

Abstract
Chronic obstructive pulmonary disease (COPD), one of the leading causes of death worldwide, is substantially influenced by genetic factors. Alpha-1 antitrypsin deficiency demonstrates that rare coding variants of large effect can influence COPD susceptibility. To identify additional rare coding variants in patients with severe COPD, we conducted whole exome sequencing analysis in 2543 subjects from two family-based studies (Boston Early-Onset COPD Study and International COPD Genetics Network) and one case–control study (COPDGene). Applying a gene-based segregation test in the family-based data, we identified significant segregation of rare loss of function variants in TBC1D10A and RFPL1 (P-value < 2x10–6), but were unable to find similar variants in the case–control study. In single-variant, gene-based and pathway association analyses, we were unable to find significant findings that replicated or were significant in meta-analysis. However, we found that the top results in the two datasets were in proximity to each other in the protein–protein interaction network (P-value = 0.014), suggesting enrichment of these results for similar biological processes. A network of these association results and their neighbors was significantly enriched in the transforming growth factor beta-receptor binding and cilia-related pathways. Finally, in a more detailed examination of candidate genes, we identified individuals with putative high-risk variants, including patients harboring homozygous mutations in genes associated with cutis laxa and Niemann–Pick Disease Type C. Our results likely reflect heterogeneity of genetic risk for COPD along with limitations of statistical power and functional annotation, and highlight the potential of network analysis to gain insight into genetic association studies.
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous and complex disease with a significant genetic component to its susceptibility (1). Using genome-wide association study (GWAS), a number of COPD susceptibility loci have been identified including FAM13A (2), HHIP (3,4) and CHRNA3/CHRNA5/IREB2 (5--7). Complementary studies have identified more than a hundred loci associated with lung function, many of which likely also affect risk of COPD (8,9). However, identified loci only explain 5–10% of the heritability of COPD or quantitative measures of lung function traits (8,10). GWAS effectively tests common variants, but the well-known examples of alpha-1 antitrypsin deficiency (11), cutis laxa (12--16), and the more recently described association between telomere-related genes (17--19) indicate that, as has been shown for other diseases (20--24), rare coding variants also contribute to COPD risk.
We previously analyzed exome sequencing data of 49 families with severe, early-onset COPD, and, although we found several candidate genes, none showed convincing evidence of replication (25,26). We further showed using simulations that genetic heterogeneity may be a major contributor to this failure to replicate (26). In this study, we applied additional sequencing and analytic strategies to increase the sample size and the power of the analysis. We applied a recently developed family-based method, gene-based segregation (GESE) (25), to a larger family-based dataset enriched for severe COPD, and also performed single-variant, as well as set-based tests using SKAT-O for both genes and pathways in the family-based and in an additional case–control study. We tested for enrichment of our results in gene expression and monogenic models of disease, and examined the overlap between case–control and family-based results using network analysis. Finally, we investigated a set of candidate genes identified in previous genetic studies, including Mendelian syndromes, for potentially deleterious rare variants.
Results
GESE test on the International COPD Genetics Network and Boston Early-Onset COPD pedigrees
Baseline characteristics of the studied subjects are shown in Table 1. Additional information on the probands can be found in Supplementary Table S1. To identify causal variants in our exome sequencing data with the characteristics of Mendelian variants for COPD (e.g. alpha-1 antitrypsin deficiency), we applied our recently described GESE test (25) to the family-based data. We focused on ultra-rare [minor allele frequency (MAF) < 0.1%] predicted loss-of-function variants. Two genes were significant after Bonferroni correction for the total of 18 268 genes: RFPL1 (P-value = 1.60e-06) and TBC1D10A (P-value = 1.10e-06). RFPL1 segregated in four families, including two singleton families and two families with affected sibling pairs of severe COPD. TBC1D10A segregated in a parent-offspring pair and a singleton family. TBC1D10A is intolerant to loss-of-function variants (ExAC intolerance probability = 0.98 (27)). The top 10 genes from this analysis are shown in Table 2. All 10 of these genes are expressed in the adult lung (see Methods, enrichment P-value = 0.17), and the expression of 5 out of 9 of those genes was associated with forced expiratory volume in 1 second (FEV1)% predicted, a measure of COPD severity, in our lung tissue data (enrichment P-value = 0.024). We further sought supportive evidence for association of these genes in the COPDGene case–control dataset. However, no subjects harbored loss of function variants in these genes. We additionally tested for evidence of higher burden of rare (MAF < 0.1%), non-synonymous variants in the cases, and did not find convincing evidence of association (RFPL1, P-value = 0.576; TBC1D10A, P-value = 0.081).
Table 1.
Baseline characteristics
| COPDGene | ICGN-BEOCOPD | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Datasets | ESP | Baylor | |||||||
| Case | Control | Case | Control | Severe Cases1 | Moderate Cases | Resistant controls1 | Other Controls | Other | |
| N | 192 | 188 | 293 | 316 | 853 | 431 | 101 | 118 | 512 |
| # Females | 92 | 103 | 117 | 146 | 412 | 199 | 53 | 68 | 30 |
| # Males | 100 | 85 | 176 | 170 | 441 | 232 | 48 | 50 | 21 |
| Age, year | 58.2 (5.1) | 69.5 (5.6) | 68 (6.4) | 61.9 (5.6) | 56.1 (12.2) | 59.4 (12.1) | 55.5 (11.3) | 37.0 (20.4) | 53.9 (24.4) |
| Pack-years | 45.0 (26.2) | 45.0 (23.5) | 51.0 (29.0) | 50.6 (19.1) | 43.8 (31.0) | 37.4 (25.6) | 30.8 (21.9) | 0.0 (7.6) | 24.0 (39.9) |
| FEV1% predicted | 30.0 (15.9) | 98.2 (12.8) | 30.2 (15.8) | 92.7 (14.3) | 30.0 (17.6) | 65.5 (14.1) | 98.0 (15.3) | 96.3 (14.6) | 77.4 (10.6) |
| FEV1/FVC | 0.33 (0.10) | 0.78 (0.07) | 0.35 (0.12) | 0.76 (0.07) | 0.33 (0.14) | 0.56 (0.13) | 0.76 (0.06) | 0.81 (0.10) | 0.71 (0.12) |
N, Number of subjects
Median (IQR) is presented for age, pack-years, FEV1% predicted, and FEV1/FVC ratio for each dataset.
1Only severe cases (GOLD Grades 3 and 4) and resistant controls (see text) were included in the GESE test of the family-based data. All subjects were included in the association analysis of the family-based data.
2A total of 51 subjects in the ICGN-BEOCOPD data had lung function values not consistent with either case or control status.
Table 2.
Results of the GESE analysis on the BEOCOPD-ICGN dataset
| GENE P-value | GESE P-value | Number of segregating families |
|---|---|---|
| TBC1D10A* | 1.1E-06 | 2 |
| RFPL1* | 1.6E-06 | 4 |
| DHODH*# | 6.9E-05 | 2 |
| CYP4F12* | 1.0E-04 | 4 |
| ANAPC7* | 1.5E-04 | 1 |
| RGS5*# | 1.5E-04 | 2 |
| CD101*# | 1.7E-04 | 5 |
| KCNMB4*# | 1.8E-04 | 1 |
| ARMC12* | 2.1E-04 | 4 |
| VPS41*# | 3.9E-04 | 5 |
Variants included are loss-of-function variants with MAF < 0.1%. The third column shows the number of families each gene is segregating in (present in all the cases and not in the controls). Genes marked with * show expression in the lung (defined as at least 50% of samples with FPKM >0.5 in the Lung Genomics Resource Consortium RNA-seq samples). Genes marked with # are differentially expressed by FEV1% predicted in lung tissue (65).
