

Abstract
Background
The cystic fibrosis transmembrane conductance regulator (CFTR) potentiator ivacaftor is approved for patients with CF with gating and residual function CFTR mutations. We report the results of an observational study investigating its effects in CF patients with non-G551D gating mutations.
Methods
Patients with non-G551D gating mutations were recruited to an open-label study evaluating ivacaftor. Primary outcomes included: lung function, sweat chloride, weight gain, and quality of life scores.
Results
Twenty-one subjects were enrolled and completed 6 months follow-up on ivacaftor; mean age was 25.6 years with 52% <18. Baseline ppFEV1 was 68% and mean sweat chloride 89.6 mEq/L. Participants experienced significant improvements in ppFEV1 (mean absolute increase of 10.9% 95% CI= [2.6,19.3], p=0.0134), sweat chloride (−48.6 95% CI= [−67.4, −29.9], p<0.0001), and weight (5.1 kilograms, 95% CI= [2.8, 7.3], p=0.0002).
Conclusions
Patients with non-G551D gating mutations experienced improved lung function, nutritional status, and quality of life. This study supports ongoing use of ivacaftor for patients with these mutations.
Keywords: ivacaftor, gating mutation, CFTR potentiator, Clinical Trials
Introduction
Cystic fibrosis (CF) is a rare, autosomal recessive genetic disorder caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene that encodes a transmembrane protein, responsible for chloride and bicarbonate conductance [1]. Mutations in CFTR result in epithelial dysfunction in a variety of tissues due to imbalance in ion and fluid homeostasis. In the lung, this imbalance results in viscous secretions that impairs normal mucociliary clearance, promoting progressive pulmonary decline and infections that result in respiratory failure and death [2].
Recent developments have resulted in a new class of drugs that target the defect in the protein, called CFTR modulators, the first of which is the CFTR potentiator ivacaftor [3–5]. As a CFTR potentiator, ivacaftor targets CFTR protein on the apical membrane of epithelial cells, to augment open probability and enhance the transport of chloride and bicarbonate anions. CFTR potentiators are particularly active in vitro for CFTR mutations that exhibit disrupted ATP-dependent channel gating, preventing efficient ion channel opening. These ‘gating’ mutations are classically grouped as molecular Class III, among the over 1900 CFTR variants, [6, 7] noting that many variants can also exhibit other cellular abnormalities [8] such as abnormal processing or reduced surface stability. [9] The most common gating mutation is G551D, found in approximately 4.4% of all CF patients in the United States. Other mutations that principally induce severe gating defects include G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, and G1349D and are quite rare, making up only 1.6% of the population in the United States [10].
In CF patients with non-G551D gating mutations, a Phase 3 trial termed KONNECTION demonstrated improved spirometry, quality of life, and SC compared to placebo, resulting in FDA approval in this mutation group [11]. In the G551D Observational Study (GOAL)-Expanded to Additional Genotypes and Extended for Long Term Follow up (GOAL-e2) to include recently approved non-G551D gating mutations, we studied the effectiveness of six months of ivacaftor in patients age 6 years and older with at least one non-G551D gating allele, expanding beyond a clinical trial in this population[12] and including additional outcomes of hospitalization events and culture results.
Methods
Study design and participants
The original GOAL longitudinal cohort study design and analysis of the first cohort of participants (those with at least one G551D allele) have been summarized elsewhere [13]. The study was subsequently expanded to include Cohort 3, an additional cohort of participants age 6 and above with at least one non-G551D gating mutation, any mutation on the second allele except G551D or R117H, and no prior exposure to ivacaftor (http://clinicaltrials.gov/ct2/show/NCT01521338). Patients with G970R, which was presumed to be a gating mutation based on initial electrophysiological investigation in Fisher rat thyroid (FRT) cells but was later identified as a splice variant in non-heterologous cells systems including primary epithelium [14], were thus excluded from further study. Study procedures were conducted as previously described [13]. Briefly, subject data were collected at baseline and at 1, 3, and 6 months after starting ivacaftor. As previously described [13], additional clinical data from the US CF Foundation Patient Registry (CFFPR) were also evaluated in this study population. Written informed consent was obtained from patients and/or guardians according to each study site’s Institutional Review Boards.
