

Introduction
Cytosolic sulfotransferases (SULTs) are important regulators of small molecule homeostasis within the body. Isoform specific roles range from defending our bodies against xenobiotic insult to maintaining adequate levels of active, locally produced, hormones such as dehydroepiandrosterone (DHEA) and estradiol (E2) (Bird et al., 1978; Falany et al., 2009; Falany et al., 1989; Pacifici et al., 1997). With the exception of one isoform, all 14 human SULTs catalyze the transfer of a sulfonate group from the “activated sulfate” cofactor, 3’-phosphoadenosine-5’-phosphosulfate (PAPS), to the recipient substrate (Falany et al., 2000; Tibbs et al., 2015). Conjugation of the sulfonate moiety to a substrate’s hydroxyl or primary amine renders the compound more polar and alters receptor binding properties while increasing the substrate’s potential for excretion from the body via urinary or biliary pathways.
SULT isoforms are named according to their sequence conservation, which moderately reflects the substrate specificity of the promiscuous enzymes (Blanchard, 2005). SULT1B1, initially termed iodothyronine sulfotransferase, is the focus of this manuscript and is classically associated with the sulfation of thyroid hormones as indicated by its alternative name (Fujita et al., 1997; Riches et al., 2009; Wang et al., 1998). SULT1B1 is expressed at highest levels throughout the human colon and small intestine but can also be found at moderate levels in human liver, kidney, and white blood cells (Riches et al., 2009; Wang et al., 1998). Unlike its closely related gene duplication sibling SULT1E1 (estrogen SULT), SULT1B1 displays no affinity for steroid hormones (Allali-Hassani et al., 2007; Meinl & Glatt, 2001; Wang et al., 1998). Instead, the enzyme prefers small phenolic compounds and planar multi-ringed chemicals such as polycyclic aromatic hydrocarbons (PAHs); therefore, SULT1B1 is one of the main SULTs investigated for the sulfation and bioactivation of the PAHs (Allali-Hassani et al., 2007; Glatt, 2000; Meinl et al., 2013).
A number of the SULT genes have been resequenced to identify common polymorphisms and characterize their functional consequences; however, SULT1B1 was not the direct focus of these resequencing studies (Carlini et al., 2001; Thomae et al., 2003; Wood et al., 1996). Many of the allelic variants identified in SULT genes are either represented at low frequency or lack obvious functional consequences. In some cases, such as the common genetic variant identified in SULT1A1 (SULT1A1*2), substantial decreases in enzyme activity and thermal stability were reported when measured in human liver or blood platelets (Lilla et al., 2005; Raftogianis et al., 1997; Raftogianis et al., 1999). Other studies have described varying cancer (e.g. breast cancer, gastric cancer, and colorectal cancer) risk differences between patients with genetic polymorphisms in the SULT1A1 gene (Bamber et al., 2001; Boccia et al., 2005; Yang et al., 2005). The results of these studies have been summarized in recent publications (Glatt & Meinl, 2004; Nowell & Falany, 2006; Sak & Everaus, 2016). In this report, we are describing a single nucleotide polymorphism (SNP) in SULT1B1 (reference SNP (rs) number 11569736) that both affects enzyme activity and is selectively represented at a significant frequency in Americans of African descent. This finding warrants further investigation of the test population to identify the group’s susceptibility to thyroid hormone imbalance and susceptibility to tissue-specific malignancies.
Materials and Methods
Cloning SULT1B1-WT and SULT1B1-L145V
Normal human endometrium was obtained from the University of Alabama at Birmingham (UAB) Tissue Procurement Center. Total RNA was isolated from tissue using RNA-STAT 60 (Tel-Test Inc., Friendswood, TX). Following RNA isolation, 4 µg of the total RNA sample served as the template for Invitrogen Superscript III reverse-transcriptase to synthesize cDNA. To amplify SULT1B1, ten percent of this reaction volume was used as the template for PCR by Pfu DNA polymerase using the sense primer 5’-CTGAACAAAGGGATTAAATTGTGAGAACAACTGTC-3’ and antisense primer 5’-GAGATTGTCTGTAGTTGATTGAAACGAGGGCA-3’ (30 cycles, manufacturer protocol). The PCR product (1021 base pairs) was subsequently purified by 1% agarose-gel electrophoresis and sequenced in the UAB Heflin Sequencing Core. Two distinct sequences, one with GTA and the other with TTA encoding the 145th amino acid were identified. This variation (rs11569736) resulted in the introduction of a new unique recognition site for the restriction enzyme HpyCH4IV (A↓CGT). Therefore, the DNA was digested with 20 units of HpyCH4IV, and the base alteration confirmed with electrophoresis.
Cloning, Expression, and Purification of Recombinant SULT1B1-WT and SULT1B1-L145V
The SULT1B1WT and SULT1B1L145V genes were amplified by PCR as described above. Restriction sites were introduced to the PCR fragment for cloning into a bacterial expression vector using a sense primer with a leading BspHI restriction site (5’-CGGTCATGATTTCCCCAAAAGATATTCTGCG-3’) and an anti-sense primer with a flanking HindIII restriction site (5’-TACCCAAGCTTGGTTTAAATCTCTGTGCGGAATTG-3’). PCR was performed with Life-technologies (Carlsbad, CA) Elongase according to the manufacturer’s protocol. The PCR product was gel purified, ligated into the pGEM-T Easy (Promega, Madison, WI) vector, and transformed into E. coli JM109. The transformed bacteria were plated on low salt LB agar containing ampicillin (Amp+, 100 µg/mL) to select for bacteria carrying the desired vector. Four of the resulting colonies were selected, grown to density in liquid culture (LB-Amp+), and their plasmids isolated using the ZYMO Research (Irvine, CA) ZR Plasmid Miniprep Classic kit. To identify colonies containing SULT1B1L145V and SULT1B1WT, the plasmids were digested by HpyCH4IV and confirmed by visualization after electrophoresis. The cDNA inserts were then digested out of pGEM-T Easy using NcoI and HindIII (New England Biolabs (NEB), Ipswich, MA) restriction enzymes, purified by electrophoresis, ligated into pKK233–2 according to the Rapid DNA ligation kit (Roche, Basel, Switzerland) protocol, and transformed into XL1-blue competent E. coli. Plasmids were isolated from positive clones, and the inserts were sequenced at the UAB Heflin Sequencing Core. After sequence confirmation, colonies were stored at −80oC in 1:1 glycerol:culture stocks.
