

Abstract
Ketamine is an N-methyl D-aspartate receptor antagonist used off label to facilitate dissociative anesthesia in children undergoing invasive procedures. Available for both intravenous and intramuscular administration, ketamine is commonly used when vascular access is limited. Pharmacokinetic (PK) data in children are sparse, and the bioavailability of intramuscular ketamine in children is unknown. We performed 2 prospective PK studies of ketamine in children receiving either intramuscular or intravenous ketamine, and combined the data to develop a pediatric population PK model using nonlinear mixed effects methods. We applied our model by performing dosing simulations targeting plasma concentrations previously associated with analgesia (>100 ng/mL) and anesthesia awakening (750 ng/mL). A total of 113 children (50 intramuscular and 63 intravenous ketamine) with a median age of 3.3 years (range: 0.02 to 17.6 years), and median weight of 14 kg (2.4 to 176.1) contributed 275 plasma samples (149 after intramuscular, 126 after intravenous ketamine). A 2-compartment model with first-order absorption following intramuscular administration and first-order elimination described the data best. Allometrically scaled weight was included in the base model for central and peripheral volume of distribution (exponent 1), and for clearance and intercompartmental clearance (exponent 0.75). Model estimated bioavailability of intramuscular ketamine was 41%. Dosing simulations suggest that doses of 2 mg/kg intravenously and 8mg/kg or 6 mg/kg intramuscularly depending on age provide adequate sedation (plasma ketamine concentrations >750 ng/mL) for procedures lasting up to 20 minutes.
Keywords: Ketamine, Children, Intramuscular, Intravenous, Bioavailability, Pharmacokinetics
Introduction
Ketamine is an N-methyl D-aspartate receptor antagonist increasingly used ‘off label’ to facilitate a dissociative anesthesia in children undergoing invasive procedures.1,2 Commercial preparations of ketamine are a racemic mixture of R- and S-enantiomers approved for use in adults as the sole anesthetic agent for diagnostic and surgical procedures and for the induction of anesthesia.3 Because administration of ketamine is associated with preservation of hemodynamics and laryngeal reflexes, it is frequently used in populations at risk for respiratory and hemodynamic compromise, such as children.2,4–6 The availability of a formulation for intramuscular (IM) administration makes it a common choice when vascular access is limited, such as in the emergency department.7 Label-recommended dosing for intravenous (IV) and IM ketamine to facilitate surgical anesthesia in adults ranges from 1 to 4.5 mg/kg and 6 to 13 mg/kg, respectively.3 Ketamine is not approved for use in children <18 years of age, but an observational study in 2007 found that when ketamine was administered to 60 children 1.5–14 years of age in the emergency department, 1.5 mg/kg IV dosing provided adequate procedural sedation for up to 10 minutes.8
In adults, ketamine exhibits low protein binding to both alpha-1 acid glycoprotein and albumin (10–30%) with high lipid solubility and extensive distribution.9 Central compartment volume (Vc) is 70 liters, and the steady state volume of distribution (Vss) is around 200 liters.9 Ketamine elimination clearance (CL) is 12–20 mL/min/kg in adults, and it is hepatic blood flow limited.9 After IV administration, the distribution of ketamine undergoes alpha and beta phases. During the alpha phase, which has a half-life of about 10–15 minutes, the drug has initial anesthetic effect.10 Ketamine then undergoes redistribution from the central compartment and hepatic metabolism by CYP3A isoenzymes, CYP2B6, and CYP2C9.8 Approximately 80% of the dose is metabolized to active norketamine, which appears in the blood 2–3 minutes after an IV ketamine bolus, and reaches peak plasma concentrations at approximately 30 minutes after administration.9 Norketamine persists in the plasma for more than 5 hours after administration, and the half-life of the beta phase of ketamine is 2.5 hours.9,10
Existing evaluations of ketamine pharmacokinetics (PK) in children are limited to smaller populations presenting to the emergency department, or children with burn injuries or cardiac disease.1,11 Ketamine PK is most commonly described by 2-compartment models with linear elimination and CL increasing with age from 26 L/hr/70 kg in infants <3 months up to 90 L/h/70 kg, in older children.8,12 Ketamine Vss decreases with age, ranging from 242 L/70 kg in infants <3 months to 53 L/70 kg by adulthood. Subsequent analyses of pediatric data have contributed valuable information on optimal sampling times, oral bioavailability (45%), and absorption half-time following oral administration (59 minutes, 95% confidence interval 29.4–109.2 minutes).1,13 Ketamine was also found to have a 36% bioavailability following intranasal administration in children ≥10 kg.14 Importantly, despite the frequent use of IM ketamine, its IM bioavailabilty has only been characterized in 6 healthy adult volunteers to date.15
In this population PK analysis, we leveraged a combined dataset from 2 prospective PK studies of ketamine in children, one with IM and the other with IV administration. In this analysis, we characterize the PK of ketamine in children including its model estimated IM bioavailability.
Materials and Methods
Patient population
PK samples used to develop the model described in this report were collected through the Pediatric Trials Network’s (PTN’s) Pharmacokinetics of Understudied Drugs Administered to Children per Standard of Care trial (clinicaltrials.gov no. NCT01431326; protocol: NICHD-2011-POP01) and the Ketamine Pharmacokinetic Study in Healthy Children Aged 2 to 5 Years Old (KPSHC2011) study (Health Sciences Authority, Singapore: HPRG/CTB 78:10/11–086). Written informed consent (and assent when applicable) was obtained from the legal guardians of all study participants in both studies. The PTN POP01 study protocol was reviewed and approved by the institutional review boards of each participating institution. The KPSHC2011 protocol was reviewed and approved by the KK Women’s and Children’s Hospital institutional review board.
The PTN POP01 trial is a multicenter, prospective, PK and safety study of understudied drugs administered to children (<21 years old) per standard of care. Children who received IV ketamine per standard of care as administered by their treating caregiver were eligible for enrollment. Exclusion criteria included failure to obtain consent or assent, or a known pregnancy as determined by interview or testing. PK samples were collected optimally with standard-of-care lab collections or at different times from standard-of-care collections if allowed per consent. Because this was a standard-of-care study, dosing and PK sample collection times varied between children. Standard-of-care laboratory assessments (e.g., basic metabolic panel) were recorded if collected within 72 hours of a study dose of the drug. Gestational age was documented in infants with a postnatal age (PNA) of less than 120 days. Children were enrolled in the study for up to 90 days.
The KPSHC2011 study is a single-center, prospective, PK, safety, and preliminary efficacy trial conducted at KK Women’s and Children’s Hospital, Singapore. Children up to 5 years old requiring IM ketamine for procedural sedation and analgesia in the emergency department were eligible for enrollment. Exclusion criteria included failure to obtain consent or assent and a history of prior anaphylaxis to ketamine. Children received IM ketamine as per standard of care, and 3 timed venous PK samples were taken following administration.
Drug dosing and sample collection
Dosing information was collected for up to 8 doses prior to the sampling dose (last dose before first biological sample collection) in the POP01 study, and for all study doses in the KPSHC2011 study. Dosing in both studies was per standard of care, but limited to the IM administration route in the KPSHC2011 study. All children enrolled in the KPSHC2011 study received a single dose of IM ketamine, while all children enrolled in the POP01 study received 1 or more bolus doses or continuous infusions of IV ketamine. For the opportunistic POP01 study, the timing of blood sample collection was dependent on standard-of-care laboratory assessments but suggested sampling times were provided (Supplemental Table S1). For the KPSHC2011study, blood samples for PK analysis were collected at the following time windows after drug administration: 10 minutes, 30–60 minutes, and 120 minutes.