Procedures and outcome measures
Study assessments conducted for all enrolled participants included: forced expiratory volume over one second (FEV1, L), FEV1 percent of predicted, weight (kilograms or percentiles for those < 20 years old), height (cm or percentiles for those < 20 years old), Body Mass Index (kg/m2 and percentiles for those < 20 years old), sweat chloride (SC, mEq/L), Cystic Fibrosis Questionnaire-Revised (CFQ-R) [15], Sino-Nasal Outcome Test-20 (SNOT-20) [16] score, and Cystic Fibrosis Respiratory Symptom Diary (CFRSD) [17] score. Hospitalization events and P. aeruginosa cultures results were ascertained via merged CFFPR data. Spirometry was performed according to American Thoracic Society standards [18] and percent of predicted values were calculated using reference equations [19, 20]. In addition, biological samples for future research (plasma, serum, buffy coat, urine, and expectorated sputum) were collected and stored at the CFFT Biorepository.
Statistical Analyses
Unless otherwise noted, continuous outcomes are summarized using paired mean changes and standard deviations [SD] from baseline with corresponding 95% confidence intervals (CI) and paired t-tests. Cross-sectional differences in proportions were compared using Fisher’s exact test and paired differences in proportions were assessed with McNemar’s test. Hospitalization rates (adjusted for participant follow-up) were compared with Wilcoxon’s sign test. Pearson’s correlation coefficient was used to explore associations between changes in SC and changes in clinical measures. Reported p-values are two-sided and a 0.05 level of significance was used. Analyses were performed using SAS (version 9.4, Cary, NC), and R (version 3.3, Vienna, Austria).
Results
Patient population
Twelve centers within the Cystic Fibrosis Therapeutics Development Network enrolled non-G551D gating mutation participants between February 2014 and January 2016. Of 24 patients with one of 8 eligible gating mutations, 23 were enrolled in the study and completed follow up. Of these, 21 were prescribed ivacaftor as part of this observational study, whereas two patients declined ivacaftor. Eighteen (86%) completed the entire study with 180 days of follow up (Figure 1); two were lost to follow-up, and one withdrew. One subject had a lung transplant approximately 3 months after initiation of ivacaftor and therefore post-transplant measurements were excluded from the analysis.
Figure 1.
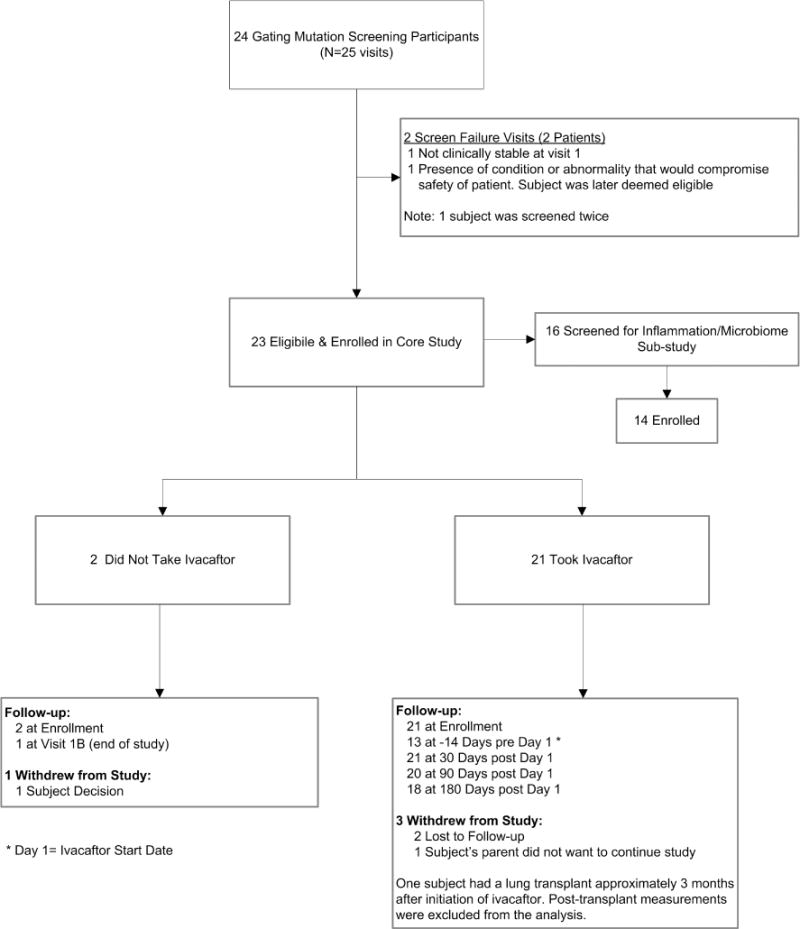
Trial Consort Diagram
Baseline characteristics and demographics of the analyzed population are described in Table 1. Ten subjects (48%) were 18 or older, and 11 subjects (52%) were 6-17 years of age. All patients had at least one non-G551D gating mutation, with the prevalence shown by CFTR mutation in trans in Table 1. Notably, three patients (one pediatric, two adult) possessed second alleles considered to have residual function which were recently approved for ivacaftor by the Food and Drug Administration; these alleles are expected to contribute to the response to ivacaftor, as well. One patient was homozygous for the S549R gating mutation and two patients had an unidentified second allele. Nine patients (43%) were Black or Hispanic, indicating a more diverse racial balance compared to other CF studies and the underlying prevalence of these rare mutations.