Single-variant association analysis in the case–control and family data
Next, we performed single-variant association analysis. We tested both rare coding variants (moderate effect by SNPEff and MAF < 5%) as well as all variants. We found no significant results (Supplementary Tables S2 and S3) in either our primary analysis using COPDGene as the discovery cohort (using a Bonferroni significance level of 1.32e-06 for non-synonymous variants with MAF < 5% and 5.07e-07 for all variants), or using the family-based data (3.55e-07 for non-synonymous variants with MAF < 5% and significance level 1.86e-07 for all variants). However, top variants in the case–control analysis included rs8040868 (MAF = 0.41) and rs1051730 (MAF = 0.35) in CHRNA3 (28) with P-value = 5.05e-05 and 7.39e-05, respectively, which reside at a previously described GWAS locus (Supplementary Table S2). Top variants in the family-based analysis included rs2232710 (MAF = 0.012; P-value = 4.05e-05) in SERPINA10 (in high D’ with the alpha-1 Z allele, which causes alpha-1 antitrypsin deficiency—note that severe alpha-1 antitrypsin deficiency, including ZZ homozygosity, was an exclusion criteria for these studies) and rs10507051 (MAF = 0.063; P-value = 1.28e-04) in VEZT (Supplementary Table S3), near a locus associated with COPD in a recent GWAS of lung function (8). We also considered whether any variants were significant in meta-analysis by combining results from the two studies [case–control status in the COPDGene data, and lung function in the Boston Early-Onset COPD–International COPD Genetics Network (BEOCOPD-ICGN) data] using the Stouffer method. Meta-analysis did not identify significant variants among the rare coding variants (significance level, 2.82e-06) or among all variants (significance level, 7.08e-07) (Supplementary Table S4, Supplementary Table S5); top results overall included variants in CHRNA3 and SERPINA10.
Gene- and pathway-based analyses in case–control and family data
Next, we performed gene-based analyses. In the analysis using SKAT-O and predicted deleterious variants with a MAF < 1%, we found no significant genes in the COPDGene data. The top 10 genes are shown in Supplementary Table S6. We found no significant enrichment of genes expressed in lung (enrichment P-value = 0.97) among the top 10 genes. However, four genes have expression associated with FEV1% predicted (P-value = 0.087), including the top two genes VNN1 and PLA1A. EGFL8, the third-ranked gene in the list, is located near the AGER locus which was previously associated with risk of COPD (8,9). In the pathway analysis using the KEGG (29) database, we found one significant pathway using the burden test, the Jak–STAT signaling pathway (P-value = 6.78e-05) (30). However, association with this pathway was not replicated in the family-based analysis (P-value = 0.54) using the burden test. Top results from family-based analyses can be found in Supplementary Table S7. We also conducted a meta-analysis of the COPDGene dataset and the BEOCOPD-ICGN dataset; however, no gene achieved significance (Supplementary Table S8).
Enrichment and network-based approach to overlap
Given our lack of significant associations using standard association tests, we sought evidence that our top case–control and family-based results were enriched for associations with COPD. We tested for enrichment of overlap of genes yielding nominal significance (i.e. P-value < 0.01) between the case–control and the family-based association results using a standard hypergeometric approach. The enrichment P-value was 1, which was consistent with our lack of overlap and meta-analysis findings.
While we did not observe overlap between the top results in the case–control analysis and the family-based analysis using a simple hypergeometric test, we were interested in studying common biological pathways shared by the two sets of top genes. Recently, network-based methods have demonstrated the ability to identify related diseases in the protein–protein interactome (31). We hypothesized that application of this method to two independent association results for COPD would (a) identify whether there were overlapping association signals, despite the lack of replication, and (b) identify genes or pathways of highest priority. We computed the network-based separation (31) defined as the normalized average shortest path between members from the two modules to see whether top genes from the case–control and family-based analyses were close to each other in the protein–protein interaction (PPI) network. For the analysis of rare and deleterious variants, we found genes with P-value < 0.01 from the case–control analysis and the family-based analysis had significantly overlapping neighborhoods with negative separation score (score = −2.29, P-value = 0.014). To explore the neighborhood of these genes and the common pathways that connect the top genes, we added the first neighbors of the top genes in the PPI. These genes (top genes, along with all of their first-degree neighbors—a total of 522 genes) formed a largest connected component (LCC) of 513 genes, which means almost all the top genes and their first neighbors were connected. Figure 1 shows the network module containing the LCC formed by the top genes from the two analyses and their first neighbors. There were 19 genes with P-value < 0.01 in the family-based data, which had 274 first-degree neighbors in the LCC network; there were 14 genes with P-value < 0.01 in the COPDGene data, which had 216 first-degree neighbors. Between the two groups of 274 and 216 first-degree neighbors, 10 overlapped, thus these genes together formed a network module of 513 genes. 14 genes at loci previously associated with COPD or lung function (out of 329 genes in the curated set, see Methods) were in this set (enrichment P-value = 0.065, Supplementary Material). Additional examination of these genes in murine models showed that the 513 genes were significantly enriched for genes associated with the respiratory system (enrichment P-value = 0.045) and were enriched for genes involved in normal murine lung development in three common inbred strains of mice (enrichment P-value = 1.35e-02, 1.96e-03 and 2.40e-03, respectively; see Methods). From this result, we postulate that there is a large disease network module exists likely including a subset of these 513 genes for severe COPD, and only part of this disease module was observed using either analysis alone due to limited power. However, since the two sections of disease module share similar function and pathways, they were significantly close to each other in the PPI network.
Figure 1.
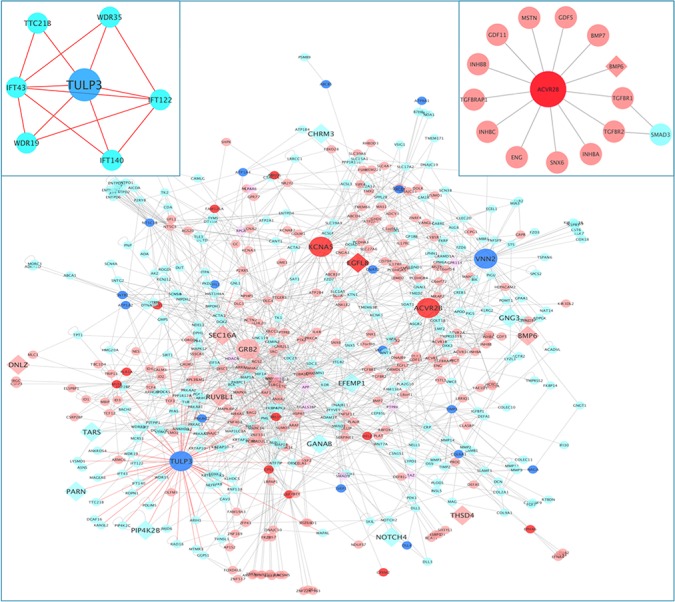
Network of the two sets of top genes with P-value < 0.01 in the case–control and family-based analyses focusing on rare, deleterious variants. The nodes in red are the top genes identified in the COPDGene case–control analysis; the nodes in pink are the first neighbors of the red nodes; the nodes in blue are the top genes identified in the BEOCOPD-ICGN family-based analysis; the nodes in light blue are the first neighbors of the blue nodes. Genes that are in both sets are colored in purple. Edges connecting genes to the largest hub TULP3 are colored in red. These genes form one large well-connected component. Larger sized nodes indicate hubs (circle) and genes reported to be associated with COPD or lung function (diamond). Hubs include TULP3, VNN2, ACVR2B, KCNA5 and GRB2, which are the top genes with the most number of degrees in this network. A total of 14 out of 513 genes are near GWAS loci for COPD or lung function (CHRM3, DNLZ, EFEMP1, EFEMP2, EGFL8, GANAB, GNG3, PARN, PIP4K2B, NOTCH4, RUVBL1, SEC16A, TARS, TEKT5, THSD4). The zoomed-in panel on the left shows the genes in the intraciliary transport particle A pathway (GO:0030991). The zoomed-in panel on the right shows the genes in the TGFB-receptor binding pathway (GO:0005160) and ACVR2B.