Analytical methods
Blood was collected (0.2 mL in children <1 year of age and 2 mL in children ≥1 year of age) in an EDTA-K2 Microtainer and was processed into plasma immediately prior to freezing at the study sites. PK samples from both studies were sent to a PTN-contracted commercial laboratory (Alturas Analytics, Inc., Moscow, ID, USA) for storage and analysis. Ketamine concentrations were quantified using validated liquid chromatography-tandem spectrometry assays.16 The chromatography system and mass spectrometer used for sample analysis were the Sciex API4000 triple Quadrupole mass spectrometer with a Synergi Polar-RP 80A 4u HPL Column from Phenomenex (Torrance, CA, USA) used to separate ketamine from the internal standard. The validation range for the assay was 10–2000 ng/L. The lower limit of quantification (LLOQ) was 10 ng/mL, and the validated dilution range was 50×. Both R- and S-ketamine were measured and the assay was not designed to differentiate between the 2 enantiomers. Accuracy and precision assessed were within the U.S. Food and Drug Administration’s bioanalytical assay validation criteria (e.g., ±15%).
Statistical analysis
Using the value at the time of first recorded dose, the median and range were calculated and presented for demographic and dosing variables. Counts and percentages are calculated and presented for categorical variables. Distribution of study variables are compared using Wilcoxon rank sum, chi square, or Fisher’s exact tests where appropriate. With the exception of the PK modeling, all statistical analyses were performed using Stata (version 14.2, College Station, TX, USA).
Population PK model development
Ketamine plasma PK data collected after IV or IM administration (Supplemental Figure S1) were analyzed with a nonlinear mixed effects modeling approach using the software NONMEM (version 7.2, Icon Development Solutions, Ellicott City, MD, USA). The first-order conditional estimation method with eta-epsilon interaction was used for all model runs. Run management was performed using Pirana (version 2.8.2).17 Bootstrap analyses were performed with Perl-speaks-NONMEM (version 3.7.6).18 Data manipulation and visualization were performed using the packages Xpose in the software R (version 3.0.3, R Foundation for Statistical Computing, Vienna, Austria), RStudio (version 0.97.551, RStudio, Boston, MA, USA), and Stata (version 14.2, College Station, TX, USA).19–21
Using the combined datasets of both studies, 1- and 2-compartment PK models with first-order absorption for the IM administration were explored with assumed linear PK.22 Bioavailability (F) of IM ketamine was estimated. Inter-individual variability (IIV) was assessed for PK model parameters using an exponential relationship (equation 1).
| (1) |
where PARij denotes the estimate of parameter j in the ith individual; θPop,j is the population value for parameter j; and ηij denotes the deviation from the average population value for parameter j in the ith individual with mean zero and variance ω2. The correlation between random effect parameters was calculated and the inclusion of a block covariance matrix in the model was evaluated but not retained in the final model.
Proportional, additive, and combined (additive plus proportional) residual error models were explored. Separate residual errors were estimated for the POP01 and KPSHC2011 study data.
Covariate analysis
Actual body weight (WT) was assumed to be a significant covariate for CL and V, and was included in the base model. The relationship between WT and PK parameters was characterized using allometric relationship for CL and V parameters (scaled to a 70-kg standardized WT), respectively. Allometric coefficients were estimated, but fixed exponents (0.75 for CL and 1 for V) were retained in the final model (equations 2 and 3).
| (2) |
| (3) |
where CLstd and Vstd represent population estimates of CL and V in a 70-kg adult and WTi denotes WT for the ith subject.
Other covariates were tested for model inclusion (Supplemental Table S2). Determination of which covariates to test for model inclusion was based on physiological relevance and by visual inspection of scatter and box plots (continuous and categorical variables, respectively) of the individual deviations from the population-typical value PK parameters (ETAs) against covariates. The following covariates were explored: extracorporeal membrane oxygenation (ECMO) support at the time of PK sampling, race, ethnicity, obese status (defined as body mass index ≥95th percentile per Centers for Disease Control reference values for children >2 years of age), postmenstrual age (PMA), PNA, and protocol under which subject was enrolled (binary variable: POP01 or KPSHC2011).23
The relationship between age and CL was characterized using linear, power, and sigmoidal Emax maturation functions (equations 4–6). Maturation function parameters were both estimated and fixed to previously published values.24–26 As a measure of age, PNA and PMA were explored.
| (4) |
| (5) |
| (6) |
For dichotomous categorical covariates (ECMO, obese status), a power relationship was used (equation 7).
| (7) |
For categorical variables with n distinct values (race, ethnicity), additive coding with n-1 indicator variables (1=yes, 0=no) was used (equation 8).
| (8) |
For all continuous covariates, missing data values were imputed by carrying forward or backward the most recent available value for each subject.
A forward inclusion (p<0.05 and change in objective function value [OFV] >3.8) and backward elimination (p<0.01 and change in OFV >6.6) approach was used to evaluate statistical significance in the covariate analysis.
Population PK model evaluation
Standard model diagnostic methods were used and included successful minimization, diagnostic plots, plausibility and precision of parameter estimates, as well as OFV and shrinkage values. Generated diagnostic plots included individual predictions and population predictions vs. observations, and conditional weighted residuals (CWRES) vs. population predictions and time after first dose.
Parameter precision for the final population PK model was evaluated using non-parametric bootstrapping (1000 replicates) to generate the 95% confidence intervals for parameter estimates. Standardized visual predictive checks (SVPCs) were performed whereby the base and final models were used to generate 1000 Monte Carlo simulation replicates per time point of ketamine concentration measurements, and simulated results were compared with those observed in the study.27 An SVPC was preferred to the commonly used visual predictive check as the primary graphical representation because of the opportunistic design of the study, resulting in variable dosage and time of PK sampling across subjects.27 However, a more commonly used typical visual predictive check was also performed with 10 bins selected to equally distribute observed concentrations over time after the last dose (Supplemental Figure S2). The percentile of each subject’s observation in the marginal distribution of model-simulated endpoints (Pi,j) as a function of time and dosing was estimated, using the subject’s individual time and dosing. Observations outside the 95% prediction interval were then estimated (equation 9).
| (9) |
where Pij is the percentile of the jth observation for the ith subject; δij,n =1 if Cobs,ij > C′obs,ij,n (otherwise, δij,n =0); Cobs,ij is the jth observed concentration for ith individual; and C′obs,ij,n is the nth simulated concentration corresponding to Cobs,ij.
The dosing and covariate values used to generate the simulations in the SVPC were the same as those used in the study population.
In addition to the SVPC, a numerical predictive check (NPC) using 1000 simulations was also performed for the final model. We calculated the percentages of outliers for each selected prediction interval (0%, 20%, 40%, 50%, 60%, 80%, 90%, 95%) in each simulation to obtain a confidence interval for the percentages of outliers, and compared the observed percentages to the empirical confidence interval.
Dosing simulations
Plasma concentrations of ketamine were simulated in virtual subjects at 25 time points after dose administration: every minute between 1–20 minutes, and at 25, 30, 40, 50, and 60 minutes. We chose 5 virtual subjects whose WTs were approximately equal to the median WT of male children in the middle of 5 age brackets covering our study population based on Centers for Disease Control and Prevention growth charts: children <6 months: 6 kg; children 6 months to <2 years: 11 kg; children 2 to <6 years: 17 kg; children 6 to <12 years: 28 kg; children ≥12 years: 56 kg. IV bolus dosing simulations were performed for scenarios on and off ECMO. Single IM dose simulations were not performed on ECMO, as this route of drug administration is typically contraindicated in this setting due to the high risk of bleeding. Fixed effects and IIV parameters were fixed for inclusion in the simulation. A total of 1000 simulations in each virtual subject were performed per dosing scenario, and the 10th percentile of simulations presented in concentration time curves plotted over previously published plasma ketamine concentration cutoffs: 100 ng/mL corresponding to analgesic effect, 750 ng/mL corresponding to awakening from anesthesia, 1000 ng/mL corresponding to arousal with verbal stimulus, and 1500 ng/mL corresponding to arousal with painful stimulus.28–31 The ketamine plasma concentration associated with unwanted toxicities has not been reported; thus, an upper limit of concentrations was not targeted in the simulations.