Table 1.
Patient demographics.
| Parameter | Subjects Taking Ivacaftor (N=21) | |
|---|---|---|
| Sex, n (%) | Female | 9 (43%) |
| Male | 12 (57%) | |
|
| ||
| Enrollment Age, mean (SD) | 25.3 (16.2) | |
| Enrollment Age, n (%) | 6-11 | 4 (19%) |
| 12-17 | 7 (33%) | |
| 18-29 | 4 (19%) | |
| 30+ | 6 (29%) | |
|
| ||
| Genotype, allele 1, n (%) | S549N | 9 (42.8%) |
| G178R | 4 (19%) | |
| S1251N | 4 (19%) | |
| S549R | 2 (9.5%) | |
| S1255P | 1 (4.8%) | |
| G551S | 1 (4.8%) | |
|
| ||
| Genotype, allele 2, n (%) | F508del | 11 (52.4%) |
| Unidentified | 2 (9.5%) | |
| 621+1G->T | 1 (4.8%) | |
| S549R | 1 (4.8%) | |
| 3272-26A->C (RF) | 1 (4.8%) | |
| L467P | 1 (4.8%) | |
| Y913X | 1 (4.8%) | |
| W1282X | 1 (4.8%) | |
| 711+3 A->G (RF) | 1 (4.8%) | |
| R352Q (RF) | 1 (4.8%) | |
|
| ||
| Race, n (%) | Black/African-American | 4 (19%) |
| Hispanic | 5 (24%) | |
| White | 12 (57%) | |
|
| ||
| FEV1 % Predicted, mean (SD) | 68.0 (28.4) | |
| FVC % Predicted, mean (SD) | 81.2 (26.4) | |
| BMI, mean (SD) | 20.8 (4.2) | |
RF=residual function
The baseline ppFEV1 of subjects enrolled who were prescribed ivacaftor (n=21) was 68.0% (Supplemental Table 1). There were some differences based on age; the baseline ppFEV1 in the pediatric group (age < 18) was 88.7 while the adult group was 45.2 (Supplemental Table 2). The baseline BMI was 20.8 Kg/m2 (26.6 percentile) and the mean baseline SC was 89.6 mmol/L. The baseline ppFEV1 values for the two subjects who enrolled but ultimately chose not to take ivacaftor (and thus were excluded from analysis) were 80.9 and 123.2.
Effect of ivacaftor on lung function
Effects on lung function at each time point are summarized for the overall cohort in Supplementary Table 1 and by age (>/=18yo or <18yo) in Supplementary Table 2. Among enrolled subjects prescribed ivacaftor, there was a mean absolute increase in ppFEV1 of 9.3% [4.2, 14.4] at one month (p=0.0011), which was sustained at three (+8.4 [1.4, 15.4], p=0.0214) and six month (+10.9 [2.6, 19.3], p=0.0134) follow-up visits (Figure 2A). Non-significant improvement in percent predicted Forced Vital Capacity (FVC) of 6.5 [−1.8, 14.8] was also observed at six months (p=0.1170; Table S2).
Figure 2. Mean change from baseline after 6 months of treatment.