To further explore the functions of this network, we also looked at the pathways enriched for these 513 genes using ToppFun in the ToppGene Suite (32), and found a large number of Gene-ontology pathways were significantly enriched. To examine more specific pathways, we examined GO pathways with fewer than 100 genes in total. The top two pathways meeting these criteria were ‘GO:0005160: transforming growth factor beta (TGFB) receptor binding’ and ‘GO:0030991: intraciliary transport particle A’. A total of 14 out of 53 genes in the TGFB receptor binding pathway were present in the network module. Multiple lines of evidence, including genetic association (which has identified TGFB2) and other genomic and mechanistic studies have implicated this pathway in risk to COPD (33--35). Twelve of these fourteen genes are expressed in human lung tissue (two genes have missing data). The right panel in Figure 1 shows the small network formed by genes in this pathway and ACVR2B, which was the top-ranked gene from this set of genes in the association analysis and was also the second largest hub in the network. For the ‘intraciliary transport particle A pathway’ (36), 7 out of 8 genes were in the network, which are shown in the left panel in Figure 1. A total of 6 out of 7 genes were expressed in human lung tissue (one gene had missing data). TULP3 was in the top-ranked genes from the family-based analysis and was the largest hub in the module. TULP3 is a known target of the Hedgehog pathway. Notably, GWASs and follow-up functional studies have identified an important role for HHIP in the development of COPD (7); TULP3 has been shown to change expression after HHIP silencing (37). Also, WDR35 and IFT140 were associated with respiratory system abnormalities in mouse models (WDR35 leads to lung hypoplasia and mutations in IFT140 produces severely misshapen lungs). Additional top results from this GO analysis can be found in Supplementary Table S9. Thus, our network results highlight ACVR2B and TULP3, which may be prioritized for further examination of functional rare variants.
Evidence of association for candidate genes
A substantial proportion of rare variants identified for complex disease are located at loci that also harbor common risk variants (38,39). In addition, several Mendelian syndromes have COPD, emphysema or obstructive lung disease as a manifestation of disease. Therefore, in addition to looking at exome-wide results, we examined a list of the 329 curated genes (see Methods, Supplementary Table S10) (1,7–9,12–19,40–45). This included regions identified from 105 SNPs from GWAS analyses (8,9) and 29 Mendelian genes with manifestations that include COPD or emphysema in their resulting syndromes (Supplementary Material). We examined functional and rare variants with MAF < 5% and found multiple genes to be nominally associated with COPD status or FEV1% predicted value, including CHRNA5, AGER and CYP2A6 (Supplementary Table S11). To identify whether there was any independent evidence of rare variant effects at these loci in the COPDGene cohort, we conditioned on the risk allele for the 104 SNPs identified by GWAS. Several genes were still nominally significant after conditioning on the GWAS SNPs, including CYP2A6 (full results shown in Table 3); whether these rare variants have independent effects on COPD susceptibility at these loci will likely need to be addressed by additional, larger studies.
Table 3.
Nominally significant gene-based results in COPDGene for 329 candidate genes after conditioning on the lead GWAS SNP
| Gene | (Conditioned on) lead GWAS SNP | #SNV | SKATO (unadjusted) | SKATO (conditional) |
|---|---|---|---|---|
| SEC16A | rs10870202 | 38 | 9.52E-04 | 1.05E-03 |
| CDC7 | rs1192404 | 5 | 5.87E-03 | 6.78E-03 |
| CCDC38 | rs12820313 | 5 | 7.20E-03 | 7.66E-03 |
| CYP2A6 | rs12459249 | 11 | 6.87E-03 | 1.15E-02 |
| TRIP11 | rs7155279 | 24 | 1.53E-02 | 2.47E-02 |
| CNGB1 | rs12447804 | 26 | 3.27E-02 | 2.89E-02 |
| PBLD | rs7095607 | 3 | 6.99E-02 | 3.43E-02 |
| RRP15 | rs10429950 | 2 | 3.75E-02 | 4.07E-02 |
| TNXB | rs2070600 | 63 | 5.95E-02 | 4.39E-02 |
| CYFIP2 | rs10515750, rs1990950 | 4 | 4.13E-02 | 4.46E-02 |
| EGFL8 | rs2070600 | 9 | 8.11E-02 | 4.60E-02 |
| CHRNA5 | rs17486278 | 5 | 1.83E-02 | 1.22E-01 |
| AGER | rs2070600 | 12 | 4.89E-03 | 1.36E-01 |
The SKAT-O tests included functional (MODERATE effect defined by SnpEff) and rare (MAF < 5%) variants in the COPDGene study.
We also looked closely at the 29 genes causing Mendelian syndromes including emphysema or obstructive lung disease as part of their syndrome. To determine whether there was enrichment in these genes in our dataset, we performed a burden test including only variants with MAF < 0.1% in ExAC and predicted deleterious by FATHMM, SIFT and CADD (>15). We found that the burden-based tests gave a P-value = 0.80 in the COPDGene case–control study, and a P-value = 0.018 for the family-based EOCOPD and ICGN data. Thus, we observed some significant accumulation of deleterious variants in these genes in the family-based data, suggesting that ultra-rare variants in these Mendelian genes contributing to lung function may be related to severe COPD risk in our family-based datasets.
To examine these variants individually, we intersected variants in these genes with Clinvar, using an annotation of significance level 4 (likely pathogenic) and above, and additionally included variants in published reports associated with respiratory disease in TERT (17–19,41–43). We found 47 of these variants in our datasets. These variants are listed in Supplementary Table S12 along with their counts among cases and controls separately. Given the strong evidence of pathogenicity for variants in SERPINA1 and telomere-related genes, these findings are shown in Table 4. We also assessed the carriers of these rare variants using a recessive model of inheritance, and those variants with homozygous genotypes present in any dataset are listed in Table 5. Among our findings for Mendelian genes were two previously identified cases from COPDGene with heterozygous TERT variants (19), and evidence for an increased burden (cases > controls) for the SERPINA1 Z and PI P (Lowell) (rs121912714) (Table 4). For recessive variants, we identified rs140130028, a splice-donor variant in NPC2, which is a gene for Niemann–Pick disease type C2, a disease previously associated with emphysema (46) (Table 5). One pair of sibs with severe COPD in the ICGN study was homozygous for this variant; two of their half-siblings carried one copy, one with severe COPD. None of these subjects had known Neimann–Pick disease. Also, variant rs61748181 in TERT was present as homozygous in seven unrelated cases in the datasets (Table 5). While this association did not reach candidate-wide significance (P-value = 0.167), this variant was experimentally demonstrated to induce telomere dysfunction (47) and predicted to be disease causing by Mutation Taster (48). For variants not annotated by Clinvar or annotated with a significance level of 3 (uncertain significance) or below, we filtered based on MAF < 0.1% in ExAC v0.3 non-Finnish Europeans and predicted deleterious effects by FATHMM, SIFT or CADD (>15). There were in total 346 such variants in our datasets. One of these variants occurred in homozygous form in a proband with severe COPD in the BEOCOPD study. This variant is an ultra-rare splice-acceptor variant in ATP6V0A2 (novel in ExAC database) (Table 4), a Mendelian gene for cutis laxa. A chest CT scan of this subject showed severe emphysema, however, no phenotypic information related to dermatological characteristics was available. In addition, 66 variants were predicted to be deleterious by all three annotations: FATHMM, SIFT and CADD (>15), and had supportive evidence in our datasets (with greater counts in cases than in controls, Supplementary Table S13). Multiple variants have supportive evidence in both case–control and family-based datasets. For example, rs141310608 in EFEMP2 is present in two cases in COPDGene study and two cases in BEOCOPD-ICGN study, while none in controls. Also, there are multiple ultra-rare variants in COL3A1 are carried by cases and none by controls. We have also listed the variants that are predicted to be deleterious by all annotations, but are present in more controls than cases; these variants are less likely to be high penetrance COPD susceptibility variants (Supplementary Table S14). We also applied a more liberal filtering criteria (MAF < 0.05, CADD >10 or predicted to be deleterious by SIFT and FATHMM) for TERT, RTEL1,CFTR and SERPINA1. Detailed information about these genes can be found in Supplementary Tables S15, S16 and S17, respectively.
