

Abstract
Chronic hepatitis B virus (HBV) infection is a major cause of chronic liver disease and cancer worldwide. The mechanisms of viral genome sensing and the evasion of innate immune responses by HBV infection are still poorly understood. Recently, the cyclic GMP-AMP synthase (cGAS) was identified as a DNA sensor. In this study, we aimed to investigate the functional role of cGAS in sensing of HBV infection and elucidate the mechanisms of viral evasion. We performed functional studies including loss- and gain-of-function experiments combined with cGAS effector gene expression profiling in an infectious cell culture model, primary human hepatocytes and HBV-infected human liver chimeric mice. Here we show that cGAS is expressed in the human liver, primary human hepatocytes and human liver chimeric mice. While naked relaxed-circular HBV DNA is sensed in a cGAS-dependent manner in hepatoma cell lines and primary human hepatocytes, host cell recognition of viral nucleic acids is abolished during HBV infection, suggesting escape from sensing, likely during packaging of the genome into the viral capsid. While the hepatocyte cGAS pathway is functionally active, as shown by reduction of viral cccDNA levels in gain-of-function studies, HBV infection suppressed cGAS expression and function in cell culture models and humanized mice.
Conclusion
HBV exploits multiple strategies to evade sensing and antiviral activity of cGAS and its effector pathways.
Keywords: antiviral, capsid, innate immunity, liver, recognition
With more than 250 million chronically infected patients, hepatitis B virus (HBV) infection is a leading cause of liver disease and hepatocellular carcinoma (1, 2). Current antiviral therapies effectively control viral load, but largely fail to cure (3). HBV is a partially double stranded DNA (dsDNA) virus infecting human hepatocytes after initial attachment to heparan sulfate proteoglycans (HSPG) and its receptor Na+/taurocholate cotransporting polypeptide (NTCP; reviewed in (4)). Following uncoating, the viral nucleocapsid is released into the cytoplasm. The viral genome is imported into the nucleus through mechanisms which are still poorly understood. The viral genome it is converted in the nucleus into a covalently closed circular DNA (cccDNA) (5). This minichromosome serves as a template for both pregenomic RNA (pgRNA) and viral mRNA transcription. While recent studies suggested sensing of the pgRNA or other HBV RNAs by either MDA5 (6) or RIG-I (7), the recognition of the viral nucleic acids by the regular pattern recognition receptors (PRRs) still remains elusive. In general, HBV does not or only marginally activate innate immune responses in cell culture models and in vivo (8-14), leading to the concept that HBV behaves like a “stealth” virus avoiding viral DNA and RNA sensing (15). Other studies have suggested an active inhibition of the innate immune responses by HBV proteins (16). Consequently, the interaction of HBV and the innate immune system of hepatocytes, and in particular the sensing of HBV DNA, is only poorly understood.
Foreign DNA recognition by cytosolic DNA sensors triggers an early antiviral innate immune response, including type I and type III IFN production (17). Recently, the cyclic GMP-AMP (cGAMP) synthase (cGAS) was identified as a DNA sensor exhibiting an antiviral activity against a broad range of DNA and RNA viruses (18-20). cGAS is encoded by MB21D1 gene and directly binds to dsDNAs inducing the production of cGAMP which is recognized by the stimulator of IFN genes (STING, encoded by TMEM173) triggering the expression of IFN-stimulated genes (ISGs) through TBK1 activation (21-23). While two studies have investigated cGAS-HBV interactions in viral replication and assembly (24, 25), the functional role of cGAS in sensing of the viral genome during natural infection of human hepatocytes remains unknown.
The understanding of HBV-host interactions, including innate immune response after infection, has been hampered for long time by the absence of robust cell culture model system for the study of viral infection (26). The development of HBV-susceptible NTCP-overexpressing hepatoma cells, such as HepG2-NTCP cells, allows the study of the full life cycle in a robust and easy-to-use cell culture model (26). HepG2 cells are capable of mounting an efficient innate immune response after infection by hepatitis C virus (27). Moreover, another study took advantage of HBV-infected HepG2-NTCP for studying the interaction between RIG-I and HBV RNA (7), suggesting that this cell line is suitable for the study of innate immune response after HBV infection. Here, we aimed to understand the functional role of cGAS for the HBV life cycle in human hepatocytes and unravel the mechanisms of viral evasion using loss- and gain-of-function experiments combined with cGAS effector gene expression profiling in human liver chimeric mice.
EXPERIMENTAL PROCEDURES
Human subjects
Human material including liver tissue from patients undergoing surgical resection or HBV-positive serum was obtained with informed consent from all patients. Protocols were approved by the Ethics Committee of the University of Strasbourg Hospitals, France (CPP 10-17 and DC-2016-2616).
Animal Experimentation
All mice were kept in a specific pathogen-free animal housing facility at Inserm U1110. The respective protocols were approved by the Ethics Committee of the University of Strasbourg Hospitals and authorized by the French Ministry of Research (number 02014120416254981AL/02/19/08/12, AL/01/18/08/1202014120416254981, 0201412051105 4408). Mice were kept in individual ventilated cages, with bedding composed of irradiated sawdust and chips from spruce and pine and enriched with cotton cocoon and aspen bricks. The animal diet consists of 25kGy irradiated RM3(E) (SDS) and mice were not fasted. Primary human hepatocytes (PHH) were transplanted into 3 week-old uPA/SCID-bg mice (male and female) by intrasplenic injection as described (28). Engraftment and viability of PHH were assessed by quantification of human serum albumin by ELISA (E80-129, Bethyl Laboratories; (28)). uPA/SCID-bg mice were then infected with serum-derived HBV and sacrificed 16 weeks after virus inoculation. Serum HBV load was determined by qPCR (Realtime HBV viral load kit, Abbott) before sacrifice. Interventions were all performed during light cycle.
Cell lines and human hepatocytes
HEK 293T (29) and HepG2-NTCP (30) cells and isolation of PHH have been described (29).
Reagents and plasmids
DMSO, PEG 8000 (polyethylene glycol), Poly (I:C) and calf thymus DNA (control dsDNA) were obtained from Sigma-Aldrich, pReceiver-Lv151 plasmid from GeneCopoeia™ and lentiCas9-Blast and lentiGuide-Puro plasmids were gifts from Feng Zhang (Addgene #52962 and #52963).
Small interfering RNAs for functional studies
Pools of ON-TARGET plus (Dharmacon) small interfering RNA (siRNA) targeting MB21D1 (cGAS), TMEM173 (STING), TBK1, and IFI16 expression were reverse-transfected into HepG2-NTCP using Lipofectamine RNAi-MAX (Invitrogen) as described (29). RNA was purified from cells harvested two days after transfection and gene expression was analyzed using qRT-PCR.
HepG2-NTCP-cGAS overexpressing and MB21D knock-out cells
Lentivirus particles were generated in HEK 293T cells by cotransfection of plasmids expressing the human immunodeficiency virus (HIV) gap-pol, the vesicular stomatitis virus glycoprotein (VSV-G) and either the human MB21D1 full open reading frame (ORF) encoding plasmid, or the MB21D1-targeting single-guide RNA (sgRNA) encoding plasmids, or the Cas9 expressing plasmid in the ratio of 10:3:10. HepG2-NTCP cells were then plated and transduced with lentivirus encoding either the human MB21D1 ORF or the eGFP ORF in pReceiver-Lv151 vector (GeneCopoeia™). After 3 days, transduced cells were selected with 200 μg/ml of neomycin (G418). The cGAS-over-expressing and control HepG2-NTCP cells were then further cultured in presence of G418 at 200 μg/ml. For the generation of MB21D1 knock-out cell lines, one MB21D1-targeting sgRNA was designed using CRISPR Design Tool (Broad Institute: http://www.genome-engineering.org/crispr/?page_id=41). The sgRNA sequence targeting the exon 1 of MB21D1 (sgcGAS 5’-CACCGCGGCCCCCATTCTCGTACGG-3’) was inserted into lentiGuide-Puro plasmid (31). We first generated Cas9 expressing HepG2-NTCP cells after transduction of cells with the lentiCas9-Blast plasmid (31). Cells were then selected with 6 μg/ml blasticidin for 10 days. HepG2-NTCP-Cas9 cells were seeded in six-well plates at 50% confluency 24 h prior to transduction with the sgcGAS-encoding plasmid. Subpopulations of cells were selected from the whole population and cultured independently. cGAS expression was controlled by Western blot. Finally, two cGAS-deficient cell lines (cGAS_KO#1 and cGAS_KO#2) were selected.
