

Abstract
Background
Ischemic heart injury activates calpains and endoplasmic reticulum (ER) stress in cardiomyocytes. This study investigated whether over-expression of calpastatin, an endogenous calpain inhibitor, protects the heart against myocardial infarction (MI) by inhibiting ER stress.
Methods
Mice over-expressing calpastatin (Tg-CAST) and littermate wild type (WT) mice were divided into four groups: WT-sham, Tg-CAST-sham, WT-MI, and Tg-CAST-MI, respectively. WT-sham and Tg-CAST-sham mice showed similar cardiac function at baseline. MI for 7 days impaired cardiac function in WT-MI mice, which was ameliorated in Tg-CAST-MI mice.
Results
Tg-CAST-MI mice exhibited significantly decreased diameter of the left ventricular cavity, scar area, and cardiac cell death compared to WT-MI mice. WT-MI mice had higher cardiac expression of C/EBP homologous protein (CHOP) and BIP, indicators of ER stress, compared to WT-sham mice, indicative of MI-induced ER stress. This increase was abolished in Tg-CAST-MI hearts. Furthermore, administration of tauroursodeoxycholic acid, an inhibitor of ER stress, reduced MI-induced expression of CHOP and BIP, scar area, and myocardial dysfunction. In an in vitro model of oxidative stress, H2O2 stimulation of H9c2 cardiomyoblasts induced calpain activation, CHOP expression, and cell death, all of which were prevented by the calpain inhibitor PD150606, as well as CHOP silencing.
Conclusions
Over-expression of calpastatin ameliorates MI-induced myocardial injury in mice. These protective effects of calpastatin are partially achieved through suppression of the ER stress/CHOP pathway.
Keywords: Calpastatin, calpain, myocardial infarction (MI), endoplasmic reticulum stress (ER stress), C/EBP homologous protein (CHOP)
Introduction
Cardiac remodeling, a process involving loss of cardiomyocytes, enlargement of infarct area, activation of fibroblasts, and deposition of fibrosis (1,2), is one of the leading mechanisms resulting in adverse cardiac function in the wake of acute myocardial infarction (MI). During cardiac remodeling, misfolded or unfolded proteins accumulate in the lumen of the endoplasmic reticulum (ER) in the myocardium, causing ER stress. ER stress initiates a highly conserved physiological activity, termed unfolded protein response (UPR), which enhances the ability of the ER to refold unfolded proteins and eliminate damaged proteins. ER stress can be triggered by activation of three ER stress sensors, including PKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) (3). During heart failure, ER stress in cardiomyocytes causes specific morphological changes, suggesting its involvement in cardiac pathophysiologic processes (4-6). These changes in ER morphology were concomitant with modifications of molecules, such as tumor necrosis factor-α (TNF-α), caspase 3, p53 upregulated modulator of apoptosis (PUMA), and glucose regulated protein 94 (GRP 94), that regulate ER stress-related pathological processes including hypoxia, production of reactive oxygen species (ROS), enhanced protein synthesis, and induction of lipotoxicity (6,7). ER stress has also been detected in both hypertrophic and failure phases induced by transverse aortic constriction in an animal model (5), and is potentially associated with the pathogenesis of human heart failure, as evidenced by the increased expression of binding immunoglobulin protein (BIP, also referred to as GRP78), an ER chaperone and signaling regulator (8), in failing human hearts (4,5). Furthermore, prolonged or excessive ER stress may induce apoptosis through c-Jun NH2-terminal kinase 1/2 (JNK1/2) and C/EBP homologous protein (CHOP) signaling pathways (9,10). Previously, we showed that hypoxia and ischemia reperfusion in cardiomyocytes triggered ER stress, increased CHOP expression, and promoted apoptosis-related cleavage of caspase 12 (11,12). All of these findings support the premise that increased ER stress accentuates cardiac injury. Indeed, inhibition of ER stress has been shown to alleviate myocardial injury (12,13). Thus, ER stress represents a potential target to prevent post-MI remodeling.
The human calpain family consists of 15 calcium-dependent thiol proteases, which execute a wide range of functions in a variety of physiological and pathophysiological processes (14,15). Among these calpain members, calpain-1 and calpain-2, whose activation depends on two distinct ranges of calcium concentrations within cells (16), are the two most extensively studied calpains and have been shown to be involved in cardiac pathophysiology. For instance, increased calpain expression and activity were observed in both infarcted animal hearts and in the myocardium of heart failure patients (17-20). Correspondingly, pharmacological inhibition of calpains alleviated cardiac injury and preserved cardiac structure and function in acute MI (21). Of note, calpain-1 and calpain-2 activity are also tightly regulated by their endogenous specific inhibitor, calpastatin (15), as evidenced by the findings that over-expression of calpastatin restrained calpain activity both in vitro (22) and in vivo (23). However, conflicting findings were reported regarding the exact role of calpastatin in MI. Global over-expression of calpastatin was shown to limit MI-induced myocardial remodeling in one transgenic mouse model (24), while in another study, ubiquitous over-expression of calpastatin impaired scar healing and accelerated death following MI by compromising immune cell recruitment and activation (25). Our previous studies demonstrated that calpain activation induced ER stress in myocardia during ischemia/reperfusion injury, and inhibition of calpain prevented ER stress and apoptosis (12). These findings prompted us to ask whether the calpain/ER stress pathway modulates myocardial injury following MI and whether calpastatin impacts MI-induced cardiac remodeling through targeting ER stress.
In this study, we first validated the protective role of calpastatin in myocardial injury following MI in mice with global calpastatin over-expression, and then investigated whether the calpain/ER stress/CHOP pathway was involved in post-MI remodeling in vivo and in vitro.
Methods
Animals
This investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011). All experimental procedures were approved by the Animal Use Subcommittee at the Western University, Canada. Transgenic mice over-expressing calpastatin (Tg-CAST) were generously provided by Dr. Laurent Baud (Institut National de la Santé et de la Recherche Médicale, Paris, France) through the European Mouse Mutant Archive (26). All adult mice used in this study were littermates of the same generation.