Table 4.
Selected set of likely pathogenic variants annotated by ClinVar in SERPINA1 and telomere-related genes
| COPDGene | BECOPD-ICGN | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GENE | SNP | Case | Cont | Case | Cont | MAF | IMPACT | CLNSIG | Disease association |
| RTEL1 | 20:62324513:T:C | 0 | 1 | . | . | 6.11E-05 | missense_variant | 5 | Telomeropathy |
| SERPINA1 | rs28929474 | 29 | 12 | 60 | 8 | 1.83E-02 | missense_variant | 5|5 | Alpha-1 antitrypsin deficiency |
| SERPINA1 | rs17580 | 53 | 29 | 148 | 32 | 3.04E-02 | missense_variant | 5 | Alpha-1 antitrypsin deficiency |
| SERPINA1 | rs28929470 | 4 | 6 | 5 | 1 | 4.95E-03 | missense_variant | 5 | Alpha-1 antitrypsin deficiency |
| SERPINA1 | rs28931570 | 2 | 4 | 8 | 2 | 1.62E-03 | missense_variant | 4|5 | Alpha-1 antitrypsin deficiency |
| SERPINA1 | rs121912714 | . | . | 3 | 0 | 7.04E-04 | missense_variant | 4 | Alpha-1 antitrypsin deficiency |
| TERT | rs61748181 | 40 | 33 | 92 | 16 | 4.97E-02 | missense_variant | 5|2 | Telomeropathy |
| TERT | 5:1278865 | 1 | 0 | . | . | 7.49E-05 | missense_variant | 5 | Telomeropathy |
| TERT | 5:1280427 | 1 | 0 | . | . | . | missense_variant | . | Telomeropathy |
| TERT | rs35719940 | 27 | 22 | . | . | 2.11E-02 | missense_variant | 5|2|2 | Telomeropathy |
| 5|5|5|3|2| | |||||||||
| TERT | rs34094720 | 4 | 7 | . | . | 1.53E-02 | missense_variant | 2 | Telomeropathy |
| TERT | rs141425941 | 1 | 1 | . | . | 2.68E-04 | missense_variant | 5 | Telomeropathy |
| TINF2 | rs142777869 | 2 | 1 | 1 | 0 | 7.25E-04 | missense_variant | 5 | Telomeropathy |
Case, Cont: number of alternative alleles carried by the cases and controls in each dataset. Note that in the family-based data, there are approximately six times more cases than controls. IMPACT, functional impact of each variant annotated by SnpEff. CLNSIG and Disease association are annotations from ClinVar; 2 = benign, 3 = likely benign, 4 = likely pathogenic, 5 = pathogenic.
Table 5.
Homozygous variants in COPD-related Mendelian genes
| COPDGene | BECOPD-ICGN | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GENE | SNP | Case | Cont | Case | Cont | MAF | IMPACT | CLNSIG | Disease association |
| ATP6V0A2 | 12:124206896* | 0 | 0 | 1 | 0 | . | Splice_acceptor_variant | . | Cutis laxa |
| CFTR | rs1800076 | 0 | 1 | 3 | 0 | 2.48E-02 | missense_variant | 2|2|5 | Cystic fibrosis |
| NPC2 | rs140130028 | . | . | 2 | 0 | 0.00551 | splice_donor_variant | 5 | Niemann-Pick disease type C2 |
| SERPINA1 | rs17580 | 1 | 1 | 4 | 1 | 3.04E-02 | missense_variant | 5 | Apha-1-antitrypsin deficiency |
| TERT | rs61748181 | 5 | 0 | 2 | 0 | 4.97E-02 | missense_variant | 5|2 | Telomeropathy |
| TERT | rs35719940 | 1 | 0 | . | . | 2.11E-02 | missense_variant | 5|2|2 | Telomeropathy |
Variants with homozygous genotypes in 29 Mendelian genes and were annotated with significance 4 and above by ClinVar, or have potential deleterious effects (MAF < 0.1% and predicted to be deleterious by FATHMM, SIFT and CADD (>15). Case, Cont: number of alternative alleles carried by the cases and controls in each dataset. Note that in the family-based data, there are approximately 6 times more cases than controls. IMPACT, functional impact of each variant annotated by SnpEff. CLNSIG and Disease association are annotations from ClinVar; 2 = benign, 3 = likely benign, 4 = likely pathogenic, 5 = pathogenic.
Discussion
COPD is a common and heterogeneous disease; under the common-disease–common-variants hypothesis, we expect that multiple common variants should contribute to a large proportion of COPD risk. However, even though a number of COPD GWAS loci have been discovered through large-scale collaborative efforts, most of the estimated heritability remains unexplained. Examples such as alpha-1 antitrypsin deficiency, cutis laxa and, more recently, telomeropathies are associated with COPD and emphysema (17–19). These results motivated us to search the entire exome for large effect variants that could represent a Mendelian subtype of COPD, in the hope of finding new treatment strategies for a subset of the patients. In this study, we examined multiple cohorts representing the largest exome sequencing study of COPD to date ascertained under an extreme phenotype approach (where samples were enriched for sever COPD and normal controls heavily exposed to smoking but with normal pulmonary function), to screen through the entire exome to identify rare coding variants controlling risk to COPD. Results failed to identify new genes, pathways or variants consistently significant across all of our analyses, suggesting that single-variant or single-gene effects of a contribution as large as alpha-1 antitrypsin deficiency are unlikely to exist (26). Yet, a network-based analysis identified a significant relationship between the two modules formed by the top results of the two analyses. These two sets of top genes, along with their first neighbors in the PPI network, form a well-connected network component. This LCC was significantly enriched in genes involved in fetal lung development in mouse models (49). Additionally, this module sheds light on related functions or pathways where such rare variants may be contributing to risk to COPD. For example, multiple studies have suggested the TGFB pathway is associated with COPD (50), and the TGFB2 locus was associated with COPD in GWAS (51). Our study identified ACVR2B as a potential candidate; of interest, ACVR1B, an activin receptor which interacts with ACVR2B (52) was identified in a network-informed genetic association study of COPD (53) and in an integrative analysis of emphysema distribution (54).
Our finding lends further support to the TGFB pathway and also suggests that rare variants related to ACVR2B may contribute to COPD risk. Similarly, the identification of TULP3 lends further support to the identification of HHIP as a causal gene at this GWAS locus and the importance of the hedgehog pathway in the development of COPD. The identification of cilia-related pathways is intriguing given the importance of cilia to lung function (55), including reports from a smaller exome study of resistant smokers (56) and reports of shortened cilia in smokers and in COPD patients (36).