To express and purify the native SULT1B1 isoforms, small-volume LB (Amp+) E. coli overnight cultures were used as starter cultures for 1 L LB (Amp+) cultures in culture flasks. The cultures were grown at 37oC, shaking 225 rpm, to an optical density (OD600) of 0.7 before induction of SULT1B1 protein expression with 2 mM IPTG (isopropyl-beta-D-thiogalactopyranoside). After 2 hours, the bacteria were harvested by centrifugation for 20 minutes at 2545 x g, and the pelleted bacteria were suspended in 60 mL of phosphate buffered saline (PBS), pH 7.4. The bacteria were incubated on ice for 20 minutes, centrifuged a second time for 20 minutes at 2545 x g, and resuspended in 5 mM sodium phosphate buffer containing dithiothreitol (DTT) (1 mM) and phenylmethanesulfonyl fluoride (PMSF) (0.1 mM) for protease inhibition. The E. coli were lysed via six 15-second sonication bursts (with 30-second cooling intervals) on ice. The cytosolic fraction was isolated by ultracentrifugation for 45 minutes at 100,000 x g. The cytosol was applied to a DEAE-Sepharose (GE Healthcare Bio-Sciences, Pittsburgh, PA) chromatography column; the column was rinsed with one bed-volume (BV) of buffer containing no salt, followed by a low salt buffer wash (10 mM NaCl, 2 BV). SULT1B1 was eluted with a 150 mL salt gradient ranging from 10–300 mM NaCl. Fractions were assayed for p-nitrophenol (PNP) sulfation activity, and PNP sulfation activity eluted at approximately 150 mM NaCl. Purity was assessed via SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis).
High purity SULT1B1 was needed to perform intrinsic fluorescence spectroscopic binding studies. To generate pure SULT1B1 using a 6xHis tagged purification procedure, both SULT1B1 isoform-containing pKK vectors were used as templates for PCR amplification using 5’- ATGCTTTCCCCAAAAGATATTCT-3’ and 5’-GCAAGCTTGCTCATCGTTTAAATCTCTGTGCGGA-3’ primers for 30 cycles with Thermo Scientific (Waltham, MA) Phusion Taq. The resulting PCR product was electrophoresed through a 1% agarose gel, and the desired band purified via Thermo Scientific’s DNA Gel Extraction Kit. The product was subjected to a 1.5 hour/37oC digestion by NEB’s HindIII restriction enzyme while the pPROEx-hta plasmid was digested with SfoI and HindIII according to the manufacturer’s protocol. The two were purified via electrophoresis and used in a 7:3, insert:plasmid, ratio for ligation with NEB’s Quick Ligase at room temperature. XL1-Blue competent E. coli were transformed with the resulting plasmids. Plasmids were isolated as described above and confirmed by sequencing at the UAB Heflin Sequencing Core facility. For protein expression, BL21-DE3-RIL competent E. coli were transformed with the plasmids and stored at −80oC in 1:1 glycerol:culture stocks.
BL21-DE3-RIL cultures were grown to density in chloramphenicol (50 μg/mL) LB-Amp+ (100 μg/mL), and protein expression was induced with addition of 0.5 mM IPTG. After 4 hours, the bacteria were harvested by centrifugation for 15 minutes at 4648 x g, and the pelleted bacteria were suspended in 15 mL of ice-cold 10 mM PBS, pH 7.4, 10% glycerol, 300 mM NaCl, 10 mM imidazole, 10 mM 2-mercaptoethanol, and PMSF (0.1 mM). E. coli were lysed via six 15-second sonication bursts (with 30-second cooling intervals) on ice, and lysate was isolated by centrifugation for 15 minutes at 10,000 x g (4oC). Lysate was applied to a matched buffer-equilibrated Thermo Scientific Ni-NTA resin column with a 750 μL BV. After application, the column was rinsed with three BV of matched buffer, followed by a low volume (16 mL total) gradient elution ranging from 10 to 300 mM imidazole. Fractions were collected and assayed for SULT1B1 activity with PNP; SULT1B1 purity was determined via SDS-PAGE. Pure active fractions were combined and dialyzed twice (5 and 16 hours) against 1 L of matched buffer lacking imidazole.
Sulfation Assays
To test the sulfation activity of each SULT isoform, a well-described sulfation assay using 1-naphthol, p-nitrophenol, or 1-hydroxypyrene was used (Falany et al., 2005). [35S]PAPS (Perkin Elmer, Waltham, MA) served as the tracer for sulfation. The substrate, 50 mM Tris (pH 7.4), 5 mM MgCl2, and the enzyme were pre-incubated at 37oC for two minutes. Reactions were initiated by addition of [35S]PAPS, mixed, and placed back into the 37oC water bath. The reactions were quenched by spotting the reaction contents directly onto thin-layer chromatography (TLC) plates after a maximum reaction time of ten minutes, ensuring <10% substrate turnover. A methylene chloride: methanol: ammonium hydroxide (85:15:5) solvent was used to resolve sulfated substrates. Product bands were visualized by autoradiography, scraped from the TLC plate, and quantified via scintillation spectroscopy. To determine the kinetic parameters with respect to substrates and cofactor, the effects of varying concentrations of each on sulfation activity were measured in duplicate sets of two independent trials. The kinetic parameters and standard error of the mean of each data set were determined using VisualEnzymics software (Softzymics, Inc., Princeton, NJ). For 1-naphthol, p-nitrophenol, and PAPS, a Michaelis-Menton fit model was used to determine the kinetic parameters, while a Sigmoid fit model was used to determine the kinetic parameters of 1-hydroxypyrene. Substrate inhibition constants were calculated using the Substrate Inhibition model in Visual Enzymics. Each reported Km/Vmax represents the apparent value at 15 μM PAPS, or in the case of PAPS, the apparent value with 6.25 μM 1-hydroxypyrene. The activity of both the untagged and 6xHis purified SULT1B1 isoforms toward 1-naphthol were measured to ensure the purification method did not affect the enzyme’s specific activity. All reported catalytic data were generated using the untagged isoform.
Binding Assays
The L145V isoform exhibited an altered Km for the obligate sulfonate donor, PAPS. This difference may be a result of an altered ability to bind or release the cofactor. To test this property via intrinsic fluorescence binding assays, 200 mM of each isoform was added to 10 mM phosphate buffer, pH 7.4, 10% glycerol, and 300 mM NaCl in quartz cuvettes with a spectral cell stir bar. While constantly stirring, PAP (3’, 5’-diphosphoadenosine), PAPS, or adenosine monophosphate (AMP) were pipetted into the cuvette incrementally in 2 μL volumes; the solution was allowed to stir for the remainder of the 15-second interval while the shutter was closed. After opening the shutter, the fluorescence (excitation 282 nm, emission 338 nm) was measured every 0.1 second for 15 seconds, at which point the shutter was closed again to begin the next titration. In these experiments, AMP, a PAPS analogue known to not bind to the SULTs, acted as a control to mediate any inner filter effects (Rens-Domiano & Roth, 1987). The 150 data points in each increment were averaged, and the change in intrinsic fluorescence was determined. The AMP control values were added into the final values to correct for photobleaching and inner filter effects. Binding trials were conducted in duplicate sets. Each Kd and standard error of the mean was determined using the one site binding [quadratic] model in the VisualEnzymics software.
