

Abstract
Morphogenetic events are driven by cell-generated physical forces and complex cellular dynamics. To improve our capacity to predict developmental effects from chemical-induced cellular alterations, we built a multi-cellular agent-based model in CompuCell3D that recapitulates the cellular networks and collective cell behavior underlying growth and fusion of the mammalian secondary palate. The model incorporated multiple signaling pathways (TGFβ, BMP, FGF, EGF, SHH) in a biological framework to recapitulate morphogenetic events from palatal outgrowth through midline fusion. It effectively simulated higher-level phenotypes (e.g., midline contact, medial edge seam (MES) breakdown, mesenchymal confluence, fusion defects) in response to genetic or environmental perturbations. Perturbation analysis of various control features revealed model functionality with respect to cell signaling systems and feedback loops for growth and fusion, diverse individual cell behaviors and collective cellular behavior leading to physical contact and midline fusion, and quantitative analysis of the TGF/EGF switch that controls MES breakdown – a key event in morphogenetic fusion. The virtual palate model was then executed with theoretical chemical perturbation scenarios to simulate switch behavior leading to a disruption of fusion following chronic (e.g., dioxin) and acute (e.g., retinoic acid, hydrocortisone) chemical exposures. This computer model adds to similar systems models toward an integrative ‘virtual embryo’ for simulation and quantitative prediction of adverse developmental outcomes following genetic perturbation and/or environmental disruption.
Keywords: Computational Biology, Morphogenetic Fusion, Predictive Toxicology, Children’s Health, Agent-Based Model, CompuCell3D, Developmental toxicity
TOC FIGURE
1. INTRODUCTION
Understanding how tissues and organs are shaped during embryogenesis is a central question in developmental biology. Morphogenetic events are driven by cell-generated physical forces and complex cellular dynamics. The key drivers are individual cell behaviors – mitosis, migration, differentiation, adhesion, shape-change, apoptosis, and extracellular matrix (ECM) remodeling – but the realized series of morphogenetic events depends on cells’ coordinated and collective behavior.1, 2 Individual cells may display incorrect or inappropriate behavior, either spontaneously or as a result of genetic and/or environmental factors, and yet gene regulatory networks exist that buffer embryos against microshifts in cell fate or specification.3 Whether such canalization ensures normal development will depend on the degree and nature of perturbation and on the resiliency of the affected systems: teratogenesis occurs when embryos encounter unexpected environmental stressors that exceed the system’s buffering limits.4 As such, our capacity to predict developmental effects from cellular alterations is limited by extant knowledge of the dynamic control and resiliency of cellular networks.
Here, we describe a multi-cellular agent-based model that recapitulates the cellular networks and collective cell behavior underlying a particular morphogenetic event – growth and fusion of the mammalian secondary palate – and use this computational (in silico) model to further mechanistic understanding and predictive modeling of developmental toxicity leading to cleft palate. The mammalian secondary palate develops from two separate bilaterally paired shelves that meet and fuse into a continuous anatomical structure separating the oral and nasal cavities.5 Disruption of these events by genetic and/or environmental factors can lead to cleft palate, an important human birth defect that affects 5 to 20 in 10,000 live births.1, 6 In animal studies and human epidemiology, cleft palate is observed following prenatal exposure to many compounds, including glucocorticoids,7–9 valproic acid,10, 11 alcohol,12, 13 and methylmercury.14 The window of vulnerability for cleft palate is 10–14 days of gestation in the mouse,15 corresponding to 5–7 weeks gestation in humans.16 In mammals, as informed by mouse studies, the morphogenetic progression spans from initial budding of palatal shelves from the maxillary processes (E12.5) to outgrowth vertically downward alongside the tongue, reorientation to a horizontal position above the tongue, contact of the shelves’ medial edge epithelium (MEE) to form a midline epithelial seam (MES), and dissolution of the MES to allow fusion of right-left palate rudiments (E15.5).17 Multiple signaling pathways are involved, including those for transforming growth factor beta (TGFβ), bone morphogenetic proteins (BMPs), fibroblast growth factors (FGFs) and sonic hedgehog (SHH).18 These pathways mediate epithelialmesenchymal interactions to drive outgrowth and fusion of the palatal processes.17−19
In an alternate emerging paradigm, computational toxicology models make predictions for apical endpoints such as cleft palate based on vast amounts of cellular and molecular effects data now on hand from profiling thousands of chemicals across hundreds of high-throughput and high-content screening assays, including human stem cells and embryonic zebrafish.20 A significant challenge is building models like the one presented here that use biological frameworks to recapitulate complex signaling networks in the embryo. Although critical aspects of in vivo palate development may await discovery, computer simulation with an agent-based model can be used to reconstruct a morphogenetic series of events from the bottom-up, cell-by-cell and interaction-by-interaction. The capacity to simulate a self-organizing biological system in silico offers a heuristic solution to predict tissue-level effects of environmental exposure(s) in a model that approximates the extant embryology and accommodates future discoveries. The model presented here for palatal development (cleft palate) adds to previous models for angiogenesis (angiodysplasia)21 and urethral fusion (hypospadias)22 toward an array of systems comprising a ‘virtual embryo’ for simulation and quantitative prediction of adverse developmental outcomes following genetic perturbation and/or environmental disruption.
2. METHODS and IMPLEMENTATION
2.a. Model scope
The virtual palate model was constructed using CompuCell3D, Version 3.7.4 (CC3D; http://CompuCell3D.org) with scripting in Python (v 2.7). CompuCell3D is an open-source environment for simulating the spatio-temporal dynamics of multicellular systems given specified cell behaviors, regulatory signals and physical properties. The model begins with an initial tissue configuration corresponding to the onset of palate shelf formation and recapitulates growth and fusion of the mammalian secondary palatal processes through midline MEE contact and MES dissolution. Specifications were developed from relevant information in the extant literature, focusing on several key signaling pathways (TGFβ, SHH, FGF, EGF, BMP), and their regulation of differentiation and cell behaviors - e.g., proliferation, apoptosis, polarization, ECM secretion and epithelial-to-mesenchymal transitions.
An appropriately abstracted model and boundary conditions were defined for gestational days E12.5 to E16.0 in the mouse to recapitulate the sequence of events spanning initial budding of palatal shelves from the maxillary processes (E12.5) to fusion of right-left palate rudiments (E15.5), timed with regards to conventional nomenclature of E0 being the onset of gestation.17 Anatomically, this progression includes mesenchymal proliferation and ECM accumulation (E12.5–13.5); shelf reorientation to a horizontal position above the tongue (E13.5–14.5); contact of the rudiments at their medial edges to form a midline epithelial seam (MES); and MES breakdown to allow right-to-left confluence of mesenchyme (E14–15.5). We modeled these stages using an idealized two-dimensional (2D) cross-section that includes right and left palatal process growing medially and surrounded by a fluid medium reflecting the primitive oral-nasal cavity (Figure 1). The model explicitly represents a coronal cross-section through the anterior region of the secondary palatal processes. It recapitulates all of the events described above except reorientation of the shelves from vertical to horizontal. Palatal shelves in the computer model grow horizontally from the start, which reflects tissue growth kinetics to bring the right and left shelves into contact, but is an abstraction of the reoriented condition that requires a third dimension and is left for future implementation.
Figure 1. Layout of the virtual palate model (pre-fusion, E13-E14) in a coronal plane.
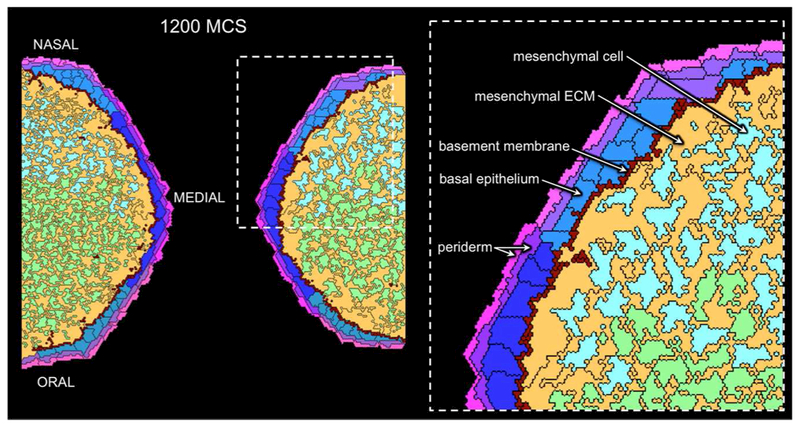
Mesenchymal cells (green and cyan) are randomly embedded in a hyaluronate-rich ECM (orange). The color-coding for mesenchymal cells denotes their origin from either the oral (green) or nasal (cyan) half of each palate shelf. The overlying ectoderm is represented as a columnar epithelium (shades of blue) covered by squamous periderm cells that are polarized with separate apical and basal compartments (shades of pink and magenta, respectively). Ectoderm cells with darker shading are those covering the medial edge. A basement membrane (dark red) lies along the ectoderm-mesenchyme border and the palatal shelves are surrounded by a fluid filling the oral-nasal cavity (black). Cell types are labeled in the expanded inset.
CompuCell3D is based on a cellular Potts model that treats individual cells as autonomous agents interacting in a shared physical environment, modeled as a discrete lattice, in which each cell occupies multiple lattice sites.23, 24 The initial configuration of the virtual palate model consists of two circular segments (radius = 46.5 lattice sites or pixels; segmenting chord positioned 19 pixels off center; 1 pixel is approximately 1 μm) that protrude from the vertical borders of a 200 × 400 hexagonal lattice. Each semi-circular segment represents the initial budding of a palatal shelf from the left or right side of the embryo with an ectodermal epithelium of 20 epithelial cells and 10 periderm cells encasing an interior 35-cell, ECM-free mesenchyme. This initial cross-section of the mid-anterior palate is roughly quarter-scaled to the biology: cross-sections of the mouse mid-anterior palate have about 150 cells and very little extracellular space at E12.5.25
Model dynamics follow a Metropolis Monte Carlo algorithm that repeatedly attempts to change which cell occupies randomly chosen lattice sites.26 The success of attempted changes is governed by corresponding changes in a pseudo-energy function (ΔE; see below for details): favorable pseudo-energy changes (ΔE ≤ 0) are always successful; unfavorable changes (ΔE > 0) are successful with probability where Teff is the fluctuation amplitude or effective temperature. This cell-specific value reflects motility of that particular cell type and adds a stochastic element to the model framework. Dynamics were discretized in Monte Carlo Steps (MCS) – each MCS corresponding to 200×400 = 80,000 attempted changes. Each simulation began with a 200-MCS equilibration period and then continued to 6000 MCS.
2.b. Cell and ECM types
The model includes three primary cell types – mesenchymal, epithelial and periderm – each with multiple sub-types (refer to Figure 1 for color schema). Mesenchymal cells can be of oral, nasal or EMT subtype based on the spatial location of the cell’s earliest predecessor; cells of EMT subtype are those that began as epithelial, but then underwent an epithelial-to-mesenchymal transition. Epithelial and periderm cells can similarly be of oral, medial or nasal subtype. In addition, periderm cells are polarized into two subcellular compartments – apical and basal – that the model treats as paired agents.
The model also includes two types of extracellular matrix – mesenchymal ECM and basement membrane (BM) – that are treated as generalized “cells” in CC3D. Elements of these types are assigned pseudo-energy terms that govern their interactions with cells (see below), but they are not granted the same agency as cells.