MI model
Wild type (WT) (C57BL/6 background) and Tg-CAST mice were divided into four groups: WT-sham, Tg-CAST-sham, WT-MI, and Tg-CAST-MI, respectively. MI was performed as previously described (20). Briefly, adult male mice, approximately 2 months old, were anesthetized intraperitoneally (i.p.) with a mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg). The mice then underwent lateral thoracotomy and a suture was placed under the left descending (LD) artery. In the MI group, the LD was permanently ligated, whereas WT mice in the control (sham) group had the same surgical procedures except that the suture placed under the LD was not tied. After surgery, buprenorphine (0.05 mg/kg per 8 h) was subcutaneously administered for 48 h. After surgery, mice were carefully monitored and survival was recorded for 7 days.
To investigate the role of ER stress in MI in vivo, tauroursodeoxycholic acid (TAUR, 150 mg/kg per day i.p.) or vehicle (ddH2O) was given daily for 7 days starting immediately after surgery. In this experimental setting, WT mice were divided into the following four groups: vehicle-sham, TAUR-sham, vehicle-MI, and TAUR-MI, respectively. Mice in the vehicle group were given the same volume of saline.
Echocardiography
Seven days after MI, animals were anesthetized with inhalation of 1% isoflurane. Echocardiography was performed as previously described (20). M-mode at the level of the papillary muscles was used to obtain cardiac functional parameters, including left ventricular end-systolic inner diameter (LVIDs), left ventricular end-diastolic inner diameter (LVIDd), LV posterior wall thickness in end-diastole (LVPWd) and in end-systole (LVPWs), ejection fraction (EF), and fractional shortening (FS).
Cell culture and siRNA treatment
Rat cardiomyoblasts, H9c2 cells (ATCC, USA), were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin/ampicillin (Gibco). Cells were pretreated with PD150606 (10 µM), TAUR (200 µM) or vehicle for 30 min followed by incubation with H2O2 (Sigma, USA, 400 µM) or ddH2O for 6 h. Cholesterol-conjugated siRNA for CHOP was purchased from Guangzhou RiboBio Co., Ltd. (China). The sequence for CHOP siRNA was: 5'CCAGCTACCTTCTGGGTAA3'. A scrambled siRNA conjugated with cholesterol served as a control. H9c2 cells were incubated with siRNA (50 nmol/L) in regular culture medium. Cholesterol-conjugated siRNAs entered H9c2 directly with an efficiency of more than 90% (7).
Measurement of caspase 3 activity
Caspase 3 activity was evaluated using a caspase 3 fluorescence assay kit (Biomol Research Laboratories, USA) as previously described (22,27-29).
Measurement of calpain activity
Calpain activity was measured by casein zymography as described in our previous study (27,30).
Determination of cell death in situ
Cardiomyocyte death in vivo and in vitro was assessed by propidium iodide (PI) and Hoechst 33342 staining was used as a nuclear counterstain as described in our previous report (31). Briefly, mice were euthanized 7 days after MI. Isolated hearts were retrogradely-perfused with Krebs-Henseleit buffer (KHB) and infused with a mixture of PI and Hoechst 33324 (50 and 1 µg/mL in KHB, respectively). Eight minutes after infusion, hearts were fixed by infusion with 4% paraformaldehyde and embedded in optimal cutting temperature (OCT) compound and stored at −80 °C. The frozen hearts were sectioned at 5 µm thickness and examined under a fluorescent microscope (Axio Scope A1, Zeiss). PI-positive nuclei (red) were counted from 20 randomly selected fields (magnification ×200) of each sample. The percentage of PI positively stained nuclei to total nuclei (Hoechst 33324 staining: blue) in the same field were calculated.
H9c2 cell death was determined as described previously (31). Briefly, cells were incubated with a mixture of PI and Hoechst 33324 in culture medium at 37 °C for 5 min. Pictures were captured from the fluorescent microscope (Axio Scope A1, Zeiss) after washing with phosphate buffered saline (PBS). At least 5 random fields (magnification ×100) of each well were scored, with each carried out in triplicate.
Western blot
The protein levels of BIP, CHOP, α-spectrin, and glyceraldehyde phosphate dehydrogenase (GAPDH) were measured by Western blot analysis as previously described using specific antibodies against BIP, CHOP, α-spectrin, and GAPDH (all at 1:1,000, Cell Signaling Technology, Inc.) (31).
Statistical analysis
Data were analyzed using GraphPad Prism 5.0 software (GraphPad, USA). All data are presented as mean ± standard deviation (SD). Two-way ANOVA was performed for multi-group comparisons followed by Newman-Keuls test. Survival curves of experimental groups were created using the Kaplan and Meier analysis and compared using the log-rank test. A value of P<0.05 was considered statistically significant.
Results
Over-expression of calpastatin is cardioprotective following MI in mice
As shown in Figure 1A, overexpression of calpastatin inhibited the upregulated calpain activity after MI. To determine the effects of over-expression of calpastatin on cardiac function, we first performed echocardiography in mice 7 days after MI in all four groups: WT-sham, Tg-CAST-sham, WT-MI, and Tg-CAST-MI. As shown in Figure 1B,C, there were no significant differences with regard to EF (%) and FS (%) between WT-sham and Tg-CAST-sham. As expected, mice in the WT-MI group exhibited significantly lower EF (%) and FS (%) compared to WT-sham mice, indicating impaired cardiac systolic function. However, these impaired cardiac systolic functional parameters were improved in Tg-CAST-MI mice, suggesting that over-expression of calpastatin relieves myocardial dysfunction. Consistent with the above observations, no differences in LVIDd and LVIDs were observed between WT-sham and Tg-CAST-sham groups (Table 1). However, LVIDd and LVIDs were higher in WT-MI mice compared to WT-sham mice, which was also evidenced by larger LV cavity size (Figure 1D). Again, these altered diastolic parameters in WT-MI mice were improved by over-expressing calpastatin (Table 1 and Figure 1D). The above protective function by calpastatin was further supported by the observation that the ratio of scar/cross-sectional area at the level of the papillary muscles was smaller in Tg-CAST-MI mice compared to WT-MI mice (Figure 1E,F). In addition, PI staining showed that MI resulted in a larger number of dead cells in non-infarcted areas of WT mouse hearts, which was significantly lower in Tg-CAST-MI mouse hearts (Figure 2A,B). These results suggest that inhibition of calpains by calpastatin prevents infarct expansion, reduces myocardial remodeling, and improves myocardial function after MI. However, the 7-day mortality rate remained comparable between WT-MI and Tg-CAST-MI mice (Figure 2C).