Determination of SULT1B1L145V Frequency in Humans
To approximate the frequency of the SULT1B1L145V allele in human populations, 37 human tissue samples (mostly endometrium) were acquired from the UAB Tissue Procurement center. In Table 1, unpaired two-tailed student t-tests were performed for statistical analysis (GraphPad Software, Inc., San Diego, CA). Tissue was homogenized in PBS using a dounce homogenizer. Insoluble contents were removed from the sample by centrifugation at 27,216 x g for 15 minutes. Genomic DNA was isolated by phenol/chloroform extraction (Stenesh, 1989). The SULT1B1 gene was amplified from each tissue by PCR with Taq DNA polymerase using the sense primer 5’- AATGTTGGAAATGACTCTCCCTGG-3’ and antisense primer 5’-AATCTCTGTGCGGAATTGAAGTGC-3’ (35 cycles, manufacturer protocol). The PCR products were gel purified, digested with 10 units of HpyCH4IV for 1 hour, subjected to electrophoresis, and visualized under UV light after treatment with ethidium bromide. If the PCR product was full length (649 base pairs), the SULT1B1 gene was assumed to be WT (L-encoding) at the 145th amino acid position. Alternatively, if the PCR product was half or completely digested, the specimen was assumed to be heterozygous or homozygous for the SULT1B1L145V allele, respectively. In addition to the tissue origin, the disease state of the tissue (normal or tumor), the age of the patient, and the ethnicity of the patient were known. Therefore, the results were analyzed with respect to each of these subpopulations in an effort to identify any significant trends.
Table 1.
Km and Vmax values calculated from SULT1B1-WT and SULT1B1-L145V’s 1-naphthol, p-nitrophenol, 1-hydroxypyrene, and PAPS variable concentration plots.
| 1-naphthola |
p-nitrophenola |
1-hydroxypyrenea |
PAPSd |
|||||
|---|---|---|---|---|---|---|---|---|
| Km(µM) |
Vmax (pmol/(min*µg)) |
Km(µM) |
Vmax (pmol/(min*µg)) |
Ks(µM)b |
Vmax (pmol/(min*µg)) |
Km(µM) |
Vmax (pmol/(min*µg)) |
|
| SULT1B1-WT | 1.4±0.6 | 10.1±1.9 | 7.2±0.7 | 14.4±0.5 | 33.5±10.8 | N/Ac | 2.2±0.4 | 1.3±0.1 |
| SULT1B1-L145Ve | 1.3±0.4 | 9.3±1.0 | 11.3±2.7 | 6.8±0.7* | 7.7±0.3 | N/Ac | 1.6±0.4 | 0.9±0.1 |
Substrate apparent values were measured using 15 µM PAPS. All errors represent SEM.
A Km estimate was not possible because of the sigmoidal curve.Instead, Ks(the nth root of K’,composed of “interaction factors” was calculated).
The sigmoidal nature of the curve and potent substrate inhibition do not allow accurate estimation of the Vmax.
PAPS apparent values were measured using 6.25 µM 1-hydroxypyrene.All errors represent SEM.
Astrisks indicate significant differences from the WT(*,P < 0.01)
Colorectal and Prostate Cancer Patient Genotyping
SULT1B1’s altered kinetic activity in conjunction with its involvement in intestinal bioactivation suggests the mutant allele may be linked to intestinal malignancies. Therefore, genomic DNA samples from colorectal cancer, prostate cancer, and normal patients were obtained from Dr. Susan Kadlubar at the University of Arkansas Medical Center for the purpose of genotyping. High-resolution melting analysis (HRM) was used to genotype the samples (N=1203). Fifty cycle PCR was conducted with Takara ExTaq (Kyoto, Japan) using the leading primer 5’- TGTTTCAGTCTCATATTACCATTTTGA-3’ and flanking primer 5’-GGAAAAGGCTGTAAATTATTCATTA-3’ to generate a small (54 base pairs) SULT1B1 amplicon. BioFire Defense’s (Salt Lake City, UT) LC Green Plus served as the fluorescent intercalating agent for the detection of amplicon melting in an Idaho Technologies LightScanner. Plasmids containing both the WT and variant sequence were used as controls. The SNP increased the amplicon’s melting temperature by 0.6 oC, resulting in easily distinguishable homozygous and heterozygous populations. First, the screen was used to confirm the observation that the SNP is only present in individuals of African descent. Once confirmed, the results of African patients were tested for genotype/disease independence using a Chi-squared test.
Results
The PCR product resulting from the amplification of total human endometrial cDNA with SULT1B1 specific primers was 1021 base pairs, an expected size for a SULT gene. However, sequencing identified two distinct nucleotides in the first position of the 145th codon, one with the sequence TTA and the other with GTA. The TTA, encoding a leucine residue, corresponded with the WT SULT1B1 sequence. Alteration of the first base to a “G” encoded a valine at the 145th amino acid position and resulted in the introduction of a new unique recognition site for the restriction enzyme HpyCH4IV (A↓CGT). The introduction of this restriction site allowed rapid identification of the L145V allele (Figure 1). Incubation of the two different SULT1B1-amplified samples with HpyCH4IV yielded two different results. In the first case (lane 2), the PCR product was unrestricted whereas the second PCR product (lane 4) was completely digested (Figure 1).
Figure 1.
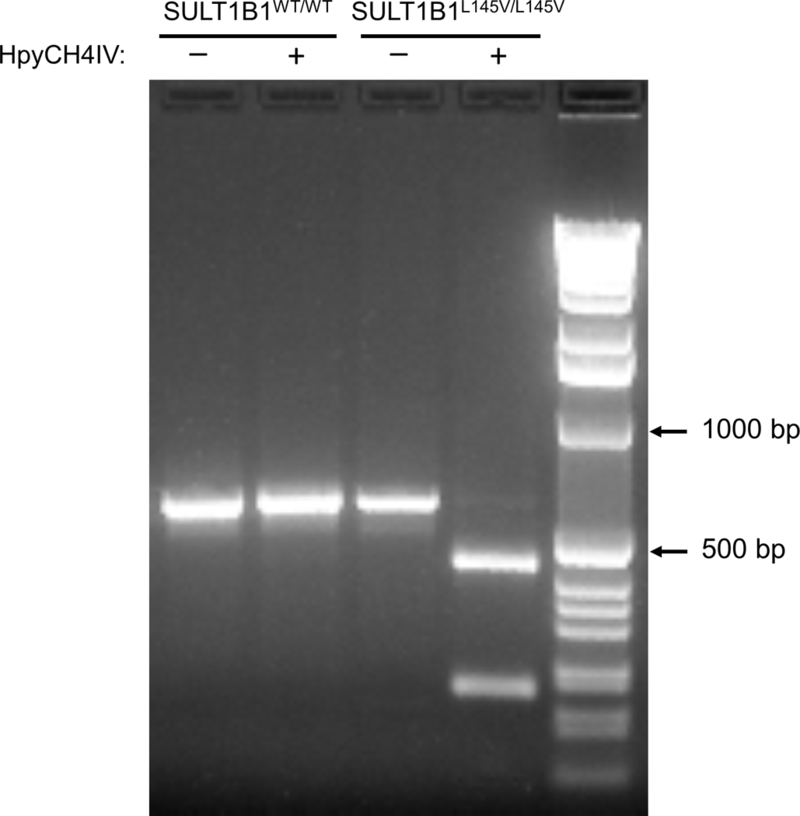
Screening human tissues for the SULT1B1L145V allele. SULT1B1 PCR products, amplified from human genomic DNA, were digested with the HpyCH4IV restriction enzyme. No apparent digestion (lane 2) suggests the patient does not carry the SULT1B1L145V allele, while complete digestion (lane 4) suggests the patient is homozygous for the SULT1B1L145V allele.