2.c. Pseudo-energy function
The pseudo-energy function governing temporal evolution of the model has terms for interfacial contact energies (Einterface), shape constraints (Eshape), and focal point plasticity (Efocal-point plasticity). The paragraphs below describe each of these terms and detail which pseudo-energy parameters are fixed and which vary dynamically as the model evolves.
The contact energy terms determine how cells, ECM and BM segregate. The pseudo-energy of any generalized cell-cell interface is proportional to its length with a proportionality constant that depends on the two contacting cell types.26 The sum over all interfaces can be written as
| (1) |
where (ρi, ρj), (σi, σj) and (τi, τj) are respectively the generalized cell IDs, cluster IDs, and cell types at neighboring lattice sites i and j; δ is the Kronecker delta function; and Jint, Jext are matrices of cell-type-dependent interfacial energies (listed in Table 1). Jext is applicable to contact between different cells or between a cell and the ECM, basement membrane or surrounding medium. Jint is applicable only for contact between subcellular compartments of a single generalized cell, e.g., the apical and basal compartments of a periderm cell, which have the same cluster ID. The primary entries in Table 1 are for Jext, but it also includes entries for the model’s only two possible intracellular contacts: Jint,Peri_A-Peri_B and Jint,BM-BM. These energies correspond biologically to a combination of cortical tension and surface adhesion between a given pair of generalized cell types: smaller energies in Table 1 denote more favorable types of contacts. The two contact energy matrices are defined in the code at the start of a simulation and remain fixed throughout.
Table 1. Matrix of contact energies in the virtual palate model.
Primary entries are for Jext with energies for the two possible intracellular contacts in Jint are listed in parentheses. Abbreviations: MESENCH, mesenchyme; ECM, extracellular matrix; BM, basement membrane, EPITH, epithelium; PERI_B, periderm basal compartment; PERI_A, periderm apical compartment; and EXT, surrounding external fluid.
| “Cell” Type | MESENCH | ECM | BM (int) | EPITH | PERI_B | PERI_A (int) | EXT |
|---|---|---|---|---|---|---|---|
| MESENCH | 3 | −1 | 6 | 16 | 30 | 30 | 100 |
| ECM | 0 | 6 | 16 | 30 | 30 | 100 | |
| BM (int) | 8 (6) | 6 | 20 | 30 | 100 | ||
| EPITH | 4 | 10 | 30 | 100 | |||
| PERI B (int) | 10 | 30 (2) | 30 | ||||
| PERI A | 10 | 6 | |||||
| EXT | 0 |
The shape constraint terms influence cell volume and shape through quadratic terms that penalize deviations from morphological target values:24, 27
| (2) |
where V, S and L are respectively the cell volume (area in 2D), surface area (perimeter in 2D), and major axis length. Each shape measure has a constant associated stiffness (λvol, λsurface or λlength) and a dynamic cell-specific target value. If a particular constraint is not used for a given cell, then that constraint’s stiffness is zero. When cell behaviors in a simulation change a cell’s target volume, the model automatically updates that cell’s target surface area and target length (if applicable) to new values that depend on the new target volume. For periderm cells, the updating relationships are set to favor elongated shapes: Starget = 0.7Vtarget and Ltarget = 0.3Vtarget for unpaired basal compartments; and Starget = 0.9Vtarget and Ltarget = 0.5Vtarget for paired apical and basal compartments. For mesenchymal and epithelial cells, the target surface area is updated to (and no length constraint is used), a relationship that favors cuboidal shapes. This general shape can be seen in the model’s epithelial cells, but its mesenchymal cells purposely deviate from cuboidal and are instead setup to favor highly ramified shapes. This is accomplished by setting the MESENCH-ECM contact energy negative, which makes the pseudo-energy lower (more favorable) when MESENCH cells adopt shapes that increase their contact length with the surrounding ECM. The generalized cells representing ECM and BM have target volumes, but not target surface areas or lengths.
The focal point plasticity terms were only used for BM elements to represent the structural elasticity of BM and help maintain it as a thin chain of elements.24, 27 A focal point plasticity link between the centroids of two BM elements acts as a breakable elastic spring. The energy term for all of these links is given by:
| (3) |
where λlink is an elastic spring constant and rm,n is the centroid-to-centroid distance between linked elements m and n. Plasticity arises because the links form probabilistically and break when stretched to rm,n > rbreak. The spring constant λlink is a global constant in our model, but the linked pairs are dynamic.
2.d. Diffusible signals
Morphogenesis in the virtual palate model, including palatal outgrowth and midline fusion, is driven by a biological network minimized to specific diffusible signals, juxtacrine signaling via cell-cell contacts, and regulated cell behaviors (Figure 2). Based on a general profile derived from extant literature,1, 17, 28–38 the model represents several key regulatory nodes explicitly. In particular, Sonic Hedgehog (SHH) and FGF10 form a key positive feedback loop for early palatal outgrowth: SHH signals from epithelium to mesenchyme to promote mesenchymal cell proliferation, and FGF10 signals go the other way (mesenchyme to epithelium) to maintain Shh expression.29, 30 Activity of this central loop is modulated by Ephrin juxtacrine signaling,34 by FGF7 (on the nasal side),39 and by the interactions among BMP2, BMP4 and Noggin.40 The other key signaling module is a mutually inhibitory EGF-TGFβ3 switch. EGF and TGFβ3 signals suppress one another’s expression and have opposite effects on ectodermal cell behaviors.41–44 In combination with additional Ephrin signals,37, 45 this switch governs whether cells in the MEE proliferate or die. The behavior of this switch can be modulated by exogenous compounds through crosstalk with pathways mediated by retinoid receptor, glucocorticoid receptor, or aryl hydrocarbon receptor (AhR) systems.43, 46–56 In addition to these explicitly modeled signals, Figure 2 shows the implicit roles of additional components in each signaling pathway, e.g., the transmission of SHH signaling through patched (Ptc1) and smoothened (Smo),30, 57 of FGF signaling through receptors (FGFR2b) in the epithelium,29 and of BMP/TGFβ signaling through SMADs.17, 58 These are not explicitly included in the model, but are highlighted to show how their mutation or inhibition would affect the model.
Figure 2. Signaling network driving the virtual palate model.
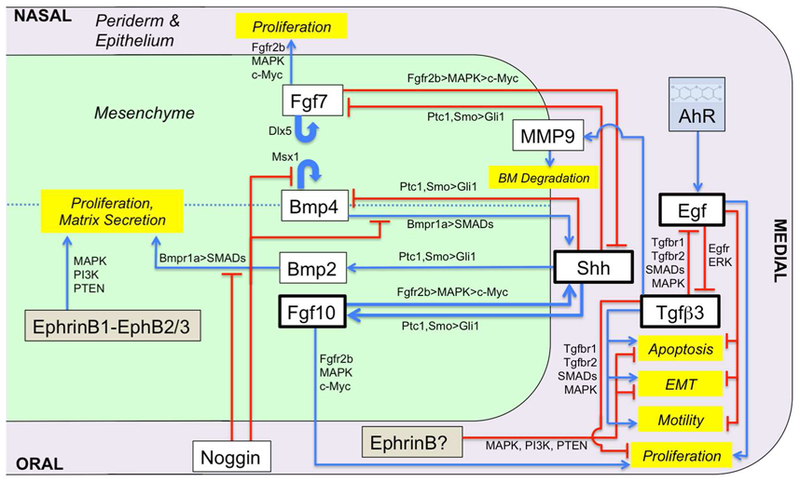
The oral side, nasal side and medial edge are indicated (not drawn to scale). A minimal signaling network that coordinates several primary pathways (SHH, FGF, BMP, TGFβ3, EGF, Noggin, EphB) was derived from extant literature. Boxes represent signaling molecules (white) and linked behaviors (yellow) that are explicitly included in the model. Intermediate molecules in the relevant pathways are noted along the connecting arrows.
Diffusion and decay parameters for the diffusible signals have not been measured in vivo, so we chose similar values for each molecule with a few notable exceptions. The global diffusion constant for signals regulating mesenchymal growth was 0.5 pixels2/MCS, but that for the ectodermal signals TGFβ3 and EGF was reduced to 20% of this value to reflect local signaling within the epithelial compartment. In addition, cell-type-specific diffusion constants were designated for FGF10, FGF7 and BMP4 to account for their sequestration by the extracellular matrix59, 60 - slowing their diffusion constant in ECM to 0.2 pixels2/MCS. Finally, the cell-type-specific diffusion constant for SHH was set to zero in the surrounding fluid and ECM to represent how palmitoylation and cholesterol esterification of SHH render the mature ligand membrane bound, thus requiring cells to create SHH gradients through reiterated uptake and secretion.61 The rate constant for decay of each signal was set to 0.005 MCS−1, except for that of FGF7, which required a reduced value of 0.001 MCS−1 for its spatial patterning in the simulations to match experimental observations.29
Most regulatory effects on cell behavior were modeled as sigmoidal functions of the diffusible signals. Signal concentrations are thus reported as multiples of each signal’s half-maximal activity concentration (AC50). The only receptor levels explicitly included in the model are those for EGF and TGFβ3 when more closely investigating how their mutual inhibition yields a switch-like biochemical circuit. Most simulations used EGFR and TGFβR levels of 1.86 and 2.14 respectively (levels relative to the AC50 for EGF and TGFβ3 effects), but a few noted exceptions used levels of 2.86 for both, which led to a more hysteretic and widely bistable switch. The strength of cellular responses to EGF and TGFβ3 signals were then calculated using standard sigmoidal functions of ligand-bound receptor levels (using Hill coefficients of 1 or 4 as detailed for each behavior below). The model did use a ligand-receptor binding affinity (KD) that was lower than the half-maximal activity concentration of bound receptor, i.e., KD = 0.71 AC50).
2.e. Model cell behaviors and their regulation
We used CompuCell3D steppable functions to implement the regulation of eight distinct cell behaviors: cell growth and proliferation, apoptosis, cell differentiation, periderm polarization, cell motility, ECM secretion, BM maintenance and signal secretion.27 Table 2 lists their assignment to the various cell types and associated effector molecules and a synopsis of their implementation. Although Python code specified the range of cell fate/state behaviors possible for each cell/agent type, their realization in a simulation was stochastic – that is, locally enabled by probabilities determined by an individual cell’s microenvironment.27
Table 2.
