

Abstract
Drought is a major abiotic stress that reduces rice development and yield. miRNAs (microRNAs) are known to mediate posttranscriptional regulation under drought stress. Although the importance of individual miRNAs has been established, the crosstalks between miRNAs and mRNAs remain unearthed. Here we performed microarray analysis of miRNAs and matched mRNA expression profiles of drought-treated rice cultivar Nipponbare. Drought-responsive miRNA-mRNA regulations were identified by a combination of a partial least square (PLS) regression approach and sequence-based target prediction. A drought-induced network with 13 miRNAs and 58 target mRNAs was constructed, and four miRNA coregulatory modules were revealed. Functional analysis suggested that drought-response miRNA targets are enriched in hormone signaling, lipid and carbohydrate metabolism, and antioxidant defense. 13 candidate miRNAs and target genes were validated by RT-qPCR, hierarchical clustering, and ROC analysis. Two target genes (DWARF-3 and P0651G05.2) of miRNA coregulatory modules were further verified by RLM-5′ RACE. Together, our integrative study of miRNA-mRNA interaction provided attractive candidates that will help elucidate the drought-response mechanisms in Oryza sativa.
1. Introduction
Rice (Oryza sativa) is one of the main staple crops that feed more than half of the global population [1]. Rice is acclimated to rain-fed or fully irrigated fields and is susceptible to drought stress [2]. The yield of rice is frequently challenged by dwindling water resources. There is a growing need to find novel drought-responsive molecules to improve the rice cultivars [3].
miRNAs (microRNAs) are short noncoding RNAs that mediate gene expression by mRNA degradation or translational repression [4]. Currently, 757 mature rice miRNA sequences have been identified in miRBase release 22 [5].
miRNAs have emerged as important players in a wide range of biological processes in plants [6]. They are involved in the resistance to environmental stresses by regulating the expression of stress-responsive genes [7–12]. Over the past decade, high-throughput technologies, e.g., microarray hybridization and RNA-seq, have accelerated the transcriptomic studies and shed light into the miRNA repertoires under drought stress conditions [13]. Drought-specific miRNAs have been investigated in different crops, including wheat [14–16], barley [17], maize [18], and Brachypodium [19], and dehydration-responsive miRNA expression patterns were detected. Several drought-related miRNA families have been identified in rice [20–24].
Drought-responsive miRNAs regulate their target mRNAs, most of which are functional proteins involved in drought stress [25]. Therefore, the function of a miRNA is often inferred via its target genes [26]. However, the identification of miRNA target genes represents a combinatorial challenge. Early miRNA target prediction methods rely on sequence complementarities, conservation, free energy, or target accessibility. However, these tools usually predict hundreds of target genes for each miRNA. Therefore, the number of false-positives is large, making it difficult to identify targets of real interest. More recent expression-based methods incorporate mRNA and miRNA expression data to predict functional miRNA-mRNA interactions. Although useful, most of the algorithms utilize the pairwise negative correlation coefficient to measure the interaction. Focusing exclusively on linear inverse correlations, they may undermine the many to many miRNA-mRNA relationships.
The aim of the present study was to detect accurate miRNA-mRNA associations in rice under drought stress. We proposed an integrative approach that combines PLS regression and sequence-based target prediction to identify potential miRNA and their targets that are important in drought response. We constructed a miRNA-mRNA regulatory network, and identified 4 miRNA-mRNA regulatory modules in the network that were associated with drought response. Furthermore, we evaluated the expression level and anticorrelation of the putative miRNA and targets. Functional analysis uncovered some drought-responsive canonical pathways that are specifically responsive to drought stress. This report demonstrates the utility of our approach to obtain new insights into posttranscriptional gene regulation at the miRNA level in drought-stressed rice. The schematic diagram of the procedures is given in Figure 1.
Figure 1.
