and total autoreceptor function remains unaltered. (d) In female rhesus monkeys, while there is no change in dopamine release, the DAT, KOR, and autoreceptor function in increased during acute withdrawal
The extensive alcohol-induced reductions in presynaptic dopamine terminal activity in the NAc would be predicted to result in differential signaling onto downstream, postsynaptic targets. In the NAc, these consist primarily of D1 and D2 receptor expressing MSNs, which are also altered in AUD models. These other aspects of alcohol-induced alterations in striatal activity are beyond the scope of the current chapter, but have been extensively characterized elsewhere and continue to be areas of intense investigation (Clarke and Adermark 2015; Engel and Jerlhag 2014; Koob 2014; Renteria et al. 2017; Soderpalm et al. 2009; Tupala and Tiihonen 2004).
3 Conclusions
It is clear that factors such as alcohol exposure length, species, route of administration, and withdrawal time-point can be important variables in determining the effects of alcohol on the dopamine system. However, in this chapter, we highlight some of the robust findings that transcend these variables and may hint at common neurobiological substrates following chronic exposure to alcohol. Primary examples of these commonalties include increased functionality/sensitivity of multiple negative regulators of dopamine signaling, such as DATs, dopamine autoreceptors, and KORs. The combination of increased clearance of dopamine from the synapse, via upregulation of DATs, with enhanced KOR and autoreceptor function, likely combines to produce hyper-inhibitory regulation of dopamine signaling. Thus, deviations from “healthy” function of the mesolimbic dopaminergic system following chronic exposure to alcohol appear to be a universal adaptation that may be driving maladaptive behaviors associated with repeated alcohol exposure.
The use of multiple species and paradigms offers insight into possible treatment strategies that could alleviate suffering in individuals with an AUD. As illustrated above, such diverse experimental designs help to “separate the wheat from the chaff” by identifying consistent, key neurobiological changes following exposure. To this end, we argue that successful pharmacological strategies may lie in a combinatorial pharmacotherapy that would quiet both DAT and KOR systems (e.g., a DAT inhibitor and KOR antagonist or partial agonist). Such a treatment could suppress two key contributors to dopamine signal reduction following chronic alcohol exposure, and may alleviate negative affective states that are pervasive in abstinent AUD patients. Resultant normalization of affect during withdrawal would be predicted to both improve quality of life and decrease likelihood of relapse in individuals striving to maintain abstinence.
Taken together, it is clear that key alterations in mesolimbic dopamine signaling are consistently observed following chronic alcohol exposure across species and administration paradigms. Although there is far more work to be done in order to fully elucidate the role of dopaminergic signaling in neural and behavioral adaptations following alcohol exposure, pharmacological strategies targeting these adaptations are a potentially important treatment avenue for alleviating suffering in individuals with an AUD.
Acknowledgments
This work was funded by NIH grants U01 AA014091, R01 AA021099, P01 AA023299 (SRJ), T32 AA007565 (CAS, ANK, KMH, JRM), F31 DA037710, F32 MH111216, Brain and Behavior Research Foundation (CAS), K01 AA023874 (ANK), and F31 AA023144 (JRM).
Footnotes
Financial Disclosure: The authors declare no competing financial interests.
Contributor Information
Cody A. Siciliano, The Picower Institute for Learning and Memory, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology (MIT), Cambridge, MA, USA
Anushree N. Karkhanis, Department of Physiology and Pharmacology, Wake Forest School of Medicine, Winston-Salem, NC, USA
Katherine M. Holleran, Department of Physiology and Pharmacology, Wake Forest School of Medicine, Winston-Salem, NC, USA
James R. Melchior, Department of Physiology and Pharmacology, Wake Forest School of Medicine, Winston-Salem, NC, USA
Sara R. Jones, Department of Physiology and Pharmacology, Wake Forest School of Medicine, Winston-Salem, NC, USA
References
- Abrahao KP, Salinas AG, Lovinger DM. Alcohol and the brain: neuronal molecular targets, synapses, and circuits. Neuron. 2017;96:1223–1238. doi: 10.1016/j.neuron.2017.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara AA, Chen V, Herring BE, Mendenhall JM, Berlanga ML. Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the rat. Brain Res. 2003;986:22–29. doi: 10.1016/s0006-8993(03)03165-2. [DOI] [PubMed] [Google Scholar]
- Allain F, Minogianis EA, Roberts DC, Samaha AN. How fast and how often: the pharmacokinetics of drug use are decisive in addiction. Neurosci Biobehav Rev. 2015;56:166–179. doi: 10.1016/j.neubiorev.2015.06.012. [DOI] [PubMed] [Google Scholar]