Safety analysis
In the POP01 study, only adverse events associated with study procedures (phlebotomy) and suspected unexpected serious adverse reactions were collected. In the KPSHC2011 study, the following adverse events of special interest were collected: (1) abnormal pulse rate, (2) low pulse oximetry, (3) need for airway interventions, and (4) need for any other interventions presumed directly related to ketamine administration.
Results
Patient characteristics
A total of 113 children, 63 from the POP01 study and 50 from the KPSHC2011 study, were included in the analysis (Table 1). Median WT and PNA did not differ between children enrolled in the 2 studies: 11 kg (range: 2 to 176) in POP01 vs. 15 kg (10 to 34) in KPSHC2011, P=.08; and 1.6 years (0.02 to 17.6) in POP01 vs. 3.7 years (1.8 to 5.9) in KPSHC2011, P=.06. However, 17 children (27%) in the POP01 study were obese, compared to only 2 children (4%) in the KPSHC2011 study (P<.001). A total of 36 children (32%) were under the age of 2 years at the time of PK sampling, and 14 children, all from the POP01 study, were supported with ECMO at the time of PK sampling. There was no significant difference in the median (range) PNA or WT between children supported with ECMO compared to those not supported with ECMO: 2.0 years (0.02 to 17) vs. 3.4 years (0.01 to 18), P=0.40; 10.7 kg (2.7 to 85.6) vs. 14.1 kg (2.4 to 176), P=0.37.
Table 1.
Clinical characteristics.
| KPSHC2011 N=50 |
POP01 N=63 |
All Subjects N=113 |
|
|---|---|---|---|
| Race | |||
| White | 2 | 36 | 38 |
| Black | 0 | 17 | 17 |
| Unknown/not reported | 0 | 5 | 5 |
| Asian | 48 | 5 | 53 |
| Male sex | 19 | 29 | 48 |
| Age, years | 3.7 (1.8–5.9) | 1.6 (0.02–17.6) | 3.3 (0.02–17.6) |
| Weight, kg | 14.9 (10.2–33.8) | 29.8 (2.4–176.1) | 14 (2.39–176.1) |
| Height, cm | 101 (82–130) | 80 (36–180) | 98 (36–180) |
| Body mass index (kg/m2) | 15 (9–20) | 17 (10–58) | 16 (9–58) |
| Obesity | |||
| Not obese | 46 | 12 | 58 |
| Obese (BMI ≥ 95th percentile) | 1 | 9 | 10 |
| Morbidly obese (BMI ≥ 99th percentile) | 1 | 8 | 9 |
| Unknown | 2 | 34 | 36 |
| On ECMO | 0 | 14 | 14 |
| Administration route | |||
| Intramuscular | 50 | 0 | 50 |
| Intravenous | 0 | 63 | 63 |
| Number of doses | 1 (1–1) | 3 (1–32) | 1 (1–32) |
| Dose, mg/kg | 4.0 (3.0–5.1) | 1.0 (0.3–205) | 3.3 (0.3–205) |
| Number of ketamine plasma concentrations: | |||
| 1 | 0 | 24 | 24 |
| 2 | 1 | 21 | 22 |
| ≥3 | 49 | 18 | 67 |
Data presented as counts or median (range). BMI, body mass index; ECMO, extracorporeal membrane oxygenation.
All children in the POP01 study received IV ketamine at a median dose of 1 mg/kg (range 0.3 to 205). This included both bolus dosing and continuous infusion of ketamine. The largest dose was administered as a continuous infusion over 114 hours. The median number of ketamine doses received by children in the POP01 study was 3 (1 to 32). Median infusion rate was 35 mg/kg/hr (0.05 to 250), and median duration of infusion was 1 minute (1 minute to 136 hours). All children in the KPSHC2011 study received a single dose of IM ketamine at a median dose of 4 mg/kg (3 to 5).
PK specimens
The median number of PK samples per subject was 2 (1 to 6) in the POP01 study, and did not differ significantly in the subset of 14 children supported with ECMO (1.5 samples (1 to 4). A single subject in the KPSHC2011 study contributed 2, while all other 49 children contributed 3 PK samples. From the total of 275 plasma PK samples drawn, 5 were excluded due to incomplete sampling time or dosing data, and 4 due to dilutions greater than the validated range (50x). Of the remaining 266 samples, 12 (<5%) were below the limit of quantification (BLQ) and dropped from the analysis, leaving a total of 254 samples—105 from the POP01 and 149 from the KPSHC2011 study—for analysis. Imputation using LLOQ/2 was evaluated, but not retained in the final model given that BLQ after the first drug dose represented <5% of the dataset and given the lack of appreciable difference in model fit or estimation results.32
Population PK model development and evaluation
A 2-compartment model with first-order absorption and elimination described the ketamine concentration vs. time data well (Figures 1–2). For the base model, we included allometrically scaled WT, normalized to a 70-kg WT, with a fixed exponent of 0.75 for CL and intercompartmental CL (Q) and 1 for central (Vc) and peripheral (Vp) volumes of distribution. Estimation of the allometric coefficients was also performed and yielded similar results to the fixed values (0.7 for CL and 0.8 for V parameters), but was not retained in the final model due to concerns about overparameterization and lack of significant effect on model fit and parameter estimates. Inclusion of a lag time to the first-order absorption model did not improve the OFV or model fit compared to the model without lag time, and was therefore not retained in the base model. Diagnostic plots identified several outliers, and the model was re-estimated after their exclusion without any significant difference in results. All concentrations were therefore retained in the final model. The IIV for absorption rate constant (KA), Vp, and F could not be accurately estimated due to excessively high shrinkage values (>50%), and these random effect parameters were therefore fixed to zero. The covariance between IIV(CL) and IIV(Vc) was not estimated as the inclusion of a block covariance matrix resulted in model instability with an unacceptably high proportion of runs with lack of successful minimization during the bootstrap analysis.
Figure 1.

Final model goodness-of-fit plots: observed concentrations vs. population predictions (A) and individual predictions (B), and conditional weighted residuals against population predictions (C) and time after first dose (D).
Figure 2.

Final model standardized visual predictive check plot. 8% of observations are outside of the 90% prediction interval.
After accounting for body size, use of linear, power, and Emax maturation functions for either PNA or PMA on CL did not significantly reduce the OFV. Similarly, inclusion of race or ethnicity using additive covariates on CL also did not reduce the OFV. However, inclusion of ECMO as a covariate on CL/F using a power relationship resulted in a significant drop in the OFV (−19.5). Thus, the final model included WT, and a power relationship of ECMO on CL (Table 2):
Table 2.