Cell behaviors and regulatory signals explicitly represented in the virtual palate mode
| Behavior | ↑ or ↓ Regulated By |
|---|---|
| Growth and Proliferation | ↑ contact with Mesench;
BMP2 ↓ Noggin(i)* |
| Apoptosis | ↓ contact with any cell or ECM |
| ECM Production | ↑ contact with Mesench; BMP2 ↓ Noggin(i) |
| Secretion of FGF10 | ↑ contact with Mesench; SHH |
| Secretion of BMP2 | ↑ contact with Mesench; SHH |
| Secretion of BMP4 | ↑ contact with Mesench;
BMP4 ↓ SHH; Noggin(i) |
| Secretion of MMPs | -- |
| Secretion of FGF7 | ↑ FGF7 ↓ SHH |
| Growth and Proliferation | ↑ FGF10; FGF7; EGF ↓ TGFβ3 |
| Apoptosis | ↑ TGFβ3 ↓ contact with any cell or ECM; contact w/ Medium or Peri; EGF |
| Differentiation to Mesench. | ↑ contact w/ Mesench or ECM;
TGFβ3 ↓ contact w/ Medium or Peri; EGF |
| Differentiation to Periderm | ↑ contact w/ Medium |
| BM Production/Maintenance | ↑ contact w/ Mesench or
ECM ↓ MMPs |
| Secretion of MMPs | ↑TGFβ3 |
| Secretion of SHH | ↑ FGF10; BMP4 ↓ FGF7; Noggin(i) |
| Secretion of TGFβ3 | ↓ EGF |
| Secretion of EGF | ↓ TGFβ3 |
| Secretion of Noggin | -- |
| Growth and Proliferation | ↑ FGF10; FGF7; EGF ↓ TGFβ3 |
| Apoptosis | ↑ TGFβ3 ↓ contact with any cell or ECM; contact w/ Medium or Peri; EGF |
| Polarization | -- |
| Motility | ↑ TGFβ3 ↓ EGF |
| Secretion of SHH | ↑ FGF10; BMP4 ↓ FGF7; Noggin(i) |
| Secretion of TGFβ3 | ↓ EGF |
| Secretion of EGF | ↓ TGFβ3 |
Noggin(i) refers to indirect regulation (i) by Noggin binding to BMP2 or BMP4 preventing activation of the BMP receptor as coded in the model.
2.e.1. Cell growth and proliferation:
The model implements cell growth and proliferation through semi-stochastic increases in cells’ target volumes; cell divisions then occur when cell volume exceeds a cell-type-specific mitosis trigger volume. All biochemical regulation of proliferation is implemented through control of cell growth. The specific amount added to each cell’s target volume is determined every 10th MCS by the cell’s regulated growth rate (Rgrowth) that is not stochastic, and growth probability (Pgrowth) that is stochastic:
| (4) |
where the function binomial[10, Pgrowth], implemented in the numpy package in Python, returns a random integer drawn from a binomial distribution for the number of successful growth steps (in the last 10) based on a single-step success probability Pgrowth. This stochastic function is used to limit the synchronicity of growth and subsequently triggered cell divisions. To keep cells well within the bounds of the simulation lattice, all growth ceases at 3000 MCS.
For mesenchymal cells, the base growth probability is regulated by juxtacrine signaling through EphrinB1-EphB2/3.34 This is implemented by increasing Pgrowth from 10% to 50% for mesenchymal cells in contact with other mesenchymal cells. The growth rate is then regulated by BMP2, BMP4 and Noggin.40 Noggin binds to BMP2 and BMP4 with a 1:1 stoichiometry and blocks their receptor binding sites.62, 63 The model assumes that binding of Noggin to BMPs is very strong and saturated so that the amount of free BMP2 (i.e., that available to bind to its receptor) is simply computed as
| (5) |
and the free BMP2 concentration then determines the growth rate via a sigmoidal Hill function:
| (6) |
Rgrowth and Pgrowth are used to calculate ΔVtarget for each cell following equation 4 above. After updating a mesenchymal cell’s target volume, its target surface area is updated accordingly.
For epithelial and periderm cells, the growth probability is always 50%. Their growth rate is upregulated by FGF10, FGF7 and EGF, and downregulated by TGFβ3.28, 29, 64 Both epithelial and periderm growth rates are determined by a similar combination of sigmoidal functions,
| (7) |
with slightly different constants: Rmax,EGF = 0.08 pixels/MCS for both, but R0 = 0.016 and 0.01 pixels/MCS and Rmax,FGF = 0.2 and 0.12 pixels/MCS for epithelial and periderm cells respectively. After updating each ectodermal cell’s target volume according to equation 4, its other shape parameters are updated accordingly.
Cell division is allowed when a cell’s volume exceeds its cell-type-specific mitosis trigger volume (Vmitosis). For most cell types, Vmitosis is set to 2x that cell type’s initial average volume. The only exceptions are medial epithelial and periderm cells for which the multiplier is instead 3x – slowing proliferation along the medial edge. When mesenchymal cells reach their trigger volume, mitosis occurs immediately, but ectodermal cells will delay mitosis until sufficient time has passed since a particular cell’s previous division. The wait time is initially set randomly to between 1 and 101 MCS (to randomize cell cycle phases), but is reset to 50 MCS after each cell division. Once a cell has committed to mitosis, all of its compartments divide and its target volumes are conserved with a random 46.5 to 53.5% split between parent and daughter cell. The division plane is random for mesenchymal cells and along the minor axis of elongated epithelial and periderm cells. After division, shape parameters of the parent and daughter cell are updated according to cell type as described above. Although ECM and basement membrane elements are encoded in the model as generalized “cells”, these do not correspond to biological cells and are thus not allowed to undergo cell division. Growth of ECM and basement membrane is instead driven by secretion from MESENCH and EPI cells (see 2.e.6–7).
2.e.2. Apoptosis:
Cells in the model commit to apoptosis (programmed cell death) stochastically. The probability for any cell to make this commitment depends on juxtacrine signaling, encoded through the identity of that cell’s neighbors, and on the local concentration of paracrine signals, in particular EGF and TGFβ3.31, 37, 43–45, 64, 65 Cellular commitment to apoptosis is irreversible. If any cell becomes isolated from all other cells, its apoptosis probability is 10% per MCS. In addition, the apoptosis probability for any epithelial cell or periderm compartment depends on the local concentration of EGF and TGFβ3 according to a pair of Hill functions:
| (8) |
where fchange is the fold-change in EGF receptor (EGFR) expression levels (e.g., induced by toxicant-driven AhR activation; see below) and Pmax is the maximum apoptosis probability determined by the nature of the cell’s microenvironment. For periderm cells, apoptosis probabilities are considered separately for their apical and basal compartments, but a decision point for apoptosis by either compartment commits the entire cell. For an apical periderm compartment, the default Pmax is 0.05% per MCS, and increases by 5% per MCS if it is not in contact with medium and another 5% per MCS if it is not in contact with basal periderm. Pmax is determined similarly for a basal periderm compartment, but the increases occur if it is not in contact with medium or apical periderm and if it is not in contact with medium or any other periderm cell. For an epithelial cell, the default Pmax is just 0.01% and increases by 1% per MCS if the cell is not in contact with medium or periderm. Apoptosis decisions are evaluated for every cell every tenth MCS (with Papo (ΔN =10) =1−(1− Papo)10).
As noted earlier, cells committed to the apoptotic pathway are not immediately removed, but are flagged and have their target volume decreased every subsequent MCS by ΔVtarget= −(1/τapo)Vtarget. As a result, the target volume of dying cells decays exponentially with time constant τapo = 120 MCS. Dying cells also round up due to a removal of their target length, if present, and by setting their target surface area to . Once Vtarget for a dying cell is below a few pixels, energy fluctuations can lead to its removal from the model. During palate fusion, when apoptosis is prevalent in the model, the two to three time constants required to completely eliminate an apoptotic cell correspond to approximately 2–6 hours.
2.e.3. Cell differentiation:
After 200 MCS, epithelial cells can stochastically differentiate into either periderm or mesenchymal cells with probabilities that depend on the local microenvironment. Differentiation to periderm can occur when an epithelial cell has lost contact with the underlying mesenchyme or its matrix. Such differentiation maintains a cell’s oral, medial or nasal designation. The probability for such an epithelial-to-periderm transition (EPT) in silico is 0.01% per MCS if the cell is already in contact with periderm and increases to 1% per MCS if it is in direct contact with the surrounding medium. Similarly, de-differentiation to mesenchyme can occur when an epithelial cell has lost contact with the overlying periderm or surrounding medium, but maintains contact with the mesenchyme or its matrix. This occurs most often as periderm cells die and small “pearls” of epithelial cells remain in the region of the MES. The base epithelial-to-mesenchymal transition (EMT) probability is 0.01% per MCS and can increase by up to 0.1% depending on the local concentration of TGFβ3 and EGF as described by a pair of Hill functions:
| (9) |
To track the prevalence of EMT, cells that undergo these transitions are designated as type MESENCH_EMT. For both types of differentiation, decisions are evaluated for every cell every 10th MCS (with PEPT or EMT (ΔN =10) =1−(1− PEPT or EMT)10).
2.e.4. Periderm polarization:
The model includes periderm polarization by allowing each periderm cell to have a single matched pair of apical and basal compartments. These are treated as separate generalized “cells” in CompuCell3D, but have the same cluster ID. The initial model configuration has no apical periderm, but any time after 50 MCS, unpaired basal compartments can nucleate apical compartments. Nucleation involves designating a location along the basal-periderm-medium border as the seed for a matching apical compartment and setting new target volumes: Vtarget,A = 1.2 (length of basal-periderm-medium border); and Vtarget,B decreased by Vtarget,A. Target surface areas and lengths are then set accordingly to favor elongated shapes for both compartments. Unpaired apical compartments can similarly nucleate new basal compartments (anywhere along their border that is not in contact with surrounding medium), but this is a rare event that occurs only when a basal periderm compartment is stochastically lost during a simulation.
2.e.5. Cell motility:
In cellular Potts models, a cell moves via stochastic fluctuations that add or remove lattice sites along its periphery. The fluctuation amplitude or effective temperature (Teff) determines how likely fluctuations are when their pseudo-energy change is unfavorable (i.e., for ) and thus determines how easy it is for a cell to move: cells with higher Teff are thus more motile. In our model, Teff is 4 for most cells and ECM elements, but can vary for periderm. After 300 MCS, the Teff for periderm cells can range from 0 to 10 as determined by Hill functions of the local concentration of TGFβ3 and EGF31:
| (10) |
Periderm cells are thus fairly quiescent until TGFβ3 signals rise and they then become highly motile.
2.e.6. ECM secretion:
Mesenchymal cells in the model also secrete ECM, so that growth of the palatal shelves is a combination of cell proliferation and ECM accumulation.17 ECM secretion and ingestion are implemented as a transfer of target volume between a mesenchymal cell and an adjacent ECM element – if one exists; if not, a new ECM element is nucleated at a random location along the cell border. The volume transferred from each cell is based on its BMP2-dependent growth rate (from Eqn 6 above) and its ECM secretion probability (Psec ECM):
| (11) |
where the transfer volume cannot exceed the cell volume. The two terms respectively represent deterministic ECM ingestion and stochastic ECM secretion. The ECM secretion probability is regulated by juxtacrine signaling implemented by increasing Psec ECM from 25% to 50% for mesenchymal cells in contact with other mesenchymal cells. Ingestion and secretion are in balance (on average) for Psec ECM = 25% in Eqn 11, but increasing this value to 50% clearly favors ECM secretion. After transferring ΔVto ECM, the shape parameters of each mesenchymal cell are updated based on its new target volume. As with cell growth, ECM secretion ceases at 3000 MCS to keep all cells and ECM well within the bounds of the simulation lattice.