Analysis of gene expression using qRT-PCR
Total RNA was extracted using ReliaPrep™ RNA Miniprep Systems (Promega) and reverse transcribed into cDNA using Maxima First Strand cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer’s instructions. Gene expression was then quantified by qPCR using a CFX96 thermocycler (Bio-Rad). Primers and TaqMan® probes for MB21D1 (cGAS), TMEM173 (STING), TBK1, IFI16, IFNB1, IFNL1, and GAPDH mRNA detection were obtained from ThermoFisher (TaqMan® Gene expression Assay, Applied Biosystems). All values were normalized to GAPDH expression.
Protein expression
The expression of cGAS, STING, and β-actin proteins was assessed by Western blot as described (30) using two polyclonal rabbit anti-cGAS antibodies (HPA031700, Sigma & NBP1-86761, Novus Biologicals, see Supporting Information), a polyclonal rabbit anti-STING antibody (19851-1-AP, Proteintech), and monoclonal anti-β-actin antibody (mAbcam8226, Abcam). Protein expression was quantified using ImageJ software.
Infection of HepG2-NTCP cells and PHH
The purification of infectious recombinant HBV particles from HepAD38 cells and infection of HepG2-NTCP cells has been described (30). Briefly, HepG2-NTCP and derived cells were plated one day prior to incubation with HBV in presence of 4% PEG at multiplicity of infection (MOI) ~ 500 genome equivalent/cell (GEq/cell) except otherwise stated. Sixteen hours after HBV inoculation, cells were washed with PBS and then cultured in 3.5% DMSO primary hepatocyte maintenance medium (PMM) for ten days. HBV infection was assessed by quantification of HBV pgRNA using qRT-PCR or HBV total DNA using qPCR as described (30), or by immunofluorescence (IF) using anti-HBsAg antibody (1044/329, Bio-Techne) and AF647-labelled goat antibody targeting mouse IgGs (115-605-003, Jackson Research) as described (30). Southern blot detection of HBV cccDNA was performed using DIG-labelled (Roche) specific probes as described (32). Total DNA from HBV-infected cells was extracted using the previously described HIRT method (33). Specific DIG-labelled probes for the detection of HBV and mitochondrial DNAs were synthetized using the PCR DIG Probe Synthesis Kit (Roche) and the primers indicated in Table S1. PHH were plated one day prior to incubation with a HBV preS1- or a control peptide for one hour at 37°C as described (30). PHH were then infected with recombinant HBV particles for ten days. HBV infection was assessed by quantification of HBV pgRNA using qRT-PCR and immunofluorescence as described above.
Sendai virus (SeV) infection
HepG2-NTCP cells and PHH were infected with SeV DI-H4 at an MOI of 10 as described (13).
Extraction of HBV rcDNA from HBV infectious particles
HBV rcDNA was extracted from HBV preparations using QiaAMP DNA MiniKit protocol (Qiagen). PEG-precipitated cell supernatants from naive HepG2-NTCP cells were used as non-virion controls. The presence of HBV DNA was confirmed by PCR and quantified by qPCR as described (30) (see Supporting Information). One μg of rcDNA or dsDNA (calf thymus DNA) was transfected in cells using Lipofectamine 2000 (Invitrogen) and CalPhos Mammalian Transfection Kit (Clonetech) according to manufacturer’s instructions. Cells transfected with HepG2-NTCP control supernatants were used as a control. Three days after transfection, total RNA was extracted and purified as described above.
Transcriptomic analysis by digital multiplexed gene profiling using nCounter NanoString
Transcriptomic analyses using nCounter NanoString were performed according to manufacturer’s instructions. Specific probes for a set of 36 innate antiviral response-related (IAR) genes (according to (20) and additional genes listed in Table S2) were obtained from the manufacturer. HepG2-NTCP cells, HepG2-NTCP derived cell lines and PHH were either infected with HBV or SeV, or were transfected with Poly (I:C) (100ng) for two days. Alternatively, HepG2-NTCP-Cas9 and HepG2-NTCP-KO_cGAS#2 cells were transfected with rcDNA (1μg) or dsDNA (calf thymus DNA, 1μg) for three days. Total RNA was then extracted and subjected to nCounter Digital Analyzer system (NanoString). Alternatively, total liver RNA was extracted from HBV-infected mice and gene expression was assessed by either qRT-PCR (MB21D1 expression) or nCounter Digital Analyzer system. The 36 genes were considered as an artificial gene set termed innate antiviral response (IAR) gene set. Its perturbation by infection, transfection or gain-/loss-of-function studies was assessed through Gene Set Enrichment Analysis (GSEA (34)). GSEA determines whether an a priori defined set of genes shows statistically significant differences between two biological states. False discovery rate (FDR) < 0.05 was considered statistically significant. Heatmaps illustrating the induction (red) or repression (blue) of the genes of the IAR compared to control were illustrated using Morpheus software (Broad Institute of MIT and Harvard, Cambridge, MA, USA). The heatmap illustrating the induction (red) or repression (blue) of individual genes in chimeric mouse livers were designed using GenePattern (Broad Institute of MIT and Harvard, Cambridge, MA, USA).
FISH analyses
Fluorescence in situ hybridization (FISH) analyses were performed as described (28, 35). Briefly, liver samples were collected from mice and then immediately embedded into optimal cutting temperature compound (OCT). OCT-embedded liver sections were cryosectioned (10 μm) using a cryostat (Leica). Upon fixation with 4% formaldehyde at 4°C, washing, and dehydration in ethanol, tissue sections were boiled at 90–95°C for 1 min in a pretreatment solution (Affymetrix-Panomics), followed by a 10 min digestion in protease QF (Affymetrix-Panomics) at 40°C. Sections were then hybridized using specific probe sets targeting HBV (target region nucleotides 483-1473 of HBV [Genotype D, GenBank V01460]) and human MD21D1 (VA1-3013492-VC, Affymetrix-Panomics). Pre-amplification, amplification and detection of bound probes were performed according to the manufacturer’s instructions. Finally, pictures were acquired by LSCM (LSM710, Carl Zeiss Microscopy) and Zen2 software.
Statistical Analysis
Except otherwise stated, cell culture experiments were performed at least three times in an independent manner. Statistical comparisons of the samples were performed using a two-tailed Mann-Whitney U test. For in vivo experiments, a two-tailed unpaired Student’s t-test was performed for comparing gene expression from non-infected and HBV-infected mice. p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) were considered significant. Significant p values are indicated by asterisks in the figures. Each digital multiplexed gene profiling experiment was performed using three biological replicates per condition and the induction or repression of the gene set was analyzed using GSEA. FDR < 0.05 was considered statistically significant.