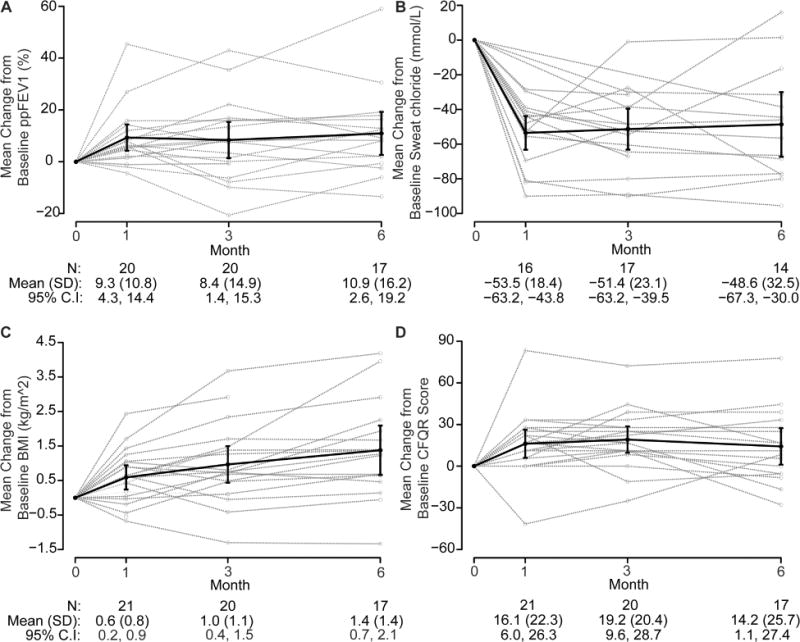
(A) percent predicted FEV1; (B) BMI; (C) sweat chloride; and (D) CFQ-R respiratory score. ppFEV1 (percent predicted FEV1); BMI (body mass index); CFQ-R (Cystic Fibrosis Questionnaire-Revised)
The absolute increase in ppFEV1 diverged in the pediatric age group compared to the adult group, although this did not reach statistical significance on direct comparison (mean difference of −10.4 [−27.4, 6.6], p= 0.2103). In pediatric subjects, there was a mean absolute increase in ppFEV1 of 6.1 [−4.1, 16.2] at six months (p=0.2058); whereas, adult subjects experienced an increase of 16.4 [1.1, 31.8] that was a statistically significant change from baseline (p=0.0393, Supplementary Table 2). Pediatric subjects reached normalized ppFEV1 (achieving a mean ppFEV1 of 94.6% at 6 months with 76% of patients exceeding 100% predicted), whereas adults had a mean ppFEV1 of 63.1%, at 6 months post ivacaftor at the time of study completion.
Effect of ivacaftor on nutritional status
Effects on nutritional status at each time point are summarized for the overall cohort in Supplementary Table 1 and by age (>/=18yo or <18yo) in Supplementary Table 2. Participants experienced a significant improvement in nutritional status, with a mean BMI increase of 1.4 kg/m2 (p<0.001, Figure 2B) and weight of 5.1 kg (p=0.0002) by six months. These results were of similar magnitude in children and adults. The pediatric group had a mean BMI gain of 1.3 kg/m2 (p=0.0516), weight gain of 5.8 kg (p=0.0123), and height z-score of 0.2 (p = 0.0179) by six months while the adults had a mean weight gain of 4.3 kg (p=0.0046) and BMI gain of 1.4 kg/m2 (p=0.0042). There was a 5-point increase in height percentile for subjects <20 years of age at 6 months (p=0.0344), while neither the weight percentiles nor the BMI percentiles exhibited statistically significant changes over the course of this study.
Effect of ivacaftor on SC
Figure 2C shows changes in SC over the course of the study and the effects on CFTR function at each time point are summarized for the overall cohort in Supplementary Table 1 and by age (>/=18yo or <18yo) in Supplementary Table 2. Overall, the SC significantly decreased by −53.5 mEq/L [−63.3, −43.7) at one month (p<0.0001); −51.4 mEq/L [−63.2, −39.5) at three months (p<0.0001); and −48.6 mEq/L [−67.4, −29.9] at six months (p<0.0001). Baseline SC among all participants was 89.6 mEq/L; in the pediatric group this was 99.0 mEq/L, and the adults 79.2 mEq/L (mean difference = 19.7, p=0.0374). Perhaps consequently, the pediatric group had a somewhat larger change in sweat chloride (SC) at all time points, as illustrated in a sustained, significant reduction at six months of −57.3 mEq/L [−85.5, −28.7] compared to −37.2 mEq/L [−67.5, −6.8] in adults (−20.1 mEq/L difference, p-value=0.2578).