Both SULT1B1WT and SULT1B1L145V were cloned into bacterial expression vectors. The native enzymes were purified to approximately 60% purity by DEAE-Sepharose chromatography while each 6xHis tagged isoform was >95% pure after elution from Ni-NTA resin (data not shown). Both isoforms were active toward classical substrates and equally reactive with a SULT1B1 polyclonal antibody (data not shown) (Teubner et al., 2007; Wang et al., 1998). The Kms for 1-naphthol, p-nitrophenol, and 1-hydroxypyrene sulfation were not significantly different between the L145V and WT isoform (Table 1 and Figure 2). Each isoform’s maximal turnover rate was nearly identical for 1-naphthol and 1-hydroxypyrene, but the WT maximal sulfation rate of p-nitrophenol was significantly different (p-value = 0.001) compared to the L145V isoform (Table 1 and Figure 2C). An accurate Km for 1-hydroxypyrene could not be calculated due to the sigmoidal nature of the curve; therefore, the pre-substrate inhibition data points were analyzed to approximate the Ks using the Adair-Pauling Simple Sequential Interaction Model (VisualEnzymics) (Figure 2B). The Ks values were determined to be 33.5 ± 10.8 µM for WT and 7.7 ± 0.3 µM for the L145V isoform (Figure 2B). Both isoforms exhibited substrate inhibition above 6.25 µM 1-hydroxypyrene. Both isoforms also exhibited substrate inhibition at 1-naphthol concentrations above 5 µM. However, the substrate inhibition of the L145V isoform was shifted to the right, with a Ki of 7.5 µM, compared to the WT isoform’s Ki of 6.3 µM (Figure 2A). This trend was consistent with the PAP(S) binding patterns of the two isoforms. Both SULT1B1 isoforms had similar Kds (within error) for PAPS (~900 nM), but the WT isoform exhibited a 4-fold more favorable Kd for PAP (Figure 4). This binding pattern was reflected in the apparent Kms of the isoforms. Surprisingly, the L145V isoform’s Km for PAPS (1.6 ± 0.4 µM) was within error of the WT isoform’s Km for PAPS (2.2 ± 0.4 µM); the WT isoform exhibited a maximal sulfation rate of 1.3 ± 0.1 pmol/(min*µg), nearly one and one half times that of the L145V isoform (0.9 ± 0.1 pmol/(min*µg)) (Table 1 and Figure 2D). SULT1B1 crystal structures are available (PDBIDs: 3CKL and 2Z5F) and were used to rationalize the observed activity differences, which is evaluated below in the discussion (Figure 4) (Dombrovski et al., 2006; Pan, 2008).
Figure 2.
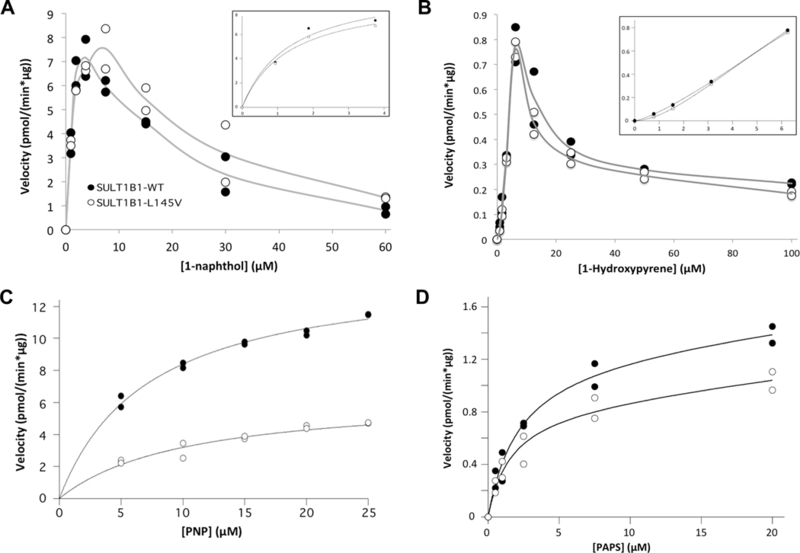
SULT1B1-L145V (white circles) displays different kinetic properties than SULT1B1-WT (opaque circles). (A) The 1-naphthol Km is unchanged while substrate inhibition is less potent for the L145V isoform. The inset provides an enhanced view of the curve preceding substrate inhibition. (B) The 1-hydroxypyrene sulfation curve is nearly identical for both isoforms, though the sigmoidal nature of the curve, as shown in the inset’s enhanced view of the curve before substrate inhibition, prevents the calculation of an accurate Km. Instead, the Ks (the nth root of K0, composed of the ‘‘interaction factors’’) for the WT isoform was calculated to be 33.5 ± 10.8 µM, while the L145V isoform exhibits a Ks of 7.7 ± 0.3 µM. (C) SULT1B1-L145V’s PNP Km is less favorable than that of the WT enzyme, while its maximal velocity is faster. (D) Finally, the L145V isoform’s PAPS Km appears to be slightly more favorable while the Vmax is significantly slower. These kinetic parameters with standard error of the mean can be found in Table 1.
Figure 4.
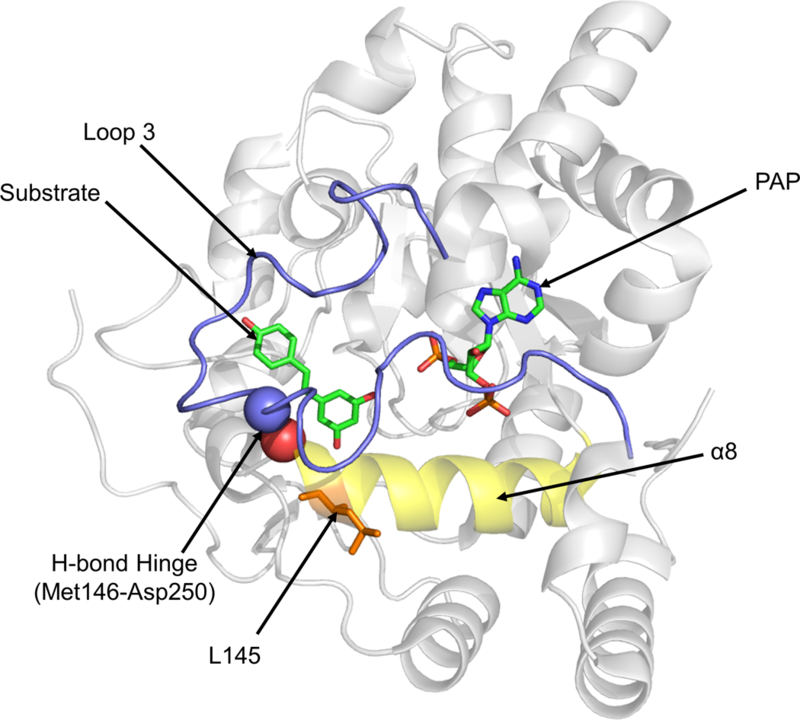
The location of L145 in the SULT1B1 crystal structure (PDBID: 3CKL). L145 is located at the end of α-helix 8 which forms the base of the enzyme’s active site. L145 is adjacent to M146, which forms a key hydrogen bond with D250, anchoring Loop 3 to the active site’s base. Loop 3 overlays both the substrate (left) and PAP (right) binding domains and has key contacts with both the substrate and the cofactor.