RESULTS
Expression of cGAS in primary human hepatocytes and an infectious HBV cell culture model
Prior to its functional characterization, we studied cGAS/MB21D1 expression in primary hepatocytes and HBV permissive cell lines. As shown in Figure 1A, cGAS protein expression was easily detectable in PHH from three independent donors. Since HBV infection of primary cells is highly variable and does not allow robust perturbation studies, we used an HBV infectious cell culture model based on differentiated hepatocyte-derived HepG2 cells overexpressing NTCP (30) – a key HBV entry factor. As shown in Figure 1A, cGAS protein is expressed in HepG2-NTCP cells. We validated the specificity of cGAS detection using a siRNA specifically targeting the MD21B1 expression (sicGAS) and Western blots applying two antibodies (Figure 1A, Figure S1). Moreover, we generated CRISPR-mediated MB21D1 knock-out (KO) cells using a specific sgRNA (Figure 1B). Two cell lines, KO_cGAS#1 and KO_cGAS#2 were selected for further studies (Figure 1B). Interestingly, the adaptor STING was also detected in HepG2-NTCP cells, suggesting a fully functional cGAS-STING pathway (Figure 1C). To test the suitability of these cells as a model to analyze cGAS-mediated innate immune response after virus infection, we stimulated cells with different analogs of viral nucleic acids. Stimulation by Poly (I:C) or dsDNA transfection elicited a dose-dependent IFNB1 expression in HepG2-NTCP cells (Figure 1D). These results suggest that the cGAS sensing machinery is present, functional, even if it is less efficient that the RNA sensing complex. Moreover, cGAS protein expression was induced by both Poly (I:C) and dsDNA stimulation confirming an efficient IFN response through the upregulation of ISGs such as MB21D1 (36) after RNA or DNA stimulation (Figure 1E). Collectively, these data show that the HepG2-NTCP model is suitable to study innate immune responses.
Figure 1. cGAS expression and function in human hepatocytes and in a cell culture model for HBV infection.
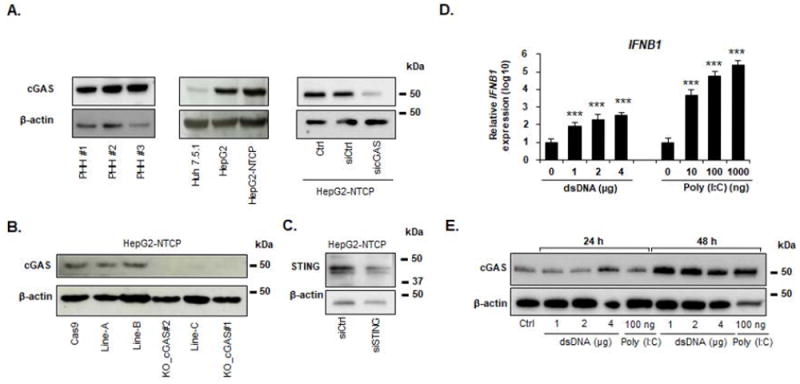
(A) Detection of endogenous cGAS protein expression in different cellular models by Western blot. Cell lysates from Huh7.5.1, HepG2, HepG2-NTCP cells, and PHH from three independent donors were used. HepG2-NTCP cells were reverse transfected with a siRNA targeting MB21D1 (sicGAS) or a non-targeting siRNA control (siCtrl) two days before cGAS detection. β-actin was used as a Western blot control. Individual representative experiments are shown. (B) Generation of MB21D1 knock out (KO) cells. MB21D1 KO HepG2-NTCP cell lines were generated via CRISPR/Cas9 technology. The absence or presence of cGAS protein was controlled by Western blot using the HPA031700 anti-cGAS antibody in Cas9-expressing HepG2-NTCP cells (Cas9) and in different cell lines after transduction with the sgRNA targeting MB21D1 (line-A, line-B, line-C, cGAS_KO#1 and cGAS_KO#2). One experiment is shown. (C) Detection of endogenous STING protein in HepG2-NTCP cells. siRNA targeting TMEM173 (siSTING) or a non-targeting siRNA (siCtrl) were reverse-transfected into HepG2-NTCP cells. Silencing efficacy was assessed by Western blot. One experiment is shown. (D-E) Poly (I:C) and dsDNA transfection induce IFNB1 and MB21D1 expression in HepG2-NTCP cells. HepG2-NTCP cells were transfected with increasing doses of Poly (I:C) or calf thymus DNA at the indicated concentrations. IFNB1 mRNA expression was quantified by qRT-PCR 24 h after transfection and cGAS protein expression was assessed by Western blot 24 h and 48 h after transfection. qRT-PCR data (D) are expressed as means ± SD relative IFNB1 expression (log10) compared to non-transfected control (0, set at 100) from four independent experiments performed in triplicate (dsDNA) or from three independent experiments performed in triplicate (Poly I:C). One representative Western blot experiment is shown (E).
HBV evades cGAS sensing
Next, we investigated whether HBV was sensed in HBV permissive cells. To address this question, we infected HepG2-NTCP cells with recombinant HBV (MOI: 500 GEq/cell) and studied the expression of IFNB1 at early time points after HBV infection. As it has been described that HBV infection may induce the expression of type III IFN (7), IFNL1 expression was also quantified. As shown in Figure 2A-B, the lack of increase in IFNB1 and IFNL1 expression in spite of efficient infection (Figure S2) indicates poor or absent detection of HBV by cellular sensors. In contrast, SeV, known to induce a strong IFN response in hepatocytes (13), strongly induced IFNB1 and IFNL1 expression (Figure 2 AB). Since cGAS has been shown to induce the expression of a large set of innate effector genes (such as OAS2 or IFI44, see (20)), the analysis of expression of a single effector gene such IFNB1 may not be sufficient to evaluate cGAS sensing. Therefore, we designed a 36 innate antiviral response gene set (named IAR), comprising 29 ISGs whose expression is modulated by cGAS activity described by Schoggins and Rice in (20) as well as 7 established innate immune response effector genes (Table S2). We then infected HepG2-NTCP cells with HBV or SeV, and measured the innate antiviral immune response at day 2 post infection by analysis of IAR gene expression using digital multiplexed gene profiling (nCounter NanoString) and GSEA-based analysis. Whereas Poly (I:C) transfection and SeV infection induced a marked modulation of cGAS effector/IAR gene expression (FDR = 0.004 and < 0.001, respectively), no significant modulation of IAR gene expression was observed in HBV-infected cells (Figure 2C), as further illustrated by the expression of IFNB1 and IFI44 (Figure 2E). To measure the impact of cGAS expression on cellular response to HBV infection, we then infected cGAS-depleted (KO_cGAS#2) and -overexpressing (cGAS_OE) HepG2-NTCP cells with HBV and analyzed the expression of the cGAS-related genes after infection. As shown in Figure 2D, no significant modulation of the IAR signature was observed in HBV-infected samples, as further illustrated by absent modulation of IFNB1 and IFI44 expression (Figure 2F).
Figure 2. Impaired cGAS-mediated sensing of HBV infection in HepG2-NTCP cells.