Finally, we identified subjects carrying homozygous genotypes of rare and deleterious variants in Mendelian genes for cutis laxa and Niemann–Pick disease, which are themselves intriguing candidates for causing severe COPD. These findings illustrate the potential relevance of using filtering-based technique for identifying syndromic forms of COPD. While we do not have enough power to individually test these or other individual rare variants here, our results may provide support for future studies in these recognized candidate genes.
COPD is known to be a highly heterogeneous disease, with varying contributions of emphysema and small airway disease. We did not examine specific subsets of COPD, as detailed phenotyping was not available in all cohorts. Multiple analysis methods are available for rare variant analysis ((57)), and the optimal methods are still not clear. Our sequencing of a large number of affected individuals in families was appropriate for methods such as GESE, which leverages a large reference dataset (ExAC); an alternative approach using association would require large-scale exome harmonization of controls with normal lung function, preferably with heavy cigarette smoke exposure. Our results highlight the importance of integration with other types of data (e.g. gene expression, PPI) to better understand the results from one data type. However, our analysis does not attempt to identify the confidence of individual genes in this network; we cannot rule out the possibility that this network includes many genes that are false positives, and our pathway analysis should be considered descriptive and exploratory. Additional investigation, including genetic studies, integration of multi-omics data, and careful functional studies will be needed to further infer biological mechanisms and potential disease causality for our identified genes.
In summary, in an exome sequencing study of COPD, we were unable to identify exome-wide significant associations, but through network analysis we identified candidate genes in related pathways and a disease module driven by rare variants. Our study is consistent with a potential contribution of multiple, heterogeneous rare variants in COPD, and demonstrates the insight that network-based methods can offer.
Materials and Methods
The COPDGene study
The COPDGene study is a multi-center epidemiologic and genetic study of 10 192 current or ex-smokers, which has been previously described (58). COPDGene subjects were sequenced in two sets. The first set sequenced as part of the NHLBI Exome Sequencing Project (ESP; named COPDGene ESP) included severe COPD cases with Global Initiative for Chronic Obstructive Lung Disease (GOLD) Grades 3 or 4 (post-bronchodilator FEV1 < 50% predicted and FEV1/FVC < 0.70), and aged < 65 years old, with substantial emphysema (>15% at −950 HU) by quantitative chest CT scan. Controls were selected to be resistant smokers with frequency-matched pack-years of cigarette smoking, normal lung function (FEV1 > 80% predicted and FEV1/FVC > 70%), aged > 65 years old and no significant emphysema (< 5% at −950 HU). The second set sequenced at Baylor (named COPDGene Baylor) included severe COPD cases (GOLD Grades 3 or 4) with no age requirement. Controls were selected to be resistant smokers with normal lung function with ages >55 years.
The BEOCOPD study and the ICGN study
The family-based data contained samples selected from the BEOCOPD (59) and the ICGN study (45). Probands from BEOCOPD were selected to be physician-diagnosed COPD cases with FEV1 ≤ 40% predicted, aged ≤ 53 years. All first-degree relatives, older second-degree relatives and additional affected family members were enrolled. Probands in the ICGN study were subjects with known COPD and were required to have FEV1 < 60% predicted, FEV1/FVC < 90% predicted at ages 45–65 years, pack-years ≥ 5 and have at least one eligible sibling. An initial set of 49 pedigrees selected from the BEOCOPD study were described and analyzed previously (26). To this sample we added 147 families from BEOCOPD and 462 families from the ICGN study. The COPDGene, BECOPD and ICGN studies all excluded subjects with severe alpha-1 antitrypsin deficiency.
Exome sequencing
We sequenced all subjects using Nimblegen capture and Illumina platforms. The COPDGene ESP, BEOCOPD and ICGN subjects were all sequenced at the University of Washington, using Nimblegen V2 exome capture; COPDGene Baylor samples used VChrome capture. Alignment, variant calling and quality control were performed using bwa, GATK and in-house pipelines, respectively. As COPDGene ESP and COPDGene Baylor used slightly different capture platforms, calling was performed on these datasets separately. All BEOCOPD and ICGN subjects were called together (joint calling) and went through the same quality control steps together to provide the final family-based data (named BEOCOPD-ICGN) for analysis. Baseline characteristics of the subjects in each of the cleaned datasets are shown in Table 1 and our overall study design is shown in Figure 2. More details can be found in the Supplementary Material.
Figure 2.
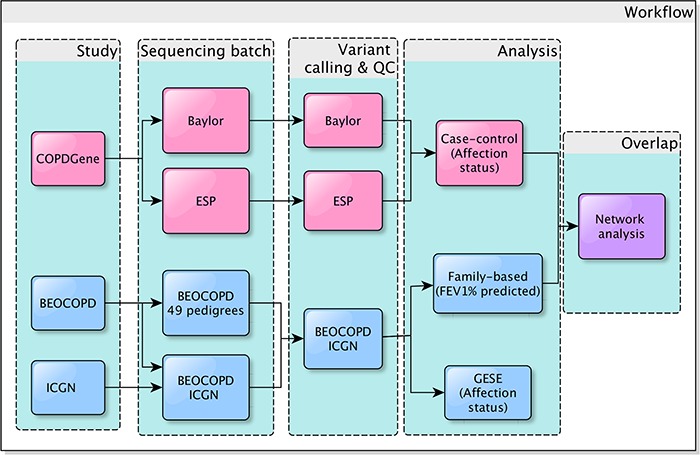
A flow chart of the study design. COPDGene (pink) samples were sequenced in two batches (Baylor and ESP, see Methods). The family-based studies (blue) included two cohorts. Forty-nine pedigrees of the Boston Early-Onset study samples were sequenced and analyzed previously (26); we combined these data with another subset of these BEOCOPD and additional samples from the ICGN study. All of these sequenced subjects from BEOCOPD and ICGN were called together, forming the BEOCOPD-ICGN dataset (blue). We applied the family-based GESE test to the most severe cases and resistant controls in the BEOCOPD-ICGN dataset. We also performed single-variant, gene-based and pathway-based association tests in COPDGene and the BEOCOPD-ICGN samples. A final network analysis was conducted to look at the topological relationship between the top results from the two datasets.
Analysis strategy
Loss of function variants using the GESE test
We first performed the GESE (25) on loss of function variants (defined by SnpEff (60)) with MAF < 0.1% in the family-based BEOCOPD-ICGN data using COPD affection status as the outcome. We included only the most severe COPD subjects (GOLD spirometry Grades 3 or 4) and resistant smoking control subjects (normal spirometry, aged > 40 years, with at le ast five pack-years of cigarette smoking). This analysis took advantage of the unique properties of a family-based strategy, including having multiple copies of rare variants, and assumes a Mendelian model with a few rare variants with very large effects. We sought supportive evidence for identified causal genes in COPDGene dataset by attempting to identify similarly deleterious variants
Association analyses
Second, we performed single-variant, gene-based and pathway-based association analyses. For all association analyses, we used Bonferroni correction based on the number of genes, pathways or variants tested. For the COPDGene case–control data, COPD affection status was used as the outcome, which was adjusted for pack-years, gender, age and ancestry-based principal components (PCs) in the COPDGene Baylor data, and the top PCs alone in the COPDGene ESP data due to the selection criteria, and as performed previously (Supplementary Material). For the family-based data, due to the low number of controls with normal lung function, but a wider range of FEV1 available through family members, we analyzed FEV1 (forced expiratory volume in one second), a lung function measure highly correlated with COPD (9) instead of COPD affection status itself. The outcome in the family-based association tests was the rank of the residuals from regressing raw post-bronchodilator FEV1 value on height, pack-years, sex, age, top 5 genetic ancestry PCs and batch indicator variable.