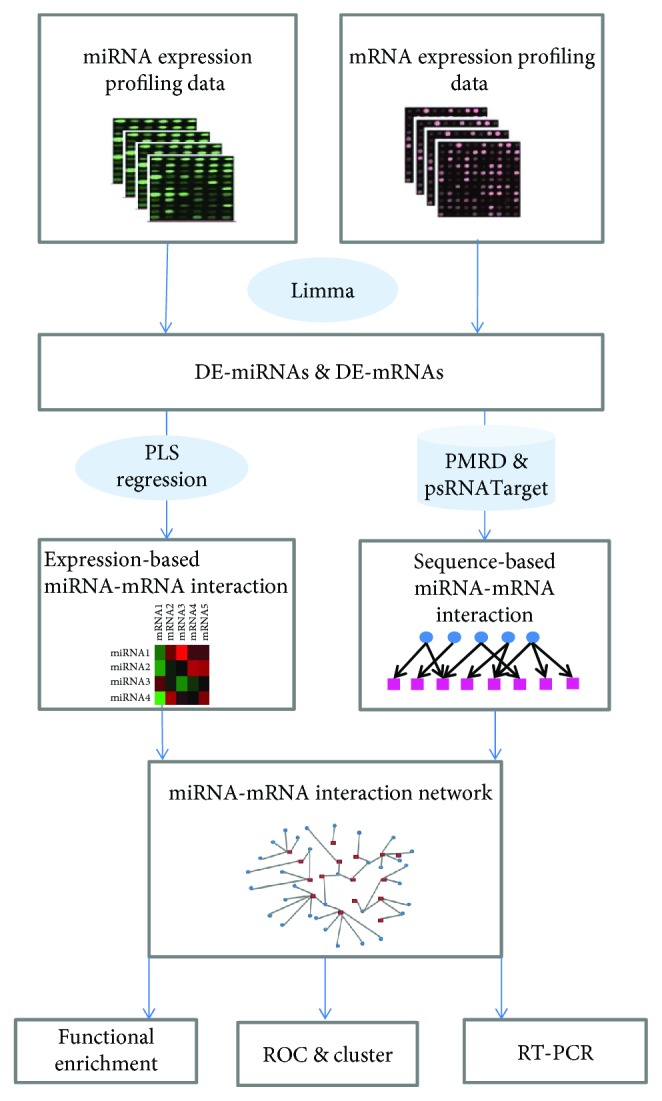
Flow chart of the general procedure of this study.
2. Materials and Methods
2.1. Plant Materials and Stress Treatment
Seeds of the rice cultivar Nipponbare were sterilized with 3% (v/v) NaClO, rinsed with distilled water, and pregerminated in moist filter paper for 48 h at 28°C. Thereafter, the seeds were transferred to sandy loam soil and grown under controlled conditions (16/8 h photoperiod, day/night air temperature of 25°C/20°C, 60% RH, and light intensity of 250 μmol m−2 s−1). Four-week-old plants were dehydrated by 20% polyethylene glycol 6000 for 48 h to simulate moderate drought stress of −0.5 MPa osmotic potential [27]. The control group was maintained under well irrigation conditions. Fifteen plants were included in each treatment and control group. After the treatment, the leaf disks (6 mmϕ) were excised from the uppermost rice leaf blades from both stress and control plants and stored in liquid nitrogen. Total RNA was isolated from 400 mg leave tissues by using the mirVana™ miRNA Isolation Kit according to the manufacturer's instructions. Three biological replicates were prepared for microarray analysis.
2.2. Microarray Analysis
miRNA microarray (LC Sciences, Houston, USA) contains 4 triplicates of 509 unique miRNA probes of 29 species in Sanger miRBase Release 19.0. Arabidopsis thaliana 5S rRNA and PUC2-20B were used as positive controls. Approximately 5 μg of the total RNA was size-fractionated, and the isolated small RNAs (~300 nt) were 3′-polyadenylated using polyA polymerase (Takara, Dalian, China) and subsequently ligated to Cy5 for fluorescent staining. Hybridization was performed at 34°C for 20 h. The mRNA expression was analyzed on Agilent-015241 Rice Gene Expression 4x44K Microarray (Feature Number version). The microarray raw data were submitted to the NCBI/GEO database and can be found under the accession number GSE99083 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99083).
2.3. Statistical Analysis
Data was processed in R programming environment v3.3.1. Raw expression files were background subtracted with the Normex method. Normalization was performed using the quantile method. Differentially expressed probe sets were identified using empirical Bayes method using the Limma v3.10. Signals with adjusted P value < 0.05 (Benjamini-Hochberg) were listed as significant.