Figure 1.

Over-expression of calpastatin protects hearts against MI in mice. (A) Calpain activity was upregulated in the infarcted myocardium in WT mice and restrained by over-expression of calpastatin in Tg-CAST mice. (B,C) Ejection fraction (EF%) and fractional shortening (FS%) were determined by echocardiography 7 days after MI. (D) Measurement of LV cavity size was performed at the papillary muscle level 7 days after MI. (E) Representative photomicrographs of Picrosirius red staining captured at the papillary muscle level 7 days after MI are shown. (F) Quantifications of scar size at the papillary muscle level were performed 7 days after MI. Data are represented as mean ± SD. n=5–6 per group. *, P<0.05 versus sham group; †, P<0.05 versus WT-MI. MI, myocardial infarction; WT, wild type.
Table 1. Over-expression of calpastatin ameliorates cardiac dysfunction following MI.
| Group | WT-sham (n=6) | Tg-CAST-sham (n=6) | WT-MI (n=6)n=6) | Tg-CAST-MI (n=6) |
|---|---|---|---|---|
| EF | 75.95±8.31 | 76.94±7.12 | 40.91±10.39* | 61.96±4.66† |
| FS | 44.66±7.96 | 43.37±3.44 | 20.24±5.86* | 33.23±4.71† |
| IVSd | 0.83±0.10 | 0.98±0.13 | 0.70±0.11* | 1.09±0.16† |
| IVSs | 1.43±0.12 | 1.51±0.12 | 1.00±0.18* | 1.51±0.18† |
| LVIDd | 3.78±0.51 | 3.39±0.35 | 4.78±0.37* | 3.94±0.33† |
| LVIDs | 2.10±0.47 | 1.96±0.29 | 3.83±0.50* | 2.66±0.21† |
| LVPWd | 0.75±0.13 | 0.62±0.07 | 0.75±0.13 | 0.9±0.11† |
| LVPWs | 1.37±0.30 | 1.06±0.26 | 0.90±0.12* | 1.43±0.08† |
| LV Mass | 105.43±14.97 | 99.25±12.98 | 144.86±32.89 | 172.92±36.89 |
| LV VOLd | 62.68±20.81 | 47.73±11.47 | 107.59±19.87* | 68.24±12.93 |
| LV VOLs | 15.55±8.14 | 12.52±4.78 | 64.57±19.87* | 25.09±5.91 |
| HR | 504±50 | 484±44 | 462±38 | 429±23 |
EF, ejection fraction; FS, fractional shortening; IVSs, interventricular septum wall thickness in end-systole; IVSd, interventricular septum wall thickness in end-diastole; LVIDd, left ventricle end-diastolic inner diameter; LVIDs, left ventricle end-systolic inner diameter; LVPWs, left ventricle posterior wall thickness in end-systole; LVPWd, left ventricle posterior wall thickness in end-diastole; LV VOLd, LV volumes at end-diastole; LV VOLs, LV volumes at end-systole; HR, heart rate. Data are presented as mean ± SD, n=6 per group. *, P<0.05 versus corresponding sham group; †, P<0.05 versus WT-MI.
Figure 2.

Over-expression of calpastatin decreases MI-induced cardiomyocyte death in mice. Cell death in non-infarcted areas was determined by PI, and Hoechst staining was used for nuclear counterstaining. (A) Representative microphotographs of Hoechst staining (blue) and PI staining (red) on sections of non-infarcted myocardia prepared from WT-MI and Tg-CAST-MI mice are shown. Arrows indicate Hoechst/PI positive cells. (B) Quantitative analysis of (A) was performed as described in “Methods” section. n=6 per group. (C) Analysis of 7-day mortality rates of WT-MI and Tg-CAST-MI mice. †, P<0.05 versus WT-MI group. MI, myocardial infarction; WT, wild type; PI, propidium iodide.
Over-expression of calpastatin prevents MI-induced ER stress in mouse hearts
ER stress can be induced in ischemic hearts (13,32,33), and induction of CHOP and BIP expression is an indicator of ER stress (34). Since our recent study reported that calpain activation induced ER stress in cardiomyocytes (7,12), we measured the protein levels of CHOP and BIP in cardiac tissues obtained from mice of four experimental groups. As shown in Figure 3A,B,C,D [original blots for panel (A) and (C) are presented in Figures S1 and S2], MI hearts had significantly higher CHOP and BIP expression in the non-infarcted areas of WT mouse hearts; in contrast, CHOP and BIP levels were much lower in Tg-CAST-MI mouse hearts. This result suggests that over-expression of calpastatin prevents ER stress induced by MI.
Figure 3.

Implication of calpains and ER stress in MI-induced cardiac injury in mice. (A,B) Representative Western blots and quantification of CHOP expression relative to GAPDH in non-infarcted areas from 2 out of 5–6 hearts from WT-sham, Tg-CAST-sham, WT-MI, and Tg-CAST-MI groups, respectively. (C,D) Representative Western blots and quantification of BIP expression relative to GAPDH from 2 out of 5–6 hearts of WT-sham, Tg-CAST-sham, WT-MI, and Tg-CAST-MI groups, respectively. (E,F) Representative Western blots and quantification of CHOP expression relative to GAPDH in non-infarcted areas from 2 out of 5–6 different hearts of vehicle-sham, TAUR-sham, vehicle-MI, and TAUR-MI groups, respectively. (G,H) Representative Western blots and quantification of BIP expression relative to GAPDH in non-infarcted areas from 2 out of 5–6 different hearts of vehicle-sham, TAUR-sham, vehicle-MI, and TAUR-MI groups, respectively. Data are presented as mean ± SD. n=5–6 per group. *, P<0.05 versus sham group; †, P<0.05 versus vehicle-MI. MI, myocardial infarction; WT, wild type; TAUR, tauroursodeoxycholic acid.
Figure S1.