Final model parameter estimates and bootstrap results
| Final Model OFV=3011.711 |
Bootstrap (n=1000)* | ||||
|---|---|---|---|---|---|
| Parameter | Estimate | RSE (%) | 2.50% | Median | 97.50% |
| KA (1/hr) | 2.51 | 38 | 1.57 | 2.49 | 4.59 |
| CL (L/hr/70kg) | 38.9 | 15 | 26.93 | 38.73 | 51.14 |
| Vc (L/70kg) | 32.8 | 35 | 19.66 | 33.66 | 58.01 |
| Q (L/hr/70kg) | 54.9 | 13 | 33.66 | 53.90 | 79.62 |
| Vp (L/70kg) | 152 | 36 | 97.12 | 152.15 | 301.99 |
| F | 0.411 | 16 | 0.31 | 0.41 | 0.57 |
| ECMO** | 2.35 | 26 | 1.46 | 2.37 | 4.02 |
| IIV (%CV) | |||||
| IIV (CL)% | 49.1 | 27 | 25.2 | 46.9 | 64.9 |
| IIV (Vc)% | 80.7 | 19 | 55.6 | 79.4 | 100.3 |
| IIV(Q)% | 57.7 | 24 | 17.3 | 53.8 | 81.1 |
| Residual variability | |||||
| Proportional error POP01 data % | 49.29 | 41 | 30.7 | 48.9 | 66.8 |
| Proportion error KPSHC2011 data % | 15.75 | 30 | 11.1 | 15.8 | 20.2 |
KA, absorption rate constant; CL, elimination clearance; Vc, central compartment volume; Q, intercompartmental CL; Vp, peripheral volume of distribution; F, bioavailability; ECMO, extracorporeal membrane oxygenation; IIV, interindividual variability.
77% of all bootstrap runs minimized successfully;
TVCL=CL*(WT/70)0.75*ECMO for subjects supported with ECMO, TVCL=CL*(WT/70)0.75 for subjects not supported with ECMO.
KA (h−1) = 2.51
CL (L/h) = (38.9)*(WT (kg)/70)0.75*2.35ECMO
Vc (L) = (32.8)*(WT (kg)/70)
Q (L/h) = (54.9)*(WT (kg)/70)0.75
Vp (L) = (152)*(WT (kg)/70)
F = 0.411
Separate proportional residual errors for the POP01 and KPSHC2011 data characterized residual variability and were estimated at 48.3% for the former and 15.8% for the latter. This difference is likely due to the opportunistic sampling design and variable dosing of the POP01 study. Shrinkage estimates for CL, Vc, and Q IIV were 15%, 31%, and 40%, respectively. Shrinkage for the POP01 and KPSC2011 proportional residual error parameters were 18% and 46%. The percentage difference between model and bootstrapped median parameter estimates was ≤4% for all parameters. Diagnostic plots for the final model showed over-prediction at higher concentrations (≥1800 ng/mL) (Figure 1). Only 8% of observed concentrations were outside of the SVPC and typical VPC 90% prediction interval, indicating good performance of the final model (Figure 2). An NPC computing prediction intervals ranging from 0% to 95% found an appropriate percentage of observed concentrations outside the confidence interval above the prediction intervals, but slightly lower than expected percentage of observed concentrations outside the confidence interval below the prediction intervals (Supplemental Table S3).
Individual empirical Bayesian estimates for PK parameters based on the final model were compared between children supported with and those not supported with ECMO (Table 3). Median empirical Bayesian estimate of CL was significantly higher in children supported with ECMO compared to those not supported with ECMO (1.70 L/h/kg vs. 0.82 L/h/kg, P<.05). As expected, median Vc and Q did not differ between both groups.
Table 3.
Individual empirical Bayesian estimates in subjects supported and not supported with ECMO
| CL (L/h/kg)* | Vc (L/kg) | Q (L/kg) | |
|---|---|---|---|
| On ECMO | 1.70 (1.01 to 4.3) | 0.49 (0.23 to 0.65) | 1.15 (0.78 to 1.74) |
| No ECMO | 0.82 (0.13 to 2.23) | 0.47 (0.11 to 2.61) | 1.13 (0.12 to 2.53) |
Median (range) presented;
p<0.05 from Wilcoxon Rank Sum test.
Simulated dose-exposure relationship
Following IM administration of doses ranging from 2 to 10 mg/kg, plasma concentrations corresponding to analgesic effects (>100 ng/mL) were reached after approximately 5 minutes and lasted for 60 minutes (Figure 3). To achieve plasma concentrations above the anesthesia awakening threshold of 750 ng/mL, doses of 8 mg/kg in children 6 and 11 kg, and 6 mg/kg in children 17, 28, and 56 kg were necessary (Supplemental Table S4). Even at these doses, approximately 10 and 15 minutes were required to reach plasma concentrations >750 ng/mL in children 6 and 11 kg, and 17–56 kg respectively. Concentrations were maintained above this threshold for 20–30 minutes. A single IM dose of 0.5 mg/kg failed to exceed the analgesic effect threshold (not shown), while a single IM dose of 1 mg/kg barely reached this threshold in all simulated cases. Following IV administration, all bolus dosing levels simulated rapidly exceeded the analgesic threshold, and doses ≥2 mg/kg resulted in plasma concentrations above the anesthesia awakening threshold of 750 ng/mL within 2 minutes of administration (Supplemental Table S4). Doses of 2 mg/kg maintained plasma concentrations above this threshold for approximately 10 minutes, while a dose of 5 mg/kg was necessary to maintain plasma concentrations >750 ng/mL for 20 minutes (Figure 4). When the typical child was simulated as supported with ECMO, similar IV doses were required to reach the threshold except for the 56-kg child, where a 1 mg/kg dose was sufficient. However, plasma concentrations fell more rapidly, and were below 750 ng/mL within less than 20 minutes even after a 5 mg/kg IV dose (Supplemental Figure S3 and Supplemental Table S4).
Figure 3.
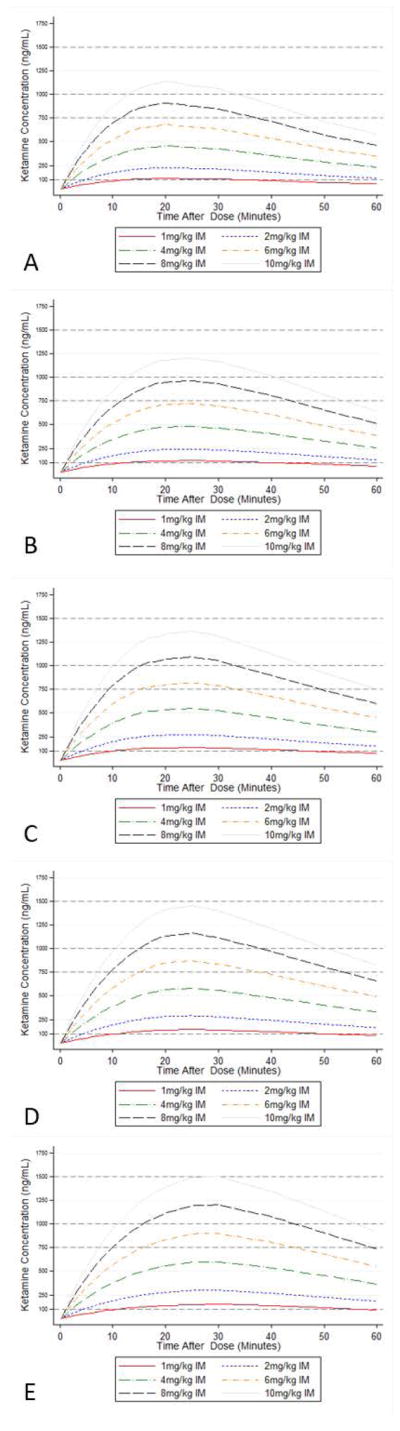
10th percentile simulated plasma concentration time curve in 5 individual children after administration of ketamine doses ranging from 1 to 10 mg/kg via intramuscular route: (A) 6-kg child; (B) 11-kg child; (C) 17-kg child; (D) 28-kg child; (E) 56-kg child.