2.4. miRNA Target Prediction
PMRD (http://bioinformatics.cau.edu.cn/PMRD/) and psRNATarget (http://plantgrn.noble.org/psRNATarget/) were searched for potential target sites. PMRD was searched with default parameters. The maximum expectation of psRNATarget was set as 2.5 to minimize the number of nonauthentic targets, and other settings remained default. Only the targets predicted by both databases were chosen to increase the prediction accuracy.
2.5. miRNA-mRNA Interaction Prediction
PLS regression can be used to measure the relationship between a given gene and other genes using their expression profiles [28, 29]. Briefly, consider a dataset of p miRNAs and m mRNAs for which N measurements are recorded. Let Y i be the expression profile for the ith mRNA, 1 < i < p. PLS first computes a number v < N of latent factors t i (l), called components for each mRNA, which represents the lth latent variable out of all orthogonal latent variables (l < <p), and then a separate regression model is fitted:
| (1) |
where e i is an n-dimensional vector of residuals.
Ordinary least squares calculate as follows:
| (2) |
The overall association score is then given by
| (3) |
PLS regression was run with 30 deregulated miRNAs and 329 mRNAs to calculate the association scores for each gene pair and recorded in a 329 × 30 matrix.
2.6. Significance Evaluation
Repeated bootstrapping (1000 times) was performed to assess the statistical significance of the miRNA-mRNA relationship (b ij). The interactions with P value < 0.01 and FDR < 0.05 were regarded as significant.
2.7. Network Construction
miRNA-mRNA interaction network was built and visualized with Cytoscape v3.4.0.
2.8. Functional Enrichment Analysis
The targets of DE-miRNAs under drought were mapped to GO and KEGG for functional enrichment using the DAVID Bioinformatics tool [30]. Terms with Benjamini-corrected P value < 0.05 were listed as significant.
2.9. Expression Validation
We employed RT-qPCR, hierarchical clustering, and ROC analysis to validate the expression levels of drought-responsive rice miRNAs.
PrimeScript one-step miRNA cDNA Synthesis Kit (Takara) was used for reverse transcribing mature miRNAs according to the manufacturer's instructions. Universal primers were provided with the kit, and the miRNA-specific primers are given in Supplementary Table S1. SYBRH Premix Ex TaqII (Takara, Japan) was used to quantify mature miRNAs. The PCR amplification parameter was set as 94°C for 2 min, then 40 cycles of 95°C for 10 s, 60°C for 30 s, and 72°C for 1 min. The melting curves were adjusted as 95°C for 1 s and 40°C for 2 min. The 2-DDCT method was used to analyze the relative expression changes. The rice UBC and U6 RNAs were used as internal standards. ef1a was used as an internal control.
Hierarchical clustering was performed with Cluster 3.0. Heat map was visualized with Java TreeView 1.1.6. ROC (receiver operating characteristic) analysis was performed by SPSS 16.0.
2.10. RNA Ligase-Mediated 5′ RACE
Total RNA was separately extracted using TRIzol reagent and subjected to RLM-5′ RACE with 5′-Full RACE Kit (Takara, Japan) following the manufacturer's instructions. Briefly, 10 μg of total RNA was ligated to the RNA adapter followed by reverse transcription using the Oligo (dT) primer and superscript reverse transcriptase. PCR was performed with 5′ adaptor primers and 3′ gene-specific primers (Supplementary Table S2). The final PCR product was purified on 2% agarose gel and cloned into a pEASY-T1 Vector, and plasmid DNA was sequenced by Huada Biotech Co.
3. Results and Discussion
Dwindling water resources frequently challenge the quality and quantity of rice and cause high economic losses [14]. miRNAs are central to posttranscriptional gene regulation in a wide range of cellular processes including stress responses [6, 31]. Over the past decade, several drought-related miRNA families in rice have been identified by either microarray hybridization or RNA-seq [20, 21, 32]. Stress-responsive miRNAs regulate their target mRNAs by targeting the gene transcripts most of which are stress-associated functional proteins, e.g., transcription factors or hormones involved under drought stress [25]. Therefore, we hoped to uncover significant miRNA-mRNA relationships that are specifically responsive to drought stress [26].