Thirty-seven human tissue samples (mostly endometrium) were screened by restriction digestion, as depicted in Figure 1. Of these specimens, 84% were homozygous for the SULT1B1WT allele, 3% were homozygous for the SULT1B1L145V allele, and 13% were heterozygous (Figure 5). No significant trends were observed with respect to tissue status (normal or tumor) or patient age; however, a significant trend emerged with ethnicity. The variant was not observed in Caucasian specimens (Figure 5B). All of those carrying this allele were of African or unknown (N=2) descent (Figure 5C). In this African ethnic group, 58% were homozygous for the WT allele, 8% were homozygous for the L145V allele, and 33% were heterozygous (Figure 5C). To achieve higher confidence in the allelic frequency, a larger scale experiment was conducted with prostate cancer and colorectal cancer genomic DNA samples; HRM analysis of these samples determined if any linkage exists between the genotype, patient’s ethnicity, and colon/prostate cancer incidence. Of the 1203 samples screened, 301 (602 alleles) were of sub-Saharan African descent (Figure 6B and Supplemental Table 1). Only a single SULT1B1L145V allele was detected in a patient who identified as Caucasian, confirming the ethnic specificity of the variant (Figure 6 and Supplemental Table 1). Within the 273 African-descent prostate cancer patients, 35 L145V carriers were non-diseased, while 17 were diseased. The allelic frequencies between the two populations, prostate cancer and healthy, were 8.02% and 10.48%, respectively; the difference was not significant (p-value = 0.24). Four of the 28 African-descent colorectal cancer patients were carriers of the L145V SNP, yielding allelic frequencies of 7.5% and 6.25% in healthy and prostate cancer patients, respectively; however, this difference in allelic frequency was also not significant (p-value = 0.84).
Figure 5.
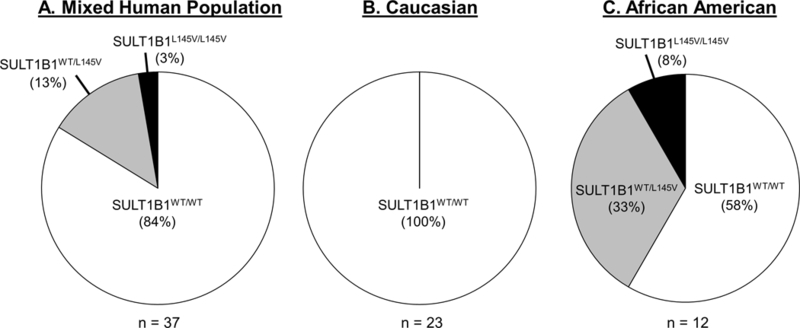
The approximate frequency of the SULT1B1WT and SULT1B1L145V alleles amongst (A) a mixed human population, (B) a Caucasian population, and (C) the black African American population in Birmingham, AL.
Figure 6.
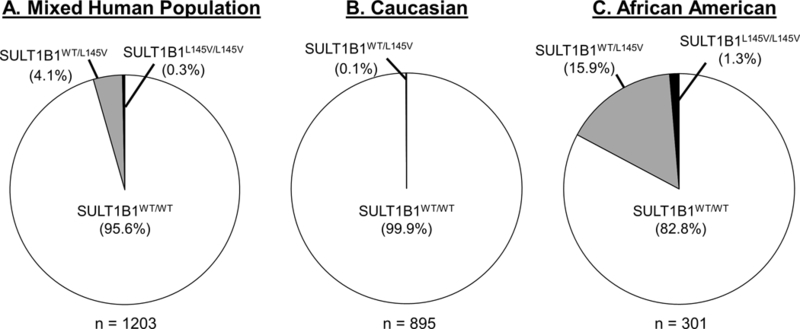
The approximate frequency of the SULT1B1WT and SULT1B1L145V alleles amongst (A) a mixed human population, (B) a Caucasian population, and (C) the black African American population in a large sample set from Arkansas Medical Center.
Discussion
An allelic variant of SULT1B1, a missense SNP resulting in the substitution of a valine for a leucine residue at the 145th amino acid position, was detected while cloning the gene from human endometrial tissue. The presence of the L145V variant prompted further investigation into its frequency in the human population as well as in-depth kinetic analysis of the two isoforms. SULT1B1’s primary role in the body is to catalyze the sulfation of drugs and hormones; therefore, we tested the two isoforms’ sulfation abilities in vitro. SULT1B1’s substrates vary in size, ranging from small (e.g. 1-naphthol and p-nitrophenol) to large (e.g. 1-hydroxypyrene). Based on the PAPS gating mechanism for substrate entrance into the active site, modest amino acid sequence alterations have been shown to alter SULT-substrate interactions in a size specific manner (Cook et al., 2015; Rohn-Glowacki & Falany, 2014). The L145V variant’s Km for 1-naphthol and p-nitrophenol were diminished, suggesting the L145V isoform’s active site is not coordinated properly to bind or orient the two small substrates in a catalytic manner. The maximal rate of p-nitrophenol sulfation was slower, whereas the 1-naphthol sulfation rate was within error and therefore indifferent.
Interestingly, the WT isoform was slightly more susceptible to substrate inhibition by 1-naphthol with a Ki of 6.3 µM (Figure 2A). Most cases of SULT substrate inhibition arise from the enzyme family’s favorable binding of the cofactor byproduct PAP (Wang et al., 2014). At high substrate concentrations, slow release of PAP favors binding of the substrate to the binary (SULT-PAP) complex, slowing the reaction rate. Therefore, we hypothesized that the WT isoform may exhibit a greater affinity for the byproduct PAP than the L145V isoform. The WT isoform had a significantly higher affinity for PAP with a Kd of 106 nM compared to the L145V variant with a Kd of 405 nM (Figure 3). Based on these results, the WT isoform’s increased susceptibility to substrate inhibition by 1-naphthol is driven by an increased affinity for PAP. Surprisingly, this increased susceptibility to substrate inhibition was not apparent with 1-hydroxypyrene (Figure 2B). Non-classical interactions exist between the enzyme and 1-hydroxypyrene, as indicated by the sigmoidal shape of the curve between 0 and 6.25 µM. The shape of the curve suggests negative cooperative binding between the substrate and PAPS, likely governed by the previously described Loop 3 gating mechanism (Cook et al., 2013b; Cook et al., 2010). The PAPS Kds for the WT and L145V isoforms were comparable within error at 872 and 980 nM, respectively. Though only small alterations, the PAP(S) dissociation constants for each isoform were altered enough to significantly change the rate of the 1-hydroxypyrene reaction with respect to PAPS concentration. The Kms of each isoform were within error (WT = 2.2 ± 0.4 µM, L145V = 1.6 ± 0.4 µM) while the maximal rate of the reaction was not within error (WT = 1.3 ± 0.1 pmol/(min*µg), L145V = 0.9 ± 0.1 pmol/(min*µg)). Together, the in vitro kinetic assays suggest the enzyme activity of the SULT1B1-L145V variant is different from the WT isoform and could be associated with specific pathologies via a number of different mechanisms: 1. Affinities for physiological and non-physiological substrates could be directly altered as with p-nitrophenol; or 2. the PAPS-concentration dependent rate may alter the metabolic pathway by which compounds progress, as illustrated by the substrate inhibition profile alterations and PAPS concentration dependent sulfation of 1-hydroxypyrene (Figure 2A and2D). The PAPS concentrations in the gastrointestinal epithelium and white blood cells are not known but could be valuable in predicting the different metabolic activities of the WT and L145V isoforms in the body.