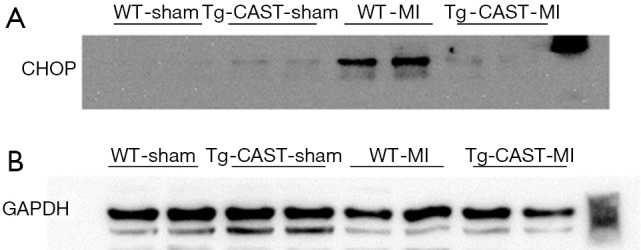
(A,B) The original blots for panel (A) presented in Figure 3 in the main text.
Figure S2.
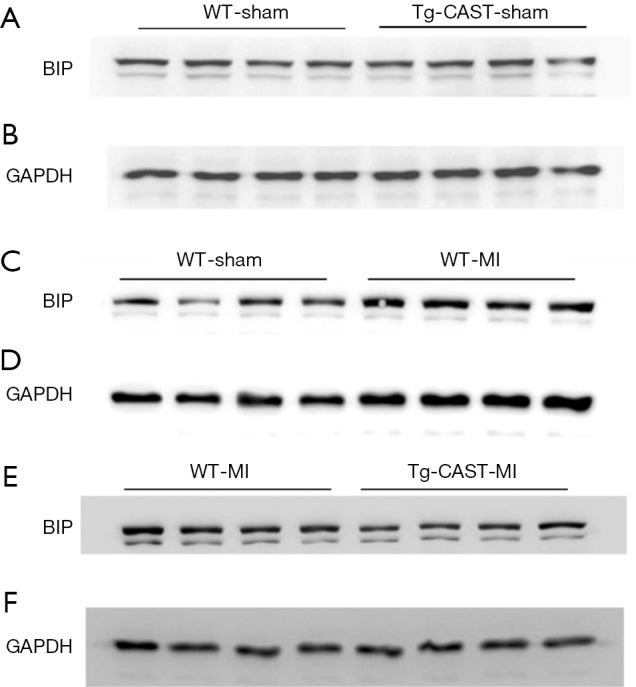
(A,B,C,D,E,F) The original blots for panel (C) presented in Figure 3 in the main text.
Inhibition of ER stress reduces myocardial injury after MI
Having shown that over-expression of calpastatin prevented ER stress in infarcted hearts, we hypothesized that inhibition of ER stress could protect the heart against MI-induced injury. To test this hypothesis, we treated sham and MI mice with either vehicle or TAUR, an inhibitor of ER stress. After MI, mice received TAUR on a daily basis for 7 days (150 µg/kg/day, i.p.). This dose of TAUR was chosen based on previous studies (35,36). Consistent with the above findings, MI substantially upregulated the expression of CHOP and BIP in vehicle-MI mouse hearts compared to vehicle-sham hearts, which was abrogated by administration of TAUR (TAUR-MI group) (Figure 3E,F,G,H) [original blots for panels (E) and (G) are presented in Figures S3 and S4], confirming the inhibition of ER stress. TAUR treatment also significantly ameliorated myocardial systolic and diastolic dysfunction after MI compared to the vehicle-MI group (Figure 4A,B, and Table 2). Altered diastolic parameters including LVIDd, LVIDs and LV cavity size in MI mice were improved by TAUR administration (Table 2 and Figure 4C).Furthermore, the ratio of scar/cross-sectional area at the level of the papillary muscles was smaller in TAUR-MI mice compared to vehicle-MI mice (Figure 4D,E). PI staining also showed that the TAUR-MI mouse hearts exhibited significantly lower cell death compared to vehicle-MI hearts (Figure 5A,B). These results support the view that inhibition of ER stress limits infarct expansion, attenuates myocardial remodeling, and improves myocardial function after MI, although the inhibition of ER stress by TAUR was not sufficient to reduce the 7-day morality rate after MI (Figure 5C).
Figure S3.
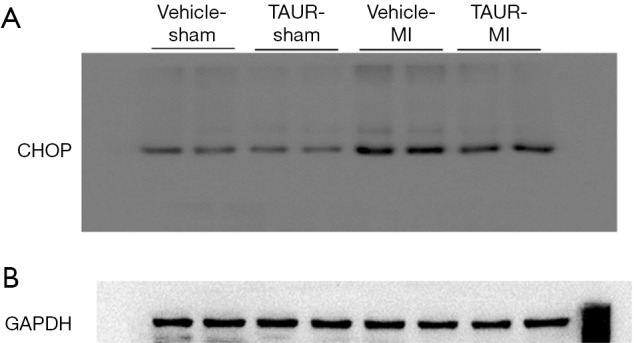
(A,B) The original blots for panel (E) presented in Figure 3 in the main text.
Figure S4.
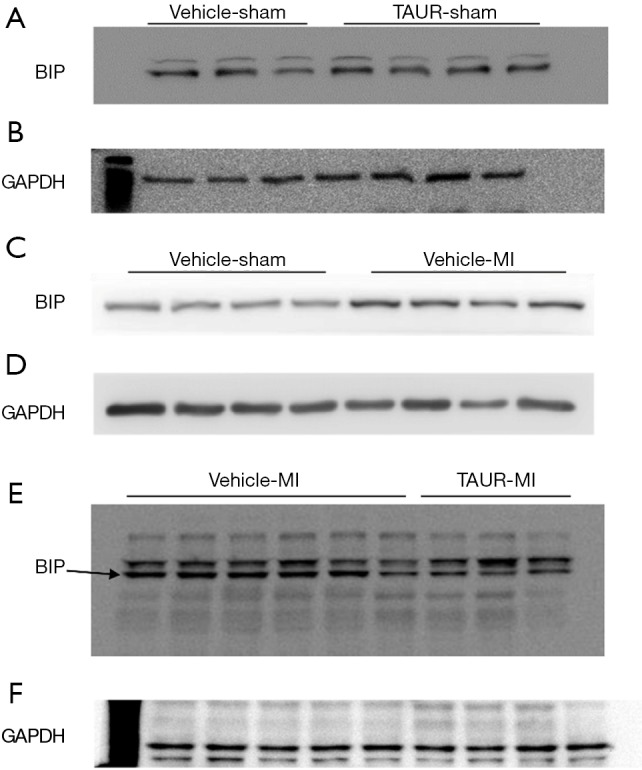
(A,B,C,D,E,F) The original blots for panel (G) presented in Figure 3 in the main text. (E,F) are the original blots of the last two blots in Figure 3G. The former 6 samples were from the vehicle MI group while the last 3 samples were from the TAUR MI group. Figures were cropped for better resolution.