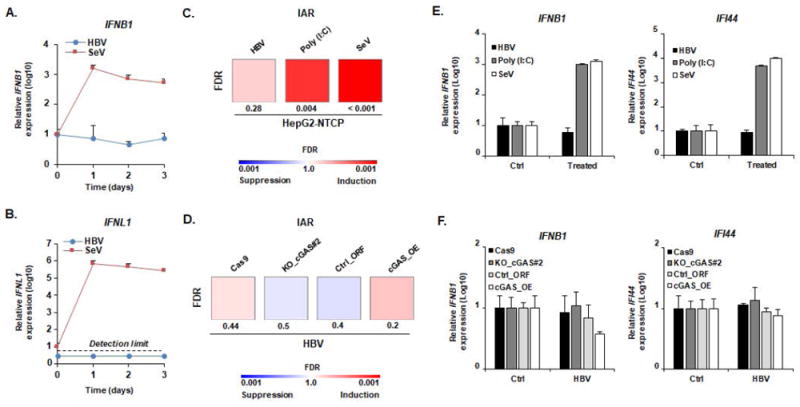
(A-B) HBV infection does not induce IFNB1 or IFNL1 expression. HepG2-NTCP cells were infected with HBV (MOI: 500) or SeV (MOI: 10) and total RNA was extracted every day for 3 days. RNA extracted from naive cells before infection was used as a control (D0). IFNB1 (A) and IFNL1 (B) expression was then assessed by qRT-PCR. Results are expressed as means ± SD IFNB1/IFNL1 relative expression (log10) compared to controls (D0, all set at 1) from three independent experiments performed at least in duplicate (SeV) or four independent experiments performed in duplicate (HBV). No robust IFNL1 expression was detected in HBV-infected samples (representative dots are presented under the “detection limit” dotted line). (C, E) cGAS-related ISGs are not affected by HBV infection. HepG2-NTCP cells were infected with HBV or SeV. Alternatively, HepG2-NTCP cells were transfected with Poly (I:C) (100ng). Two days after infection or transfection, total RNA was extracted. Gene expression of IAR signature was then analyzed using multiplexed gene profiling. Results were analyzed by GSEA enrichment compared to non-transfected or non-infected controls (C) or by IFNB1 and IFI44 gene expression (log10) compared to non-transfected or non-infected controls (set at 1) (E). One experiment performed in triplicate is shown. (D, F) cGAS expression level does not affect the cellular response to HBV infection. HepG2-NTCP-Cas9 (Cas9), HepG2-NTCP-KO_cGAS#2 (Ko_cGAS#2), HepG2-NTCP-Ctrl_ORF (Ctrl_ORF), and HepG2-NTCP-cGAS_OE (cGAS_OE) were infected with HBV. Two days after infection, total RNA was extracted. Gene expression of IAR signature gene set was then analyzed using multiplexed gene profiling. Results were analyzed by GSEA enrichment compared to non-transfected or non-infected controls (D) or by IFNB1 and IFI44 gene expression (log10) compared to non-transfected or non-infected controls (set at 1) (F). One experiment performed in triplicate is shown. IAR: innate antiviral response gene set.
To exclude the possibility that low HBV infection could be the reason for the absence of IFN induction, we performed additional experiments using increasing MOIs. As shown in Figure 3A-C, no induction of IFNB1 (Figure 3A) was observed even at a MOI 10000, despite very high infection efficiency as shown by quantitation of pre-genomic RNA and immunofluorescence of HBsAg (Figure 3B-C). To investigate whether IFN induction occurs potentially at a very early step of viral infection and was missed in the experimental design shown above, we performed a time course studying IFN response to HBV within the first 24 hours post infection. As shown in Figure 3D, HBV infection did not induce a measurable IFN response during early steps of HBV infection. In contrast, SeV, an established inducer of IFN showed robust induction of IFN responses in HepG2-NTCP cells (Figure 3D). Taken together, these data suggest an absence of sensing of HBV infection by the cGAS-STING pathway in HepG2-NTCP cells.
Figure 3. Impaired sensing of HBV infection at high MOIs or early time points of infection.
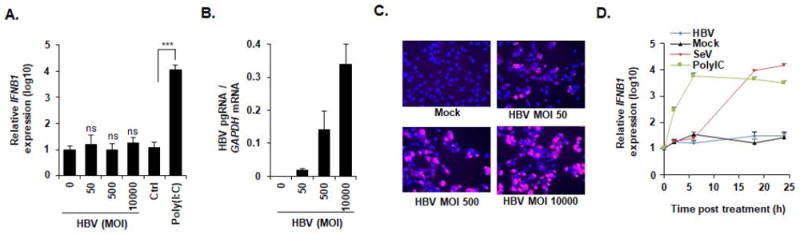
(A-C) HepG2-NTCP cells were infected with HBV at increasing MOIs (0, 50, 500, and 10000 GEq/cell) or transfected with Poly (I:C) (100 ng). Two days after infection or transfection, cells were lysed, total RNA was extracted, and IFNB1 expression (A) as well as HBV pgRNA levels (B) were quantified by qRT-PCR. (A) Results are expressed as means ± SD relative IFNB1 expression (log10) compared to mock infected cells (MOI 0, set at 1) from three independent experiments performed in triplicate. (B) Results are expressed means ± SD relative HBV pgRNA levels compared to mock infected cells (MOI 0, set at 100%) from three independent experiments performed in triplicate. Alternatively, HBV infection was assessed at 10 days post infection by IF of HBsAg (C). One representative experiment is shown. (D) HBV infection does not induce IFNB1 expression at early time points. HepG2-NTCP cells were either infected by HBV or SeV, or transfected with Poly (I:C). Total RNA was extracted at 2 h, 6 h, 18 h, and 24 h post infection/transfection and IFNB1 expression was assessed by qPCR. Results are expressed as means ± SD relative IFNB1 expression (log10) compared to naive cells (0, set at 1) from three independent experiments performed in triplicate.
As the HBV genome is packaged into the nucleocapsid (37), we investigated whether packaging shields virion DNA from cGAS recognition. We purified HBV genomic rcDNA from HBV infectious particles (Figure S3) and transfected the naked viral genome into HepG2-NTCP cells (1 μg, corresponding to approximately 106-107 HBV DNA copies/μL). As shown in Figure 4A, a significant (FDR = 0.02) induction of the IAR signature (illustrated by IFNB1 and IFI44 expression, Figure 4B) was observed after both rcDNA and dsDNA transfection, suggesting sensing of the naked HBV genome. Interestingly, the amount of cellular HBV DNA copies was higher in HBV-infected cells compared to rcDNA transfected cells (Figure 4C), confirming that the levels of HBV DNA in HBV infected cells are sufficient to trigger IFN signaling and the absence of HBV sensing in infected cells was not due to low MOIs. Moreover, the induction of the IAR gene expression was absent in HepG2-NTCP-KO_cGAS#2 cells, suggesting a cGAS-specific activation of innate immunity by both dsDNA and rcDNA transfection in our model.
Figure 4. Sensing of HBV rcDNA in HepG2-NTCP cells.
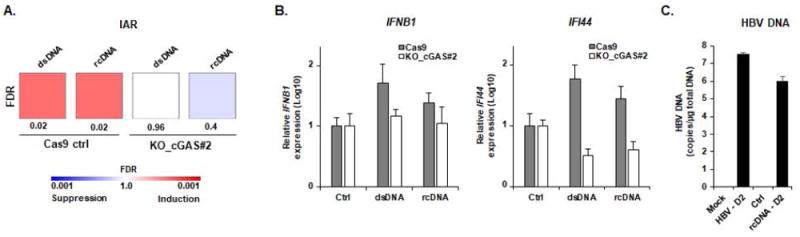
(A-B) Transfection of purified HBV rcDNA genome induces a cGAS-mediated innate immune response. HBV rcDNA was extracted from recombinant HBV virions as described in Experimental Procedures and quantified by qPCR (Figure S3). HBV rcDNA (1 μg) and positive control dsDNA (1 μg) were transfected into HepG2-NTCP-Cas9 and HepG2-NTCP-KO_cGAS#2 cells. Three days after transfection, total RNA was extracted. Gene expression of IAR set was then analyzed using multiplexed gene profiling. The transcripts were analyzed by GSEA enrichment compared to non-transfected control (A) or by IFNB1 and IFI44 gene expression (log10) compared to non-transfected control (set at 1) (B). One experiment in triplicate is shown. (C) HBV DNA in HepG2-NTCP cells after rcDNA transfection and HBV infection are similar. HepG2-NTCP were infected with HBV or transfected with HBV rcDNA (1μg). At day 2 after transfection/infection, DNA was extracted and total HBV DNA was quantified by qPCR. Results are expressed as means ± SD total DNA copies per μg of total DNA (log10) from three independent experiments performed in triplicate (HBV infection) and two independent experiments performed in triplicate (HBV rcDNA transfection). IAR: innate antiviral response gene set.