- Anderson RI, Becker HC. Role of the dynorphin/kappa opioid receptor system in the motivational effects of ethanol. Alcohol Clin Exp Res. 2017;41:1402–1418. doi: 10.1111/acer.13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RI, Lopez MF, Becker HC. Forced swim stress increases ethanol consumption in C57BL/6J mice with a history of chronic intermittent ethanol exposure. Psychopharmacology. 2016a;233:2035–2043. doi: 10.1007/s00213-016-4257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RI, Lopez MF, Becker HC. Stress-induced enhancement of ethanol intake in C57BL/6J mice with a history of chronic ethanol exposure: involvement of kappa opioid receptors. Front Cell Neurosci. 2016b;10:45. doi: 10.3389/fncel.2016.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SB, Liu Z, McElvain MA, Brodie MS. Ethanol excitation of dopaminergic ventral tegmental area neurons is blocked by quinidine. J Pharmacol Exp Ther. 2003;306:437–446. doi: 10.1124/jpet.103.050963. [DOI] [PubMed] [Google Scholar]
- Ariansen JL, Heien ML, Hermans A, Phillips PE, Hernadi I, Bermudez MA, Schultz W, Wightman RM. Monitoring extracellular pH, oxygen, and dopamine during reward delivery in the striatum of primates. Front Behav Neurosci. 2012;6:36. doi: 10.3389/fnbeh.2012.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal P, Dvir H, Choe S, Slesinger PA. A discrete alcohol pocket involved in GIRK channel activation. Nat Neurosci. 2009;12:988–995. doi: 10.1038/nn.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EJ, Farro J, Gonzales S, Helms C, Grant KA. Chronic alcohol self-administration in monkeys shows long-term quantity/frequency categorical stability. Alcohol Clin Exp Res. 2014;38:2835–2843. doi: 10.1111/acer.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EJ, Walter NA, Salo A, Rivas Perea P, Moore S, Gonzales S, Grant KA. Identifying future drinkers: behavioral analysis of monkeys initiating drinking to intoxication is predictive of future drinking classification. Alcohol Clin Exp Res. 2017;41:626–636. doi: 10.1111/acer.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazov I, Kononenko O, Watanabe H, Kuntic V, Sarkisyan D, Taqi MM, Hussain MZ, Nyberg F, Yakovleva T, Bakalkin G. The endogenous opioid system in human alcoholics: molecular adaptations in brain areas involved in cognitive control of addiction. Addict Biol. 2013;18:161–169. doi: 10.1111/j.1369-1600.2011.00366.x. [DOI] [PubMed] [Google Scholar]
- Belin D, Everitt BJ. Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57:432–441. doi: 10.1016/j.neuron.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noain D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, Rubinstein M. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci. 2011;14:1033–1038. doi: 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaine SK, Sinha R. Alcohol, stress, and glucocorticoids: from risk to dependence and relapse in alcohol use disorders. Neuropharmacology. 2017;122:136–147. doi: 10.1016/j.neuropharm.2017.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Alcohol-induced neuronal loss in developing rats: increased brain damage with binge exposure. Alcohol Clin Exp Res. 1990;14:107–118. doi: 10.1111/j.1530-0277.1990.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Bradberry CW. Dose-dependent effect of ethanol on extracellular dopamine in mesolimbic striatum of awake rhesus monkeys: comparison with cocaine across individuals. Psychopharmacology. 2002;165:67–76. doi: 10.1007/s00213-002-1233-9. [DOI] [PubMed] [Google Scholar]
- Brodie MS. Increased ethanol excitation of dopaminergic neurons of the ventral tegmental area after chronic ethanol treatment. Alcohol Clin Exp Res. 2002;26:1024–1030. doi: 10.1097/01.ALC.0000021336.33310.6B. [DOI] [PubMed] [Google Scholar]
- Brodie MS, Shefner SA, Dunwiddie TV. Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain Res. 1990;508:65–69. doi: 10.1016/0006-8993(90)91118-z. [DOI] [PubMed] [Google Scholar]
- Brodie MS, Pesold C, Appel SB. Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin Exp Res. 1999;23:1848–1852. [PubMed] [Google Scholar]
- Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology. 2010;210:137–147. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budygin EA, John CE, Mateo Y, Daunais JB, Friedman DP, Grant KA, Jones SR. Chronic ethanol exposure alters presynaptic dopamine function in the striatum of monkeys: a preliminary study. Synapse. 2003;50:266–268. doi: 10.1002/syn.10269. [DOI] [PubMed] [Google Scholar]
- Budygin EA, Oleson EB, Mathews TA, Lack AK, Diaz MR, McCool BA, Jones SR. Effects of chronic alcohol exposure on dopamine uptake in rat nucleus accumbens and caudate putamen. Psychopharmacology. 2007;193:495–501. doi: 10.1007/s00213-007-0812-1. [DOI] [PubMed] [Google Scholar]
- Calipari ES, Ferris MJ, Zimmer BA, Roberts DC, Jones SR. Temporal pattern of cocaine intake determines tolerance vs sensitization of cocaine effects at the dopamine transporter. Neuropsychopharmacology. 2013;38:2385–2392. doi: 10.1038/npp.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Chartoff EH. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat Protoc. 2007;2:2987–2995. doi: 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]