Effect of ivacaftor on patient-reported quality-of-life instruments
In this study, the CFQR-Respiratory, CFRSD, and SNOT-20 were used to assess patient reported symptoms (Figure 2D). Effects on quality of life at each time point are summarized for the overall cohort in Supplementary Table 1 and by age (>/=18yo or <18yo) in Supplementary Table 2. While score improvements (indicating reduced symptoms) in all three instruments were reported at one month (CFQ-R-Respiratory improvement of +16.1 points, p=0.0035; CFRSD reduction of −11.4, p=0.0154; SNOT-20 reduction of −0.4, p=0.0098), only improvement in CFQ-R was statistically significant at six months (improvement of 14.2 points, p=0.0366).
The response in patient reported outcomes varied by age group. At baseline the pediatric and adult CFQ-R scores were discrepant, reflecting the overall better health of the pediatric population. The pediatric baseline score was 74.0 whereas the adult score was 48.9. The improvement above baseline reported by the adult group was larger at all time points (Supplementary Table 2). Similar trends were seen in the CFRSD, with a lower baseline score in children compared to adults, and a larger, although not statistically significant score reduction by six months in adults (Supplementary Table 2). The ivacaftor response to SNOT-20 scores, in contrast, did not vary as widely between adults and children (Supplementary Table 2).
Effect of ivacaftor use on hospitalization frequency and isolation of Pseudomonas aeruginosa (Pa)
Data from the CFFPR were extracted to evaluate the effect of ivacaftor use on hospitalization rates and respiratory Pa isolation during the 6 month follow-up period as compared to findings prior to ivacaftor use. There was a mean of 16.7 months of follow-up data for subjects taking ivacaftor. Because of variation in clinical follow up (compared to the research visits) and data entry into CFFPR, not all subjects are included in all time-points. While the percent of participants hospitalized after ivacaftor initiation decreased following ivacaftor use (Figure 3A), these changes were not statistically significant. In the immediate 6 months prior to starting ivacaftor, 38% of participants were hospitalized; this decreased to 10% in the first 6 months after initiation of ivacaftor, a finding that was sustained for an additional 6 months. When adjusted for differential follow up, there was a statistically significant decrease in hospitalization rates post-ivacaftor initiation. On average, there were 1.05 hospitalizations/year before ivacaftor, 0.34 post-ivacaftor, and a paired rate reduction of 0.71 hospitalizations per participant-year (95% CI = [0.19-1.24], p-value = 0.004)
Figure 3. Hospitalization Rates and P. Aeruginosa Isolation.
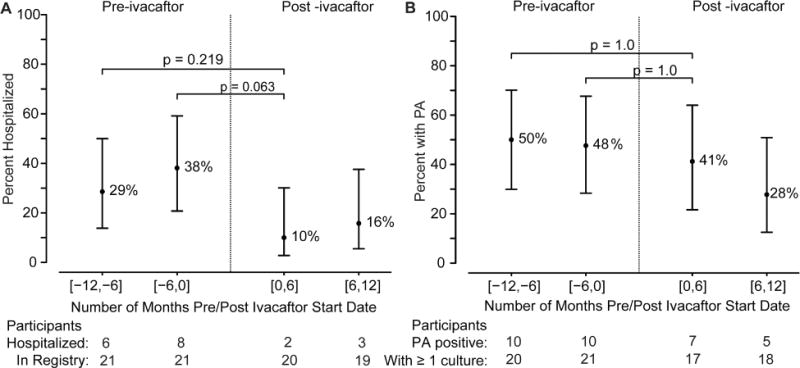
Data was extracted from CFF Patient Registry for the subjects in the time period 12 months before initiation of ivacaftor and in the time period after initiation of ivacaftor. (A) Percent of subjects hospitalized in the indicated time periods; (B) Percent of subjects with P. Aeruginosa (PA) in the indicated time period.