Figure 3.
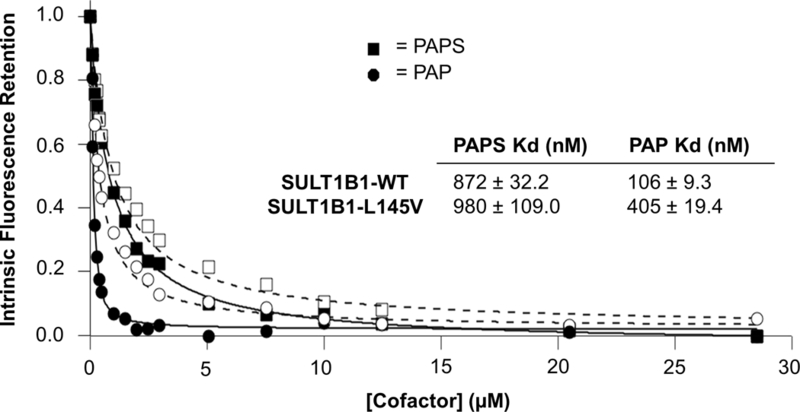
Binding affinities of SULT1B1-WT and L145V for PAP and PAPS. The SULT1B1-WT (opaque shapes) and L145V (hollow shapes) dissociation constants for PAPS (squares) and PAP (circles) were measured by intrinsic fluorescence. Both isoforms exhibit a similar Kd for PAPS (~900 nM), while PAP binds less potently to the L145V isoform (405 nM as opposed to 106 nM).
L145 is located at the end of the conserved helix (α8) spanning the base of the active site (Allali-Hassani et al., 2007; Pan, 2008). In SULT1B1, L145’s R-group is facing away from the interior of the pore; therefore, activity alteration must not be a result of the residue’s direct interactions with the substrates or cofactor. The change in R-group size could destabilize α8, changing the helix’s contribution to pocket shape, or have indirect effects on another portion of the pocket, such as PAPS binding domain. This destabilization may disrupt hydrogen bonding between M146, directly adjacent to L145, and aspartic acid (D) 250, anchoring the active site “lid” (Loop 3) to the “floor”, helping to preserve Loop 3’s location (Cook et al., 2013b). This bond acts much like a hinge for the Loop 3 lid, separating it into two portions: one portion overlays the substrate binding domain and the other overlays the PAPS binding domain (Cook et al., 2013a; Tibbs & Falany, 2015). Alteration of the M146-Loop 3 hydrogen bond by mutation of an adjacent residue could feasibly alter both the PAPS and substrate affinities, as was observed in the L145V isoform’s kinetics (Pan, 2008).
Conveniently, the SNP resulted in the establishment of a restriction enzyme recognition site for HpyCH4IV; this site provided a means to rapidly screen human tissues after amplification of their SULT1B1 genes. Using this screening method, seven of 74 alleles (37 patients) were the SULT1B1L145V, with an allelic frequency of 9% (Figure 5). Interestingly, no variant alleles were detected in any of the 23 Caucasian samples. Twelve of the 14 remaining patients were confirmed to be of African descent, while two were unknown. From our limited sample size, the allelic frequency for SULT1B1L145V in black African Americans was determined to be 25% (Figure 5). The National Center for Biotechnology Information (NCBI)/Ensembl genome browser reported the SULT1B1L145V allelic frequency to be 2.5% out of 5008 human samples (Table 2) (Sherry et al., 2001). In agreement with our data, the Ensembl data confirmed the SULT1B1L145V allele is largely specific to individuals of African descent (8.7% allele frequency), with a low frequency (1.6–2.4%) in South American populations. Among Africans, SULT1B1L145V frequency ranged from 5.6% in Yoruba in Ibadan, Nigeria to 11.5% in the Southwestern region of the United States. In agreement with our data, the variant SULT1B1L145V allele was not present in 1000 European samples. Furthermore, the allele was not present in approximately 2000 South Asians or Eastern Asians. Our results indicate a higher SULT1B1L145V frequency (25%) than those reported on Ensembl (8.7%). This discrepancy was likely caused by our limited sample size (N=12); however, further studies may reveal that these results represent local population differences.
Table 2.
Summary of SULT1B1L145V allele frequency in various populations around the world.