- Cervera-Juanes R, Wilhem LJ, Park B, Lee R, Locke J, Helms C, Gonzales S, Wand G, Jones SR, Grant KA, et al. MAOA expression predicts vulnerability for alcohol use. Mol Psychiatry. 2016;21:472–479. doi: 10.1038/mp.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B, Dopico AM, Lemos JR, Treistman SN. Ethanol potentiation of calcium-activated potassium channels reconstituted into planar lipid bilayers. Mol Pharmacol. 1998;54:397–406. doi: 10.1124/mol.54.2.397. [DOI] [PubMed] [Google Scholar]
- Church DM, Goodstadt L, Hillier LW, Zody MC, Goldstein S, She X, Bult CJ, Agarwala R, Cherry JL, DiCuccio M, et al. Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 2009;7:e1000112. doi: 10.1371/journal.pbio.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Adermark L. Dopaminergic regulation of striatal interneurons in reward and addiction: focus on alcohol. Neural Plast. 2015;2015:814567. doi: 10.1155/2015/814567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools R, Frank MJ, Gibbs SE, Miyakawa A, Jagust W, D’Esposito M. Striatal dopamine predicts outcome-specific reversal learning and its sensitivity to dopaminergic drug administration. J Neurosci. 2009;29:1538–1543. doi: 10.1523/JNEUROSCI.4467-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ, Rice ME. DAncing past the DAT at a DA synapse. Trends Neurosci. 2004;27:270–277. doi: 10.1016/j.tins.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Cuzon Carlson VC, Seabold GK, Helms CM, Garg N, Odagiri M, Rau AR, Daunais J, Alvarez VA, Lovinger DM, Grant KA. Synaptic and morphological neuroadaptations in the putamen associated with long-term, relapsing alcohol drinking in primates. Neuropsychopharmacology. 2011;36:2513–2528. doi: 10.1038/npp.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danjo T, Yoshimi K, Funabiki K, Yawata S, Nakanishi S. Aversive behavior induced by optogenetic inactivation of ventral tegmental area dopamine neurons is mediated by dopamine D2 receptors in the nucleus accumbens. Proc Natl Acad Sci U S A. 2014;111:6455–6460. doi: 10.1073/pnas.1404323111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Goldstein RB, Grant BF. Rates and correlates of relapse among individuals in remission from DSM-IV alcohol dependence: a 3-year follow-up. Alcohol Clin Exp Res. 2007;31:2036–2045. doi: 10.1111/j.1530-0277.2007.00536.x. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Goldstein RB, Chou SP, Ruan WJ, Grant BF. Age at first drink and the first incidence of adult-onset DSM-IV alcohol use disorders. Alcohol Clin Exp Res. 2008;32:2149–2160. doi: 10.1111/j.1530-0277.2008.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana M, Pistis M, Carboni S, Gessa GL, Rossetti ZL. Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: electrophysiologi-cal and biochemical evidence. Proc Natl Acad Sci U S A. 1993;90:7966–7969. doi: 10.1073/pnas.90.17.7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MR, Christian DT, Anderson NJ, McCool BA. Chronic ethanol and withdrawal differentially modulate lateral/basolateral amygdala paracapsular and local GABAergic synapses. J Pharmacol Exp Ther. 2011;337:162–170. doi: 10.1124/jpet.110.177121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrossy MD, Furlanetti LL, Coenen VA. Electrical stimulation of the medial forebrain bundle in pre-clinical studies of psychiatric disorders. Neurosci Biobehav Rev. 2015;49:32–42. doi: 10.1016/j.neubiorev.2014.11.018. [DOI] [PubMed] [Google Scholar]
- Dziegielewski SF. DSM-IV-TR in action. 2. Wiley; Hoboken: 2010. [Google Scholar]
- Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH. Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology. 2010;210:241–252. doi: 10.1007/s00213-010-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge MA, Lerchner W, Saunders RC, Kaneko H, Krausz KW, Gonzalez FJ, Ji B, Higuchi M, Minamimoto T, Richmond BJ. Chemogenetic disconnection of monkey orbitofrontal and rhinal cortex reversibly disrupts reward value. Nat Neurosci. 2016;19:37–39. doi: 10.1038/nn.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel JA, Jerlhag E. Alcohol: mechanisms along the mesolimbic dopamine system. Prog Brain Res. 2014;211:201–233. doi: 10.1016/B978-0-444-63425-2.00009-X. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. From the ventral to the dorsal striatum: devolving views of their roles in drug addiction. Neurosci Biobehav Rev. 2013;37:1946–1954. doi: 10.1016/j.neubiorev.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Exley R, Cragg SJ. Presynaptic nicotinic receptors: a dynamic and diverse cholinergic filter of striatal dopamine neurotransmission. Br J Pharmacol. 2008;153(Suppl 1):S283–S297. doi: 10.1038/sj.bjp.0707510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk JL. Schedule-induced polydipsia as a function of fixed interval length. J Exp Anal Behav. 1966;9:37–39. doi: 10.1901/jeab.1966.9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris MJ, Calipari ES, Yorgason JT, Jones SR. Examining the complex regulation and drug-induced plasticity of dopamine release and uptake using voltammetry in brain slices. ACS Chem Neurosci. 2013;4:693–703. doi: 10.1021/cn400026v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris MJ, Espana RA, Locke JL, Konstantopoulos JK, Rose JH, Chen R, Jones SR. Dopamine transporters govern diurnal variation in extracellular dopamine tone. Proc Natl Acad Sci U S A. 2014;111:E2751–E2759. doi: 10.1073/pnas.1407935111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg BA, Nimmerjahn A, Cocker ED, Mukamel EA, Barretto RP, Ko TH, Burns LD, Jung JC, Schnitzer MJ. High-speed, miniaturized fluorescence microscopy in freely moving mice. Nat Methods. 