In this study, a multistep approach combining microarray miRNA and mRNA expression profile and bioinformatics analysis was adopted to identify the drought-responsive miRNA-mRNA regulatory network. First, we simultaneously analyzed miRNA and mRNA expression profiles in the same rice leaf sample and identified 76 differentially expressed miRNAs and 2916 mRNAs in drought-stressed samples. Secondly, 13 deregulated miRNAs and their 58 target mRNAs (69 miRNA-mRNA pairs) were identified by a combination of PLS regression approach and sequence-based target prediction. Their differential expression and regulation were further validated experimentally by RT-qPCR and RLM-5′ RACE.
Most of the existing algorithms for miRNA target prediction are based on the sequence complementarity, and the results vary widely. A rule of thumb to address this problem is to intersect the results of multiple prediction algorithms to reduce the false-positive. However, this procedure improves accuracy at the cost of many bona fide targets and therefore leads to poor sensitivity. The research pipeline in this study combines the sequence features and the expression of miRNAs and mRNAs simultaneously. Thus, our method precludes artifacts and improves prediction accuracy, thereby facilitating a more targeted experimental validation.
3.1. Identification of Drought-Responsive miRNAs and mRNAs in Rice
Microarray-based miRNA and mRNA expression profiles for Oryza sativa under/without drought stress were simultaneously investigated. As a result, 76 miRNAs (FDR < 0.05) were identified to be differentially expressed between drought-stressed and control leaves, corresponding to 38 rice miRNA families. The list of 76 differentially expressed miRNAs is provided in Supplementary Table S3. Of these, 9 were upregulated and 67 were downregulated. Using the same criteria, 2916 genes were identified to be differentially expressed.
3.2. PLS Reveals Significant miRNA-mRNA Associations
We first filtered possible miRNA target genes using miRNA target databases PMRD [33] and psRNATarget [34] and obtained 346 miRNA-mRNA pairs among 30 DE-miRNAs and 329 DE-mRNAs.
Next, we used the PLS regression method to remove false-positive discoveries among the 346 miRNA-mRNA pairs in order to find the most likely miRNA-mRNA associations.
The 30 DE-miRNAs were input as independent variables while the 329 DE-mRNAs as dependent variables for PLS regression. The association score b ij was calculated for each miRNA-mRNA pair, and repeated bootstrapping (1000 times) was subsequently performed to identify significant associations. Repeated bootstrapping (1000 times) was performed to assess the statistical significance of the miRNA-mRNA relationship. The interactions with b ij > 0.8, P value < 0.01, and FDR < 0.05 were regarded as significant.
This filtering step reduced considerably the number of potential candidate interactions, resulting in 69 significant miRNA-mRNA pairs among 13 DE-miRNAs and 58 DE-mRNAs, as listed in Table 1. Among them, 11 miRNAs were significantly downregulated and 2 miRNAs were upregulated. The maximum number of targets was 11 for osa-miR156b-3p.
Table 1.
Drought-responsive miRNAs and their target genes predicted by PLS.
| miRNA | Family | Response | Target no. | Targets predicted by PLS | Literature |
|---|---|---|---|---|---|
| osa-miR1430 | MIR169 | Down | 3 | MYB30, NFYA, UGE-4 | |
| osa-miR156b-3p | MIR156 | Down | 11 | 49D11.4, LCAT3, NRAMP5, RPSM, SPL15, SPL16, SPL2, SPL3, SPL4, SPL9, NCED1 | [21, 24] |
| osa-miR159a.2 | MIR159 | Down | 3 | 49D11.4, MYB4, MYB39 | [21, 24] |
| osa-miR164c | MIR164 | Down | 4 | CML22, CUC, NAC22, TCP8 | [35, 36] |
| osa-miR169d | MIR169 | Down | 6 | CBF, DREB, HAP2D, NAAT2, NFYA, UGE-4 | [32] |
| osa-miR172d-5p | MIR172 | Down | 7 | AP2D2, AP2D23, AP2D4, ARF9, bZIP, GT43A, JMJ705 | [21] |
| osa-miR319a-3p | MIR159 | Down | 5 | 49D11.4, MYB4, MYB59, TCP14, TCP7 | [21] |
| osa-miR395a | MIR395 | Down | 4 | ATPS3, P0651G05.2, RRF, URH2 | [21] |
| osa-miR396g | MIR396 | Down | 9 | 6PGL 1, P0651G05.2, PMP22, RGA2, RHL1, RPM1, RRM_SF, TPT | [21] |
| osa-miR444b.1 | MIR444 | Down | 3 | MADS2, MADS27, UEL-1 | [36] |
| osa-miR528-3p | MIR528 | Up | 5 | AO, DCL1, DWARF-3, IAR, SODCC.1 | [24] |
| osa-miR529a | MIR529 | Down | 4 | ACOX4, NUS1, SPL2, SPL9 | [21] |
| osa-miR812q | MIR812 | Up | 3 | CIPK10, DWARF-3, ERF |
Among the 13 differentially expressed miRNAs, some are known to respond to drought, including miR156b-3p [21], miR159a.2 [21], miR164c [35], miR169d [32], miR172d-5p [21], miR319a-3p [21], miR395a [21], miR396g [21], miR444b.1 [36], miR528-3p [24], and miR529a [21]. Two novel miRNAs (miR812q and miR1430) are reported for the first time to be involved in drought stress in rice, and their role remains unknown.