In the year prior to initiation of ivacaftor, approximately 50% of subjects had at least one culture positive for P. aeruginosa (Figure 3B). After initiation, 41% of participants had at least one positive culture in the first 6 months, while only 28% were positive 6-12 months after initiation of ivacaftor, although these changes were not statistically significant. The change at 6-12 months post initiation represents a 41.6% relative reduction in P. aeruginosa isolation compared to the same interval one-year prior. In G551D patients a 32.7% reduction was previously observed.18 Expectorated sputum gradually made up a smaller proportion of the respiratory cultures over time, and consequently the use of oropharyngeal swabs increased during the study. 83% of samples were expectorated in -12 to -6 months pre-ivacaftor, and this sequentially diminished for each additional 6 month interval: 76%, 72%, and finally 60% in 6-12 months after initiation of ivacaftor, respectively; this may have been due to reductions in productive cough in the cohort with prolonged CFTR modulator therapy, as also experienced in G551D participants.[13, 21]
Discussion
In this six-month observational study, the effects of ivacaftor on patients with non-G551D gating mutations were beneficial and sustained over the course of 180 days. There were overall improvements in lung function, SC, weight, growth (height), quality of life, and number of hospitalizations (adjusted for differential follow up). The magnitude of response appears generally comparable to the G551D participants of the GOAL study [22], and the conclusions substantiate prior randomized controlled trials [12, 23, 24]. Supporting data previously observed in G551D participants, there was also a reduction in the prevalence of detectable Pa infection, although this did not achieve statistical significance.
The beneficial response to ivacaftor in non-G551D gating patients was not uniform, and appears to be age-related, in part, likely related in part to differences in baseline functional status of younger vs. older participants. The short-term improvement in lung function among all subjects in this study was greater (+9.3% at one month) than that reported in the KONNECTION (7.5% at 8 weeks) [12] study. When the adult participants of this study were analyzed separately, the improvement as compared to all subjects in KONNECTION was even larger (+16.4 at 6 months compared to +13.5 in the KONNECTION open label extension). As has been reported previously, the change in lung function among the pediatric group was less substantial, and may reflect the already high ppFEV1 among that group. As a comparison, the ENVISION trial [23] enrolled patients with at least one G551D allele who were 6-11 years of age. These patients had a mean baseline ppFEV1 of 84.7, and the mean absolute increase from baseline was 12.6 percentage points. In our study, the baseline ppFEV1 was slightly higher (88.7), and the improvement in ppFEV1 was slightly lower (6.1%).
The effect of ivacaftor in children <18y is less pronounced for lung function, CFQ-R, and CFRSD, possibly due to a ceiling effect on these parameters, while effects on SC are much greater. This emphasizes that SC has important limitations as a clinical biomarker to predict effects on ppFEV1 across the entire spectrum of the CF patient population, and cannot be used on an individual basis for all patients [25–28]. In children, other modalities of lung function assessment, such as the lung clearance index (LCI), may be more applicable since this measure has been shown to be more sensitive to changes in lung function in children [29–31].
The differences in the two age groups are also pronounced with respect to changes in sweat chloride. When compared to other clinical trials in pediatric gating mutation patients, the baseline sweat chloride and change in sweat chloride with ivacaftor treatment were comparable to sweat chloride values observed in other ivacaftor trials [23, 29, 32]. In contrast, the adult patients had a relatively lower baseline sweat chloride, probably due to the small sample size in the study. While this differs from previous controlled trials, we nevertheless observed that the sweat chloride achieved following ivacaftor treatment as similar to previously published values, suggesting a similar degree of CFTR modulation [5]. We also speculate that differences in the change in sweat chloride between age groups may be due to variable pharmacokinetics of CFTR modulators between children and adults.
Our study was limited by a small sample size, reflecting the paucity of non-G551D gating mutations amongst patients with CF. Three subjects had a residual function mutation on the alternate allele that are known to respond to ivacaftor, and one subject was homozygous for a non-G551D gating mutation, which may also affect results, potentially augmenting response beyond the single gating allele. Small sample size, for example, is probably responsible for a relatively low baseline sweat chloride in the adult subjects studied and apparent differences in changes in sweat chloride marking response to CFTR modulation. The study is an observational study, and all data are derived from patients who were on ivacaftor – there is no placebo group for comparison. Importantly, 64.6% of pediatric participants are in the pubertal range (12-17 years), which may impact the growth velocity during this time frame, but would be expected to be enhanced by ivacaftor use, as reported previously [33]. All participants were unblinded and aware that they were on the study drug, which could influence subjective responses on the CFQR, SNOT-20 and CFSRD scores. Notably, the response rates in the ENVISION study (children, 5.9) [23]; PERSIST (maximum of 10.8 from all groups) [24]; and KONNECTION (adults, 9.6) [12] were all lower than the improvement seen in this study among similar patients, which may reflect this bias.