2008;5:935–938. doi: 10.1038/nmeth.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience. 2014;282:13–22. doi: 10.1016/j.neuroscience.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Richardson HN, Cole M, Koob GF. Vapor inhalation of alcohol in rats. Curr Protoc Neurosci. 2008;Chapter 9(Unit 9):29. doi: 10.1002/0471142301.ns0929s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience. 1983;10:301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007;30:220–227. doi: 10.1016/j.tins.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Grant KA, Bennett AJ. Advances in nonhuman primate alcohol abuse and alcoholism research. Pharmacol Ther. 2003;100:235–255. doi: 10.1016/j.pharmthera.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Grant KA, Leng X, Green HL, Szeliga KT, Rogers LS, Gonzales SW. Drinking typography established by scheduled induction predicts chronic heavy drinking in a monkey model of ethanol self-administration. Alcohol Clin Exp Res. 2008;32:1824–1838. doi: 10.1111/j.1530-0277.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Goldstein RB, Saha TD, Chou SP, Jung J, Zhang H, Pickering RP, Ruan WJ, Smith SM, Huang B, et al. Epidemiology of DSM-5 alcohol use disorder: results from the national epidemiologic survey on alcohol and related conditions III. JAMA Psychiat. 2015;72:757–766. doi: 10.1001/jamapsychiatry.2015.0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM. The basal ganglia. Trends Neurosci. 1995;18:60–62. [PubMed] [Google Scholar]
- Graybiel AM. Habits, rituals, and the evaluative brain. Annu Rev Neurosci. 2008;31:359–387. doi: 10.1146/annurev.neuro.29.051605.112851. [DOI] [PubMed] [Google Scholar]
- Green AS, Grahame NJ. Ethanol drinking in rodents: is free-choice drinking related to the reinforcing effects of ethanol? Alcohol. 2008;42:1–11. doi: 10.1016/j.alcohol.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Lopez MF, Yanke AB, Middaugh LD, Becker HC. Repeated cycles of chronic intermittent ethanol exposure in mice increases voluntary ethanol drinking and ethanol concentrations in the nucleus accumbens. Psychopharmacology. 2009;201:569–580. doi: 10.1007/s00213-008-1324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagman BT, Cohn AM. Toward DSM-V: mapping the alcohol use disorder continuum in college students. Drug Alcohol Depend. 2011;118:202–208. doi: 10.1016/j.drugalcdep.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin DS, Stinson FS, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830–842. doi: 10.1001/archpsyc.64.7.830. [DOI] [PubMed] [Google Scholar]
- Healey JC, Winder DG, Kash TL. Chronic ethanol exposure leads to divergent control of dopaminergic synapses in distinct target regions. Alcohol. 2008;42:179–190. doi: 10.1016/j.alcohol.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricson AW, Thomas MP, Lippmann MJ, Morrisett RA. Suppression of L-type voltage-gated calcium channel-dependent synaptic plasticity by ethanol: analysis of miniature synaptic currents and dendritic calcium transients. J Pharmacol Exp Ther. 2003;307:550–558. doi: 10.1124/jpet.103.055137. [DOI] [PubMed] [Google Scholar]
- Hernandez G, Trujillo-Pisanty I, Cossette MP, Conover K, Shizgal P. Role of dopamine tone in the pursuit of brain stimulation reward. J Neurosci. 2012;32:11032–11041. doi: 10.1523/JNEUROSCI.1051-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries MD, Prescott TJ. The ventral basal ganglia, a selection mechanism at the crossroads of space, strategy, and reward. Prog Neurobiol. 2010;90:385–417. doi: 10.1016/j.pneurobio.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Hwa LS, Chu A, Levinson SA, Kayyali TM, DeBold JF, Miczek KA. Persistent escalation of alcohol drinking in C57BL/6J mice with intermittent access to 20% ethanol. Alcohol Clin Exp Res. 2011;35:1938–1947. doi: 10.1111/j.1530-0277.2011.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperato A, Di Chiara G. Preferential stimulation of dopamine release in the nucleus accumbens of freely moving rats by ethanol. J Pharmacol Exp Ther. 1986;239:219–228. [PubMed] [Google Scholar]
- Karkhanis AN, Rose JH, Huggins KN, Konstantopoulos JK, Jones SR. Chronic intermittent ethanol exposure reduces presynaptic dopamine neurotransmission in the mouse nucleus accumbens. Drug Alcohol Depend. 2015;150:24–30. doi: 10.1016/j.drugalcdep.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis AN, Huggins KN, Rose JH, Jones SR. Switch from excitatory to inhibitory actions of ethanol on dopamine levels after chronic exposure: role of kappa opioid receptors. Neuropharmacology. 2016;110:190–197. doi: 10.1016/j.neuropharm.2016.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashem MA, Ahmed S, Sarker R, Ahmed EU, Hargreaves GA, McGregor IS. Long-term daily access to alcohol alters dopamine-related synthesis and signaling proteins in the rat striatum. Neurochem Int. 2012;61:1280–1288. doi: 10.1016/j.neuint.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Kissler JL, Sirohi S, Reis DJ, Jansen HT, Quock RM, Smith DG, Walker BM. The one-two punch of alcoholism: role of central amygdala dynorphins/kappa-opioid receptors. Biol Psychiatry. 2014;75:744–782. doi: 10.1016/j.biopsych.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhnke MD, Batra A, Kolb W, Köhnke AM, Lutz U, Schick S, Gaertner I. Association of the dopamine transporter gene with alcoholism. Alcohol Alcohol. 2005;40(5):339–342. doi: 10.1093/alcalc/agh179. [DOI] [PubMed] [Google Scholar]