Single-variant association analysis
For single-variant analyses, we applied the Stouffer method to meta-analyze the results from the hybrid method in SKATBinary_Single function (SKAT package) in the COPDGene case–control data, since the two cohorts selected from the COPDGene study were sequenced and called separately. The hybrid method in SKATBinary_Single function selects the most appropriate approach to compute P-values for each variant. For single-variant analysis in family-based data, we applied the variant-based generalized linear mixed model association test (GMMAT (61)). In addition to using COPDGene as discovery and BEOCOPD-ICGN as replication, we also examined using BEOCOPD-ICGN and both datasets as discovery by meta-analyzing the results from the COPDGene case–control data and the BEOCOPD-ICGN data using the Stouffer method. For single-variant analyses, we tested all variants, and also the subset with moderate effect with MAF < 5%.
Gene- and pathway-based association analysis
For both the gene- and pathway-based analyses, we applied SKAT-O tests. In the COPDGene case–control datasets, we applied the hybrid method in the SKATBinary function, implemented in the SKAT package to each of the datasets, and meta-analyzed the two datasets using Fisher’s method. For the BEOCOPD-ICGN family-based data, we applied MONSTER (62), which is a generalized version of SKAT-O for family-based studies. We also meta-analyzed all results (case–control and family-based results) using Fisher’s method. Our primary gene- and pathway-based association analyses focused on deleterious variants defined using FATHMM (57,63) with MAF < 1% in the association analysis. In one study of amyotrophic lateral sclerosis (ALS), FATHMM was found to give the best power to identify known causal genes for ALS in gene-based association tests (57). Our secondary analyses included association testing on functional variants with moderate effects (defined by SnpEff (60)) with MAF < 5%. This is a less stringent filtering criterion on the variants to prevent missing signals in this set of variants. Pathways were defined using KEGG pathways (29) and the c2 collection of curated gene sets from the Molecular Signatures Database (MsIGdb) in GSEA (64).
Identification of enrichment in gene expression
To help determine whether the identified genes were relevant for our phenotypes, we used publicly available FPKM (per kilobase of gene model per million mapped reads) results from gene expression data from the Lung Genomics Research Consortium (http://www.lung-genomics.org) to identify whether any gene was expressed in the lung (using a cutoff of 0.5) (26). We also used the results of differential expression for lung function and COPD case–control status in an independent set of lung tissue from severe COPD subjects and controls (65). In addition, enrichment for genes associated with respiratory system in mouse was carried out using a curated set of genes associated with respiratory phenotype in the Mouse Genome Database (http://www.informatics.jax.org/marker) (66). Gene expression information in human and normal murine lung development for three common inbred strains of mice were obtained from the GEO dataset (GSE14334 and GSE74243), and genes involved in fetal lung development were obtained using methods described in (49).
Network-based analysis
Finally, we applied the network-based separation measure defined in (31) to examine how closely connected the top genes from the two independent analyses are in the PPI network. This measure has been shown to predict pathobiological similarity of two sets of disease genes (31). In our application here, since the two outcomes analyzed for the COPDGene and BEOCOPD-ICGN dataset are highly correlated, genes that are causal for these outcomes should have much shorter network-based distance. Therefore, a significant result tells us that at least a subset of the top genes from the two analyses is topologically overlapping and exerts some effect on risk of COPD.
Examination of previously identified genetic associations with COPD
To examine loci previously described to be associated with risk of COPD or lung function itself in GWAS or harboring Mendelian variants related to COPD, we curated a set of 329 genes for closer examination (Supplementary Table S12) (8,9). At COPD GWAS loci, we identified all variants in a European reference population with an r2 > 0.8 with the lead variant, and then expanded these borders by 100 kb. For Mendelian syndromes, we included connective tissue disorders such as cutis laxa (12--16), as well as telomere-related genes including TERT, TERC, RTEL1, and NAF1 (17,18,41–43). We looked for supportive evidence of association for these genes using several methods. First, we examined the association results in both primary and secondary analyses as described above. Since 104 of the previously described lead SNPs based on GWAS of lung function or COPD were also available for the COPDGene subjects, we additionally performed conditional analyses for these genes by conditioning on the GWAS SNPs in proximity in an attempt to identify independent rare variants contributing to COPD susceptibility. For both the marginal association analyses and conditional analyses, COPD affection status was the outcome in the COPDGene case–control analyses and FEV1 was the outcome in the family-based analyses. Finally, we examined Mendelian genes for evidence of pathogenic variants using Clinvar and other public annotation resources.
Supplementary Material
Acknowledgements
The COPDGene project (NCT00608764) is supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Novartis, Pfizer, Siemens and Sunovion. Authors would like acknowledge Victor M Pinto-Plata (Baystate Medical Center, Springfield, MA), Nathaniel Marchetti (Temple University School of Medicine, Philadelphia, PA), Raphael Bueno(Brigham and Women’s Hospital and Harvard Medical School, Boston, MA), Bartolome R. Celli (Brigham and Women’s Hospital and Harvard Medical School, Boston, MA), Gerald J. Criner (Temple University School of Medicine, Philadelphia, PA) and Dawn Demeo (Brigham and Women’s Hospital and Harvard Medical School, Boston, MA) for providing the lung tissue samples and their support of the project. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Conflict of Interest statement. Dr. Silverman has received honoraria and consulting fees from Merck, grant support and consulting fees from GlaxoSmithKline, and honoraria from Novartis. Dr. Hersh has been a consultant for CSL Behring and Mylan. Dr. Cho has received grant support from GSK.
Funding
NHLBI (R01 HL089856 to E.K.S., R01 HL089897 to J.D.C., R01 HL113264 to M.H.C. and E.K.S., P01 HL105339 to E.K.S., PO1 114501 to E.K.S, K01 HL129039 to D.Q., K07 CA181480 to Y.L.); GlaxoSmithKline; National Human Genome Research Institute and the National Heart, Lung and Blood Institute (grant 1U54HG006493).
References
- 1. Hersh C.P., Demeo D.L. and Silverman E.K. (2005) Chronic obstructive pulmonary disease; In Silverman EK, Shapiro SD, Lomas DA, Weiss ST, editors, Respiratory genetics Hodder Arnold, New York, 253–296.