Figure 4.
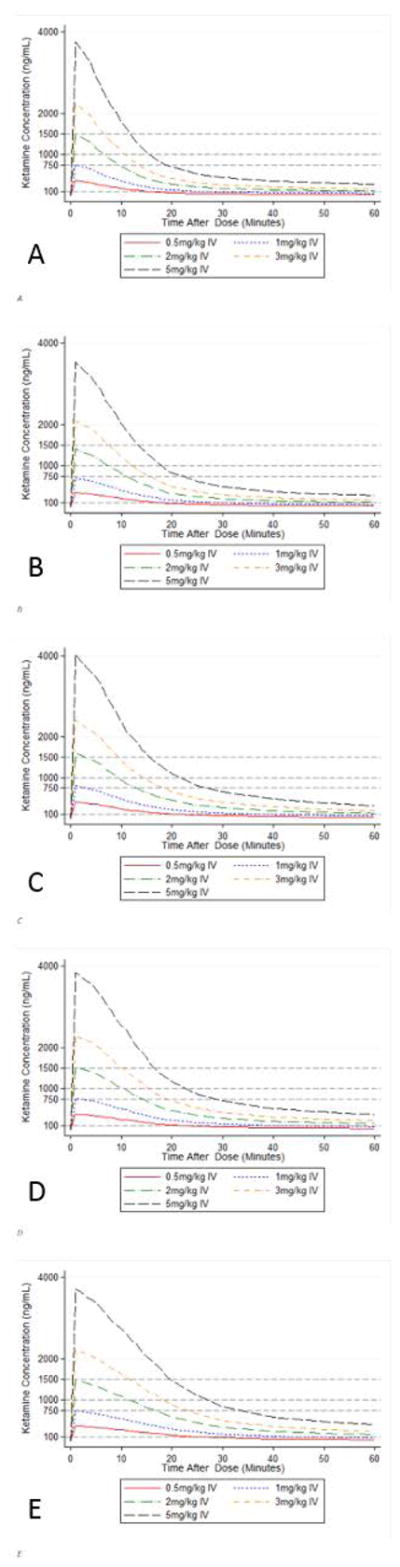
10th percentile simulated plasma concentration time curve in 5 individual children not supported with ECMO after administration of ketamine doses ranging from 0.5 to 5 mg/kg via intravenous route: (A) 6-kg child; (B) 11-kg child; (C) 17-kg child; (D) 28-kg child; (E) 56-kg child.
Safety
No study procedure-related adverse events were reported in the 63 children enrolled in the POP01 study. In the KPSHC2011 study, one of the 50 children required airway repositioning and suctioning following ketamine administration.
Discussion
We developed a pediatric population PK model combining plasma concentrations of ketamine measured after IV and IM administration. Similar to published population PK models of ketamine in children and adults, a 2-compartment model best characterized the data. Our model is the first to estimate the bioavailability of IM ketamine in children, and the first to report on the effect of ECMO on ketamine CL. Our model is applicable to pediatric clinical practice by simulating optimal ketamine dosing for IM and IV administration, and for children supported with ECMO.
The population estimates for ketamine Vc and Vp scaled to a 70-kg adult WT (32.8 L and 152 L) and the resulting Vss (=Vc+Vp=184.8 L) were 41% higher than previously reported in a cohort of 54 children with a mean age of 8.3 years (range 1.5–14) receiving IV ketamine for procedural sedation in the emergency department.8 Vss of ketamine is known to decrease with age, ranging from 242 L/70 kg in infants < 3 months to 53 L/70 kg by adulthood. Our estimate (185 L/70 kg) is consistent with the younger median age of 3.3 years in our cohort.12,33
The population estimate for ketamine CL scaled to a 70-kg adult WT (38.9 L/hr) was comparable to the value (38.7 L/hr) previously derived in a cohort of 10 infants 1 week to 30 months of age receiving ketamine via continuous infusion while recovering from cardiac surgery.28 Our estimate was lower (51%) than the CL previously reported in 54 children with a mean age of 8.3 years (range 1.5–14) receiving ketamine in the emergency department (90 L/h/70 kg), although the authors pointed out that the latter value may have been overestimated by the truncated sampling scheme of the study, which was shorter than the sampling scheme available in our study (especially for the POP01 study).8 Our estimate is also 24–74% lower than CL estimated in adults, with the highest adult CL being reported in critically ill patients.34,35 Ketamine CL is known to be decreased in infants <3 months of age (26 L/hr/70 kg), but approaches adult values by 3–12 months (86 L/hr/70 kg).12 As a result, estimation of CL from a pediatric cohort that includes infants < 3 months would be expected to yield lower values, as was the case in our study and the study of infants recovering from cardiac surgery.28 An estimate of CL in an older pediatric cohort should in turn result in higher CL values, as was the case in the report from the emergency department or a subsequent re-analysis of the same dataset.1
Ketamine CL was 2.3-fold higher among children supported with ECMO in our cohort, while volume of distribution was not significantly affected. The IIV on CL was only modestly decreased following inclusion of ECMO as a covariate and remained high (51% to 49%), but the OFV was significantly lower.
Our study is the first to report on the PK of ketamine in children supported with ECMO, but previous studies have identified PK alterations of multiple drugs in children during ECMO support. Several potential pathophysiologic mechanisms responsible for these alterations have been described: drug extraction by the ECMO circuit, hemodilution, and physiological changes related to the support of critical illness.36 We can only postulate as to the mechanism responsible for the increased CL of ketamine during ECMO support observed in our study. Hepatic N-demethylation is the primary elimination pathway of ketamine, with CL limited by liver blood flow.9 ECMO may ameliorate hepatic function through improved organ blood flow, particularly when compared to other critically ill children. An increase in CL among children supported with ECMO has previously been reported for micafungin, and was presumed to be related to lower albumin concentration following hemodilution on ECMO.37 This is less likely to be the case in our cohort, as albumin concentrations were generally high in all children (>2.1 g/dL), ketamine exhibits low protein binding, and its CL has been shown to be blood flow limited.9 Higher ketamine elimination with ECMO may also be due to nonspecific and irreversible drug adsorption by the circuit if adsorption is an ongoing process.38,39 Significant drug adsorption typically results in increased V, and is most often seen with lipophilic drugs.39 Because ketamine is more hydrophilic, adsorption may be less severe and occur more gradually over time, resulting in increased CL without affecting V.
Our analysis leveraged a combined dataset from 2 clinical trials to estimate the bioavailability of IM ketamine using a population PK model. To our knowledge, the previously published bioavailability estimate of IM ketamine was calculated from the ratio of the trapezoidal area under the concentration time curve after IM and IV injection of ketamine in 6 healthy adult volunteers.15 In this population, the bioavailability ranged from 85.9% to 97.2%, with a mean of 93%. Our estimate derived in a larger pediatric population using a population PK modeling approach is significantly lower (41%). The value does, however, reflect current dosing recommendations for induction of anesthesia with IM ketamine, which are 2–3 times higher than recommended IV doses for children ≥3 months to < 16 years: 5–10 mg/kg IM or 1–3 mg/kg IV.40–42 A similar dosing ratio is recommended for procedural sedation without concomitant use of propofol: 4–5 mg/kg IM or 1–2 mg/kg IV.43,44 The majority of these dosing recommendations were derived primarily from evaluating the dose-response relationship rather than characterizing exposure.