The regulatory edges in the miRNA-mRNA network well reflect the existing evidence described in the literature, indicating a high predictive value of our integrative method in identifying genuine miRNA-mRNA interactions.
Prominent drought-responsive miRNA targets were found to encode well-known stress-associated transcription factors or kinases. For example, MADS-box transcription factors targeted by miR444b.1. participate in diverse stress conditions, such as cold stress [37]. SBP-box transcription factor family members targeted by miR156b-3p and miR529a are involved in vegetative phase change induced by various environmental stimuli [38].
ATP-sulfurylase targeted by miR395a is involved in sulfate assimilation and allocation. miR169d targets NFYA regulates the expression of drought-responsive genes, e.g., GT and POD, and is crucial for plant adaptation to environmental stress [39]. miR319a targets two TCP family members (TCP7, 14) that inhibit cell proliferation as well as MYB4 and MYB59 mRNAs. miR164c targets NAC transcription factors, e.g., NAC22 and CUC, which determines the cellular levels of free auxin [40]. miR812q targets CIPK10, which interacts with CBL to trigger the CBL-CIPK-signaling pathway-modulating environmental stress tolerance [41].
3.3. Analysis of Drought-Responsive miRNA Coregulatory Network
Using the statistically significant PLS association score between the miRNA and mRNA, we built interaction networks out of the significant DE-miRNAs and their DE-mRNA targets. Edges are formed between miRNA-mRNA pairs with b ij > 0.8. The resulting miRNA-mRNA regulatory network is essentially a bipartite graph including two disjoint sets of vertices, miRNAs and mRNAs. The network consists of 69 regulations between 13 miRNAs and 58 mRNAs.
Then we analyzed the out-degree and in-degree distribution of the network (Figures 2(b) and 2(c)). On average, each miRNA has 3–5 outgoing edges and is loosely connected with each other, with a few exceptions such as the highly conserved miR156b-3p with 11 target genes, most of which belong to the SPL family, and the miR172d-5p with 3 target genes belonging to APD family. The out-degree distribution fits the power law with a slope of −0.8399 and R 2 = 0.9618. The in-degree distribution reveals an exponential distribution with an exponent of −0.967 and R 2 = 0.9183. It is obvious that as with many biological networks, the miRNA-mRNA network is not random but features a core set of organizing principles in structure.
Figure 2.

The layout of the miRNA-mRNA network and its structural features. (a) miRNA-mRNA network constructed by PLS. (b) Out-degree distribution of the miRNA-mRNA network. (c) In-degree distribution of the miRNA-mRNA network.
3.4. PLS Reveals miRNA-mRNA Coregulatory Modules
The PLS regression method successfully models the complex interrelationship of multiple miRNAs regulating one mRNA and identified miRNA-mRNA coregulatory modules, i.e., groups of miRNAs that regulate groups of mRNAs in concert. As shown in Figure 2(a), miRNAs regulate at least 3 targets, and 13.8% of mRNAs are coregulated by multiple miRNAs, indicating target multiplicity and miRNA cooperation in the drought response of rice.
One mRNA, 49D11.4 is common target of 3 significantly downregulated miRNAs, miR156b-3p, miR159a.2, and miR319a-3p. The coregulation of miR159 and miR319 was expected, since mature miR159 and miR319 have sequence similarity at 17 out of 21 nucleotides and belong to the same family [42]. The miRNAs within the same family are often derived from recent duplication events and functionally related. Likewise, miR529a showed 14 nucleotide similarities with miR156a-3p and is a shared common target of the SPL family (SPL2, 9) [43]. Moreover, miR1430 is similar to miR169d in sequence and shares the target of NFYA and UGE4 [44].