- Kokkinidis L, McCarter BD. Postcocaine depression and sensitization of brain-stimulation reward: analysis of reinforcement and performance effects. Pharmacol Biochem Behav. 1990;36:463–471. doi: 10.1016/0091-3057(90)90242-a. [DOI] [PubMed] [Google Scholar]
- Koller BH, Smithies O. Altering genes in animals by gene targeting. Annu Rev Immunol. 1992;10:705–730. doi: 10.1146/annurev.iy.10.040192.003421. [DOI] [PubMed] [Google Scholar]
- Koob GF. Neurocircuitry of alcohol addiction: synthesis from animal models. Handb Clin Neurol. 2014;125:33–54. doi: 10.1016/B978-0-444-62619-6.00003-3. [DOI] [PubMed] [Google Scholar]
- Koyama S, Brodie MS, Appel SB. Ethanol inhibition of m-current and ethanol-induced direct excitation of ventral tegmental area dopamine neurons. J Neurophysiol. 2007;97:1977–1985. doi: 10.1152/jn.00270.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin A, Chefer V, Bazov I, Meis J, Ogren SO, Shippenberg T, Bakalkin G. Upregulated dynorphin opioid peptides mediate alcohol-induced learning and memory impairment. Transl Psychiatry. 2013;3:e310. doi: 10.1038/tp.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine TP, Ahonen A, Torniainen P, Heikkila J, Pyhtinen J, Rasanen P, Niemela O, Hillbom M. Dopamine transporters increase in human brain after alcohol withdrawal. Mol Psychiatry. 1999;4:189–191. 104–185. doi: 10.1038/sj.mp.4000514. [DOI] [PubMed] [Google Scholar]
- Lam MP, Marinelli PW, Bai L, Gianoulakis C. Effects of acute ethanol on opioid peptide release in the central amygdala: an in vivo microdialysis study. Psychopharmacology. 2008;201:261–271. doi: 10.1007/s00213-008-1267-8. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le AD, Quan B, Juzytch W, Fletcher PJ, Joharchi N, Shaham Y. Reinstatement of alcohol-seeking by priming injections of alcohol and exposure to stress in rats. Psychopharmacology. 1998;135:169–174. doi: 10.1007/s002130050498. [DOI] [PubMed] [Google Scholar]
- Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Maggio R, Brann MR, Ciliax BJ. Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci U S A. 1993;90:8861–8865. doi: 10.1073/pnas.90.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TK, Lumeng L, McBride WJ, Waller MB. Progress toward a voluntary oral consumption model of alcoholism. Drug Alcohol Depend. 1979;4:45–60. doi: 10.1016/0376-8716(79)90040-1. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic ethanol actions: potential roles in ethanol seeking. In: Grant KA, editor. Neuropharmacology of alcohol, Handbook of experimental pharmacology. Springer; Heidelberg: 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey DJ, Schulteis G, Heinrichs SC, Koob GF. Time-dependent quantifiable withdrawal from ethanol in the rat: effect of method of dependence induction. Alcohol. 1996;13:163–170. doi: 10.1016/0741-8329(95)02030-6. [DOI] [PubMed] [Google Scholar]
- Majchrowicz E, Mendelson JH. Blood concentrations of acetaldehyde and ethanol in chronic alcoholics. Science. 1970;168:1100–1102. doi: 10.1126/science.168.3935.1100. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Lam M, Bai L, Quirion R, Gianoulakis C. A microdialysis profile of dynorphin A(1-8) release in the rat nucleus accumbens following alcohol administration. Alcohol Clin Exp Res. 2006;30:982–990. doi: 10.1111/j.1530-0277.2006.00112.x. [DOI] [PubMed] [Google Scholar]
- Martinez D, Gil R, Slifstein M, Hwang DR, Huang Y, Perez A, Kegeles L, Talbot P, Evans S, Krystal J, et al. Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biol Psychiatry. 2005;58:779–786. doi: 10.1016/j.biopsych.2005.04.044. [DOI] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Damask SP, Watson SJ., Jr Differential expression of autoreceptors in the ascending dopamine systems of the human brain. Proc Natl Acad Sci U S A. 1994;91:8297–8301. doi: 10.1073/pnas.91.17.8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior JR, Jones SR. Chronic ethanol exposure increases inhibition of optically targeted phasic dopamine release in the nucleus accumbens core and medial shell ex vivo. Mol Cell Neurosci. 2017;85:93–104. doi: 10.1016/j.mcn.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Spiga S, Diana M. The dopamine hypothesis of drug addiction: hypodopaminergic state. Int Rev Neurobiol. 2005;63:101–154. doi: 10.1016/S0074-7742(05)63005-X. [DOI] [PubMed] [Google Scholar]
- Meller E, Bohmaker K, Goldstein M, Basham DA. Evidence that striatal synthesis-inhibiting autoreceptors are dopamine D3 receptors. Eur J Pharmacol. 1993;249:R5–R6. doi: 10.1016/0014-2999(93)90674-7. [DOI] [PubMed] [Google Scholar]
- Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA. 2004;291:1238–1245. doi: 10.1001/jama.291.10.1238. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Morrisett RA. Ethanol action on dopaminergic neurons in the ventral tegmental area: interaction with intrinsic ion channels and neurotransmitter inputs. Int Rev Neurobiol. 2010;91:235–288. doi: 10.1016/S0074-7742(10)91008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisot N, Ron D. Alcohol-dependent molecular adaptations of the NMDA receptor system. Genes Brain Behav. 2017;16:139–148. doi: 10.1111/gbb.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeshima T, Katoh A, Wada M, Kameyama T. Stress-induced changes in brain met-enkephalin, Leu-enkephalin and dynorphin concentrations. Life Sci. 1992;51:211–217. doi: 10.1016/0024-3205(92)90077-3. [DOI] [PubMed] [Google Scholar]
- Nealey KA, Smith AW, Davis SM, Smith DG, Walker BM. Kappa-opioid receptors are implicated in the increased potency of intra-accumbens nalmefene in ethanol-dependent rats. Neuropharmacology. 2011;61:35–42. doi: 10.1016/j.neuropharm.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Miller LL. Intracranial self-stimulation to evaluate abuse potential of drugs. Pharmacol Rev. 2014;66:869–917. doi: 10.1124/pr.112.007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM. The flexible approach hypothesis: unification of effort and cue-responding hypotheses for the role of nucleus accumbens dopamine in the activation of reward-seeking behavior. J Neurosci. 2010;30:16585–16600. doi: 10.1523/JNEUROSCI.3958-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimitvilai S, You C, Arora DS, McElvain MA, Vandegrift BJ, Brodie MS, Woodward JJ. Differential effects of toluene and ethanol on dopaminergic neurons of the ventral tegmental area. Front Neurosci. 2016;10:434. doi: 10.3389/fnins.2016.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimitvilai S, Uys JD, Woodward JJ, Randall PK, Ball LE, Williams RW, Jones BC, Lu L, Grant KA, Mulholland PJ. Orbitofrontal neuroadaptations and cross-species synaptic biomarkers in heavy-drinking macaques. J Neurosci. 2017;37:3646–3660. doi: 10.1523/JNEUROSCI.0133-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien C. Addiction and dependence in DSM-V. Addiction. 2011;106:866–867. doi: 10.1111/j.1360-0443.2010.03144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Harnett MT, Morikawa H. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J Neurophysiol. 2006;95:619–626. doi: 10.1152/jn.00682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn PE, McBride WJ, Lumeng L, Gaff TM, Li TK. Neurochemical and operant behavioral studies of a strain of alcohol-preferring rats. Pharmacol Biochem Behav. 1978;8:475–481. doi: 10.1016/0091-3057(78)90087-4. [DOI] [PubMed] [Google Scholar]
- Phillips PE, Stuber GD, Heien ML, Wightman RM, Carelli RM. Subsecond dopamine release promotes cocaine seeking. Nature. 2003;422:614–618. doi: 10.1038/nature01476. [DOI] [PubMed] [Google Scholar]
- Pickens RW, Hatsukami DK, Spicer JW, Svikis DS. Relapse by alcohol abusers. Alcohol Clin Exp Res. 1985;9:244–247. doi: 10.1111/j.1530-0277.1985.tb05744.x. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Crawford CA, Nonneman AJ, Mattingly BA, Bardo MT. Effect of forebrain dopamine depletion on novelty-induced place preference behavior in rats. Pharmacol Biochem Behav. 1990;36:321–325. doi: 10.1016/0091-3057(90)90411-a. [DOI] [PubMed] [Google Scholar]
- Pleil KE, Lowery-Gionta EG, Crowley NA, Li C, Marcinkiewcz CA, Rose JH, McCall NM, Maldonado-Devincci AM, Morrow AL, Jones SR, et al. Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology. 2015a;99:735–749. doi: 10.1016/j.neuropharm.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleil KE, Rinker JA, Lowery-Gionta EG, Mazzone CM, McCall NM, Kendra AM, Olson DP, Lowell BB, Grant KA, Thiele TE, et al. NPY signaling inhibits extended amygdala CRF neurons to suppress binge alcohol drinking. Nat Neurosci. 2015b;18:545–552. doi: 10.1038/nn.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrino LJ, Lyons D, Smith HR, Daunais JB, Nader MA. Cocaine self-administration produces a progressive involvement of limbic, association, and sensorimotor striatal domains. J Neurosci. 2004;24:3554–3562. doi: 10.1523/JNEUROSCI.5578-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Grabner CP, Johnson M, Pierce RC, Bardo MT. Transient increases in catechol-aminergic activity in medial prefrontal cortex and nucleus accumbens shell during novelty. Neuroscience. 1997;76:707–714. doi: 10.1016/s0306-4522(96)00382-x. [DOI] [PubMed] [Google Scholar]
- Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223–2233. doi: 10.1016/S0140-6736(09)60746-7. [DOI] [PubMed] [Google Scholar]
- Renteria R, Buske TR, Morrisett RA. Long-term subregion-specific encoding of enhanced ethanol intake by D1DR medium spiny neurons of the nucleus accumbens. Addict Biol. 2017 doi: 10.1111/adb.12526. [DOI] [PMC free article] [PubMed]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Ford MM, Yu CH, Brown LL, Finn DA, Garland T, Jr, Crabbe JC. Mouse inbred strain differences in ethanol drinking to intoxication. Genes Brain Behav. 2007;6:1–18. doi: 10.1111/j.1601-183X.2006.00210.x. [DOI] [PubMed] [Google Scholar]
- Rimondini R, Sommer W, Heilig M. A temporal threshold for induction of persistent alcohol preference: behavioral evidence in a rat model of intermittent intoxication. J Stud Alcohol. 2003;64:445–449. doi: 10.15288/jsa.2003.64.445. [DOI] [PubMed] [Google Scholar]