We applied our model to ketamine dosing simulations following IM and IV bolus dosing in children with and without ECMO support. We designed our simulations to primarily reflect ketamine use for procedural sedation, given the prevalence of this indication in multiple clinical settings (intensive care unit, emergency department), and because it was an inclusion criteria for participation in the KPSHC2011 study. Our recommended doses for children not supported with ECMO are consistent with previous reports for both IV and IM administration of ketamine for procedural sedation, and with the ketamine FDA label.3,40,43,45–47 These reports primarily recommended doses based on safety profiles and clinical success rates of sedation and achievement of adequate conditions to complete procedures. Our simulation complements these reports by confirming that doses of 8 mg/kg and 6 mg/kg IM in children 6–11 kg and 17–56 kg, respectively, and 2 mg/kg IV exceed plasma threshold concentrations previously associated with analgesia and arousability. When compared to the most recent and largest pediatric study of IV ketamine for procedural sedation that measured plasma concentrations, our simulations yielded similar exposures, with doses of 2 mg/kg resulting in plasma concentration <750 ng/mL (level associated with arousal) after 10 minutes, while doses >3 mg/kg exceeded this level for 20 minutes. An important difference between our IM and IV dosing simulations is the time to achievement of plasma concentrations > 750 ng/mL. Our results suggest that for children not supported with ECMO, IM doses of 8 mg/kg in children 6–11 kg and 6 mg/kg in children 17–56 kg will provide adequate sedation approximately 10 and 15 minutes after administration for procedures lasting up to 20 minutes. Doses of 2 mg/kg and 3 mg/kg IV will provide adequate sedation after approximately 2 minutes for up to 10 and 15 minutes, respectively. For children supported with ECMO, IV doses of 2 mg/kg will provide adequate levels of sedation (>750 ng/mL) but for shorter duration. For procedures lasting ≥10 minutes, doses ≥ 5 mg/kg would be required to maintain plasma concentrations >750 ng/mL.
Despite its strengths, our study has several important limitations. Our model diagnostics suggest some model misspecification, specifically overprediction of higher ketamine plasma concentrations (mostly ≥1800 ng/mL). The limited number of PK samples with high concentrations likely contributed to this bias. While we believe our report of IM bioavailability to be of clinical relevance given the frequent use of the drug in this population, we note that it is estimated from a population PK model with variable, sparse sampling schemes of distinct IV and IM dosing cohorts, and not estimates of area under the concentration vs. time curve calculated in the same subjects following both IV and IM administration. This includes a notable lack of sampling times >120 minutes after IM dosing, which is particularly relevant given the potential for longer absorption time after IM administration, which may limit our ability to truly describe the elimination phase. In addition, we recognize that merging of PK datasets should be undertaken with caution as unrecognized sources of systemic bias may exist. We have attempted to address this possibility by estimating separate residual errors models and have tested numerous covariate relationships that may have differed between datasets (including age and race). We also attempted to fit a model with separate CL estimated for each study, but minimization problems likely related to individual study sample size and sampling scheme precluded the reporting of reliable estimates. Despite these limitations, we believe that the challenges associated with conducting a traditional bioavailability study of ketamine in children warrant the reporting of our results as long as sufficient caution is applied in their interpretation. The sparsity of our data also did not allow us to provide interindividual variability estimates of the IM bioavailability, which is likely to be substantial in children. We acknowledge that inclusion of IIV in these terms would increase the clinical value of our simulation results. Further, the opportunistic design of our study resulted in a wide spectrum of indications and dosing regimens. In part because of this limitation, we focused our simulations on achieving lower plasma concentration targets required for procedural sedation, and believe our model is useful in optimizing ketamine dosing for this indication in children with and without ECMO support even though our study is not a true PK/pharmacodynamic design and sedation scores were not obtained. The higher concentrations observed after larger doses should be interpreted cautiously. Other limitations of our model include the relatively large residual error, particularly for children in the POP01 study. Again, the opportunistic study design with variable PK sampling times and large dosing range likely contribute to this effect. In the KPSHC2011 data, where dosing and PK sampling times were specified by the protocol, residual variability was lower. IIV also remained high for all 3 parameters for which it could be estimated, even after including ECMO as a covariate on CL in our final model. As previously noted by others, high IIV is a consistent feature across multiple pediatric population PK models of ketamine, although the exact reason remains unclear.8 In both studies used in our analysis, several clinical characteristics that may affect ketamine exposure were not collected, limiting our ability to test all potentially relevant covariates. This includes pharmacogenomic differences in enzymatic activity of ketamine metabolizing enzymes including CYP3A isoenzymes, CYP2C9, and CYP2B6. We evaluated race as a surrogate for pharmacogenomic differences, given previously described racial differences in allelic distribution particularly of CYP2B6, which was undetectable in 70% of Japanese adults in 1 study.34,48 We were unable to identify a significant relationship between age and ketamine CL, despite the known ontogeny of CYP3A isoenzymes in children <2 years of age. The limited number of PK samples collected for this subpopulation may, at least in part, explain this finding. It is further possible that CYP2C9 and CYP2B6 may play a greater role in ketamine metabolism, in our population. Ketamine has an important pharmacologically active metabolite, norketamine, which was not measured in the plasma samples of either study. We therefore cannot comment on how norketamine plasma concentrations may affect our dosing recommendations. Finally, our evaluation of ketamine safety was limited by the fact that only study procedure-related adverse events were collected in the POP01 study, and only one event was recorded in the KPSHC2011 study. The observed event (suctioning and repositioning) is consistent with previous reports that excessive salivation and the need for airway suctioning are the most common adverse effects of ketamine, occurring in 13–33% of children.49 Unfortunately, we are unable to comment on the prevalence of other important adverse events such as the occurrence of emergence delirium or any long term consequences of ketamine use.50
Conclusions
In conclusion, we used combined data from 2 PK studies to develop a pediatric population PK model of IM- and IV-administered ketamine. Our model is the first to estimate the bioavailability of IM ketamine of 41%, consistent with the ratio of currently recommended IM to IV dosing of ketamine for multiple indications. We also identified ECMO exposure as a significant covariate associated with higher ketamine CL. Using our model for dosing simulations, we recommend that children not supported with ECMO receive IV and IM doses of 2 mg/kg and 6 or 8 mg/kg, respectively, for sedation for procedures lasting up to 20 minutes. An important difference between the 2 administration routes is the time to onset of sedation, which is up to 10 minutes after IM administration. For children supported with ECMO, doses ≥ 5 mg/kg are required for procedures lasting ≥ 10 minutes. As with all PK models, additional ketamine PK samples collected in future studies may be used to iteratively improve our model performance and maximize its clinical value.
Supplementary Material
Acknowledgments
Source of Funding:
C.P.H receives support for research from the National Institute for Child Health and Human Development (NICHD) (K23HD090239). D.G. receives support for research from the NICHD (K23HD083465). Portions of this work were funded under NICHD contract HHSN275201000003I for the Pediatric Trials Network (PI Danny Benjamin). M.C.W. receives support for research from the National Institutes of Health (1R01-HD076676-01A1), National Institute of Allergy and Infectious Diseases (HHSN272201500006I and HHSN272201300017I), NICHD (HHSN275201000003I), the Biomedical Advanced Research and Development Authority (HHSO100201300009C), and industry for drug development in adults and children. All disclosures are available at www.dcri.duke.edu/research/coi.jsp.
The assay measuring ketamine plasma concentrations was developed and performed at Alturas Analyticals Inc. (Moscow, ID, USA).
The Best Pharmaceuticals for Children Act – Pediatric Trials Network Publication Committee: Gary Furda, Duke Clinical Research Institute, Durham, NC; Danny Benjamin, Duke Clinical Research Institute, Durham, NC; Edmund Capparelli, University of California San Diego, San Diego, CA; Gregory L. Kearns, Arkansas Children’s Hospital Research Institute, Little Rock, AR; Ian M. Paul, Penn State College of Medicine, Hershey, PA; Christoph Hornik, Duke Clinical Research Institute, Durham, NC; Kelly Wade, Children’s Hospital of Philadelphia, Philadelphia, PA.