To validate these observations in a more physiological model, we infected PHH with HBV and control cGAS gene expression after two days of infection. Interestingly, while SeV strongly induced IAR gene expression (Figure 5B), a highly efficient HBV infection (Figure 5A) did not induce the induction of the expression of innate antiviral response genes (Figure 5B). In contrast, the transfection of rcDNA and dsRNA into PHH from four different donors robustly triggered the expression of IFNB1 and IFNL1 (Figure 5C), suggesting a robust sensing of viral DNA in human hepatocytes. Collectively, these data suggest that non-encapsidated HBV DNA is sensed by cGAS, but this sensing is impaired during HBV infection.
Figure 5. Sensing of naked HBV rcDNA but not infectious HBV virions in primary human hepatocytes.
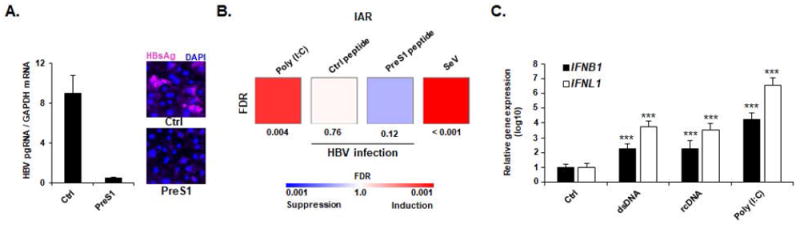
(A-B) HBV infection does not induce ISG expression in PHH. PHH were treated with a preS1 peptide (PreS1) or a scrambled peptide (Ctrl) for one hour before infection with HBV. As positive controls, PHH were transfected with Poly (I:C) (100ng) or infected with SeV (MOI ~ 10). HBV infection of PHH was assessed 10 days post infection by qRT-PCR (A, left panel) or immunofluorescence (A, right panel). qRT-PCR results are expressed as means ± SD ratio HBV pgRNA RNA / GAPDH mRNA from one experiment corresponding to the gene profiling experiment and performed in duplicate. Two days after infection, total RNA was extracted and gene expression of IAR was then analyzed using multiplexed gene profiling. Results were analyzed by GSEA enrichment compared to non-transfected control (B). One experiment in triplicate is shown. (C) Induction of IFBN1 and IFNL1 expression in PHH. PHH were transfected with dsDNA (4 μg) or HBV rcDNA (4 μg). IFNB1- and IFNL1 expression was assessed 24 h after transfection by qRT-PCR. Results are expressed as means ± SD log10 IFNB1 or IFNL1 expression compared to non-transfected control cells (Ctrl, set at 1) from five independent experiments performed at least in duplicate. IAR: innate antiviral response gene set.
The cGAS-STING pathway exhibits robust antiviral activity against HBV infection with reduction of cccDNA levels
As cGAS exhibits an antiviral activity against a broad range of DNA and RNA viruses (meaning even in absence of direct viral sensing) (20), we then investigated the antiviral effect of the cGAS-STING signaling pathway in HBV infection. We silenced the expression MB21D1, TMEM173 (encoding the STING protein), TBK1 and IFI16 (encoding the gamma-interferon-inducible protein 16, another cytoplasmic DNA sensor able to directly activate STING (17)) in HepG2-NTCP cells prior to infection with HBV. As shown in Figure 6A-B, silencing of MB21D1, TMEM173 and TBK1 expression induced a marked increase in HBV infection. In contrast, the silencing of IFI16 had no effect on HBV infection. CRISPR/Cas9-mediated KO or overexpression of cGAS protein resulted in a marked increase or decrease in HBV infection and HBV cccDNA levels – the key viral nucleic acid responsible for viral persistence (Figure 6C-E). Notably, the overexpression of cGAS did not affect NTCP expression at the cell surface, suggesting that the susceptibilities of the different cell lines to HBV infection are equivalent (Figure S4). Taken together, our results suggest that cGAS is functional and exerts antiviral activity in HBV permissive cells.
Figure 6. Antiviral activity of cGAS results in reduction of HBV cccDNA.
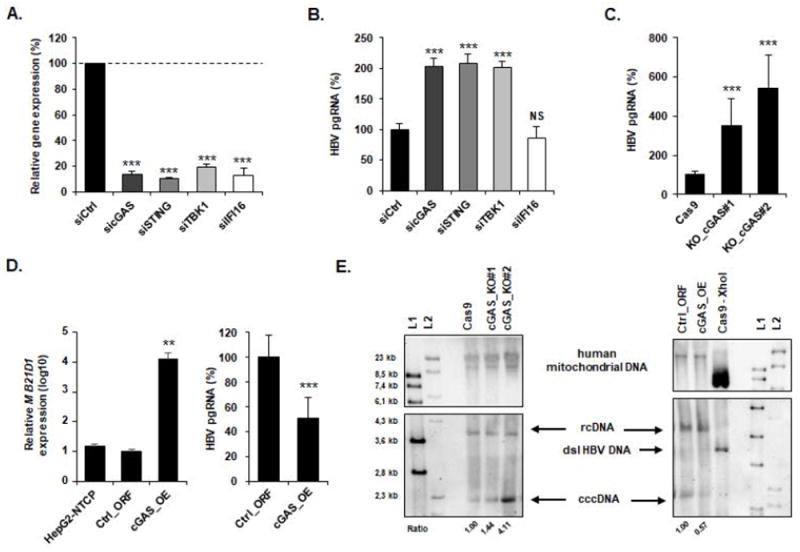
(A-B) Silencing of cGAS-related gene expression increases HBV infection. siRNA targeting MB21D1 (sicGAS), TMEM173 (siSTING), TBK1 (siTBK1), IFI16 (siIFI16) or a non-targeting siRNA (siCtrl) were reverse-transfected into HepG2-NTCP cells 2 days prior to HBV infection. Silencing efficacy was assessed by qRT-PCR 2 days after transfection (A). Results are expressed as means ± SD % gene expression relative to siCtrl (set at 100%) from four independent experiments performed in technical duplicate. HBV infection was assessed by quantification of HBV pgRNA by qRT-PCR 10 days after infection (B). Results are expressed as means ± SD % HBV pgRNA expression relative to siCtrl (set at 100%) from four independent experiments performed in technical duplicate. (C) KO of MB21D1 gene increases HBV infection. cGAS_KO#1, cGAS_KO#2, and the control Cas9 cells were then infected with HBV and viral infection was assessed 10 days after infection as described above. Results are expressed as means ± SD % HBV pgRNA expression relative to control cell line (Cas9, set at 100%) from three independent experiments performed in triplicate. (D) cGAS overexpression reduces HBV infection. HepG2-NTCP cells were transduced with lentivirus encoding either a control plasmid (Ctrl_ORF) or a plasmid encoding the full length MB21D1 ORF (cGAS_OE). MB21D1 expression was assessed by qRT-PCR (left panel). Results are expressed as means ± SD % relative MB21D1 expression (log10) relative to control cell line (Ctrl_ORF, set at 1) from three independent experiments performed in duplicate. HepG2-NTCP, Ctrl_ORF, and cGAS_OE cells were then infected with HBV for ten days and HBV infection was assessed as described above. Results are expressed as means ± SD % HBV pgRNA expression relative to control cell line (Ctrl_ORF, set at 100%) from three independent experiments performed in triplicate. (E) Detection of HBV cccDNA by Southern blot. HepG2-NTCP-derived cGAS_KO- or cGAS_overexpressing cell lines were infected for 10 days with HBV. Total DNA from indicated HBV infected cells was extracted and HBV DNA were detected by Southern blot. Two different DNA ladders (L1 & L2) were used. XhoI digestion of DNA extracted from HBV-infected HepG2-NTCP-Cas9 cells was used as a control and resulted in a single 3.2 kb band (dsl HBV DNA). Mitochondrial DNA (mt DNA) was detected as a loading control. One experiment is shown.