| SULT1B1WT |
SULT1B1L145V |
N |
P-Value |
||
|---|---|---|---|---|---|
| All population |
0.975 | 0.025 | 5008 | ||
| African | 0.913 | 0.087 | 1322 | 9.20E-26 | |
| African Caribbean in Barbados | 0.911 | 0.089 | 192 | 9.57E-08 | |
| African Ancestry in Southwest US | 0.885 | 0.115 | 122 | 1.50E-09 | |
| Esan in Nigeria | 0.919 | 0.081 | 198 | 1.96E-06 | |
| Luhya in Webuye,Kenya | 0.919 | 0.081 | 198 | 1.96E-06 | |
| Mandinka in the Gambia | 0.093 | 0.097 | 226 | 1.96E-10 | |
| Mende in the sierra leone | 0.894 | 0.106 | 170 | 2.40E-10 | |
| Yoruba in lbandan,Nigeria | 0.944 | 0.056 | 216 | 5.29E-03 | |
| American | 0.987 | 0.013 | 694 | 5.07E-02 | |
| Columbian in Medellin | 0.984 | 0.016 | 188 | 4.35E-01 | |
| Mexican Ancestry in Los Angels,CA | 1.000 | 128 | 7.01E-02 | ||
| Peruvian in Lima,Peru | 0.994 | 0.006 | 170 | 1.14E-01 | |
| Puerto Rican in Puerto Rico | 0.976 | 0.024 | 208 | 9.28E-01 | |
| East Asian | 1.000 | 1008 | 3.92E-07 | ||
| Chinese Dai in Xishuangbanna,China | 1.000 | 186 | 2.90E-02 | ||
| Han Chinese in Bejing,China | 1.000 | 206 | 2.16E-02 | ||
| Southern Han Chinese,China | 1.000 | 210 | 2.04E-02 | ||
| Japanese in Tokyo, japan | 1.000 | 208 | 2.10E-02 | ||
| Kinh in Ho Chi Minh City, Vietnam | 1.000 | 198 | 2.43E-02 | ||
| European | 1.000 | 1006 | 4.02E-07 | ||
| Utah residents,N.and.European ancestry |
1.000 | 198 | 2.43E-02 | ||
| Finnish in Finland | 1.000 | 198 | 2.43E-02 | ||
| British in England and Scotland | 1.000 | 182 | 3.08E-02 | ||
| Lberian populations in Spain | 1.000 | 214 | 1.92E-02 | ||
| Tosconi in italy | 1.000 | 214 | 1.92E-02 | ||
| South Asian | 1.000 | 978 | 5.82E-07 | ||
| Bengali in Bangladesh | 1.000 | 172 | 3.58E-02 | ||
| Gujarati indian in Houston,TX | 1.000 | 206 | 2.16E-02 | ||
| Indian Telugu in UK | 1.000 | 204 | 2.23E-02 | ||
| Punjabi in Lhore,Pakistan | 1.000 | 192 | 2.66E-02 | ||
| Sri Lankan Tamil in the UK | 1.000 | 204 | 2.23E-02 | ||
P-Values were calculate using a chi-squared test for independence, comparing the allelic frequencies Of each independent population to “all populations.” P-values < 5E-02 indicate significant differences
SULT1B1 is expressed throughout the gastrointestinal tract, kidneys, and white blood cells; therefore, the L145V variant enzyme may directly contribute to pathologies in these tissues via carcinogen bioactivation. However, the effects of bioactivated carcinogens are not limited to the tissue of origin (Teubner et al., 2007). Genomic DNA samples from colorectal and prostate cancer patient samples were obtained from Dr. Susan Kadlubar (University of Arkansas Medical Center) to determine if the SULT1B1L145V allele is associated with risk of developing the two types of cancer. In this sample set, the SULT1B1L145V allelic frequency amongst individuals of African-descent (9.52%) was in agreement with the allelic frequency reported by NCBI (Figure 6 and Table 2). Only one individual who identified as Caucasian carried the SULT1B1L145V allele, suggesting admixture. The colorectal cancer patient analysis lacked power, with only 28 individuals of African descent. Regardless, no significant difference in SULT1B1L145V allelic frequency was observed between patients with and without colorectal cancer. With 273 African-descent individuals, the prostate cancer analysis was more powerful. The SULT1B1L145V allelic frequency of individuals with prostate cancer was 8.02% and 10.48% in healthy individuals; however, this difference was not statistically significant (p-value = 0.24). If this result holds true in the future, the variant enzyme may provide protection arising from altered substrate specificity toward PAHs.
SULT1B1 performs a number of diffuse roles in the body; however, the presence of a highly specific role for SULT1B1 remains to be identified. If the L145V isoform’s primary role is indeed the metabolism of thyroid hormones, we predict carriers to be susceptible to thyroid hormone imbalance (Wang et al., 1998). Based on the metabolic rate of the variant enzyme and previous SULT1A1 variant studies, SULT1B1L145V carriers may display a different propensity to metabolize pharmaceuticals and aid in the elimination of dietary compounds, such as potentially carcinogenic PAHs (Edavana et al., 2012; Ung & Nagar, 2007; Zhang et al., 2012; Zheng et al., 2010). With an overall goal of personalized medicine, the clinical implications of the SULT1B1L145V allelic variant within the given test group should be the focus of future studies.
Conclusion
In conclusion, this study identified and enzymatically characterized a human SULT1B1 allelic variant that is selectively present in Americans of African descent. Key regions of SULT1B1’s protein structure are destabilized by the amino acid modification (L145V) produced by this variant. These changes in the protein’s structure resulted in several alterations to the enzyme’s kinetic properties in comparison to WT SULT1B1. The identification of this high frequency dysfunctional isoform prompted linkage analysis with colorectal cancer and prostate cancer patient samples. Although there was no significant correlation between the SULT1B1 variant and cancer patient samples, SULT1B1L145V carriers may have an unpredictable clinical issue which is not associated with colorectal or prostate cancer.
Supplementary Material
References
- Allali-Hassani A, Pan PW, Dombrovski L, et al. (2007). Structural and chemical profiling of the human cytosolic sulfotransferases. PLoS Biol 5: e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamber DE, Fryer AA, Strange RC, et al. (2001). Phenol sulphotransferase sult1a1*1 genotype is associated with reduced risk of colorectal cancer. Pharmacogenetics 11: 679–685. [DOI] [PubMed] [Google Scholar]
- Bird CE, Murphy J, Boroomand K, et al. (1978). Dehydroepiandrosterone: Kinetics of metabolism in normal men and women. J Clin Endocrinol Metab 47: 818–822. [DOI] [PubMed] [Google Scholar]
- Blanchard RL. (2005). Nomenclature and molecular biology of the human sulfotransferase family. In: Pacifici GMC, Michael WH, ed. Human cytosolic sulfotransferases 1 ed. Boca Raton, FL: CRC Press; 1–26. [Google Scholar]
- Boccia S, Persiani R, La Torre G, et al. (2005). Sulfotransferase 1a1 polymorphism and gastric cancer risk: A pilot case-control study. Cancer Lett 229: 235–243. [DOI] [PubMed] [Google Scholar]
- Carlini EJ, Raftogianis RB, Wood TC, et al. (2001). Sulfation pharmacogenetics: Sult1a1 and sult1a2 allele frequencies in caucasian, chinese and african-american subjects. Pharmacogenetics 11: 57–68. [DOI] [PubMed] [Google Scholar]
- Cook I, Wang T, Almo SC, et al. (2013a). The gate that governs sulfotransferase selectivity. Biochemistry 52: 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook I, Wang T, Falany CN, et al. (2013b). High accuracy in silico sulfotransferase models. J Biol Chem 288: 34494–34501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook I, Wang T, Leyh TS. (2015). Sulfotransferase 1a1 substrate selectivity: A molecular clamp mechanism. Biochemistry 54: 6114–6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook IT, Leyh TS, Kadlubar SA, et al. (2010). Structural rearrangement of sult2a1: Effects on dehydroepiandrosterone and raloxifene sulfation. Horm Mol Biol Clin Investig 1: 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotterchio M, Boucher BA, Manno M, et al. (2008). Red meat intake, doneness, polymorphisms in genes that encode carcinogen-metabolizing enzymes, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev 17: 3098–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrovski L, Dong A, Bochkarev A, et al. (2006). Crystal structures of human sulfotransferases sult1b1 and sult1c1 complexed with the cofactor product adenosine-3’- 5’-diphosphate (pap). Proteins 64: 1091–1094. [DOI] [PubMed] [Google Scholar]