The Eunice Kennedy Shriver National Institute of Child Health and Human Development: David Siegel and Perdita Taylor-Zapata.
The EMMES Corporation (Data Coordinating Center): Ravinder Anand and Gina Simone.
Ketamine Pharmacokinetic Study in Healthy Children Aged 2 to 5 Years Old (KPSHC2011) study team: Drs. Lai Peng Tham, Suraj Manickam, Rachael Gaudry and Ms. Dianna Sri Dewi.
Pediatric Trials Network’s Pharmacokinetics of Understudied Drugs Administered to Children per Standard of Care Study Team and Study Coordinators
Duke Clinical Research Institute: Kevin Watt (PI), Samantha Wrenn (SC), Christi Milleson (SC)
Children’s National Medical Center: John van den Anker (PI), Elaine Williams (SC)
Medical University of South Carolina Children’s Hospital: Andrew Atz (PI), Hibah Al Nasiri (SC), Patricia Infinger (SC)
Ann and Robert H Lurie Children’s Hospital of Chicago: Ram Yogev (PI), Laura Fearn (SC),
Riley Hospital for Children at Indiana University: Brenda Poindexter (PI), Susan Gunn (SC), Dianne Herron (SC), Shirley Wright-Coltart (SC), Jeffrey Joyce (SC)
KK Women’s and Children’s Hospital, Singapore: Jan Hau Lee (PI), Kathy Liaw (SC), Cecilia Chanda (SC)
Wesley Medical Center: Paula Delmore (PI), Barry Bloom (Sub-I)
Case Western: David Speicher (PI), Sue Bergant (SC)
Children’s Hospital Colorado: Peter Mourani (PI), Kimberly Ralston (SC), Matthew Steinbeiss (SC)
Oregon Health and Science University: Amira Al-UZri (PI), Kira Clark (SC)
University of Arkansas Medical Center: Laura James (PI), Dawn Hansberry (SC), Michelle Hart (SC), Lee Howard (SC)
Alfred I. DuPont Hospital for Children: Marisa Meyer (PI), Glen Stryjewski (PI), Kimberly Klipner (SC), Ramany John (SC)
University of Virginia Children’s Hospital: Michelle Adu-Darko (PI), Robin Kelly (SC)
References
- 1.Brunette KE, Anderson BJ, Thomas J, Wiesner L, Herd DW, Schulein S. Exploring the pharmacokinetics of oral ketamine in children undergoing burns procedures. Paediatr Anaesth. 2011;21(6):653–662. doi: 10.1111/j.1460-9592.2011.03548.x. [DOI] [PubMed] [Google Scholar]
- 2.Grunwell JR, Travers C, McCracken CE, et al. Procedural sedation outside of the operating room using ketamine in 22,645 children: a report from the Pediatric Sedation Research Consortium. Pediatr Crit Care Med. 2016;17(12):1109–1116. doi: 10.1097/PCC.0000000000000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.U.S. National Library of Medicine. DailyMed. [Accessed August 31, 2017];Label: ketamine hydrochloride-ketamine hydrochloride injection. Available at https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=14b54219-f5fd-42fa-8784-719f9785497f&audience=professional.
- 4.Flores-Gonzalez JC, Lechuga Sancho AM, Saldana Valderas M, et al. Respiratory adverse events during upper digestive endoscopies in children under ketamine sedation. Minerva Pediatr. 2017 Feb 7; doi: 10.23736/S2724-5276.16.04758-7. doi:10.23736/S0026-4946.16.04758-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 5.Patterson AC, Wadia SA, Lorenz DJ, Stevenson MD. Changes in blood pressure and heart rate during sedation with ketamine in the pediatric ED. Am J Emerg Med. 2017;35(2):322–325. doi: 10.1016/j.ajem.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 6.Suryaprakash S, Tham LP. Predictors of emesis in children undergoing procedural sedation with intramuscular ketamine in a paediatric emergency department. Singapore Med J. 2016 Dec 9; doi: 10.11622/smedj.2016187. doi:10.11622/smedj.2016187. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deasy C, Babl FE. Intravenous vs intramuscular ketamine for pediatric procedural sedation by emergency medicine specialists: a review. Paediatr Anaesth. 2010;20(9):787–796. doi: 10.1111/j.1460-9592.2010.03338.x. [DOI] [PubMed] [Google Scholar]
- 8.Herd D, Anderson BJ. Ketamine disposition in children presenting for procedural sedation and analgesia in a children’s emergency department. Paediatr Anaesth. 2007;17(7):622–629. doi: 10.1111/j.1460-9592.2006.02145.x. [DOI] [PubMed] [Google Scholar]
- 9.Mion G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings) CNS Neurosci Ther. 2013;19(6):370–380. doi: 10.1111/cns.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pappas A, Shankaran S, Hansen NI, et al. Outcome of extremely preterm infants (<1,000 g) with congenital heart defects from the National Institute of Child Health and Human Development Neonatal Research Network. Pediatric Cardiol. 2012;33(8):1415–1426. doi: 10.1007/s00246-012-0375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elkomy MH, Drover DR, Hammer GB, Galinkin JL, Ramamoorthy C. Population pharmacokinetics of ketamine in children with heart disease. Int J Pharm. 2015;478(1):223–231. doi: 10.1016/j.ijpharm.2014.11.026. [DOI] [PubMed] [Google Scholar]
- 12.Anderson BJ, McKee AD, Holford NH. Size, myths and the clinical pharmacokinetics of analgesia in paediatric patients. Clin Pharmacokinet. 1997;33(5):313–327. doi: 10.2165/00003088-199733050-00001. [DOI] [PubMed] [Google Scholar]
- 13.Sherwin CM, Stockmann C, Grimsrud K, Herd DW, Anderson BJ, Spigarelli MG. Development of an optimal sampling schedule for children receiving ketamine for short-term procedural sedation and analgesia. Paediatr Anaesth. 2015;25(2):211–216. doi: 10.1111/pan.12521. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen BN, Friis SM, Romsing J, et al. Intranasal sufentanil/ketamine analgesia in children. Paediatr Anaesth. 2014;24(2):170–180. doi: 10.1111/pan.12268. [DOI] [PubMed] [Google Scholar]
- 15.Clements JA, Nimmo WS, Grant IS. Bioavailability, pharmacokinetics, and analgesic activity of ketamine in humans. J Pharm Sci. 1982;71(5):539–542. doi: 10.1002/jps.2600710516. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez D, Melloni C, Poindexter BB, et al. Simultaneous determination of trimethoprim and sulfamethoxazole in dried plasma and urine spots. Bioanalysis. 2015;7(9):1137–1149. doi: 10.4155/bio.15.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keizer RJ, van Benten M, Beijnen JH, Schellens JH, Huitema AD. Pirana and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101(1):72–79. doi: 10.1016/j.cmpb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 18.Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit--a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Sarkar D. Lattice: Multivariate Data Visualization with R. 1. New York, NY: Springer; 2008. [Google Scholar]
- 20.Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer; 2009. [Google Scholar]
- 21.Jonsson EN, Karlsson MO. Xpose--an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 22.Jelliffe RW, Gomis P, Tahani B, Ruskin J, Sattler FR. A population pharmacokinetic model of trimethoprim in patients with pneumocystis pneumonia, made with parametric and nonparametric methods. Ther Drug Monit. 1997;19(4):450–459. doi: 10.1097/00007691-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 23.Centers for Disease Control and Prevention. [Accessed October 1, 2017];About child & teen BMI. Available at https://www.cdc.gov/healthyweight/assessing/bmi/childrens_bmi/about_childrens_bmi.html.