We acknowledge limitations in the data collection for rates of exacerbations and Pa culture positivity. As these data were derived from CFFPR, inherent bias toward treatment effect in the setting of unblinded treatment may have falsely reduced hospitalizations. Nevertheless, the findings were consistent with a previous cohort of G551D patients, and were sustained over one year of observation. While changes in Pa status were not statistically significant, probably due to the small sample size, the proportional reduction in Pa positivity was highly consistent with our previous report in G551D patients [22] and is supported by mechanistic data indicating reduced bacterial survival upon ivacaftor administration.[34]
In this study, ivacaftor resulted in improvements in multiple parameters in our patient population with non-G551D gating mutations. These improvements were sustained and significant over the course of the entire observation period, supporting the continued use of ivacaftor in both adult and pediatric patients with these alleles. In particular, the treatment of young children suggests the possibility of significantly modifying their lifelong disease progression.
Supplementary Material
Highlights.
Twenty-one subjects were followed for 6 months on ivacaftor in an open label trial.
This is the largest study of participants taking ivacaftor with non-G551D gating mutations
Participants experienced significant improvements in ppFEV1, sweat chloride, and BMI.
Positive responses in these endpoints were observed in pediatric and adult participants
Pediatric subjects had higher baseline lung function and smaller improvement in ppFEV1
Acknowledgments
None
SOURCES OF FUNDING
The authors gratefully acknowledge funding support for this work from the NIH (UL1 TR001417, P30 DK072482, to S.M.R and KL2 to G.M.S.; NIH/NCATS Colorado CTSA Grant Number UL1 TR001082., P30 NIDDDK 089507 to S.D.S) and the CFF Foundation (GOAL11K1, GOAL13K0, Clancy 09Y0 to G.M.S.).
Abbreviations
- CFTR
Cystic Fibrosis Transmembrane conductance regulator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.M.R., S.D.S., and S.L.H. designed the study. A.B., S.L.H. analyzed the data. G.M.S., J.S.G., S.M.R., A.B., and S.L.H. interpreted the data. J.S.G., A.B., S.L.H. G.M.S. and S.M.R. prepared the manuscript. All authors reviewed the manuscript and approved of the final version of the manuscript before submission.
CONFLICTS OF INTEREST
SMR serves as an investigator and consultant to Vertex Pharmaceuticals, the manufacturer of ivacaftor, on the design and conduct of CF clinical trials. GMS serves as an investigator on studies conducted by Vertex Pharmaceuticals.
References
- 1.Collawn JF, Matalon S. CFTR and lung homeostasis. Am J Physiol Lung Cell Mol Physiol. 2014;307(12):L917–23. doi: 10.1152/ajplung.00326.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 3.Van Goor F, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106(44):18825–30. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Accurso FJ, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsey BW, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quon BS, Rowe SM. New and emerging targeted therapies for cystic fibrosis. BMJ. 2016;352:i859. doi: 10.1136/bmj.i859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotha K, Clancy JP. Ivacaftor treatment of cystic fibrosis patients with the G551D mutation: a review of the evidence. Ther Adv Respir Dis. 2013;7(5):288–96. doi: 10.1177/1753465813502115. [DOI] [PubMed] [Google Scholar]
- 8.Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16(1):45–56. doi: 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oliver KE, et al. Transformative therapies for rare CFTR missense alleles. Curr Opin Pharmacol. 2017;34:76–82. doi: 10.1016/j.coph.2017.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foundation, C.F. Patient Registry Annual Data Report. 2016 [Google Scholar]
- 11.US Food and Drug Administration Approves KALYDECO™ (ivacaftor) for Use in Eight Additional Mutations that Cause Cystic Fibrosis. Vertex Pharmaceuticals, Inc.; 2014. U.S. Food and Drug Administration Approves KALYDECO™ (ivacaftor) for Use in Eight Additional Mutations that Cause Cystic Fibrosis. http://investors.vrtx.com/ [Google Scholar]