2.e.7. BM maintenance:
The model includes an explicit basement membrane (BM) that underlies and is maintained by epithelial cells. Individual BM elements are treated as generalized “cells” with a small target volume (3.5) and a large volume stiffness (40x that of epithelial cells). The large volume stiffness is needed to limit the rate at which BM elements are lost to stochastic fluctuations and the half-integer target volume is purposely chosen to allow these stiff elements to still move over the model’s lattice – i.e., there is no energy penalty for changing a BM “cell’s” volume from 3 to 4, or vice versa, but a very large energy penalty for lowering the volume to 2 or less. To create an elongated BM chain (as seen in cross-section), BM elements are nucleated in pairs with matching cluster IDs and each element is allowed to form one intra-cluster and one inter-cluster focal-point plasticity (FPP) link. FPP links are elastic connections between the centroids of BM elements that form stochastically (when allowed) and that break when stretched beyond a maximum length (see Eqn 3). The BM is maintained by a balance between nucleation of new BM elements – which occurs when an epithelial cell is in direct contact with the mesenchyme or its matrix – and BM degradation through the action of MMPs. This degradation occurs stochastically in proportion to the local concentration of MMPs (P = 0.5% [MMP]) and is implemented by setting the target volume of a degraded BM element to zero. If a BM element becomes unpaired due to the loss of its partner from fluctuation or degradation, it is then also degraded by immediately setting its target volume to zero.
2.e.8. Signal secretion:
The final cell behavior is regulated secretion of diffusible signals. Since concentrations and secretion rates of these signaling molecules are normalized by their respective half-maximal activity, AC50 values in the above equations can generally be treated as equal to 1. The one exception arises from the direct binding of Noggin to BMP2 and BMP4,62 which requires identical normalization for all three concentrations. We normalize these to the minimum AC50 found in the model for any BMP2- or BMP4-regulated process (self-activation of BMP4 secretion). With this choice, the relevant AC50 is instead equal to 2 for BMP4-regulated secretion of SHH (Table 3) and for BMP2-regulated mesenchymal growth and ECM secretion (Eqn 6).
Table 3. Regulated signal secretion in the virtual palate model.
Negative sigmoid coefficients represent down-regulation with overall secretion rates limited to a minimum of zero. Note that the rate at which FGF10, BMP2 and BMP4 are secreted depends on whether the secreting mesenchymal cell is in contact with other mesenchymal cells.
| Epithelium and Periderm | FGF10 |
| BMP4 (free) | |
| FGF7 | |
| Mesenchyme | SHH |
| Nasal Mesenchyme | FGF7 |
| SHH | |
| Mesenchyme | SHH |
| Mesenchyme | BMP4 (free) |
| SHH | |
| Oral Epithelium and Periderm | basal |
| Medial Epithelium and Periderm | basal |
| EGF | |
| Medial Epithelium and Periderm | basal |
| TGFβ3 | |
| Mesenchyme | basal |
| Epithelium | basal |
| TGFβ3 |
The secretion rate for a particular diffusible signal can be regulated by multiple other signals. Mathematically, the rate is determined by a sum of sigmoidal functions (Hill coefficient = 1), with one sigmoid for each regulating signal. Regulated secretion in the model is summarized in Table 3 with a listing of the coefficients of these sigmoidal functions.
2.f. Modeling teratogenic perturbations of the TGFβ3/EGF switch:
As noted in Figure 2 and Table 3, the actions of TGFβ3 and EGF comprise a mutually inhibitory switch. To enable simulations of chemical exposures, we modeled this switch in greater detail as follows: (1) EGF binding to its receptor (EGFR) leads to a decreased rate of TGFβ3 expression; (2) TGFβ3 binding to its receptor (TGFβR) leads to a decrease of EGF expression; (3) these ligand-receptor bindings are fast processes compared to transcriptional rates; and (4) transcription-degradation processes of both pathways combine to yield a net cellular response. Ligand-binding of either growth factor [GF] to its cognate receptor [RGF] is thus described by
with transcription and degradation combining for each growth factor to yield
The symmetry between EGF and TGFβ3 is maintained for most parameter values: dissociation constant Kd = 0.71; Michaelis constant Km = 1; max transcription rate vm = 0.214; Hill coefficient h = 4; and degradation rate kd = 0.07. The symmetry was broken by either having different cognate receptor levels or different initial growth factor concentrations. The teratogenic effects simulated below were modeled by modifying EGF receptor levels.
3. RESULTS
3.a. Morphogenetic outgrowth and fusion
Simulations of growth and fusion in an idealized cross-section of the anterior palate ran for 6000 MCS, corresponding to mouse gestation days E12.5 to E16. Earlier pre-patterning events (E11.5 to E12.5) were imposed as initial conditions. Model outputs of the cell field for normal fusion are shown at various stages in Figure 3; model outputs for the signaling gradients are shown as snapshots at the midline fusion stage in Figure 4. The full time-dependent signaling gradients are shown alongside the cell field in Supplemental Movie S1. As expected for a Monte Carlo simulation, the relationship between developmental stage and MCS is not linear, with the simulation slowing as development progresses. Based on the landmarks of first contact and the completion of fusion between E14.5 and E15.5, the simulation corresponds to gestational days E13, E14, E15 and E16 at approximately 25, 500, 2000 and 5000 MCS, respectively.
Figure 3. Outgrowth, midline contact and fusion of the right-left palatal process simulated with the virtual palate model.
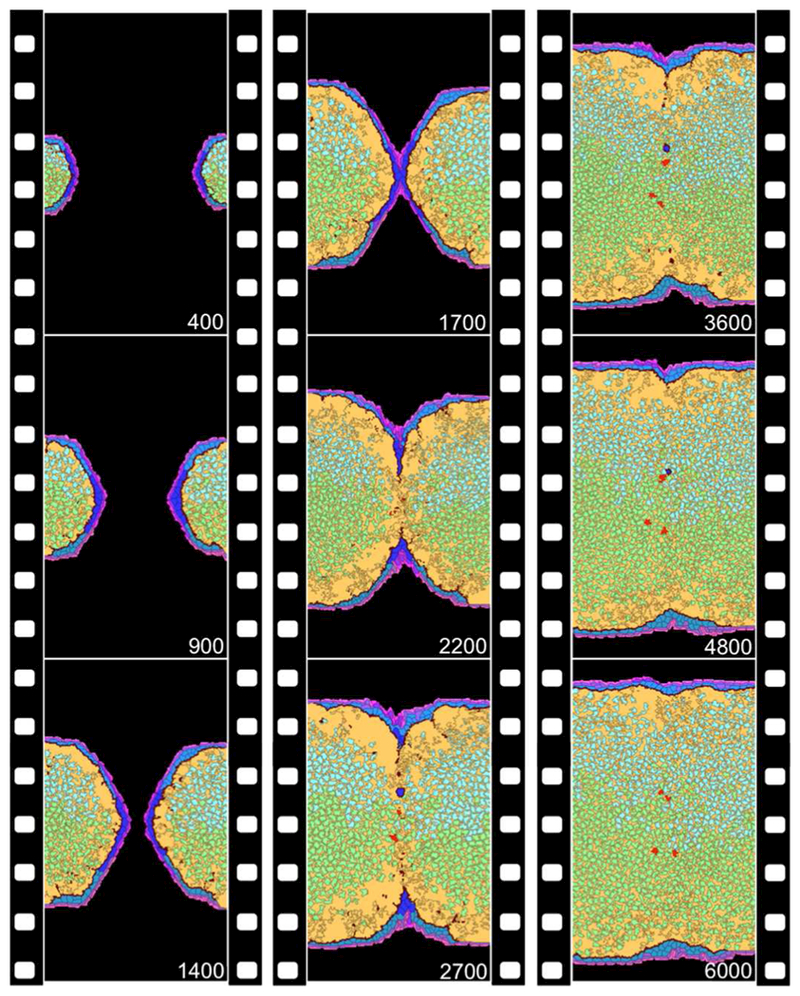
Orientation and cell types are defined and color-coded as in Figure 1. Simulation time (in MCS) is noted at the bottom right of each frame, and progresses from top to bottom along the three panels. Real time slows as the model progresses: E14 ~ 500 MCS, E15 ~ 2000 MCS and E16 ~ 5000 MCS. Note MES formation and breakdown – leaving behind residual pearls (blue) and EMT-derived mesenchyme (red) – as well as limited movement of interior mesenchymal cells. A complete set of time-lapse frames is available as Supplemental Movie S1.
Figure 4. Biochemical signal gradients in the virtual palate model.
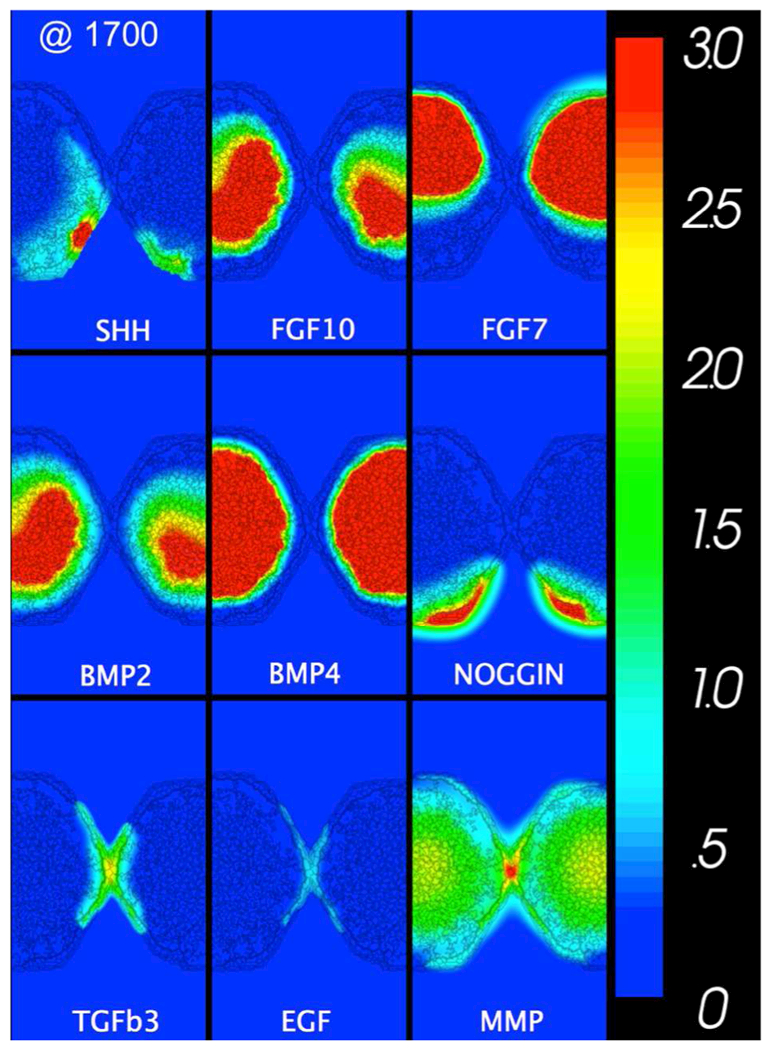
All gradient fields as labeled at the midline fusion stage (1700 MCS in Figure 3). The network driving the simulation is shown in Figure 2. The concentration of each effector molecule has been scaled relative to its minimum AC50 for biological effects (color bar to the right). A complete set of time-lapse gradients is available in Supplemental Movie S1.
Prefusion outgrowth was driven primarily by internal expansion of the mesenchyme, initially via cell growth and proliferation, but increasingly through ECM secretion at later times. Although sub-epithelial accumulation of ECM in the model implicitly represents sulfated proteoglycans, hydration-induced expansion of the hyaluronate-rich matrix is not represented in the model but is left for future development.25 Signaling between mesenchyme and ectoderm allowed the epithelial and periderm layers to match mesenchymal growth, maintaining complete barrier coverage, but not over-proliferating.