- 2. Cho M.H., Boutaoui N., Klanderman B.J., Sylvia J.S., Ziniti J.P., Hersh C.P., DeMeo D.L., Hunninghake G.M., Litonjua A.A., Sparrow D. et al. (2010) Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet., 42, 200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou X., Baron R.M., Hardin M., Cho M.H., Zielinski J., Hawrylkiewicz I., Sliwinski P., Hersh C.P., Mancini J.D., Lu K. et al. (2012) Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum. Mol. Genet., 21, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilk J.B., Chen T.H., Gottlieb D.J., Walter R.E., Nagle M.W., Brandler B.J., Myers R.H., Borecki I.B., Silverman E.K., Weiss S.T. et al. (2009) A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet., 5, e1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hardin M., Zielinski J., Wan E.S., Hersh C.P., Castaldi P.J., Schwinder E., Hawrylkiewicz I., Sliwinski P., Cho M.H. and Silverman E.K. (2012) CHRNA3/5, IREB2, and ADCY2 are associated with severe chronic obstructive pulmonary disease in Poland. Am. J. Resp. Cell. Mol., 47, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeMeo D.L., Mariani T., Bhattacharya S., Srisuma S., Lange C., Litonjua A., Bueno R., Pillai S.G., Lomas D.A., Sparrow D. et al. (2009) Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am. J. Hum. Genet., 85, 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pillai S.G., Ge D., Zhu G., Kong X., Shianna K.V., Need A.C., Feng S., Hersh C.P., Bakke P., Gulsvik A. et al. (2009) A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet., 5, e1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wain L.V., Shrine N., Artigas M.S., Erzurumluoglu A.M., Noyvert B., Bossini-Castillo L., Obeidat M., Henry A.P., Portelli M.A., Hall R.J. et al. (2017) Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat. Genet., 49, 416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hobbs B.D., Jong K., Lamontagne M., Bosse Y., Shrine N., Artigas M.S., Wain L.V., Hall I.P., Jackson V.E., Wyss A.B. et al. (2017) Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet., 49, 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou J.J., Cho M.H., Castaldi P.J., Hersh C.P., Silverman E.K. and Laird N.M. (2013) Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am. J. Respir. Crit. Care Med., 188, 941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laurell C.B. and Eriksson S. (2013) The electrophoretic alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. 1963. COPD, 10(suppl 1), 3–8. [DOI] [PubMed] [Google Scholar]
- 12. Urban Z., Gao J., Pope F.M. and Davis E.C. (2005) Autosomal dominant cutis laxa with severe lung disease: synthesis and matrix deposition of mutant tropoelastin. J. Invest. Dermatol., 124, 1193–1199. [DOI] [PubMed] [Google Scholar]
- 13. Hucthagowder V., Sausgruber N., Kim K.H., Angle B., Marmorstein L.Y. and Urban Z. (2006) Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am. J. Hum. Genet., 78, 1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hucthagowder V., Morava E., Kornak U., Lefeber D.J., Fischer B., Dimopoulou A., Aldinger A., Choi J., Davis E.C., Abuelo D.N. et al. (2009) Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum. Mol. Genet., 18, 2149–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Q., Loeys B.L., Coucke P.J., De Paepe A., Mecham R.P., Choi J., Davis E.C. and Urban Z. (2006) Fibulin-5 mutations: mechanisms of impaired elastic fiber formation in recessive cutis laxa. Hum. Mol. Genet., 15, 3379–3386. [DOI] [PubMed] [Google Scholar]
- 16. Callewaert B., Su C.T., Van Damme T., Vlummens P., Malfait F., Vanakker O., Schulz B., Mac Neal M., Davis E.C., Lee J.G. et al. (2013) Comprehensive clinical and molecular analysis of 12 families with type 1 recessive cutis laxa. Hum. Mutat., 34, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stanley S.E., Merck S.J. and Armanios M. (2016) Telomerase and the genetics of emphysema susceptibility. Implications for pathogenesis paradigms and patient care. Ann. Am. Thorac. Soc., 13, S447–S451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stanley S.E., Gable D.L., Wagner C.L., Carlile T.M., Hanumanthu V.S., Podlevsky J.D., Khalil S.E., DeZern A.E., Rojas-Duran M.F., Applegate C.D. et al. (2016) Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci. Transl. Med., 8, 351ra107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stanley S.E., Chen J.J., Podlevsky J.D., Alder J.K., Hansel N.N., Mathias R.A., Qi X., Rafaels N.M., Wise R.A., Silverman E.K. et al. (2015) Telomerase mutations in smokers with severe emphysema. J. Clin. Invest., 125, 563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Do R., Stitziel N.O., Won H.H., Jorgensen A.B., Duga S., Angelica Merlini P., Kiezun A., Farrall M., Goel A., Zuk O. et al. (2015) Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature, 518, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ellinghaus D., Zhang H., Zeissig S., Lipinski S., Till A., Jiang T., Stade B., Bromberg Y., Ellinghaus E., Keller A. et al. (2013) Association between variants of PRDM1 and NDP52 and Crohn's disease, based on exome sequencing and functional studies. Gastroenterology, 145, 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu T.W., Chahrour M.H., Coulter M.E., Jiralerspong S., Okamura-Ikeda K., Ataman B., Schmitz-Abe K., Harmin D.A., Adli M., Malik A.N. et al. (2013) Using whole-exome sequencing to identify inherited causes of autism. Neuron, 77, 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gonzaga-Jauregui C., Harel T., Gambin T., Kousi M., Griffin L.B., Francescatto L., Ozes B., Karaca E., Jhangiani S.N., Bainbridge M.N. et al. (2015) Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell. Rep., 12, 1169–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lange L.A., Hu Y., Zhang H., Xue C., Schmidt E.M., Tang Z.Z., Bizon C., Lange E.M., Smith J.D., Turner E.H. et al. (2014) Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am. J. Hum. Genet., 94, 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qiao D., Lange C., Laird N.M., Won S., Hersh C.P., Morrow J., Hobbs B.D., Lutz S.M., Ruczinski I., Beaty T.H. (2017) Gene-based segregation method for identifying rare variants in family-based sequencing studies. Genet. Epidemiol., 41, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qiao D., Lange C., Beaty T.H., Crapo J.D., Barnes K.C., Bamshad M., Hersh C.P., Morrow J., Pinto-Plata V.M., Marchetti N. et al. (2016) Exome sequencing analysis in severe, early-onset chronic obstructive pulmonary disease. Am. J. Resp. Crit. Care. Med., 193, 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsson H., Soderhall C., Einarsdottir E., Lamontagne M., Gudmundsson S., Backman H., Lindberg A., Ronmark E., Kere J., Sin D. et al. (2016) Targeted high-throughput sequencing of candidate genes for chronic obstructive pulmonary disease. BMC Pulm. Med., 16, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanehisa M., Furumichi M., Tanabe M., Sato Y. and Morishima K. (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res., 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yew-Booth L., Birrell M.A., Lau M.S., Baker K., Jones V., Kilty I. and Belvisi M.G. (2015) JAK-STAT pathway activation in COPD. Eur. Respir. J., 46, 843–845. [DOI] [PubMed] [Google Scholar]
- 31. Menche J., Sharma A., Kitsak M., Ghiassian S.D., Vidal M., Loscalzo J. and Barabasi A.L. (2015) Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science, 347, 1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J., Xu H., Aronow B.J. and Jegga A.G. (2007) Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics, 8, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zandvoort A., Postma D.S., Jonker M.R., Noordhoek J.A., Vos J.T., Geld Y.M. and Timens W. (2006) Altered expression of the Smad signalling pathway: implications for COPD pathogenesis. Eur. Respir. J., 28, 533–541. [DOI] [PubMed] [Google Scholar]
- 34. Baraldo S., Bazzan E., Turato G., Calabrese F., Beghe B., Papi A., Maestrelli P., Fabbri L.M., Zuin R. and Saetta M. (2005) Decreased expression of TGF-beta type II receptor in bronchial glands of smokers with COPD. Thorax, 60, 998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ezzie M.E., Crawford M., Cho J.H., Orellana R., Zhang S., Gelinas R., Batte K., Yu L., Nuovo G., Galas D. et al. (2012) Gene expression networks in COPD: microRNA and mRNA regulation. Thorax, 67, 122–131. [DOI] [PubMed] [Google Scholar]