On the other hand, miRNAs within the same families also have exclusive targets. As shown in Figure 2(a), miR159 and miR319 share MYB targets, but miR159 fails to guide cleavage of TCPs as miR319. This case argues that a single nucleotide difference is sufficient to dictate miRNA target binding.
Interestingly, coregulation occurs also between phylogenetically unrelated miRNAs, i.e., miRNA belonging to different families, e.g., DWARF-3 by miR528-3p and miR8121 and P0651G05.2 by miR396g and miR395a, representing a stress-specific functional redundancy.
Coordinate miRNA regulation has previously been demonstrated in Drosophila and mammals. let-7/lin-4 is the first miRNA pair to be experimentally verified in Drosophila [45]. miR-375/miR-124/let-7b were found to jointly regulate Mtpn in mammals [46]. In this study, we provide evidence for miRNA synergism in drought-stressed rice. It is proposed that synergistic gene regulation is less dependent on miRNA abundance so that limited miRNAs are able to regulate more genes. Moreover, through synergism, miRNAs may exert reinforced repression effects and therefore enhance their regulatory efficacy. The identification of coregulatory miRNA modules would facilitate researchers to make more-informative choices of multiple mutant combinations to circumvent redundancy of miRNA regulation under drought stress.
3.5. Hierarchical Clustering and ROC Analysis
Hierarchical clustering and ROC were performed to validate the differential miRNA expression profiles. A heat map is illustrated in Figure 3.
Figure 3.

Heat map of the 13 DE-miRNAs in drought-stressed and control rice leaves.
The AUC (areas under the ROC curve), sensitivity, specificity, and accuracy for the 13 candidate miRNAs are listed in Table 2, and the ROC curves are provided as Supplementary Figure S1. ROC is a way of evaluating the performance of binary classifiers in classifying two classes, e.g., drought vs. control. A ROC curve is a plot of the true positive rates (sensitivity) vs. false-positive rates (1 − specificity). ROC curves can be used to evaluate the quality of classifiers for different discrimination thresholds. The closer the ROC curve to the upper left corner, the greater the AUC and the better the discrimination accuracy. The AUC for the 13 miRNAs are 0.870–0.986, with an average accuracy of 80.76%. The results showed that the candidate DE-miRNAs are able to differentiate drought-stressed samples from the controls. ROC analysis demonstrated that the miRNAs identified in this study are both sensitive and specific in sample discrimination, indicating that they are authentic miRNAs under drought stress.
Table 2.
ROC analysis of drought-responsive miRNAs.
| miRNA | AUC | Sensitivity | Specificity | Accuracy |
|---|---|---|---|---|
| osa-miR1430 | 0.930 | 0.900 | 0.900 | 0.800 |
| osa-miR156b-3p | 0.978 | 0.900 | 1.000 | 0.900 |
| osa-miR159a.2 | 0.908 | 0.800 | 1.000 | 0.800 |
| osa-miR164c | 0.902 | 0.900 | 0.800 | 0.700 |
| osa-miR169d | 0.945 | 0.850 | 0.950 | 0.800 |
| osa-miR172d-5p | 0.930 | 0.950 | 0.900 | 0.850 |
| osa-miR319a-3p | 0.950 | 0.950 | 0.950 | 0.900 |
| osa-miR395a | 0.957 | 0.850 | 1.000 | 0.850 |
| osa-miR396g | 0.913 | 0.800 | 0.950 | 0.700 |
| osa-miR444b.1 | 0.870 | 0.900 | 0.850 | 0.750 |
| osa-miR528-3p | 0.890 | 0.800 | 0.900 | 0.700 |
| osa-miR529a | 0.931 | 0.850 | 0.950 | 0.800 |
| osa-miR812q | 0.986 | 0.900 | 0.950 | 0.850 |
3.6. RT-qPCR Validated the Expression Trends of Drought-Responsive miRNAs and Coregulated Target Genes
We conducted RT-qPCR analysis to validate the expression level of 13 drought-responsive miRNAs and their anticorrelation with coregulated target genes. The results were consistent with the microarray data (Supplementary Figure S2). The relative expression trends of all of the 13 miRNAs were confirmed as differentially expressed under drought stress and were negatively correlated with their target genes.