- Ron D, Barak S. Molecular mechanisms underlying alcohol-drinking behaviours. Nat Rev Neurosci. 2016;17:576–591. doi: 10.1038/nrn.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JH, Karkhanis AN, Chen R, Gioia D, Lopez MF, Becker HC, McCool BA, Jones SR. Supersensitive kappa opioid receptors promotes ethanol withdrawal-related behaviors and reduce dopamine signaling in the nucleus accumbens. Int J Neuropsychopharmacol. 2016:19. doi: 10.1093/ijnp/pyv127. [DOI] [PMC free article] [PubMed]
- Rossetti ZL, Melis F, Carboni S, Diana M, Gessa GL. Alcohol withdrawal in rats is associated with a marked fall in extraneuronal dopamine. Alcohol Clin Exp Res. 1992;16:529–532. doi: 10.1111/j.1530-0277.1992.tb01411.x. [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Isola D, De Vry J, Fadda F. Effects of nimodipine on extracellular dopamine levels in the rat nucleus accumbens in ethanol withdrawal. Neuropharmacology. 1999;38:1361–1369. doi: 10.1016/s0028-3908(99)00039-8. [DOI] [PubMed] [Google Scholar]
- Rothblat DS, Rubin E, Schneider JS. Effects of chronic alcohol ingestion on the mesostriatal dopamine system in the rat. Neurosci Lett. 2001;300:63–66. doi: 10.1016/s0304-3940(01)01548-8. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Phillips TJ, Bunzow JR, Falzone TL, Dziewczapolski G, Zhang G, Fang Y, Larson JL, McDougall JA, Chester JA, et al. Mice lacking dopamine D4 receptors are supersen-sitive to ethanol, cocaine, and methamphetamine. Cell. 1997;90:991–1001. doi: 10.1016/s0092-8674(00)80365-7. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Markou A, Cole M, Koob GF. Decreased brain reward produced by ethanol withdrawal. Proc Natl Acad Sci U S A. 1995;92:5880–5884. doi: 10.1073/pnas.92.13.5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Schultz W. Updating dopamine reward signals. Curr Opin Neurobiol. 2013;23:229–238. doi: 10.1016/j.conb.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Seress L. Comparative anatomy of the hippocampal dentate gyrus in adult and developing rodents, non-human primates and humans. Prog Brain Res. 2007;163:23–41. doi: 10.1016/S0079-6123(07)63002-7. [DOI] [PubMed] [Google Scholar]
- Shnitko TA, Robinson DL. Regional variation in phasic dopamine release during alcohol and sucrose self-administration in rats. ACS Chem Neurosci. 2015;6:147–154. doi: 10.1021/cn500251j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Cuzon Carlson VC, Helms CM, Lovinger DM, Grant KA, Jones SR. Voluntary ethanol intake predicts kappa-opioid receptor supersensitivity and regionally distinct dopaminergic adaptations in macaques. J Neurosci. 2015a;35:5959–5968. doi: 10.1523/JNEUROSCI.4820-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Ferris MJ, Jones SR. Adaptations of presynaptic dopamine terminals induced by psychostimulant self-administration. ACS Chem Neurosci. 2015b;6:27–36. doi: 10.1021/cn5002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Yorgason JT, Lovinger DM, Mateo Y, Jimenez VA, Helms CM, Grant KA, Jones SR. Increased presynaptic regulation of dopamine neurotransmission in the nucleus accumbens core following chronic ethanol self-administration in female macaques. Psychopharmacology. 2016a;233:1435–1443. doi: 10.1007/s00213-016-4239-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Yorgason JT, Mateo Y, Helms CM, Lovinger DM, Grant KA, Jones SR. Chronic ethanol self-administration in macaques shifts dopamine feedback inhibition to predominantly D2 receptors in nucleus accumbens core. Drug Alcohol Depend. 2016b;158:159–163. doi: 10.1016/j.drugalcdep.2015.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Locke JL, Mathews TA, Lopez MF, Becker HC, Jones SR. Dopamine synthesis in alcohol drinking-prone and -resistant mouse strains. Alcohol. 2017;58:25–32. doi: 10.1016/j.alcohol.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg M, Silberberg R. Factors modifying the lifespan of mice. Am J Phys. 1954;177:23–26. doi: 10.1152/ajplegacy.1954.177.1.23. [DOI] [PubMed] [Google Scholar]
- Soderpalm B, Lof E, Ericson M. Mechanistic studies of ethanol’s interaction with the mesolimbic dopamine reward system. Pharmacopsychiatry. 2009;42(Suppl 1):S87–S94. doi: 10.1055/s-0029-1220690. [DOI] [PubMed] [Google Scholar]
- Stauffer WR, Lak A, Yang A, Borel M, Paulsen O, Boyden ES, Schultz W. Dopamine neuron-specific Optogenetic stimulation in rhesus macaques. Cell. 2016;166:1564–1571. e1566. doi: 10.1016/j.cell.2016.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR. Cocaine-induced c-fos messenger RNA is inversely related to dynorphin expression in striatum. J Neurosci. 1993;13:5066–5081. doi: 10.1523/JNEUROSCI.13-12-05066.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR. Dynorphin regulates D1 dopamine receptor-mediated responses in the striatum: relative contributions of pre- and postsynaptic mechanisms in dorsal and ventral striatum demonstrated by altered immediate-early gene induction. J Comp Neurol. 1996;376:530–541. doi: 10.1002/(SICI)1096-9861(19961223)376:4<530::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Svingos AL, Chavkin C, Colago EE, Pickel VM. Major coexpression of kappa-opioid receptors and the dopamine transporter in nucleus accumbens axonal profiles. Synapse. 2001;42:185–192. doi: 10.1002/syn.10005. [DOI] [PubMed] [Google Scholar]