- 36. Hessel J., Heldrich J., Fuller J., Staudt M.R., Radisch S., Hollmann C., Harvey B.G., Kaner R.J., Salit J., Yee-Levin J. et al. (2014) Intraflagellar transport gene expression associated with short cilia in smoking and COPD. PLoS One, 9, e85453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou X., Qiu W., Sathirapongsasuti J.F., Cho M.H., Mancini J.D., Lao T., Thibault D.M., Litonjua A.A., Bakke P.S., Gulsvik A. et al. (2013) Gene expression analysis uncovers novel hedgehog interacting protein (HHIP) effects in human bronchial epithelial cells. Genomics, 101, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peloso G.M., Auer P.L., Bis J.C., Voorman A., Morrison A.C., Stitziel N.O., Brody J.A., Khetarpal S.A., Crosby J.R., Fornage M. et al. (2014) Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am. J. Hum. Genet., 94, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beaudoin M., Goyette P., Boucher G., Lo K.S., Rivas M.A., Stevens C., Alikashani A., Ladouceur M., Ellinghaus D., Torkvist L. et al. (2013) Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet., 9, e1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stuart B.D., Choi J., Zaidi S., Xing C., Holohan B., Chen R., Choi M., Dharwadkar P., Torres F., Girod C.E. et al. (2015) Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet., 47, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Diaz de Leon A., Cronkhite J.T., Katzenstein A.L., Godwin J.D., Raghu G., Glazer C.S., Rosenblatt R.L., Girod C.E., Garrity E.R., Xing C. et al. (2010) Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One, 5, e10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Petrovski S., Todd J.L., Durheim M.T., Wang Q., Chien J.W., Kelly F.L., Frankel C., Mebane C.M., Ren Z., Bridgers J. et al. (2017) An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am. J. Respir. Crit. Care. Med., 196, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsakiri K.D., Cronkhite J.T., Kuan P.J., Xing C., Raghu G., Weissler J.C., Rosenblatt R.L., Shay J.W. and Garcia C.K. (2007) Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA, 104, 7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bertuch A.A. (2016) The molecular genetics of the telomere biology disorders. RNA Biology, 13, 696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu G., Warren L., Aponte J., Gulsvik A., Bakke P., Anderson W.H., Lomas D.A., Silverman E.K., Pillai S.G. and International, C.G.N.I (2007) The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. Am. J. Respir. Crit. Care. Med., 176, 167–173. [DOI] [PubMed] [Google Scholar]
- 46. Elleder M., Houstkova H., Zeman J., Ledvinova J. and Poupetova H. (2001) Pulmonary storage with emphysema as a sign of Niemann-Pick type C2 disease (second complementation group). Report of a case. Virchows Arch., 439, 206–211. [DOI] [PubMed] [Google Scholar]
- 47. Zhang Y., Calado R., Rao M., Hong J.A., Meeker A.K., Dumitriu B., Atay S., McCormick P.J., Garfield S.H., Wangsa D. et al. (2014) Telomerase variant A279T induces telomere dysfunction and inhibits non-canonical telomerase activity in esophageal carcinomas. PLoS One, 9, e101010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dumanski J.P., Rasi C., Bjorklund P., Davies H., Ali A.S., Gronberg M., Welin S., Sorbye H., Gronbaek H., Cunningham J.L. et al. (2017) A MUTYH germline mutation is associated with small intestinal neuroendocrine tumors. Endocr. Relat. Cancer, 24, 427–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kho A.T., Chhabra D., Sharma S., Qiu W., Carey V.J., Gaedigk R., Vyhlidal C.A., Leeder J.S., Tantisira K.G. and Weiss S.T. (2016) Age, sexual dimorphism, and disease associations in the developing human fetal lung transcriptome. Am. J. Respir. Cell Mol. Biol., 54, 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beghe B., Bazzan E., Baraldo S., Calabrese F., Rea F., Loy M., Maestrelli P., Zuin R., Fabbri L.M. and Saetta M. (2006) Transforming growth factor-beta type II receptor in pulmonary arteries of patients with very severe COPD. Eur. Respir. J., 28, 556–562. [DOI] [PubMed] [Google Scholar]
- 51. Cho M.H., McDonald M.L., Zhou X., Mattheisen M., Castaldi P.J., Hersh C.P., Demeo D.L., Sylvia J.S., Ziniti J., Laird N.M. et al. (2014) Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir. Med., 2, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. De Winter J.P., De Vries C.J., Van Achterberg T.A., Ameerun R.F., Feijen A., Sugino H., De Waele P., Huylebroeck D., Verschueren K. and Van Den Eijden-Van Raaij A.J. (1996) Truncated activin type II receptors inhibit bioactivity by the formation of heteromeric complexes with activin type I. receptors. Exp. Cell Res., 224, 323–334. [DOI] [PubMed] [Google Scholar]
- 53. McDonald M.L., Mattheisen M., Cho M.H., Liu Y.Y., Harshfield B., Hersh C.P., Bakke P., Gulsvik A., Lange C., Beaty T.H. et al. (2014) Beyond GWAS in COPD: probing the landscape between gene-set associations, genome-wide associations and protein-protein interaction networks. Hum. Hered., 78, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boueiz A., Chase R., Lamb A., Lee S., Naing Z.Z.C., Cho M.H., Parker M.M., Hersh C.P., Crapo J.D., Stergachis A.B. et al. (2017) Integrative genomics analysis identifies ACVR1B as a candidate causal gene of emphysema distribution in non-alpha 1-antitrypsin deficient smokers. bioRxiv, doi.org/10.1101/189100. [DOI] [PMC free article] [PubMed]
- 55. Tilley A.E., Walters M.S., Shaykhiev R. and Crystal R.G. (2015) Cilia dysfunction in lung disease. Annu. Rev. Physiol., 77, 379–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wain L.V., Sayers I., Soler Artigas M., Portelli M.A., Zeggini E., Obeidat M., Sin D.D., Bosse Y., Nickle D., Brandsma C.A. et al. (2014) Whole exome re-sequencing implicates CCDC38 and cilia structure and function in resistance to smoking related airflow obstruction. PLoS Genet., 10, e1004314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kenna K.P., Doormaal P.T., Dekker A.M., Ticozzi N., Kenna B.J., Diekstra F.P., Rheenen W., Eijk K.R., Jones A.R., Keagle P. et al. (2016) NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet., 48, 1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Regan E.A., Hokanson J.E., Murphy J.R., Make B., Lynch D.A., Beaty T.H., Curran-Everett D., Silverman E.K. and Crapo J.D. (2010) Genetic epidemiology of COPD (COPDGene) study design. COPD, 7, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Silverman E.K., Chapman H.A., Drazen J.M., Weiss S.T., Rosner B., Campbell E.J., O'Donnell W.J., Reilly J.J., Ginns L., Mentzer S. et al. (1998) Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am. J. Respir. Crit. Care Med., 157, 1770–1778. [DOI] [PubMed] [Google Scholar]
- 60. Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X. and Ruden D.M. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen H., Wang C., Conomos M.P., Stilp A.M., Li Z., Sofer T., Szpiro A.A., Chen W., Brehm J.M., Celedon J.C. et al. (2016) Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. Am. J. Hum. Genet., 98, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jiang D. and McPeek M.S. (2014) Robust rare variant association testing for quantitative traits in samples with related individuals. Genet. Epidemiol., 38, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shihab H.A., Gough J., Cooper D.N., Stenson P.D., Barker G.L., Edwards K.J., Day I.N. and Gaunt T.R. (2013) Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat., 34, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Morrow J., Qiu W., DeMeo D.L., Houston I., Pinto Plata V.M., Celli B.R., Marchetti N., Criner G.J., Bueno R. and Washko G.R. (2015) Network analysis of gene expression in severe COPD lung tissue samples (abstract). Am. J. Respir. Crit. Care Med., 191, A1253; (published conference abstract from: A30. Big Data: Harvesting Fruits COPD and Lung Cancer, May 1, 2015, A1253–A1253). [Google Scholar]
- 66. Blake J.A., Eppig J.T., Kadin J.A., Richardson J.E., Smith C.L., Bult C.J., the Mouse Genome Database and G. (2017) Mouse genome database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res., 45, D723–D729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.