- Edavana VK, Dhakal IB, Yu X, et al. (2012). Sulfation of 4-hydroxy toremifene: Individual variability, isoform specificity, and contribution to toremifene pharmacogenomics. Drug Metab Dispos 40: 1210–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falany CN, He D, Li L, et al. (2009). Regulation of hepatic sulfotransferase (sult) 1e1 expression and effects on estrogenic activity in cystic fibrosis (cf). J Steroid Biochem Mol Biol 114: 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falany CN, Strom P, Swedmark S. (2005). Sulphation of o-desmethylnaproxen and related compounds by human cytosolic sulfotransferases. Br J Clin Pharmacol 60: 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falany CN, Vazquez ME, Kalb JM. (1989). Purification and characterization of human liver dehydroepiandrosterone sulphotransferase. Biochem J 260: 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falany CN, Xie X, Wang J, et al. (2000). Molecular cloning and expression of novel sulphotransferase-like cdnas from human and rat brain. Biochem J 346 Pt 3: 857–864. [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Nagata K, Ozawa S, et al. (1997). Molecular cloning and characterization of rat st1b1 and human st1b2 cdnas, encoding thyroid hormone sulfotransferases. J Biochem 122: 1052–1061. [DOI] [PubMed] [Google Scholar]
- Glatt H (2000). Sulfotransferases in the bioactivation of xenobiotics. Chem Biol Interact 129: 141–170. [DOI] [PubMed] [Google Scholar]
- Glatt H, Meinl W. (2004). Pharmacogenetics of soluble sulfotransferases (sults). Naunyn Schmiedebergs Arch Pharmacol 369: 55–68. [DOI] [PubMed] [Google Scholar]
- Lilla C, Risch A, Kropp S, et al. (2005). Sult1a1 genotype, active and passive smoking, and breast cancer risk by age 50 years in a german case-control study. Breast Cancer Res 7: R229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinl W, Glatt H. (2001). Structure and localization of the human sult1b1 gene: Neighborhood to sult1e1 and a sult1d pseudogene. Biochem Biophys Res Commun 288: 855–862. [DOI] [PubMed] [Google Scholar]
- Meinl W, Tsoi C, Swedmark S, et al. (2013). Highly selective bioactivation of 1- and 2-hydroxy-3-methylcholanthrene to mutagens by individual human and other mammalian sulphotransferases expressed in salmonella typhimurium. Mutagenesis 28: 609–619. [DOI] [PubMed] [Google Scholar]
- Nowell S, Falany CN. (2006). Pharmacogenetics of human cytosolic sulfotransferases. Oncogene 25: 1673–1678. [DOI] [PubMed] [Google Scholar]
- Pacifici GM, Giulianetti B, Quilici MC, et al. (1997). (−)-salbutamol sulphation in the human liver and duodenal mucosa: Interindividual variability. Xenobiotica 27: 279–286. [DOI] [PubMed] [Google Scholar]
- Pan PW, Tempel W, Dong A, Loppnau P, Kozieradzki I, Edwards AM, Arrowsmith CH, Weigelt J, Bountra C, Bochkarev A, Min J. 2008. Crystal structure of human cytosolic sulfotransferase sult1b1 in complex with pap and resveratrol
- Raftogianis RB, Wood TC, Otterness DM, et al. (1997). Phenol sulfotransferase pharmacogenetics in humans: Association of common sult1a1 alleles with ts pst phenotype. Biochem Biophys Res Commun 239: 298–304. [DOI] [PubMed] [Google Scholar]
- Raftogianis RB, Wood TC, Weinshilboum RM. (1999). Human phenol sulfotransferases sult1a2 and sult1a1. Biochem Pharmacol 58: 605–616. [DOI] [PubMed] [Google Scholar]
- Rens-Domiano SS, Roth JA. (1987). Inhibition of m and p phenol sulfotransferase by analogues of 3’-phosphoadenosine-5’-phosphosulfate. J Neurochem 48: 1411–1415. [DOI] [PubMed] [Google Scholar]
- Riches Z, Stanley EL, Bloomer JC, et al. (2009). Quantitative evaluation of the expression and activity of five major sulfotransferases (sults) in human tissues: The sult “pie”. Drug Metab Dispos 37: 2255–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn-Glowacki KJ, Falany CN. (2014). The potent inhibition of human cytosolic sulfotransferase 1a1 by 17alpha-ethinylestradiol is due to interactions with isoleucine 89 on loop 1. Horm Mol Biol Clin Investig 20: 81–90. [DOI] [PubMed] [Google Scholar]
- Sak K, Everaus H. (2016). Sulfotransferase 1a1 as a biomarker for susceptibility to carcinogenesis: From molecular genetics to the role of dietary flavonoids. Curr Drug Metab 17: 528–541. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, et al. (2001). Dbsnp: The ncbi database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenesh J (1989). Dictionary of biochemistry and molecular biology 2nd edition. New York: John Wiley & Sons. [Google Scholar]
- Teubner W, Meinl W, Florian S, et al. (2007). Identification and localization of soluble sulfotransferases in the human gastrointestinal tract. Biochem J 404: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomae BA, Rifki OF, Theobald MA, et al. (2003). Human catecholamine sulfotransferase (sult1a3) pharmacogenetics: Functional genetic polymorphism. J Neurochem 87: 809–819. [DOI] [PubMed] [Google Scholar]
- Tibbs ZE, Falany CN. (2015). Dimeric human sulfotransferase 1b1 displays cofactor-dependent subunit communication. Pharmacol Res Perspect 3: e00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbs ZE, Rohn-Glowacki KJ, Crittenden F, et al. (2015). Structural plasticity in the human cytosolic sulfotransferase dimer and its role in substrate selectivity and catalysis. Drug Metab Pharmacokinet 30: 3–20. [DOI] [PubMed] [Google Scholar]
- Ung D, Nagar S. (2007). Variable sulfation of dietary polyphenols by recombinant human sulfotransferase (sult) 1a1 genetic variants and sult1e1. Drug Metab Dispos 35: 740–746. [DOI] [PubMed] [Google Scholar]
- Wang J, Falany JL, Falany CN. (1998). Expression and characterization of a novel thyroid hormone-sulfating form of cytosolic sulfotransferase from human liver. Mol Pharmacol 53: 274–282. [DOI] [PubMed] [Google Scholar]
- Wang T, Cook I, Falany CN, et al. (2014). Paradigms of sulfotransferase catalysis: The mechanism of sult2a1. J Biol Chem 289: 26474–26480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood TC, Her C, Aksoy I, et al. (1996). Human dehydroepiandrosterone sulfotransferase pharmacogenetics: Quantitative western analysis and gene sequence polymorphisms. J Steroid Biochem Mol Biol 59: 467–478. [DOI] [PubMed] [Google Scholar]
- Yang G, Gao YT, Cai QY, et al. (2005). Modifying effects of sulfotransferase 1a1 gene polymorphism on the association of breast cancer risk with body mass index or endogenous steroid hormones. Breast Cancer Res Treat 94: 63–70. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang M, Blair IA, et al. (2012). Detoxication of benzo[a]pyrene-7,8-dione by sulfotransferases (sults) in human lung cells. J Biol Chem 287: 29909–29920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Sha X, Liu J, et al. (2010). Association of human cytochrome p450 1a1 (cyp1a1) and sulfotransferase 1a1 (sult1a1) polymorphisms with differential metabolism and cytotoxicity of aminoflavone. Mol Cancer Ther 9: 2803–2813. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.