Physical contact between right-left palatal processes occurred in the midline leading to formation of a transitory MES (Figure 3). Across dozens of simulation runs, first contact and MES formation occurred 1600–2000 MCS, corresponding in real time to E14.8 to E15.0. By 3000 MCS, breakdown of the MES was marked by a few remaining pearls of ectoderm. Such pearls are observed in cross-sectional images of mouse palates taken between E15–15.5.18 By 5000 MCS (E16) mesenchymal confluence was normally complete.
Morphological changes in the MEE commenced with increased motility of periderm cells and programmed cell death. Both changes were semi-autonomous, following increases in TGFβ3 levels, but not requiring physical contact between opposing palatal shelves.33 Once contact did occur, periderm and then epithelial cells underwent increased rates of cell death. This initial MES breakdown was followed by migration of much of the residual MEE to triangular regions near the oral and nasal surfaces. Such epithelial triangles are regularly observed in vivo.18, 66 In the model, the migrating cells were not following a chemical gradient. Instead, migration to the epithelial triangles was an emergent phenomenon driven by differential cell-cell adhesion. By this point, the remnants of the MES were just a few pearls of epithelial cells. As the surrounding basement membrane was degraded by secreted MMPs, these MES pearls were eventually eliminated through a combination of cell death and EMT (red cells in Figure 3). With the probabilities chosen for each process in the model, approximately 70–85% of MEE cells were eventually cleared via cell death (many after initially migrating to the epithelial triangles). The remaining handful of cells were either incorporated into the oral/nasal ectoderm or underwent EMT to join the mesenchyme.
As the MES broke down and the mesenchyme became confluent, there was a slight mixing among mesenchymal cells from the right and left shelves. A similar degree of mixing is evident along the boundary of oral and nasal mesenchyme, but there was little movement by interior mesenchyme. Growth and proliferation in the mesenchyme also slowed as the MES broke down and fewer cells were available to secrete SHH. Despite this slowing, the few surviving MEE cells could secrete enough SHH to drive local mesenchymal growth. The model thus ceased all growth processes at 3000 MCS (~ E15.4), an artificial constraint imposed only to keep the final cell configuration from squeezing against the bounds of the modeled lattice. By the time this constraint took effect, fusion was essentially complete. The final period in the simulation (5000–6000 MCS) was not designed to recapitulate normal post-fusion biology, but was instead present to permit discriminate between inhibition versus delay of MES breakdown following various pre-fusion disturbances (described below).
3.b. Biochemical gradients
Diffusion gradients of the model’s explicit biochemical signals are shown as snapshots at the midline fusion stage in Figure 4 and dynamically for the entire simulation in Supplemental Movie S1. The patterns varied slightly between simulations and were similar but non-identical when comparing the right and left processes in a single simulation. This highlights the model’s inherent degree of stochasticity. The main driver of outgrowth was a positive feedback loop between SHH secreted from the MEE and FGF10 from the underlying mesenchyme. Differences in the spatial distribution of these signals’ sources and in their relative diffusion kinetics created a broader paracrine domain for FGF10. As SHH expression decayed with dissolution of the MEE, so too did FGF10 expression. The model further captured the mutual inhibitory loop between SHH-FGF7 as well as the interaction between SHH and BMP signals. Note that mesenchymal proliferation and ECM production were influenced by the concentration of free BMP, i.e., that not bound to Noggin. Dissolution of the MES was regulated by the mutual inhibition of EGF and TGFβ3, signals co-expressed in the MEE. As shown at first contact, TGFβ3 signals were a few times stronger than EGF signals. This disparity increased during midline fusion as the mutually inhibitory switch fell into a TGFβ3-high state. Matrix metalloproteinases (MMPs) are represented generically in the model. Some (e.g., MMP-13) are known to be highly expressed in the murine MEE and palatal mesenchyme in a manner dependent on TGFβ3 function.67 In simulations, MMP was ubiquitously expressed at basal levels, but was enhanced at sites of high TGFβ3 signaling. The spatial patterns of these key signals qualitatively matched those observed in coronal sections of mouse palates through a combination of immunostaining, in situ hybridization and LacZ reporters: FGF10, FGF7;29 SHH;30 TGFβ3;33, 68 Noggin;36 SHH, FGF10, BMP2, BMP4;40 MMP;67 EGF, TGFβ.69
3.c. Hacking the control network
To test functionality in the control network, simulations were run for in silico knockouts – i.e., by individually shutting off the secretion of each effector molecule after 200 MCS. This permitted the model to initiate normally and then simulate loss of function phenotypes. The final morphologies of the tested knockouts are shown in Figure 5.
Figure 5. Growth and fusion defects for in silico knockouts.
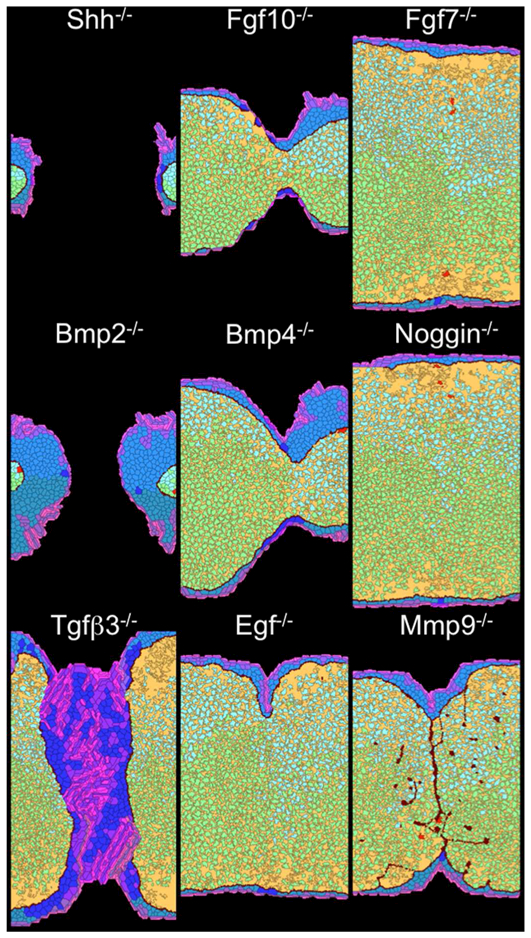
Each image shows model output at 6000 MCS (after E16) for loss of function of the indicated effector molecule. A time-lapse movie of the TGFβ3 knockout is available as Supplemental Movie S2.
Loss of SHH or BMP2 function severely disrupted outgrowth and resulted in failure to achieve midline contact (Figure 5). Weaker phenotypes were seen in simulations with lost BMP4 or FGF10 function, which partially disrupted outgrowth, but did not completely prevent contact or fusion. These phenotypes are consistent with an SHH-FGF10 loop acting through BMP2 and modulated by BMP4 as the major in silico drivers of outgrowth.29, 30, 40 In contrast, loss of function for FGF7 and Noggin had little to no impact on growth or fusion, yielding only slight differences in the relative proliferation of oral and nasal mesenchyme.
Loss of TGFβ3 function led to excessive MEE proliferation and a failure of tissue fusion. In contrast, loss of EGF function had little effect save a small midline indentation. These phenotypes are consistent with an EGF-TGFβ3 negative feedback loop being the key regulator of MES dissolution.41–44 These signals not only inhibited one another, but they influenced MEE cell behaviors in a diametrically opposed manner: EGF signaling promoted MEE growth and survival, and TGFβ3 signaling promoted epithelial apoptosis, EMT, and motility (Figure 2). The two signals act like a switch (see below). Loss of EGF function only pushes the switch to its normal TGFβ3-high state, but loss of TGFβ3 function pushes the switch the other way and prevents the processes necessary for MES breakdown. Downstream of TGFβ3, loss of MMP function also prevents complete MES breakdown through its control of BM degradation.
3.d. Simulating the teratogenic response to environmental exposures
To investigate the predictive capacity of the virtual palate model to simulate adverse effects of environmental insult, we attempted to recapitulate the teratogenic action of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in a chronic exposure scenario, and of all-trans retinoic acid (ATRA) in an acute exposure scenario.54 Among the components of the EGF and TGFβ3 pathways that regulate MES dissolution, TCDD or ATRA exposure leads to an overexpression of the EGF receptor (EGFR), which usually decreases in the MEE around E13.46 We thus investigated the mutual inhibition of EGF and TGFβ3 in more detail using a model of EGF-TGFβ3 interactions described above in Methods and shown in Figure 6A. Overall, the EGF-TGFβ3 switch is modeled by bound TGFβ3:TGFβR suppressing EGF secretion and bound EGF:EGFR suppressing TGFβ3 secretion. Changes in receptor levels can be induced by exogenous agents to alter the switch’s bias.
Figure 6. Bifurcation plots for the EGF-TGFβ3 circuit implemented in the virtual palate model.
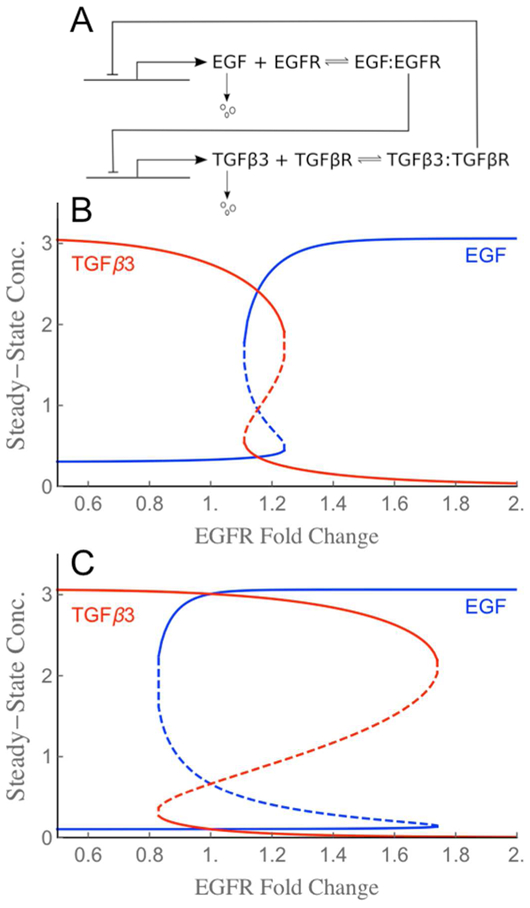
(A) Schematic of the mutual inhibitory circuit. Symmetry between EGF and TGFβ3, as well as the structure of the bistable zone can be broken through parameter choices. (B) Steady-state behavior for base receptor levels that yield a narrowly bistable switch with low hysteresis. For any EGFR fold-change in the bistable zone from 1.1 to 1.3, the circuit has three steady state solutions (i.e., three solutions for EGF and three for TGFβ3): two of the solutions are stable (solid lines) and one unstable (dashed line). (C) Increasing and equating the base receptor levels yields a switch with a wider bistable zone (0.8 to 1.8) and higher hysteresis (unstable solutions dashed).