- Tigges G, Gordon T, McClure H, Hall E, Peters A. Survival rate and life span of rhesus monkeys at the Yerkes regional primate research center. Am J Primatol. 1988;15:263–273. doi: 10.1002/ajp.1350150308. [DOI] [PubMed] [Google Scholar]
- Tiihonen J, Kuikka J, Bergstrom K, Hakola P, Karhu J, Ryynanen OP, Fohr J. Altered striatal dopamine re-uptake site densities in habitually violent and non-violent alcoholics. Nat Med. 1995;1:654–657. doi: 10.1038/nm0795-654. [DOI] [PubMed] [Google Scholar]
- Tupala E, Tiihonen J. Dopamine and alcoholism: neurobiological basis of ethanol abuse. Prog Neuro-Psychopharmacol Biol Psychiatry. 2004;28:1221–1247. doi: 10.1016/j.pnpbp.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Tupala E, Halonen P, Tiihonen J. Visualization of the cortical dopamine transporter in type 1 and 2 alcoholics with human whole hemisphere autoradiography. Eur Neuropsychopharmacol. 2006;16:552–560. doi: 10.1016/j.euroneuro.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Twining RC, Wheeler DS, Ebben AL, Jacobsen AJ, Robble MA, Mantsch JR, Wheeler RA. Aversive stimuli drive drug seeking in a state of low dopamine tone. Biol Psychiatry. 2015;77:895–902. doi: 10.1016/j.biopsych.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venniro M, Caprioli D, Shaham Y. Animal models of drug relapse and craving: from drug priming-induced reinstatement to incubation of craving after voluntary abstinence. Prog Brain Res. 2016;224:25–52. doi: 10.1016/bs.pbr.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Vivian JA, Green HL, Young JE, Majerksy LS, Thomas BW, Shively CA, Tobin JR, Nader MA, Grant KA. Induction and maintenance of ethanol self-administration in cynomolgus monkeys (Macaca fascicularis): long-term characterization of sex and individual differences. Alcohol Clin Exp Res. 2001;25:1087–1097. [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemann R, Ding YS, Pappas N, Shea C, Piscani K. Decreases in dopamine receptors but not in dopamine transporters in alcoholics. Alcohol Clin Exp Res. 1996;20:1594–1598. doi: 10.1111/j.1530-0277.1996.tb05936.x. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley SJ, Pappas N, et al. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature. 1997;386:827–830. doi: 10.1038/386827a0. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Jayne M, Ma Y, Pradhan K, Wong C. Profound decreases in dopamine release in striatum in detoxified alcoholics: possible orbitofrontal involvement. J Neurosci. 2007;27:12700–12706. doi: 10.1523/JNEUROSCI.3371-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BM, Zorrilla EP, Koob GF. Systemic kappa-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addict Biol. 2011;16:116–119. doi: 10.1111/j.1369-1600.2010.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Willuhn I, Clark JJ, Phillips PE. Phasic dopamine release in appetitive behaviors and drug addiction. Curr Drug Abuse Rev. 2009;2:195–213. doi: 10.2174/1874473710902020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss F, Lorang MT, Bloom FE, Koob GF. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther. 1993;267:250–258. [PubMed] [Google Scholar]
- Werling LL, Frattali A, Portoghese PS, Takemori AE, Cox BM. Kappa receptor regulation of dopamine release from striatum and cortex of rats and guinea pigs. J Pharmacol Exp Ther. 1988;246:282–286. [PubMed] [Google Scholar]
- Wightman RM. Voltammetry with microscopic electrodes in new domains. Science. 1988;240:415–420. doi: 10.1126/science.240.4851.415. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Strope E, Plotsky PM, Adams RN. Monitoring of transmitter metabolites by voltammetry in cerebrospinal fluid following neural pathway stimulation. Nature. 1976;262:145–146. doi: 10.1038/262145a0. [DOI] [PubMed] [Google Scholar]
- Yen CH, Yeh YW, Liang CS, Ho PS, Kuo SC, Huang CC, Chen CY, Shih MC, Ma KH, Peng GS, et al. Reduced dopamine transporter availability and neurocognitive deficits in male patients with alcohol dependence. PLoS One. 2015;10:e0131017. doi: 10.1371/journal.pone.0131017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen CH, Shih MC, Cheng CY, Ma KH, Lu RB, Huang SY. Incongruent reduction of dopamine transporter availability in different subgroups of alcohol dependence. Medicine (Baltimore) 2016;95:e4048. doi: 10.1097/MD.0000000000004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim HJ, Schallert T, Randall PK, Gonzales RA. Comparison of local and systemic ethanol effects on extracellular dopamine concentration in rat nucleus accumbens by microdialysis. Alcohol Clin Exp Res. 1998;22:367–374. [PubMed] [Google Scholar]
- Yorgason JT, Ferris MJ, Steffensen SC, Jones SR. Frequency-dependent effects of ethanol on dopamine release in the nucleus accumbens. Alcohol Clin Exp Res. 2014;38:438–447. doi: 10.1111/acer.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, Rose JH, McIntosh JM, Ferris MJ, Jones SR. Greater ethanol inhibition of presynaptic dopamine release in C57BL/6J than DBA/2J mice: role of nicotinic acetylcholine receptors. Neuroscience. 2015;284:854–864. doi: 10.1016/j.neuroscience.2014.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]