- 24.Bjorkman S, Redke F. Clearance of fentanyl, alfentanil, methohexitone, thiopentone and ketamine in relation to estimated hepatic blood flow in several animal species: application to prediction of clearance in man. J Pharm Pharmacol. 2000;52(9):1065–1074. doi: 10.1211/0022357001774985. [DOI] [PubMed] [Google Scholar]
- 25.Salem F, Johnson TN, Abduljalil K, Tucker GT, Rostami-Hodjegan A. A re-evaluation and validation of ontogeny functions for cytochrome P450 1A2 and 3A4 based on in vivo data. Clin Pharmacokinet. 2014;53(7):625–636. doi: 10.1007/s40262-014-0140-7. [DOI] [PubMed] [Google Scholar]
- 26.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45(9):931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 27.Wang DD, Zhang S. Standardized visual predictive check versus visual predictive check for model evaluation. J Clin Pharmacol. 2012;52(1):39–54. doi: 10.1177/0091270010390040. [DOI] [PubMed] [Google Scholar]
- 28.Hartvig P, Larsson E, Joachimsson PO. Postoperative analgesia and sedation following pediatric cardiac surgery using a constant infusion of ketamine. J Cardiothorac Vasc Anesth. 1993;7(2):148–153. doi: 10.1016/1053-0770(93)90207-2. [DOI] [PubMed] [Google Scholar]
- 29.Herd DW, Anderson BJ, Keene NA, Holford NH. Investigating the pharmacodynamics of ketamine in children. Paediatr Anaesth. 2008;18(1):36–42. doi: 10.1111/j.1460-9592.2007.02384.x. [DOI] [PubMed] [Google Scholar]
- 30.Idvall J, Ahlgren I, Aronsen KR, Stenberg P. Ketamine infusions: pharmacokinetics and clinical effects. Br J Anaesth. 1979;51(12):1167–1173. doi: 10.1093/bja/51.12.1167. [DOI] [PubMed] [Google Scholar]
- 31.Leung LY, Baillie TA. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J Med Chem. 1986;29(11):2396–2399. doi: 10.1021/jm00161a043. [DOI] [PubMed] [Google Scholar]
- 32.Keizer RJ, Jansen RS, Rosing H, et al. Incorporation of concentration data below the limit of quantification in population pharmacokinetic analyses. Pharmacol Res Perspect. 2015;3(2):e00131. doi: 10.1002/prp2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook DR. Paediatric anaesthesia: pharmacological considerations. Drugs. 1976;12(3):212–221. doi: 10.2165/00003495-197612030-00004. [DOI] [PubMed] [Google Scholar]
- 34.Hijazi Y, Bodonian C, Bolon M, Salord F, Boulieu R. Pharmacokinetics and haemodynamics of ketamine in intensive care patients with brain or spinal cord injury. Br J Anaesth. 2003;90(2):155–160. doi: 10.1093/bja/aeg028. [DOI] [PubMed] [Google Scholar]
- 35.Domino EF, Domino SE, Smith RE, et al. Ketamine kinetics in unmedicated and diazepam-premedicated subjects. Clin Pharmacol Ther. 1984;36(5):645–653. doi: 10.1038/clpt.1984.235. [DOI] [PubMed] [Google Scholar]
- 36.Watt K, Li JS, Benjamin DK, Jr, Cohen-Wolkowiez M. Pediatric cardiovascular drug dosing in critically ill children and extracorporeal membrane oxygenation. J Cardiovasc Pharmacol. 2011;58(2):126–132. doi: 10.1097/FJC.0b013e318213aac2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Autmizguine J, Hornik CP, Benjamin DK, Jr, et al. Pharmacokinetics and safety of micafungin in infants supported with extracorporeal membrane oxygenation. Pediatr Infect Dis J. 2016;35(11):1204–1210. doi: 10.1097/INF.0000000000001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shekar K, Roberts JA, McDonald CI, et al. Protein-bound drugs are prone to sequestration in the extracorporeal membrane oxygenation circuit: results from an ex vivo study. Crit Care. 2015;19:164. doi: 10.1186/s13054-015-0891-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wildschut ED, Ahsman MJ, Allegaert K, Mathot RA, Tibboel D. Determinants of drug absorption in different ECMO circuits. Intensive Care Med. 2010;36(12):2109–2116. doi: 10.1007/s00134-010-2041-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin C, Durieux ME. Ketamine and kids: an update. Paediatr Anaesth. 2005;15(2):91–97. doi: 10.1111/j.1460-9592.2005.01475.x. [DOI] [PubMed] [Google Scholar]
- 41.Sungur Ulke Z, Kartal U, Orhan Sungur M, Camci E, Tugrul M. Comparison of sevoflurane and ketamine for anesthetic induction in children with congenital heart disease. Paediatr Anaesth. 2008;18(8):715–721. doi: 10.1111/j.1460-9592.2008.02637.x. [DOI] [PubMed] [Google Scholar]
- 42.Cote CJ, Lerman J, Anderson BJ. A Practice of Anesthesia for Infants and Children. 5. Philadelphia, PA: Elsevier; 2013. [Google Scholar]
- 43.Asadi P, Ghafouri HB, Yasinzadeh M, Kasnavieh SM, Modirian E. Ketamine and atropine for pediatric sedation: a prospective double-blind randomized controlled trial. Pediatr Emerg Care. 2013;29(2):136–139. doi: 10.1097/PEC.0b013e31828058b2. [DOI] [PubMed] [Google Scholar]
- 44.McGlone RG, Howes MC, Joshi M. The Lancaster experience of 2.0 to 2.5 mg/kg intramuscular ketamine for paediatric sedation: 501 cases and analysis. Emerg Med J. 2004;21(3):290–295. doi: 10.1136/emj.2002.003772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berkenbosch JW, Graff GR, Stark JM. Safety and efficacy of ketamine sedation for infant flexible fiberoptic bronchoscopy. Chest. 2004;125(3):1132–1137. doi: 10.1378/chest.125.3.1132. [DOI] [PubMed] [Google Scholar]
- 46.Koruk S, Mizrak A, Gul R, Kilic E, Yendi F, Oner U. Dexmedetomidine-ketamine and midazolam-ketamine combinations for sedation in pediatric patients undergoing extracorporeal shock wave lithotripsy: a randomized prospective study. J Anesth. 2010;24(6):858–863. doi: 10.1007/s00540-010-1023-1. [DOI] [PubMed] [Google Scholar]
- 47.Koruk S, Mizrak A, Kaya Ugur B, Ilhan O, Baspinar O, Oner U. Propofol/dexmedetomidine and propofol/ketamine combinations for anesthesia in pediatric patients undergoing transcatheter atrial septal defect closure: a prospective randomized study. Clin Ther. 2010;32(4):701–709. doi: 10.1016/j.clinthera.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Yanagihara Y, Ohtani M, Kariya S, et al. Plasma concentration profiles of ketamine and norketamine after administration of various ketamine preparations to healthy Japanese volunteers. Biopharm Drug Dispos. 2003;24(1):37–43. doi: 10.1002/bdd.336. [DOI] [PubMed] [Google Scholar]
- 49.Gutstein HB, Johnson KL, Heard MB, Gregory GA. Oral ketamine preanesthetic medication in children. Anesthesiology. 1992;76(1):28–33. doi: 10.1097/00000542-199201000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Gutstein HB. Potential physiologic mechanism for ketamine-induced emergence delirium. Anesthesiology. 1996;84(2):474. doi: 10.1097/00000542-199602000-00032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.