As then shown in the bifurcation plots in Figure 6B-C, the receptor expression levels determine the behavior of this biochemical circuit. For the base receptor levels used in all simulations above (1.86 and 2.14 for EGFR and TGFβR respectively), the switch has a single steady state with EGF low and TGFβ3 high. This can be seen by looking at the steady state values for an EGFR fold-change of one in Figure 6B. If the EGFR level is then changed, the circuit has only TGFβ3-high steady states for fold-changes < 1.1, only EGF-high (TGFβ3-low) steady states for fold-changes > 1.3, and a bistable region in between. EGFR fold-changes in that bistable region could yield steady states with either TGFβ3 or EGF high. The one adopted by the system is history dependent. If the base receptor levels are increased (2.86 for both), the switch has a much wider bistable zone – spanning EGFR fold-changes from 0.8 to 1.8 as shown in Figure 6C. Given the central role of this biochemical switch in palate fusion, we investigate below how the symmetry between EGF-TGFβ3 pathways and the width of the switch’s bistable zone impacts normal and toxicant-perturbed fusion in the palate model.
TCDD-induced cleft palate in the mouse is mediated by AhR with over-proliferation of cells in the MEE leading to a failure of palate fusion.54 To simulate the effect of chronic TCDD exposure scenarios on palate fusion, we parameterized TCDD-induced activation of AhR in terms of EGFR fold-changes, i.e., TCDD exposure was modeled synthetically as a direct increase in the expression of EGFR imposed by the degree of AhR activation (Figure 7A-B and Supplemental Movies S3 and S4). Modeling the low-hysteresis switch described in Figure 6B (and used in all simulations above) showed a critical effect on MEE hyperplasia and MES breakdown as the EGFR fold-change increased from 1.1x to 1.2x (Figure 7A). This result is consistent with the bifurcation plot computed for the isolated biochemical switch (Figure 6B). Modeling the high-hysteresis switch (described in Figure 6C) yielded similar consequences at slightly higher EGFR fold-changes (Figure 7B). Interestingly, the high-hysteresis switch also produced more cases with partial fusion (Table 4). This shows that bistability in the switch can be manifest as adjacent regions of the MES alternately adopting TGFβ3-high or EGF-high states.
Figure 7. Simulations of toxicant-induced fusion defects in the virtual palate model.
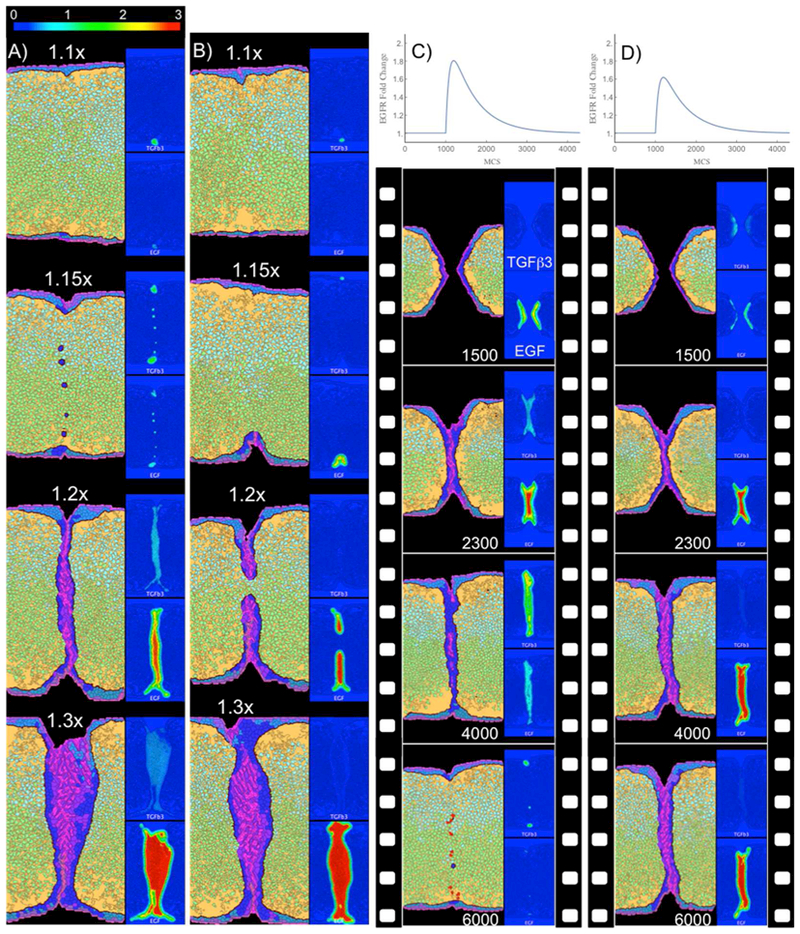
(A-B) Final states for simulations of chronic TCDD exposure. Scenarios were parameterized via AhR-mediated fold-changes in EGFR (as indicated from 1.1x to 1.3x) for (A) the low-hysteresis version of the EGF-TGFβ3 switch (n=54, tipping point ~1.2x) and (B) the high-hysteresis version (n=34, tipping point ~1.2x). Insets show corresponding EGF and TGFβ3 signal gradients. The phenotype of a thickened MES preventing mesenchymal confluence is similar in appearance to the histology of palatal shelves from a TGFβ3 knockout (with or without additional Alk mutations)44. (C-D) Time-lapse images from simulations of transient acute exposure to ATRA parameterized as time-dependent EGFR fold-changes (top). Palate fusion was delayed using the low-hysteresis switch (n=24, tipping point >1.8x) (C), but failed using the high-hysteresis switch (n=16, tipping point 1.5x) (D) despite being subjected to a smaller maximum EGFR fold-change. Complete time-lapse image sets are available for specific examples as Supplemental Movies S3 (panel A, 1.15x), S4 (panel B, 1.2x), S5 (panel C), and S6 (panel D).
Table 4.
Palatal Fusion Outcomes from Simulated Exposures.
| low | 1.10x | 15 (100%) | 0 |
| 1.15x | 15 (100%) | 0 | |
| 1.20x | 0 | 0 | |
| ≥1.30x | 0 | 0 | |
| high | 1.0x | 15 (100%) | 0 |
| 1.2x | 2 (17%) | 9 (75%) | |
| 1.3x | 0 | 0 | |
| 1.4x | 0 | 0 | |
| low | 1.2x | 6 (100%) | 0 |
| 1.3x | 8 (100%) | 0 | |
| 1.6x | 2 (100%) | 0 | |
| 1.8x | 8 (100%) | 0 | |
| high | 1.2x | 4 (100%)* | 0 |
| 1.3x | 3 (100%)* | 0 | |
| 1.6x | 0 | 1 (25%) | |
| 1.8x | 0 | 0 |
minor indentations noted
In a second set of scenarios, we modeled acute exposure to ATRA. Similar to TCDD, ATRA induces an increase in EGFR.54 Negative crosstalk between retinoid and TGFβ3 signaling is thus imposed synthetically in the model by an ATRA-mediated effect on EGF signaling that in turn suppresses TGFβ3 secretion (Figure 7C-D and Supplemental Movies S5 and S6). The imposed time-dependent EGFR fold-changes are shown at the top of each panel, with expression levels increasing rapidly just after 1000 MCS and decaying back to normal with a time constant of 600 MCS. In simulations using the low-hysteresis switch (Figure 7C), an initial flip from TGFβ3-high to EGF-high occurred around 1100 MCS as the EGFR fold-change peaked (near 1.8x) and a switch back to TGFβ3-high occurred when the EGFR fold-change dropped back down below 1.1. Although fusion was delayed, it was eventually complete in 8 of 8 such simulations (Table 4). On the other hand, simulations using the high-hysteresis switch (Figure 7D) were unable to switch back to a TGFβ3-high state (despite a lower maximum EGFR fold-change of just 1.6x) and failed to fuse in 4 of 4 cases. This is a hysteresis effect inherent in the bistability of the switch. Transient exposure simulations with the high-hysteresis switch were only able to complete fusion when the maximum EGFR fold-change was sufficiently low (1.2 or 1.3) that the circuit never entered an EGF-high state.
4. DISCUSSION
Computer simulations that predictively model morphogenetic fusion provide a resource to investigate the interplay of developmental pathways and processes, and assess the impact of environmental stressors on these processes. The virtual tissue model constructed here for the mouse secondary palate effectively simulated higher-level phenotypes (e.g., midline contact, MES breakdown, mesenchymal confluence, fusion defects) in response to genetic or environmental perturbations. An in silico perturbation analysis of various control features revealed model functionality with respect to cell signaling systems and feedback loops for growth and fusion, diverse individual cell behaviors and collective cellular behaviors leading to physical contact and midline fusion, and quantitative analysis of the TGF/EGF switch that controls MES breakdown – a key event in morphogenetic fusion. This computer model adds to similar systems models toward a growing ‘virtual embryo’ toolbox for simulation and quantitative prediction of adverse developmental outcomes following genetic perturbation and/or environmental disruption.21, 22 Simulating a complex embryological system in this manner has potential applications for high-throughput hypothesis-based testing and translating chemical-biological interactions into tissue-level predictions.
Several strengths of the virtual palate model include the ability to forward-engineer biological circuits underlying growth and fusion defects of various types following inactivation of specific elements of the signaling network. This included quantitative analysis of cell signaling systems for growth and fusion, including positive (SHH-FGF10) and negative (EGF-TGFβ3) feedback loops mediating epithelial-mesenchymal interactions. In silico knockouts or ‘cyber-morphs’ provide an indication of the specificity of the underlying signaling systems as they are currently understood. SHH-FGF signaling, for example, is an important mediator of epithelialmesenchymal interactions during outgrowth of the palatal processes,19 and this circuit was represented in the molecular network. On the other hand, a novel SHH-FOXF-FGF18 circuit in the palate development molecular network was recently demonstrated.70 That circuit regulates FOXF-dependent palatal mesenchymal growth downstream of SHH signaling, partly by repressing mesenchymal expression of Fgf18, which is an inhibitor of Shh expression. This newer information is not yet implemented in the virtual palate model. The model can, however, be continually refined to incorporate new molecular information and knowledge – making it a ‘living document’ that gets updated as our understanding of the biology improves.
Individual cellular behaviors such as differential adhesion, cell motility, proliferation, EMT, and apoptosis coded into the model led to collective cellular behaviors that enable physical contact and midline fusion as higher order (emergent) properties. These emergent behaviors can then be traced back to the underlying molecular control circuits, for example, the TGF/EGF switch that controls MES degeneration.54 The computational model simulated the critical balance in relative levels of TGFβ3 and EGF signaling in the MEE, and MES breakdown. Flipping the TGFb3/EGF switch on or off in the MEE provided a singular heuristic approximation for an MES-breakdown phenotype. This speaks to the unique advantage of cell agent-based modeling for systems toxicology in predicting the tissue-level impact of a localized molecular lesion. Furthermore, as demonstrated by the panel of ‘cybermorphs’ derived from more generally hacking the control network, in silico manipulation of the biological network exposes the system to different ‘what-if’ scenarios that can be cell type-specific and/or stage-dependent, turning genes up or down and in different combinations to probe the sensitivity of of the system to potential molecular initiating events that could disrupt palatal growth and fusion.