Figure 4.

TAUR ameliorates MI-induced cardiac dysfunction. (A,B) Ejection fraction (EF%) and fractional shortening (FS%) were determined by echocardiography 7 days after MI. (C) Measurement of LV cavity size was performed at the papillary muscle level 7 days after MI. (D) Representative photomicrographs of Picrosirius red staining at the papillary muscle level 7 days after MI are shown. (E) Quantification of scar size at the papillary muscle level was performed 7 days after MI. Data are presented as mean ± SD. n=5–6 per group. *, P<0.05 versus sham group; †, P<0.05 versus vehicle-MI group. MI, myocardial infarction; WT, wild type; TAUR, tauroursodeoxycholic acid.
Table 2. TAUR treatment improves cardiac function after MI.
| Group | Vehicle-sham (n=6) | TAUR-sham (n=6) | Vehicle-MI (n=6) | TAUR-MI (n=6) |
|---|---|---|---|---|
| EF | 75.95±8.31 | 65.08±6.84 | 43.44±7.86* | 55.93±4.28† |
| FS | 44.66±7.96 | 35.27±5.27 | 21.62±4.55* | 29.17±2.76† |
| IVSd | 1.43±0.12 | 1.53±0.26 | 1.08±0.13* | 1.06±0.23 |
| IVSs | 0.83±0.10 | 0.97±0.14 | 0.73±0.08* | 0.70±0.12 |
| LVIDd | 3.78±0.51 | 3.74±0.43 | 4.86±0.46* | 4.62±0.32 |
| LVIDs | 2.10±0.47 | 2.42±0.37 | 3.81±0.48* | 3.27±0.31† |
| LVPWd | 0.75±0.13 | 0.81±0.10 | 0.72±0.09 | 0.78±0.16 |
| LVPWs | 1.37±0.30 | 1.16±0.17 | 0.94±0.11* | 1.02±0.24 |
| LV Mass | 105.43±14.97 | 123.47±27.68 | 144.65±32.79 | 138.56±39.78 |
| LV VOLd | 62.68±20.81 | 60.86±16.08 | 111.92±24.5* | 98.82±16.51 |
| LV VOLs | 15.55±8.14 | 21.47±8.29 | 64.00±19.1* | 43.88±9.71 |
| HR | 504±50 | 474±40 | 449±42 | 459±43 |
EF, ejection fraction; FS, fractional shortening; IVSs, interventricular septum wall thickness in end-systole; IVSd, interventricular septum wall thickness in end-diastole; LVIDd, left ventricle end-diastolic inner diameter; LVIDs, left ventricle end-systolic inner diameter; LVPWs, left ventricle posterior wall thickness in end-systole; LVPWd, left ventricle posterior wall thickness in end-diastole; LV VOLd, LV volumes at end-diastole; LV VOLs, LV volumes at end-systole; HR, heart rate. Data are presented as mean ± SD, n=6 per group. *, P<0.05 versus corresponding sham group; †, P<0.05 versus vehicle-MI group.
Figure 5.

TAUR reduces MI-induced cardiomyocyte death in mice. Cell death in non-infarcted areas was determined by PI, and Hoechst staining was used for nuclear counterstaining. (A) Representative microphotographs of Hoechst staining (blue) and PI staining (red) on sections of non-infarcted myocardia prepared from vehicle-MI and TAUR-MI mice are shown. Arrows indicate Hoechst/PI positive cells. (B) Quantification of (A) was performed as described in “Methods” section. n=6 per group. (C) Analysis of mortality rates of vehicle-MI and TAUR-MI mice 7 days after MI. †, P<0.05 versus vehicle-MI mice. MI, myocardial infarction; PI, propidium iodide; TAUR, tauroursodeoxycholic acid.
Inhibition of calpains prevents apoptosis and ER stress in H9c2 cells induced by oxidative stress
To provide direct evidence to support the role of calpains in apoptotic cell death and ER stress, we used H2O2 to induce oxidative stress and mimic MI conditions in cultured H9c2 cardiomyoblasts. Incubation with H2O2 induced calpain activation as indicated by the higher cleaved fragment of α-spectrin (150/140 kDa, Figure 6A) [original blot for panel (A) is presented in Figure S5], which coincided with increased cell death and caspase 3 activity (Figure 6B,C,D). H2O2 treatment also up-regulated CHOP protein expression, indicative of ER stress (Figure 6E,F). All of these H2O2-induced pathological changes were attenuated by the calpain inhibitor, PD150606. Collectively, these findings demonstrate that calpain activation contributes to ER stress and apoptosis in H9c2 cells in response to oxidant stress.
Figure 6.

Inhibition of calpain activation and CHOP expression reduces cell death in response to oxidative stress. H9c2 cells were incubated with H2O2 (400 µM) or ddH2O (vehicle) in the presence of the calpain inhibitor PD150606 (10 µM) or vehicle. (A) A representative Western blot of cleaved fragments of α-spectrin after H2O2 exposure is shown. (B) Representative microphotographs of Hoechst and PI staining in H9c2 cells treated with vehicle or Pd150606 after H2O2 exposure. (C) Quantification of (B) was performed as described in “Methods” section. (D) Determination of caspase 3 activity in H9c2 cells treated with vehicle or Pd150606. (E) A representative Western blot for evaluation of the expression levels of CHOP and GAPDH from three independent cultures is shown. (F) Quantification of CHOP expression relative to GAPDH in (E) was performed as described in “Methods” section. Data are presented as mean ± SD from three independent assays. *, P<0.05 versus ddH2O; †, P<0.05 versus vehicle. CHOP, C/EBP homologous protein.
Figure S5.

(A) The original blots for panel (A) presented in Figure 6 in the main text. One of the gels of Figure 6 was cropped from the picture shown above. The expression of α-spectrin was detected 6 h, 5 min and 1 h after H2O2 exposure. Since only 6 h data were needed, the corresponding images were cropped and used. (B,C) The original blots for panel (E) presented in Figure 6 in the main text.