HBV infection induces repression of cGAS and its effector gene expression in cell culture and in liver chimeric mice
As several reports have suggested that HBV proteins can inhibit IFN-signaling pathways (16), we next investigated whether HBV infection interferes with the expression of cGAS-related gene by quantifying MB21D1/cGAS mRNA and protein expression (Figure 7A-B). Interestingly, cGAS protein expression (Figure 7B) as well as the expression of MB21D1, TMEM173, and TBK1 mRNA (Figure 7C) were significantly inhibited in HBV-infected cells. To confirm this observation in vivo, we then investigated the expression of human MB21D1 expression in HBV-infected human liver chimeric mice. MB21D1 was expressed at low but detectable levels (Figure 7D). As shown in Figure 7E, MB21D1 expression was significantly (p = 0.013) downregulated in HBV-infected mice compared to non-infected control mice, confirming our results in the cell culture model. Importantly, MB21D1 expression levels did not correlate with HBV genotype (Table 1). An absent correlation of human serum albumin with either MB21D1 expression (Table 1, Figure 7E) or status of HBV infection (Table 1, t-test HBV versus Ctrl: p = 0.26) largely excludes that the observed differences in cGAS expression are due to different human hepatocyte repopulation levels or due to a decrease of human hepatocyte cell viability in individual animals. To investigate whether HBV modulates cGAS effector function, we analyzed virus-induced changes on cGAS effector gene expression using gene expression profiling in three control mice and the three HBV-infected mice exhibiting the lowest levels of MB21D1 expression (Table 1). As shown in Figure 7F, HBV infection resulted in a significant (FDR = 0.047) down-regulation of the expression of cGAS effector genes in human hepatocytes in chimeric mice. The data showed that HBV represses expression of cGAS and its effector genes in vivo.
Figure 7. HBV infection suppresses the expression of the cGAS-related genes cell culture and humanized liver chimeric mice in vivo.
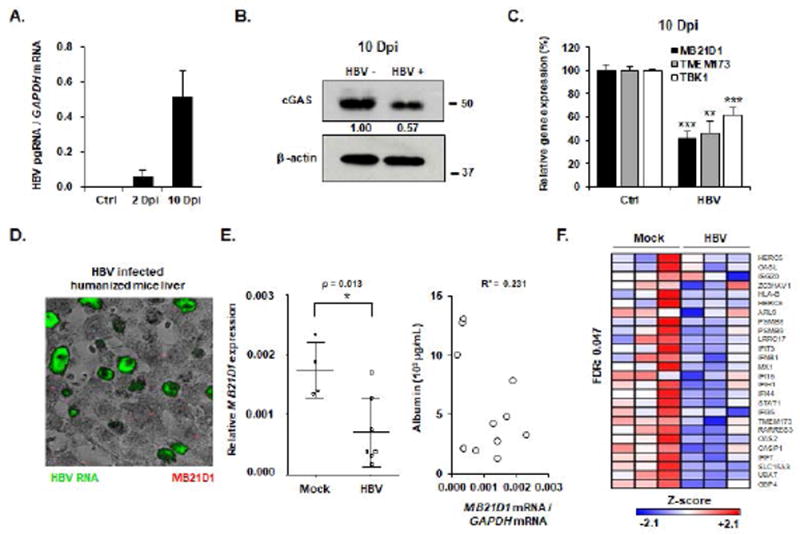
(A-C) HepG2-NTCP cells were infected with HBV for 10 days. HBV infection was assessed by quantification of HBV pgRNA by qRT-PCR. Results are expressed as means ± SD from three experiments performed in triplicate. cGAS protein expression was assessed 10 days after infection (B, one experiment is shown). Gene expression relative to non-infected control cells of MB21D1, TMEM173 and TBK1 were assessed by qRT-PCR at day 10 after infection (C). Results are expressed as means ± SEM from three independent experiments performed in triplicate. (D-E) MB21D1- and IAR gene set expression is impaired in HBV-infected mice. uPA-SCID mice were infected with HBV for 16 weeks. Mice were then sacrificed and HBV infection was assessed by HBV RNA specific in situ hybridization (D) and quantification of HBV viral load in the serum (Table 1). Human MB21D1 expression was detected in human hepatocytes by FISH from one HBV-infected mouse (D) and by qRT-PCR from 7 HBV-infected mice and 4 control mice (E, left panel). Results are expressed as the ratio MB21D1 mRNA / GAPDH mRNA. All individual mice are presented as well as means ± SD for each group (Mock- and HBV-infected mice). The level of MB21D1 expression was independent of the viability of engrafted human hepatocytes as indicated by an absent correlation between MB21D1 expression and the human serum albumin expression in humanized mice (R² = 0.231, E, right panel). (F) The IAR gene set was analyzed using the nCounter NanoString in mice 6472, 6251, and 6254 (Mock-infected mice, Table 1) and 4766, 4771, and 4847 (HBV-infected mice, Table 1). A significant downregulation (FDR = 0.047) of the gene set was observed in HBV-infected mice compared to control mice. Individual Z-scores for the genes significantly modulated between the two groups according to GSEA analysis are presented. Negative Z-score (blue) and positive Z-score (red) correspond to repression and induction of the indicated genes, respectively. Dpi: days post infection.
Table 1. cGAS expression in HBV-infected human liver chimeric mice.
Levels of human albumin-, HBV viral load-, and MB21D1 levels in liver tissue from HBV-infected and control mice are shown. MB21D1 expression is normalized to GAPDH expression.
| Mouse | Albumin (μg/ml) | HBV (IU/ml) | MB21D1 mRNA | ||
|---|---|---|---|---|---|
| Ctrl | 6410 | 1280 | - | 1.40E-03 | |
| 6472 | 2720 | - | 1.40E-03 | ||
| 6251 | 3240 | - | 2.30E-03 | ||
| 6254 | 7870 | - | 1.90E-03 | ||
| HBV | Gt E | 4770 | 4200 | 2.9E+07 | 1.30E-03 |
| 4773 | 4800 | 1.5E+08 | 1.70E-03 | ||
| 4766 | 12760 | 3.6E+06 | 3.20E-04 | ||
| 4771 | 13120 | 6.2E+05 | 3.70E-04 | ||
| Gt D | 4846 | 2127 | 5.5E+05 | 3.80E-04 | |
| 4847 | 10045 | 1.6E+08 | 1.70E-04 | ||
| 4848 | 1992 | 6.7E+06 | 7.50E-04 | ||
The HBV genotype (Gt) is indicated. Bold: mice used for multiplexed gene profiling
DISCUSSION
The interaction between HBV and the innate immune system is a complex process still remaining elusive and controversial (15). Collectively, our data demonstrate that in human hepatocytes (i) naked HBV genomic rcDNA is sensed in a cGAS-dependent manner whereas the packaged HBV genome appears not to be recognized during viral infection; (ii) cGAS-STING pathway exhibits antiviral activity against HBV infection including reduction of viral cccDNA levels; (iii) HBV infection suppresses both cGAS expression and function in cell culture and humanized liver chimeric mice.