Signaling through the EGFR is a key event in at least some forms of teratogen-induced cleft palate.54 Functional EGFR receptors are expressed in the mouse palatal epithelium as early as E12 and sensitize the MEE to hyperplasia instead of breakdown.71 Chemicals that increase EGFR expression, induce MEE proliferation, and disrupt fusion include retinoic acid, hydrocortisone, and TCDD.28, 46, 69, 72 For example, litters dosed with TCDD (24 μg/kg) or ATRA on E12 had fetuses where the palatal processes came in contact with one another, but MEE cells continued to express high levels of EGFR and the palatal processes failed to fuse.46 New insights into the EGF-TGFβ3 switch governing clearance of the MES can be gauged from the linkage of the cell agent-based simulations with a formal hysteresis switch for transient exposures to teratogens. This could guide the design of experimental studies to measure and reengineer the molecular and cellular parameters governing the complexity of this switch in the context of systems toxicology and synthetic biology.73 Multiple runs of both the low- and high-hysteresis models predicted a tipping point in the system controlling MES breakdown when chronic TCDD exposure induced as little as a 1.2-fold increase in EGFR expression. In contrast, the impact of acute ATRA exposure (through EGF-mediated functional inhibition of TGFβ3 signaling) had a higher tipping point. The greater tolerance to transiently increased EGF signaling was especially evident in the low-hysteresis switch model where its reversibility pushed the tipping point above a 1.8-fold increase in EGFR.
The capacity of the computational model to quantitatively output both low-level (e.g., cell fate and behavior) and high-level (e.g., growth dynamics and emergent behaviors) features of the system provides a direct linkage to address how much change in different pathways would elevate risk for cleft palate. The virtual palate model identified the critical roles of a number of the effector molecules as cybermorph phenotypes that recapitulated in vivo models. It also identified critical points of imbalance in the control network driving palatal development that serve as potential molecular initiating events for adverse outcome pathways (AOPs) leading to cleft palate. Nonetheless, there may be additional AOPs leading to cleft palate that are not captured by the model – either because the present two-dimensional model does not address some known aspects of the physical biology, e.g., reorientation, post-fusion osteogenic differentiation, or regional differences in mesenchymal cell orientation between oral-nasal sides or anterior-posterior axes,74, 75 or because alterations of the cellular/molecular mechanisms leading to palate fusion may be compensated by unknown interactions that prevent manifestation of a morphological defect. As such, future improvement in the virtual palate model should strive toward a more realistic representation of the ECM and mechanical forces in 3D, and carefully compare how perturbations alter fusion in the model versus in vivo. Quantifying those differences will be the key to identifying key compensatory mechanisms.
Understanding how tissues and organs are shaped during embryogenesis is a central question in developmental biology and a challenge for predicting developmental toxicity. The virtual palate model adds to similar systems-based models that ultimately lead to a ‘virtual embryo’ for computer simulation and quantitative prediction of adverse developmental outcomes. Although the current computational model was constructed from published information in the mouse, it can be used as a heuristic approximation to translate in vitro data generated from human cell-based studies such as ToxCast. This helps integrate the very large body of research at the cellular-molecular scale, and the equally large body of knowledge at the organ-embryo scale both clinically, and more recently through physics-based investigation utilizing novel bioengineering, biomechanics, and microphysiological systems.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the EPA’s Virtual Tissue Models team for their helpful input into this manuscript, and to Drs. Nicole Kleinstreuer (National Toxicology Program, NIEHS) and Barbara Abbott (National Health and Environmental Effects Research Laboratory – US EPA) for their insightful contributions to the manuscript.
This work supported by: EPA/ORD Chemical Safety for Sustainability Research Program, Virtual Tissue Models Project (MCKL, NCB, TBK), the Environmental Modeling and Visualization Laboratory (RMS) and a co-operative agreement under U.S. EPA-Science To Achieve Results (STAR) Program grant # 83573601 (MSH).
FUNDING INFORMATION
This research was supported by EPA’s Office of Research and Development under the Chemical Safety for Sustainability Research Program (MCKL, NCB, TBK), the Environmental Modeling and Visualization Laboratory (RMS) and a co-operative agreement under U.S. EPA-Science To Achieve Results (STAR) Program grant # 83573601 (MSH). The authors declare that no conflicts of interest exist.
ABBREVIATIONS
- 2D
two-dimensional
- 3D
three-dimensional
- AC50
half-maximal activity concentration
- AhR
aryl hydrocarbon receptor
- AOP
adverse outcome pathway
- ATRA
all-trans retinoic acid
- BM
basement membrane
- BMP
bone morphogenetic protein
- CC3D
CompuCell3D software
- ΔE
change in pseudo-energy function
- δ
Kronecker delta function
- ΔN
duration in MCS
- E11.5 to E16.0
embryonic days 11.5 to 16.0 in mouse
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EGFR
EGF receptor
- Einterface, Eshape, Efocal-point plasticity
terms in pseudo-energy function
- EMT
epithelial-to-mesenchymal transition
- EphB
ephrin receptor B
- EPITH
epithelial cell type
- EPT
epithelial-to-periderm transition
- EXT
external fluid
- fchange
fold-change
- FGF
fibroblast growth factor
- FGFR2b
fibroblast growth factor receptor 2b
- FOXF
forkhead box protein F
- FPP
focal-point plasticity
- Jint, Jext
matrices of cell-type-dependent interfacial energies
- Kd
ligand-receptor binding affinity
- L
cell’s major axis length
- λlink
stiffness parameter for focal-point plasticity links
- λvol, λsurface, λlength
stiffness parameters of shape energy terms
- MCS
Monte Carlo steps
- MEE
medial edge epithelium
- MES
medial edge seam
- MESENCH
mesenchymal cell type
- MMP
matrix metalloproteinase
- P
probability
- PERI_A, PERI_B
periderm cell type - apical or basal compartment
- Ptc1
patched (receptor for SHH)
- r
centroid-to-centroid distance
- Rgrowth
growth rate
- R0, Rmax
basal and maximal growth rates
- ρ
generalized cell ID
- σ
cluster ID
- S
cell surface area
- SHH
sonic hedgehog
- Smo
smoothened (downstream effector of SHH signaling)
- τapo
time constant for apoptotic cell death
- τ
cell type index
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- Teff
fluctuation amplitude or effective temperature
- TGFβ
transforming growth factor beta
- TGFβR
transforming growth factor beta receptor
- V
cell volume
Footnotes
Publisher's Disclaimer: Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
SUPPORTING INFORMATION
Six supplemental movies are available showing cellular dynamics for simulations covering normal palate fusion (S1), disrupted fusion caused by TGFβ3 knockout (S2), and effects of exposure to sub- or supra-critical chronic exposure to TCDD (S3, S4) and sub- or supra-critical transient exposure to ATRA (S5, S6). In addition, the supporting information includes an archive of complete python code for the model. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1). Ray HJ, and Niswander L (2012) Mechanisms of tissue fusion during development. Development 139, 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2). Scarpa E, and Mayor R (2016) Collective cell migration in development. J. Cell Biol 212, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3). Staller MV, Fowlkes CC, Bragdon MD, Wunderlich Z, Estrada J, and DePace AH (2015) A gene expression atlas of a bicoid-depleted Drosophila embryo reveals early canalization of cell fate. Development 142, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4). Hamdoun A, and Epel D (2007) Embryo stability and vulnerability in an always changing world. Proc. Natl. Acad. Sci. U. S. A 104, 1745–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5). Ferguson MW (1988) Palate development. Development 103 Suppl, 41–60. [DOI] [PubMed] [Google Scholar]
- (6). Forrester MB, and Merz RD (2004) Descriptive epidemiology of oral clefts in a multiethnic population, Hawaii, 1986–2000. Cleft Palate. Craniofac. J 41, 622–628. [DOI] [PubMed] [Google Scholar]
- (7). Walker BE (1967) Induction of cleft palate in rabbits by several glucocorticoids. Proc. Soc. Exp. Biol. Med 125, 1281–1284. [DOI] [PubMed] [Google Scholar]
- (8). Zimmerman EF, Andrew F, and Kalter H (1970) Glucocorticoid inhibition of RNA synthesis responsible for cleft palate in mice: a model. Proc. Natl. Acad. Sci. U. S. A 67, 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9). Carmichael SL, Shaw GM, Ma C, Werler MM, Rasmussen SA, Lammer EJ, and National Birth Defects Prevention Study. (2007) Maternal corticosteroid use and orofacial clefts. Am. J. Obstet. Gynecol 197, 585 e581–587; discussion 683–584, e581–587. [DOI] [PubMed] [Google Scholar]
- (10). Paulson GW, and Paulson RB (1981) Teratogenic effects of anticonvulsants. Arch. Neurol 38, 140–143. [DOI] [PubMed] [Google Scholar]
- (11). Alsdorf R, and Wyszynski DF (2005) Teratogenicity of sodium valproate. Expert Opin. Drug. Saf 4, 345–353. [DOI] [PubMed] [Google Scholar]
- (12). Sharma A, and Rawat AK (1986) Teratogenic effects of lithium and ethanol in the developing fetus. Alcohol 3, 101–106. [DOI] [PubMed] [Google Scholar]
- (13). DeRoo LA, Wilcox AJ, Drevon CA, and Lie RT (2008) First-trimester maternal alcohol consumption and the risk of infant oral clefts in Norway: a population-based case-control study. Am. J. Epidemiol 168, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14). Olson FC, and Massaro EJ (1977) Effects of methyl mercury on murine fetal amino acid uptake, protein synthesis and palate closure. Teratology 16, 187–194. [DOI] [PubMed] [Google Scholar]
- (15). Gebhardt DO, and Schade GJ (1969) Is there an optimal stage for the induction of cleft palate in the mouse? Cleft Palate J 6, 373–380. [PubMed] [Google Scholar]
- (16). Freni SC, and Zapisek WF (1991) Biologic basis for a risk assessment model for cleft palate. Cleft Palate. Craniofac. J 28, 338–346. [DOI] [PubMed] [Google Scholar]
- (17). Bush JO, and Jiang R (2012) Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18). Gritli-Linde A (2007) Molecular control of secondary palate development. Dev. Biol 301, 309–326. [DOI] [PubMed] [Google Scholar]
- (19). Lipinski RJ, Song C, Sulik KK, Everson JL, Gipp JJ, Yan D, Bushman W, and Rowland IJ (2010) Cleft lip and palate results from Hedgehog signaling antagonism in the mouse: Phenotypic characterization and clinical implications. Birth Defects Res. Part A-Clin. Mol. Teratol 88, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20). Sturla SJ, Boobis AR, FitzGerald RE, Hoeng J, Kavlock RJ, Schirmer K, Whelan M, Wilks MF, and Peitsch MC (2014) Systems toxicology: from basic research to risk assessment. Chem. Res. Toxicol 27, 314–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21). Kleinstreuer N, Dix D, Rountree M, Baker N, Sipes N, Reif D, Spencer R, and Knudsen T (2013) A computational model predicting disruption of blood vessel development. PloS Comput. Biol 9, e1002996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22). Leung MC, Hutson MS, Seifert AW, Spencer RM, and Knudsen TB (2016) Computational modeling and simulation of genital tubercle development. Reprod. Toxicol 64, 151–161. [DOI] [PubMed] [Google Scholar]