Inhibition of the ER stress/CHOP pathway protects H9c2 cells against oxidative stress-induced injury
To further determine whether ER stress contributed to oxidant stress-linked injury, we first inhibited ER stress in H9c2 cells with TAUR (100 µM) and then exposed the cells to H2O2. As shown in Figure 7A,B, pretreatment with TAUR significantly attenuated caspase 3 activity and cell death induced by H2O2, although TAUR did not return these to normal levels compared to the control group, suggesting that an ER stress-independent pathway may also be involved in H2O2-induced cell death.
Figure 7.

TAUR or CHOP silencing reduces cell death induced by oxidative stress. (A) TAUR treatment reduced caspase 3 activity in H9c2 cells in response to H2O2 stimulation. (B) TAUR treatment reduced H9c2 cell death induced by H2O2 stimulation. (C) Representative Western blots showing CHOP expression after CHOP silencing. (D) CHOP silencing reduced caspase 3 activity in H9c2 cells induced by H2O2 stimulation. (E) CHOP silencing reduced H9c2 cell death induced by H2O2 stimulation. Determination of caspase 3 activity and scoring of PI/Hoechst staining positive cells were performed as described in “Methods” section. Data are presented as mean ± SD from three independent cell cultures. *, P<0.05 versus vehicle; †, P<0.05 versus vehicle. CHOP, C/EBP homologous protein; TAUR, tauroursodeoxycholic acid.
To examine whether CHOP is involved in H2O2-induced cell death, we performed loss-of-function experiments by silencing CHOP in H9c2 cells with a specific siRNA targeting rat CHOP. H9c2 cells were incubated with the CHOP siRNA (50 nM) or a scrambled siRNA as a control and then exposed to H2O2. CHOP siRNA efficiently depleted H2O2 induced CHOP expression in H9c2 cells (Figure 7C) [original blots for panel (C) are presented in Figure S6] and substantially decreased H2O2 induced caspase 3 activity and cell death (Figure 7D,E). Thus, these observations support the notion that the calpain-ER stress/CHOP pathway mediates H2O2-induced death of rat cardiomyoblasts.
Figure S6.
(A,B) The original blots for panel (C) presented in Figure 7 in the main text.
Discussion
Calpains have been implicated in myocardial injury as well as subsequent cardiac remodeling (17,20,30,37-39). In this study, we confirmed an important role of calpains in MI-induced myocardial injury and demonstrated that increased expression of calpastatin reduced myocardial injury and protected myocardial function following MI. The protective effects exerted by over-expressed calpastatin may be partially mediated through the inhibition of ER stress and prevention of CHOP activation.
Calpain activation has been observed in infarcted hearts (20,38,40,41) and several mechanisms have been proposed to contribute to calpain activation in the diseased heart. For example, angiotensin and norepinephrine (NE) can bind to G-protein-coupled receptors (GPCRs) and activate the phospholipase C/inositol triphosphate receptor (PLC/IP3R) pathway, consequently leading to calpain activation (41). Previously, we showed that activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase increased ROS in cardiomyocytes (27), which enhanced calpain activation in response to NE, H2O2, and lipopolysaccharide (27,28). It was also reported that ROS induced translocation of calpain-2 from the cytoplasm to the nucleus, where it promoted cardiomyocyte apoptosis in tail-suspended rats (42). Given that ROS production is increased and oxidative stress significantly contributes to myocardial injury in infarcted hearts (43-45), we used H2O2 to induce oxidative stress in cardiomyoblasts to mimic an MI condition in vitro, and confirmed that H2O2 induced calpain activation in cardiomyoblasts in vitro.
Accumulating evidence has implicated calpain activation in cardiomyocyte injury and death. For instance, calpain activation directly promotes caspase 3 activation (7,46), activation of mitochondrial calpain-1 cleaves apoptosis-inducing factor (AIF) (47), and the cleaved AIF trans-locates to the nucleus where it induces apoptosis (48). Also, calpains hydrolyze α-fodrin and ankyrin, leading to sarcolemmal rupture and cell death (38). In addition, calpains can also cleave troponin, thus causing myofibrillar degradation and loss of sarcomeric structure (23). Correspondingly, inhibition of calpain has been shown to protect cardiomyocytes against stress, as evidenced by our recent findings showing that cardiomyocyte-specific deletion of calpain-4 reduced myocardial remodeling post MI (20). Poncelas et al. reported that both intraperitoneal and oral administration of a calpain inhibitor attenuated post-infarction remodeling (49). Consistent with a recent report (24), the present study provides evidence supporting the premise that inhibition of calpains by over-expression of calpastatin also reduces myocardial injury and improves myocardial function following MI in mice. In the present study, transgenic over-expression of calpastatin did not change the 7-day mortality after MI, which was different from a recent report where an increased mortality 7 days after MI was observed in transgenic mice over-expressing calpastatin (25). Currently, the exact causes of this disparity between these observations are not clear. However, various levels of transgene expression in two different gain-of-function mouse models might partially account for these discrepant findings.
Our recent study showed that calpain activation induced ER stress in cardiomyocytes following hypoxia/re-oxygenation and in mouse hearts after ischemia/reperfusion (12). The present study extends this observation to MI. Furthermore, the present study shows that inhibition of ER stress reduces myocardial injury and improves myocardial function following MI. In the in vitro model of oxidative stress-induced injury in H9c2 cells, H2O2 induced CHOP expression and cell death, which were prevented by inhibiting either calpain or ER stress. Moreover, silencing CHOP inhibited H2O2-induced cell death. Thus, we believe that oxidative stress induces cell death in cardiomyocytes through ER stress and CHOP expression. Although the present study did not examine how CHOP facilitates cell death in cardiomyocytes, it is well-known that CHOP acts as a transcriptional factor that regulates expression of many pro- and anti-apoptotic genes. For example, over-expression of CHOP inhibits the expression of anti-apoptotic factor Bcl-2 and up-regulates the expression of pro-apoptotic factors including BAX, Bcl-x and caspase 3 (50). In addition, CHOP also cooperates with ATF4, promoting ATP depletion, oxidative stress, and cell death during ER stress (51,52).