The detection of HBV DNA by the cellular sensors within infected cells is still poorly understood and remains controversial. In vitro and in vivo data strongly suggest that HBV behaves like a stealth virus unable to trigger any innate immune response (8, 9, 11). Other studies have suggested that HBV-derived dsDNA fragments (25) and viral nucleocapsid destabilization and disassembly (38, 39) could induce innate immune responses. Our results demonstrate, that in human hepatocytes - the natural target cell of HBV infection - the exposure of the naked HBV genome leads to the activation of innate antiviral immune responses. In contrast, sensing is largely absent during HBV infection, most likely due to packaging into the viral capsid. These results extend a previous observation in hepatoma cell lines transfected with replication-competent HBV DNA that the HBV genome itself can be recognized by the classical sensors (25).
Interestingly, the capsid of HIV-1 also prevents the sensing of HIV cDNA by cGAS following reverse-transcription up to integration, whereas HIV-2 capsid may unmask the cDNA leading to a stronger sensing by cGAS and a lower pathogenicity of the strain (40).
Another explanation of this absence of sensing would be the lack a functional STING protein in hepatocyte, as it has been recently reported (41). In our study, rcDNA and dsDNA were sensed in a cGAS-dependent manner and were able to activate the cGAS-mediated antiviral response in HepG2-NTCP cells (Figure 2). Moreover, we detected STING at the protein level in accordance with a recent study (42) and specific silencing of TMEM173 (STING) expression was associated with a significant increase in HBV infection (Figure 6). Consequently, it is likely that STING is functionally active in HepG2 cells. The observed HBV DNA sensing in PHH (Figure 5) suggests that the foreign DNA detection pathways are active in PHH as well. This observed innate immune response in spite of a weak STING expression may suggest a STING-independent activity of cGAS as it has been recently reported (43), including in hepatocytes (44). To understand the impact of the cGAS and STING expression on innate immune response to HBV infection, it would be of further interest to analyze the HBV-induced modulation of gene expression in Kupffer cells following phagocytosis, as they exhibit higher STING- and cGAS levels compared to hepatocytes (41, 45) and respond to HBV infection (11). In the same vein, cGAMP has been shown to be packaged in viral particles (46). It would be of interest to determine whether HBV particles can incorporate cGAMP during viral assembly and to test their ability to stimulate other cell types through this indirect pathway.
Moreover, our results show conclusive evidence that cGAS basal expression has antiviral activity against HBV infection including reduction of viral cccDNA. This finding extends a previous studies showing that cGAS exhibited an antiviral activity against a broad range of RNA and DNA viruses (20) and that the cGAS/STING pathway can impair HBV replication and assembly in transfection studies (24, 25). Schoggins and colleagues have proposed that the expression of cGAS may be responsible for the establishment of a basal antiviral level in the cells through its activation by an unknown ligand. cGAS-depleted cells may then be more susceptible to viral infections through the downregulation of the basal level of innate antiviral genes (20).
Given its antiviral function, cGAS is a target of choice for viruses in order to evade immune responses. It has been reported that the Kaposi’s sarcoma-associated herpesvirus negatively regulated cGAS-dependent signaling pathway (47, 48). In the same vein, HBV viral proteins have been shown to interfere with the JAK-STAT signaling pathway (16). Our data suggest that HBV can repress the expression of the cGAS and its related genes, such as MB21D1, TMEM17 and TBK1. More interestingly, MB21D1 expression was downregulated in the liver of HBV-infected mice, validating the relevance of these findings in vivo. It still needs to be determined whether HBV can directly target cGAS and cGAS-related factors for an active inhibition of this signaling pathway. A recent study elegantly demonstrated an active inhibition of cGAS pathway by Dengue virus through NS2B protein (49). On the other hand, MB21D1 (as a classical member of the ISGs (20, 36)) downregulation may be the consequence of the global inhibition of the canonical IFN pathways by HBV as suggested by some investigators (16, 50), but not by others (11, 13, 14). Given the antiviral activity of the cGAS-signaling pathway against HBV including reduction of HBV cccDNA (Figure 5, (24, 25)) the virus-mediated restriction of MB21D1 expression may play an additional role in HBV immune evasion.
Supplementary Material
Acknowledgments
We thank D. Garcin (University of Geneva, Switzerland) for providing the SeV inoculum; D. Calabrese, S. Wieland and M. Heim (University of Basel, Switzerland) for the FISH analyses and helpful discussions; F. Zhang (Broad Institute of MIT and Harvard, Cambridge, MA, USA) for lentiCas9-Blast and lentiGuide-Puro plasmids, C. Thumann, N. Brignon, and M. Oudot (Inserm U1110) for excellent technical support, and J.M. Rousée from Laboratoire Schuh-groupement Bio 67 (Strasbourg) for the quantification of HBV loads in sera of humanized mice.
Financial support: This work was supported by the European Union (ERC-AdG-2014-671231-HEPCIR, EU H2020-667273-HEPCAR, EU-InfectEra HepBccc), the National Institute of Health (NIAID 1R03AI131066-01A1, NCI R21 CA209940 and NIAID U19AI123862), the Agence Nationale de Recherches sur le Sida et les Hepatites Virales (ANRS, 2015/1099), the Fondation ARC pour la Recherche sur le Cancer (TheraHCC IHUARC IHU201301187), GACR 17-15422S, and a PhD fellowship of the Région Alsace, France. The work has been published under the framework of the LABEX ANR-10-LABX-0028_HEPSYS and benefits from funding from the state managed by the French National Research Agency as part of the Investments for the future program. E.R.V. was supported by an ANRS fellowship (ECTZ50121).
List of Abbreviations
- HBV
hepatitis B virus
- cGAS
cyclic GMP-AMP synthase
- HSPG
heparan sulfate proteoglycan
- NTCP
Na+/taurocholate cotransporting polypeptide
- rcDNA
relaxed circular DNA
- cccDNA
covalently closed circular DNA
- pgRNA
pregenomic RNA
- PRR
pattern recognition receptors
- cGAMP
cyclic GMP-AMP
- IFN
interferon
- STING
stimulator of IFN genes
- ISG
IFN-stimulated gene
- PHH
primary human hepatocyte
- PEG
polyethylene glycol
- siRNA
small interfering RNA
- HIV
human immunodeficiency virus
- VSV-G
vesicular stomatitis virus glycoprotein
- sgRNA
single-guide RNA
- PMM
primary hepatocyte maintenance medium
- dslDNA
double stranded linear DNA
- dsDNA
double stranded DNA
- GSEA
gene set enrichment analysis
- FDR
false discovery rate
- FISH
fluorescence in situ hybridization
- KO
knock-out
- SeV
Sendai virus
- IAR
innate antiviral response gene set
- Dpi
days post infection
Footnotes
Author contribution. TFB initiated the study. TFB, ERV and CS designed and supervised research. ERV, SAY, LH, CB, VTL, SD, LM, JL, TC and DD performed the experiments. ERV, SAY, LH, CB, VTL, JL, DD, MBZ, NP, CS and TFB analyzed the data. PP provided liver resections for PHH isolation. NM, IH and NP made substantive intellectual contributions. ERV, SAY, CS and TFB wrote the paper.
Conflict of interest: The authors declare no competing financial interests.