- (23). Stott EL, Britton NF, Glazier JA, and Zajac M (1999) Stochastic simulation of benign avascular tumour growth using the Potts model. Math. Comp. Modelling 30, 183–198. [Google Scholar]
- (24). Ouchi NB, Glazier JA, Rieu JP, Upadhyaya A, and Sawada Y (2003) Improving the realism of the cellular Potts model in simulations of biological cells. Physica A 329, 451–458. [Google Scholar]
- (25). Knudsen TB, Bulleit RF, and Zimmerman EF (1985) Histochemical localization of glycosaminoglycans during morphogenesis of the secondary palate in mice. Anat. Embryol. (Berl) 173, 137–142. [DOI] [PubMed] [Google Scholar]
- (26). Graner F, and Glazier JA (1992) Simulation of biological cell sorting using a 2-dimensional extended Potts-model. Phys. Rev. Lett 69, 2013–2016. [DOI] [PubMed] [Google Scholar]
- (27). Swat MH, Thomas GL, Belmonte JM, Shirinifard A, Hmeljak D, and Glazier JA (2012) Multi-scale modeling of tissues using CompuCell3D. Methods Cell Biol 110, 325–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28). Abbott BD, Adamson ED, and Pratt RM (1988) Retinoic acid alters EGF receptor expression during palatogenesis. Development 102, 853–867. [DOI] [PubMed] [Google Scholar]
- (29). Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, and Rice DPC (2004) Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Invest 113, 1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30). Rice R, Connor E, and Rice DPC (2006) Expression patterns of Hedgehog signalling pathway members during mouse palate development. Gene Expr. Patterns 6, 206–212. [DOI] [PubMed] [Google Scholar]
- (31). Casey LM, Lan Y, Cho E-S, Maltby KM, Gridley T, and Jiang R (2006) Jag2-Notch1 signaling regulates oral epithelial differentiation and palate development. Dev. Dyn 235, 1830–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32). Jin J-Z, and Ding J (2006) Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development 133, 3341–3347. [DOI] [PubMed] [Google Scholar]
- (33). Jin J-Z, Li Q, Higashi Y, Darling DS, and Ding J (2008) Analysis of Zfhx1a mutant mice reveals palatal shelf contact-independent medial edge epithelium differentiation during palate fusion. Cell Tissue Res 333, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34). Bush JO, and Soriano P (2010) Ephrin-B1 forward signaling regulates craniofacial morphogenesis by controlling cell proliferation across Eph-ephrin boundaries. Genes Dev 24, 2068–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35). Greene RM, and Pisano MM (2010) Palate morphogenesis: Current understanding and future directions. Birth Defects Res. C 90, 133–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36). He F, Xiong W, Wang Y, Matsui M, Yu X, Chai Y, Klingensmith J, and Chen Y (2010) Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev. Biol 347, 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37). Dravis C, and Henkemeyer M (2011) Ephrin-B reverse signalin controls septation events at the embryonic midline through separate tyrosine phosphorylation-independent signaling avenues. Dev. Biol 355, 138–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38). Potter AS, and Potter SS (2015) Molecular Anatomy of Palate Development. PLoS One 10, e0132662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39). Han J, Mayo J, Xu X, Li J, Bringas P Jr., Maas RL, Rubenstein JL, and Chai Y (2009) Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development 136, 4225–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40). Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, and Chen Y (2002) Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 129, 4135–4146. [DOI] [PubMed] [Google Scholar]
- (41). Gehris AL, Pisano MM, Nugent P, and Greene RM (1994) Regulation of TGF beta 3 gene expression in embryonic palatal tissue. In Vitro Cell. Dev. Biol. Anim 30A, 671–679. [DOI] [PubMed] [Google Scholar]
- (42). del Rio A, Barrio MC, Murillo J, Maldonado E, Lopez-Gordillo Y, Martinez-Sanz E, Martinez ML, and Martinez-Alvarez C (2011) Analysis of the presence of cell proliferation-related molecules in the Tgf-beta3 null mutant mouse palate reveals misexpression of EGF and Msx-1. Cells Tissues Organs 193, 135–150. [DOI] [PubMed] [Google Scholar]
- (43). Abbott BD, and Pratt RM (1987) Retinoids and epidermal growth factor alter embryonic mouse palatal epithelial and mesenchymal cell differentiation in organ culture. J. Craniofac. Genet. Dev. Biol 7, 219–240. [PubMed] [Google Scholar]
- (44). Dudas M, Nagy A, Laping NJ, Moustakas A, and Kaartinen V (2004) Tgf-β3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev. Biol 266, 96–108. [DOI] [PubMed] [Google Scholar]
- (45). San Miguel S, Serrano MJ, Sachar A, Henkemeyer M, Svoboda KKH, and Benson MD (2011) Ephrin reverse signaling controls palate fusion via a PI3 kinase-dependent mechanism. Dev. Dyn 240, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46). Abbott BD, and Birnbaum LS (1989) TCDD alters medial edge epithelial cell differentiation during palatogenesis. Toxicol. Appl. Pharmacol 99, 276–286. [DOI] [PubMed] [Google Scholar]
- (47). Abbott BD, and Birnbaum LS (1990) TCDD-induced altered expression of growth factors may have a role in producing cleft palate and enhancing the incidence of clefts after coadministration of retinoic acid and TCDD. Toxicol. Appl. Pharmacol 106, 418–432. [DOI] [PubMed] [Google Scholar]
- (48). Abbott BD, and Birnbaum LS (1989) Cellular alterations and enhanced induction of cleft palate after coadministration of retinoic acid and TCDD. Toxicol. Appl. Pharmacol 99, 287–301. [DOI] [PubMed] [Google Scholar]
- (49). Abbott BD, Harris MW, and Birnbaum LS (1989) Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology 40, 533–553. [DOI] [PubMed] [Google Scholar]
- (50). Miettinen PJ, Chin JR, Shum L, Slavkin HC, Shuler CF, Derynck R, and Werb Z (1999) Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat. Genet 22, 69–73. [DOI] [PubMed] [Google Scholar]
- (51). Song CZ, Tian X, and Gelehrter TD (1999) Glucocorticoid receptor inhibits transforming growth factor-beta signaling by directly targeting the transcriptional activation function of Smad3. Proc. Natl. Acad. Sci. U. S. A 96, 11776–11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52). Bolkenius U, Hahn D, Gressner AM, Breitkopf K, Dooley S, and Wickert L (2004) Glucocorticoids decrease the bioavailability of TGF-beta which leads to a reduced TGF-beta signaling in hepatic stellate cells. Biochem. Biophys. Res. Commun 325, 1264–1270. [DOI] [PubMed] [Google Scholar]
- (53). Miettinen HM, Huuskonen H, Partanen AM, Miettinen P, Tuomisto JT, Pohjanvirta R, and Tuomisto J (2004) Effects of epidermal growth factor receptor deficiency and 2,3,7,8-tetrachlorodibenzo-p-dioxin on fetal development in mice. Toxicol. Lett 150, 285–291. [DOI] [PubMed] [Google Scholar]
- (54). Abbott BD (2010) The etiology of cleft palate: a 50-year search for mechanistic and molecular understanding. Birth Defects Res. B 89, 266–274. [DOI] [PubMed] [Google Scholar]
- (55). Liu X, Zhang H, Gao L, Yin Y, Pan X, Li Z, Li N, Li H, and Yu Z (2014) Negative interplay of retinoic acid and TGF-beta signaling mediated by TG-interacting factor to modulate mouse embryonic palate mesenchymal-cell proliferation. Birth Defects Res. B 101, 403–409. [DOI] [PubMed] [Google Scholar]
- (56). Xu Q, and Kopp JB (2012) Retinoid and TGF-beta families: crosstalk in development, neoplasia, immunity, and tissue repair. Semin. Nephrol 32, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57). Lan Y, and Jiang R (2009) Sonic hedgehog signaling regulates reciprocal epithelialmesenchymal interactions controlling palatal outgrowth. Development 136, 1387–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58). Plouhinec J-L, Zakin L, and De Robertis EM (2011) Systems control of BMP morphogen flow in vertebrate embryos. Curr. Opin. Genet. Dev 21, 696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59). Ornitz DM (2000) FGFs, heparan sulfate and FGFRs: complex interactions essential for development. BioEssays 22, 108–112. [DOI] [PubMed] [Google Scholar]
- (60). Wang X, Harris RE, Bayston LJ, and Ashe HL (2008) Type IV collagens regulate BMP signalling in Drosophila. Nature 455, 72–77. [DOI] [PubMed] [Google Scholar]
- (61). Etheridge LA, Crawford TQ, Zhang S, and Roelink H (2010) Evidence for a role of vertebrate Disp1 in long-range Shh signaling. Development 137, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62). Zimmerman LB, De Jesus-Escobar JM, and Harland RM (1996) The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell 86, 599–606. [DOI] [PubMed] [Google Scholar]
- (63). Smith WC, and Harland RM (1992) Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell 70, 829–840. [DOI] [PubMed] [Google Scholar]
- (64). Hassell JR, and Pratt RM (1977) Elevated levels of cAMP alters the effect of epidermal growth factor in vitro on programmed cell death in the secondary palatal epithelium. Exp. Cell Res 106, 55–62. [DOI] [PubMed] [Google Scholar]
- (65). Xu X, Han J, Ito Y, Bringas P Jr., Urata MM, and Chai Y (2006) Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev. Biol 297, 238–248. [DOI] [PubMed] [Google Scholar]
- (66). Cuervo R, and Covarrubias L (2004) Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development 131, 15–24. [DOI] [PubMed] [Google Scholar]
- (67). Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, and Kaartinen V (2001) TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol. Biol. Cell 12, 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68). He F, Xiong W, Wang Y, Li L, Liu C, Yamagami T, Taketo MM, Zhou C, and Chen Y (2011) Epithelial Wnt/β-catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. Dev. Biol 350, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69). Abbott BD, Probst MR, Perdew GH, and Buckalew AR (1998) AH receptor, ARNT, glucocorticoid receptor, EGF receptor, EGF, TGF alpha, TGF beta 1, TGF beta 2, and TGF beta 3 expression in human embryonic palate, and effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Teratology 58, 30–43. [DOI] [PubMed] [Google Scholar]
- (70). Xu J, Liu H, Lan Y, Aronow BJ, Kalinichenko VV, and Jiang R (2016) A Shh-Foxf-Fgf18-Shh molecular circuit regulating palate development. PLoS Genet 12, e1005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71). Tyler MS, and Pratt RM (1980) Effect of epidermal growth factor on secondary palatal epithelium in vitro: tissue isolation and recombination studies. J. Embryol. Exp. Morphol 58, 93–106. [PubMed] [Google Scholar]
- (72). Abbott BD, Best DS, and Narotsky MG (2005) Teratogenic effects of retinoic acid are modulated in mice lacking expression of epidermal growth factor and transforming growth factor-alpha. Birth Defects Res A Clin Mol Teratol 73, 204–217. [DOI] [PubMed] [Google Scholar]
- (73). Brophy JA, and Voigt CA (2014) Principles of genetic circuit design. Nat Methods 11, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74). Bulleit RF, and Zimmerman EF (1985) The influence of the epithelium on palate shelf reorientation. J. Embryol. Exp. Morphol 88, 265–279. [PubMed] [Google Scholar]
- (75). Yu K, and Ornitz DM (2011) Histomorphological study of palatal shelf elevation during murine secondary palate formation. Dev. Dyn 240, 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76). Frisch SM, and Francis H (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol 124, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77). Richardson RJ, Hammond NL, Coulombe PA, Saloranta C, Nousiainen HO, Salonen R, Berry A, Hanley N, Headon D, Karikoski R, and Dixon MJ (2014) Periderm prevents pathological epithelial adhesions during embryogenesis. J. Clin. Invest 124, 3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78). O’Brien LE, Zegers MM, and Mostov KE (2002) Opinion: Building epithelial architecture: insights from three-dimensional culture models. Nature reviews. Molecular cell biology 3, 531–537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.