Some limitations in the present study need to be acknowledged. First, we used H9C2 cells to investigate the relationship between calpain and apoptosis. Considering the differences of pathophysiological pathways between cardiomyoblasts H9c2 and adult cardiomyocytes, the findings obtained from this study might not fully apply to adult mature cardiomyocytes. Also, our MI model was induced by permanent coronary ligation instead of ischemia/reperfusion, the latter of which is believed to more closely represent the clinical setting.
In summary, we demonstrate an important role of the calpain/ER stress/CHOP pathway in MI-induced injury and the cardioprotective effects of calpastatin following MI. Hence, calpains and their associated ER stress might be an effective therapeutic target for MI.
Acknowledgements
Funding: This work was supported by a Grant-In-Aid from the Heart and Stroke Foundation of Canada (G-17-0018361) (to TQ Peng), National Natural Science Foundation of China (Grant 81300177) (to J Ma), Natural Science Foundation of Shanghai (Grant 13ZR1459200) (to J Ma) and grant from the Scientific Research Fund of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital (Grant ynlc201718) (to S Li).
Ethical Statement: All experimental procedures were approved by the Animal Use Subcommittee at the Western University, Canada.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Gaudron P, Eilles C, Kugler I, et al. Progressive left ventricular dysfunction and remodeling after myocardial infarction. Potential mechanisms and early predictors. Circulation 1993;87:755-63. 10.1161/01.CIR.87.3.755 [DOI] [PubMed] [Google Scholar]
- 2.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 2000;101:2981-8. 10.1161/01.CIR.101.25.2981 [DOI] [PubMed] [Google Scholar]
- 3.Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: programmed to conserve. J Mol Cell Cardiol 2013;55:85-91. 10.1016/j.yjmcc.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dally S, Monceau V, Corvazier E, et al. Compartmentalized expression of three novel sarco/endoplasmic reticulum Ca2+ATPase 3 isoforms including the switch to ER stress, SERCA3f, in non-failing and failing human heart. Cell Calcium 2009;45:144-54. 10.1016/j.ceca.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 5.Okada K, Minamino T, Tsukamoto Y, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004;110:705-12. 10.1161/01.CIR.0000137836.95625.D4 [DOI] [PubMed] [Google Scholar]
- 6.Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol 2010;48:1105-10. 10.1016/j.yjmcc.2009.10.026 [DOI] [PubMed] [Google Scholar]
- 7.Li S, Zhang L, Ni R, et al. Disruption of calpain reduces lipotoxicity-induced cardiac injury by preventing endoplasmic reticulum stress. Biochim Biophys Acta 2016;1862:2023-33. 10.1016/j.bbadis.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005;35:373-81. 10.1016/j.ymeth.2004.10.010 [DOI] [PubMed] [Google Scholar]
- 9.Logue SE, Cleary P, Saveljeva S, et al. New directions in ER stress-induced cell death. Apoptosis 2013;18:537-46. 10.1007/s10495-013-0818-6 [DOI] [PubMed] [Google Scholar]
- 10.Verma G, Datta M. The critical role of JNK in the ER-mitochondrial crosstalk during apoptotic cell death. J Cell Physiol 2012;227:1791-5. 10.1002/jcp.22903 [DOI] [PubMed] [Google Scholar]
- 11.Terai K, Hiramoto Y, Masaki M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol 2005;25:9554-75. 10.1128/MCB.25.21.9554-9575.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng D, Wang G, Li S, et al. Calpain-1 induces endoplasmic reticulum stress in promoting cardiomyocyte apoptosis following hypoxia/reoxygenation. Biochim Biophys Acta 2015;1852:882-92. 10.1016/j.bbadis.2015.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo T, Kim JK, Chen B, et al. Attenuation of ER stress prevents post-infarction-induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis. Chem Biol Interact 2015;225:90-8. 10.1016/j.cbi.2014.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sorimachi H, Suzuki K. The structure of calpain. J Biochem 2001;129:653-64. 10.1093/oxfordjournals.jbchem.a002903 [DOI] [PubMed] [Google Scholar]
- 15.Suzuki K, Hata S, Kawabata Y, et al. Structure, activation, and biology of calpain. Diabetes 2004;53 Suppl 1:S12-8. 10.2337/diabetes.53.2007.S12 [DOI] [PubMed] [Google Scholar]
- 16.Khorchid A, Ikura M. How calpain is activated by calcium. Nat Struct Biol 2002;9:239-41. 10.1038/nsb0402-239 [DOI] [PubMed] [Google Scholar]
- 17.Yang D, Ma S, Tan Y, et al. Increased expression of calpain and elevated activity of calcineurin in the myocardium of patients with congestive heart failure. Int J Mol Med 2010;26:159-64. 10.3892/ijmm_00000448 [DOI] [PubMed] [Google Scholar]
- 18.Takahashi M, Tanonaka K, Yoshida H, et al. Possible involvement of calpain activation in pathogenesis of chronic heart failure after acute myocardial infarction. J Cardiovasc Pharmacol 2006;47:413-21. [DOI] [PubMed] [Google Scholar]
- 19.Patterson C, Portbury AL, Schisler JC, et al. Tear me down: role of calpain in the development of cardiac ventricular hypertrophy. Circ Res 2011;109:453-62. 10.1161/CIRCRESAHA.110.239749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma J, Wei M, Wang Q, et al. Deficiency of Capn4 gene inhibits nuclear factor-kappaB (NF-kappaB) protein signaling/inflammation and reduces remodeling after myocardial infarction. J Biol Chem 2012;287:27480-9. 10.1074/jbc.M112.358929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani SK, Balasubramanian S, Zavadzkas JA, et al. Calpain inhibition preserves myocardial structure and function following myocardial infarction. Am J Physiol Heart Circ Physiol 2009;297:H1744-51. 10.1152/ajpheart.00338.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thorp E, Li G, Seimon TA, et al. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell Metab 2009;9:474-81. 10.1016/j.cmet.2009.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maekawa A, Lee JK, Nagaya T, et al. Overexpression of calpastatin by gene transfer prevents troponin I degradation and ameliorates contractile dysfunction in rat hearts subjected to ischemia/reperfusion. J Mol Cell Cardiol 2003;35:1277-84. 10.1016/S0022-2828(03)00238-4 [DOI] [PubMed] [Google Scholar]