- 12.De Boeck K, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–80. doi: 10.1016/j.jcf.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Rowe SM, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190(2):175–84. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee M, et al. Systematic Computational Identification of Variants That Activate Exonic and Intronic Cryptic Splice Sites. Am J Hum Genet. 2017;100(5):751–765. doi: 10.1016/j.ajhg.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quittner AL, et al. Development and validation of The Cystic Fibrosis Questionnaire in the United States: a health-related quality-of-life measure for cystic fibrosis. Chest. 2005;128(4):2347–54. doi: 10.1378/chest.128.4.2347. [DOI] [PubMed] [Google Scholar]
- 16.Piccirillo JF, Merritt MG, Jr, Richards ML. Psychometric and clinimetric validity of the 20-Item Sino-Nasal Outcome Test (SNOT-20) Otolaryngol Head Neck Surg. 2002;126(1):41–7. doi: 10.1067/mhn.2002.121022. [DOI] [PubMed] [Google Scholar]
- 17.Goss CH, et al. Patient-reported respiratory symptoms in cystic fibrosis. J Cyst Fibros. 2009;8(4):245–52. doi: 10.1016/j.jcf.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Standardization of Spirometry, 1994 Update. American Thoracic Society. Am J Respir Crit Care Med. 1995;152(3):1107–36. doi: 10.1164/ajrccm.152.3.7663792. [DOI] [PubMed] [Google Scholar]
- 19.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159(1):179–87. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, et al. Pulmonary function between 6 and 18 years of age. Pediatr Pulmonol. 1993;15(2):75–88. doi: 10.1002/ppul.1950150204. [DOI] [PubMed] [Google Scholar]
- 21.Heltshe SL, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis. 2015;60(5):703–12. doi: 10.1093/cid/ciu944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heltshe SL, et al. Pseudomonas aeruginosa in Cystic Fibrosis Patients With G551D-CFTR Treated With Ivacaftor. Clin Infect Dis. 2014 doi: 10.1093/cid/ciu944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies JC, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187(11):1219–25. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKone EF, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST) Lancet Respir Med. 2014;2(11):902–10. doi: 10.1016/S2213-2600(14)70218-8. [DOI] [PubMed] [Google Scholar]
- 25.Accurso FJ, et al. Sweat chloride as a biomarker of CFTR activity: proof of concept and ivacaftor clinical trial data. J Cyst Fibros. 2014;13(2):139–47. doi: 10.1016/j.jcf.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barry PJ, et al. Sweat chloride is not a useful marker of clinical response to Ivacaftor. Thorax. 2014;69(6):586–7. doi: 10.1136/thoraxjnl-2013-204532. [DOI] [PubMed] [Google Scholar]
- 27.Durmowicz AG, et al. Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience. Chest. 2013;143(1):14–8. doi: 10.1378/chest.12-1430. [DOI] [PubMed] [Google Scholar]
- 28.Fidler MC, et al. Correlation of sweat chloride and percent predicted FEV1 in cystic fibrosis patients treated with ivacaftor. J Cyst Fibros. 2017;16(1):41–44. doi: 10.1016/j.jcf.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Davies J, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med. 2013;1(8):630–8. doi: 10.1016/S2213-2600(13)70182-6. [DOI] [PubMed] [Google Scholar]
- 30.Stanojevic S, et al. Progression of Lung Disease in Preschool Patients with Cystic Fibrosis. Am J Respir Crit Care Med. 2017;195(9):1216–1225. doi: 10.1164/rccm.201610-2158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lenherr N, et al. Ivacaftor in a young boy with the rare gating mutation S549R–use of lung clearance index to track progress: a case report. BMC Pulm Med. 2015;15:123. doi: 10.1186/s12890-015-0120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies JC, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4(2):107–15. doi: 10.1016/S2213-2600(15)00545-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stalvey MS, et al. Growth in Prepubertal Children With Cystic Fibrosis Treated With Ivacaftor. Pediatrics. 2017;139(2) doi: 10.1542/peds.2016-2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pohl K, et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood. 2014;124(7):999–1009. doi: 10.1182/blood-2014-02-555268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.