References
- 1.Trepo C, Chan HL, Lok A. Hepatitis B virus infection. Lancet. 2014;384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 2.Zeisel MB, Lucifora J, Mason WS, Sureau C, Beck J, Levrero M, Kann M, et al. Towards an HBV cure: state-of-the-art and unresolved questions-report of the ANRS workshop on HBV cure. Gut. 2015;64:1314–1326. doi: 10.1136/gutjnl-2014-308943. [DOI] [PubMed] [Google Scholar]
- 3.Levrero M, Testoni B, Zoulim F. HBV cure: why, how, when? Curr Opin Virol. 2016;18:135–143. doi: 10.1016/j.coviro.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Verrier ER, Colpitts CC, Sureau C, Baumert TF. Hepatitis B virus receptors and molecular drug targets. Hepatol Int. 2016;10:567–573. doi: 10.1007/s12072-016-9718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nassal M. Hepatitis B virus cccDNA - viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 6.Lu HL, Liao F. Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J Immunol. 2013;191:3264–3276. doi: 10.4049/jimmunol.1300512. [DOI] [PubMed] [Google Scholar]
- 7.Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, Watanabe T, et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity. 2015;42:123–132. doi: 10.1016/j.immuni.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 8.Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A. 2004;101:6669–6674. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol. 2005;79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fletcher SP, Chin DJ, Ji Y, Iniguez AL, Taillon B, Swinney DC, Ravindran P, et al. Transcriptomic analysis of the woodchuck model of chronic hepatitis B. Hepatology. 2012;56:820–830. doi: 10.1002/hep.25730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng X, Xia Y, Serti E, Block PD, Chung M, Chayama K, Rehermann B, et al. Hepatitis B virus evades innate immunity of hepatocytes but activates macrophages during infection. Hepatology. 2017;66:1779–1793. doi: 10.1002/hep.29348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luangsay S, Gruffaz M, Isorce N, Testoni B, Michelet M, Faure-Dupuy S, Maadadi S, et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J Hepatol. 2015;63:1314–1322. doi: 10.1016/j.jhep.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 13.Suslov A, Boldanova T, Wang X, Wieland S, Heim MH. Hepatitis B Virus Does Not Interfere with Innate Immune Responses in the Human Liver. Gastroenterology. 2018 doi: 10.1053/j.gastro.2018.01.034. In press. [DOI] [PubMed] [Google Scholar]
- 14.Mutz P, Metz P, Lempp FA, Bender S, Qu B, Schoneweis K, Seitz S, et al. HBV Bypasses the Innate Immune Response and Does not Protect HCV From Antiviral Activity of Interferon. Gastroenterology. 2018 doi: 10.1053/j.gastro.2018.01.044. In press. [DOI] [PubMed] [Google Scholar]
- 15.Ferrari C. HBV and the immune response. Liver Int. 2015;35(Suppl 1):121–128. doi: 10.1111/liv.12749. [DOI] [PubMed] [Google Scholar]
- 16.Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut. 2012;61:1754–1764. doi: 10.1136/gutjnl-2011-301073. [DOI] [PubMed] [Google Scholar]
- 17.Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol. 2016;14:360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao TS, Fitzgerald KA. The cGAS-STING pathway for DNA sensing. Mol Cell. 2013;51:135–139. doi: 10.1016/j.molcel.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Shi H, Wu J, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He J, Hao R, Liu D, Liu X, Wu S, Guo S, Wang Y, et al. Inhibition of hepatitis B virus replication by activation of the cGAS-STING pathway. J Gen Virol. 2016;97:3368–3378. doi: 10.1099/jgv.0.000647. [DOI] [PubMed] [Google Scholar]
- 25.Dansako H, Ueda Y, Okumura N, Satoh S, Sugiyama M, Mizokami M, Ikeda M, et al. The cyclic GMP-AMP synthetase-STING signaling pathway is required for both the innate immune response against HBV and the suppression of HBV assembly. FEBS J. 2016;283:144–156. doi: 10.1111/febs.13563. [DOI] [PubMed] [Google Scholar]
- 26.Verrier ER, Colpitts CC, Schuster C, Zeisel MB, Baumert TF. Cell Culture Models for the Investigation of Hepatitis B and D Virus Infection. Viruses. 2016;8 doi: 10.3390/v8090261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Israelow B, Narbus CM, Sourisseau M, Evans MJ. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology. 2014;60:1170–1179. doi: 10.1002/hep.27227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mailly L, Xiao F, Lupberger J, Wilson GK, Aubert P, Duong FH, Calabrese D, et al. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat Biotechnol. 2015;33:549–554. doi: 10.1038/nbt.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verrier ER, Colpitts CC, Bach C, Heydmann L, Weiss A, Renaud M, Durand SC, et al. A targeted functional RNAi screen uncovers Glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology. 2016;63:35–48. doi: 10.1002/hep.28013. [DOI] [PubMed] [Google Scholar]
- 31.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucifora J, Salvetti A, Marniquet X, Mailly L, Testoni B, Fusil F, Inchauspe A, et al. Detection of the hepatitis B virus (HBV) covalently-closed-circular DNA (cccDNA) in mice transduced with a recombinant AAV-HBV vector. Antiviral Res. 2017;145:14–19. doi: 10.1016/j.antiviral.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 33.Gao W, Hu J. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol. 2007;81:6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wieland S, Makowska Z, Campana B, Calabrese D, Dill MT, Chung J, Chisari FV, et al. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology. 2014;59:2121–2130. doi: 10.1002/hep.26770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blondot ML, Bruss V, Kann M. Intracellular transport and egress of hepatitis B virus. J Hepatol. 2016;64:S49–59. doi: 10.1016/j.jhep.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Cui X, Clark DN, Liu K, Xu XD, Guo JT, Hu J. Viral DNA-Dependent Induction of Innate Immune Response to Hepatitis B Virus in Immortalized Mouse Hepatocytes. J Virol. 2015;90:486–496. doi: 10.1128/JVI.01263-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui X, Luckenbaugh L, Bruss V, Hu J. Alteration of Mature Nucleocapsid and Enhancement of Covalently Closed Circular DNA Formation by Hepatitis B Virus Core Mutants Defective in Complete-Virion Formation. J Virol. 2015;89:10064–10072. doi: 10.1128/JVI.01481-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, El Marjou A, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 41.Thomsen MK, Nandakumar R, Stadler D, Malo A, Valls RM, Wang F, Reinert LS, et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology. 2016;64:746–759. doi: 10.1002/hep.28685. [DOI] [PubMed] [Google Scholar]
- 42.Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I, Park H, et al. Lipotoxicity induces hepatic protein inclusions through TBK1-mediated p62/SQSTM1 phosphorylation. Hepatology. 2017 doi: 10.1002/hep.29742. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114:E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lei Z, Deng M, Yi Z, Sun Q, Shapiro RA, Xu H, Li T, et al. cGAS-mediated autophagy protects the liver from ischemia/reperfusion injury independent of STING. Am J Physiol Gastrointest Liver Physiol. 2018 doi: 10.1152/ajpgi.00326.2017. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leong CR, Oshiumi H, Okamoto M, Azuma M, Takaki H, Matsumoto M, Chayama K, et al. A MAVS/TICAM-1-independent interferon-inducing pathway contributes to regulation of hepatitis B virus replication in the mouse hydrodynamic injection model. J Innate Immun. 2015;7:47–58. doi: 10.1159/000365113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gentili M, Kowal J, Tkach M, Satoh T, Lahaye X, Conrad C, Boyron M, et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015;349:1232–1236. doi: 10.1126/science.aab3628. [DOI] [PubMed] [Google Scholar]
- 47.Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe. 2015;18:333–344. doi: 10.1016/j.chom.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Avey D, Fu B, Wu JJ, Ma S, Liu X, Zhu F. Kaposi’s Sarcoma-Associated Herpesvirus Inhibitor of cGAS (KicGAS), Encoded by ORF52, Is an Abundant Tegument Protein and Is Required for Production of Infectious Progeny Viruses. J Virol. 2016;90:5329–5342. doi: 10.1128/JVI.02675-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, Fredericks AC, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol. 2017;2:17037. doi: 10.1038/nmicrobiol.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ortega-Prieto AM, Skelton JK, Wai SN, Large E, Lussignol M, Vizcay-Barrena G, Hughes D, et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat Commun. 2018;9:682. doi: 10.1038/s41467-018-02969-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.