- 24.Ye T, Wang Q, Zhang Y, et al. Over-expression of calpastatin inhibits calpain activation and attenuates post-infarction myocardial remodeling. PLoS One 2015;10:e0120178. 10.1371/journal.pone.0120178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan F, Letavernier E, Le Saux CJ, et al. Calpastatin overexpression impairs postinfarct scar healing in mice by compromising reparative immune cell recruitment and activation. Am J Physiol Heart Circ Physiol 2015;309:H1883-93. 10.1152/ajpheart.00594.2015 [DOI] [PubMed] [Google Scholar]
- 26.Peltier J, Bellocq A, Perez J, et al. Calpain activation and secretion promote glomerular injury in experimental glomerulonephritis: evidence from calpastatin-transgenic mice. J Am Soc Nephrol 2006;17:3415-23. 10.1681/ASN.2006050542 [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Arnold JM, Pampillo M, et al. Taurine prevents cardiomyocyte death by inhibiting NADPH oxidase-mediated calpain activation. Free Radic Biol Med 2009;46:51-61. 10.1016/j.freeradbiomed.2008.09.025 [DOI] [PubMed] [Google Scholar]
- 28.Li X, Li Y, Shan L, et al. Over-expression of calpastatin inhibits calpain activation and attenuates myocardial dysfunction during endotoxaemia. Cardiovasc Res 2009;83:72-9. 10.1093/cvr/cvp100 [DOI] [PubMed] [Google Scholar]
- 29.Zhu H, Yang Y, Wang Y, et al. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc Res 2011;92:75-84. 10.1093/cvr/cvr145 [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Ma J, Zhu H, et al. Targeted Inhibition of Calpain Reduces Myocardial Hypertrophy and Fibrosis in Mouse Models of Type 1 Diabetes. Diabetes 2011;60:2985-94. 10.2337/db10-1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Zheng D, Wei M, et al. Over-expression of calpastatin aggravates cardiotoxicity induced by doxorubicin. Cardiovasc Res 2013;98:381-90. 10.1093/cvr/cvt048 [DOI] [PubMed] [Google Scholar]
- 32.Thuerauf DJ, Marcinko M, Gude N, et al. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res 2006;99:275-82. 10.1161/01.RES.0000233317.70421.03 [DOI] [PubMed] [Google Scholar]
- 33.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res 2010;107:1071-82. 10.1161/CIRCRESAHA.110.227819 [DOI] [PubMed] [Google Scholar]
- 34.Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 2011;490:71-92. 10.1016/B978-0-12-385114-7.00004-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galán M, Kassan M, Choi SK, et al. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension 2012;60:71-80. 10.1161/HYPERTENSIONAHA.112.192500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kassan M, Galan M, Partyka M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol 2012;32:1652-61. 10.1161/ATVBAHA.112.249318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Letavernier E, Perez J, Bellocq A, et al. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res 2008;102:720-8. 10.1161/CIRCRESAHA.107.160077 [DOI] [PubMed] [Google Scholar]
- 38.Letavernier E, Zafrani L, Perez J, et al. The role of calpains in myocardial remodelling and heart failure. Cardiovasc Res 2012;96:38-45. 10.1093/cvr/cvs099 [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Li Y, Feng Q, et al. Calpain activation contributes to hyperglycaemia-induced apoptosis in cardiomyocytes. Cardiovasc Res 2009;84:100-10. 10.1093/cvr/cvp189 [DOI] [PubMed] [Google Scholar]
- 40.Perrin C, Ecarnot-Laubriet A, Vergely C, et al. Calpain and caspase-3 inhibitors reduce infarct size and post-ischemic apoptosis in rat heart without modifying contractile recovery. Cell Mol Biol (Noisy-le-grand) 2003;49 Online Pub:OL497-505. [PubMed]
- 41.Sandmann S, Yu M, Unger T. Transcriptional and translational regulation of calpain in the rat heart after myocardial infarction--effects of AT(1) and AT(2) receptor antagonists and ACE inhibitor. Br J Pharmacol 2001;132:767-77. 10.1038/sj.bjp.0703860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang H, Sheng JJ, Zhang L, et al. ROS-Induced Nuclear Translocation of Calpain-2 Facilitates Cardiomyocyte Apoptosis in Tail-Suspended Rats. J Cell Biochem 2015;116:2258-69. 10.1002/jcb.25176 [DOI] [PubMed] [Google Scholar]
- 43.von Harsdorf R, Li PF, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 1999;99:2934-41. 10.1161/01.CIR.99.22.2934 [DOI] [PubMed] [Google Scholar]
- 44.Grieve DJ, Byrne JA, Cave AC, et al. Role of oxidative stress in cardiac remodelling after myocardial infarction. Heart Lung Circ 2004;13:132-8. 10.1016/j.hlc.2004.02.008 [DOI] [PubMed] [Google Scholar]
- 45.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res 2009;81:457-64. 10.1093/cvr/cvn335 [DOI] [PubMed] [Google Scholar]
- 46.Tan Y, Wu C, De Veyra T, et al. Ubiquitous calpains promote both apoptosis and survival signals in response to different cell death stimuli. J Biol Chem 2006;281:17689-98. 10.1074/jbc.M601978200 [DOI] [PubMed] [Google Scholar]
- 47.Chen Q, Paillard M, Gomez L, et al. Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun 2011;415:533-8. 10.1016/j.bbrc.2011.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daugas E, Susin SA, Zamzami N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J 2000;14:729-39. 10.1096/fasebj.14.5.729 [DOI] [PubMed] [Google Scholar]
- 49.Poncelas M, Inserte J, Aluja D, et al. Delayed, oral pharmacological inhibition of calpains attenuates adverse post-infarction remodelling. Cardiovasc Res 2017;113:950-61. 10.1093/cvr/cvx073 [DOI] [PubMed] [Google Scholar]
- 50.Li Y, Guo Y, Tang J, et al. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai) 2014;46:629-40. 10.1093/abbs/gmu048 [DOI] [PubMed] [Google Scholar]
- 51.Han J, Back SH, Hur J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 2013;15:481-90. 10.1038/ncb2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004;11:381-9. 10.1038/sj.cdd.4401373 [DOI] [PubMed] [Google Scholar]