3.7. RLM-5′ RACE Confirmed Direct Targets of Coregulatory miRNAs
To confirm the direct targets of coregulatory miRNAs, we performed RLM-5′ RACE to determine the exact miRNA cleavage sites on target genes. Two mRNAs (DWARF-3 and P0651G05.2) that are coregulated by miRNAs which do not belong to the same family were chosen for verification.
After sequencing the RLM-5′ RACE PCR products, we found that both DWARF-3 and P0651G05.2 had specific cleavage sites in the complementary miRNA sequences (Figures 4(c) and 4(d)). DWARF-3 was cleaved by miR528-3p and miR812q at positions 776 and 1150, respectively. Both cleavage sites are located at the positions 12–13 of miRNAs from the 5′ end. P0651G05.2 can be cleaved at position 486 by miR395a. The cleavage site was mapped to the paired miR395a sequence between the nucleotides 10 and 11. Moreover, P0651G05.2 could be simultaneously cleaved at the two sites by miR396g. The cleavage sites are located between nucleotides 10 and 11 and between 12 and 13, indicating that the cleavage sites of miR396g may be diverse.
Figure 4.

(a) Expression level of coregularory miR528-3p/miR812q and their target DWARF-3 by microarray and RT-qPCR. (b) Expression level of coregularory miR395a/miR396g and their target P0651G05.2 by microarray and RT-qPCR. The expression level is expressed as the mean fold changes of 3 replicates. Error bars depict the standard error (n = 3). (c) miR528-3p and miR812q cleavage sites on DWARF-3 identified by RLM-5′ RACE. (d) miR395a and miR396g cleavage sites on P0651G05.2 identified by RLM-5′ RACE. Arrows represent the 5′ termini of mRNA fragments, and the numbers beside them denote the cleavage frequency of cloned PCR products at the exact miRNA cleavage sites. The vertical dashes indicate matched RNA base pairs and circles represent GU mismatch whereas no dashes represent other types of mismatch.
The results of RLM-5′ RACE generally confirmed the coregulation of miR528-3p and miR812q on DWARF-3, as well as the coregulation of miR395a and miR396g on P0651G05.2, indicating a high reliability of the PLS analysis.
miR812q and miR528-3p were found to target auxin receptor F-box protein DWARF-3, which is essential for auxin-mediated signaling and plant development [47]. When auxin binds to DWARF-3, it promotes the ubiquitination and degradation of auxin transcriptional repressors, thereby activating auxin-responsive genes [48]. As a coreceptor for phytohormone, e.g., strigolactones and polar auxin, DWARF-3 has a pleiotropic effect on stalk height, leaf angle, and root growth in plants [49, 50]. Under drought stress conditions, upregulated miR812q and miR528-3p would decrease the expression of DWARF-3, and the miRNA-mediated DWARF-3 suppression would lead to attenuation of auxin signaling and inhibition of lateral root growth. We assume that the miR528-3p/miR812q-DWARF-3 coregulatory module might play a central role in development-related gene transcription and plant adaptation to drought stress.
In this study, we only validated two targets, DWARF-3 and P0651G05.2 by RACE experiment, because they are coregulatory hub nodes in the miRNA target network. Degradome sequencing is also a primary and effective tool to prove the miRNA targets. It can directly detect cleaved miRNA targets without predictions or overexpression. Degradome sequencing has been performed in rice by several studies and revealed endogenous small RNA targets under specific tissues or treatments [51, 52]. In the future, a large-scale target validation can be performed based on degradome sequencing data.
3.8. Functional Annotation of Drought-Stressed miRNAs
MiRNAs and their predicted target mRNAs were mapped to Gene Ontology and KEGG pathways to explore their biological significance.
According to GO enrichment, the target of drought-responsive miRNAs belonged to five important biological processes (BP): response to temperature stimulus, response to heat, response to abiotic stimulus, carbohydrate metabolic process, and response to water deprivation. Enriched molecular function (MF) terms included hormone binding, iron-sulfur cluster binding, hydrolase activity, peroxidase activity, and antioxidant activity. Plastid, chloroplast, anchored component of membrane, intrinsic component of plasma membrane, and plant-type cell wall were most significant among the cellular components (CC). A detailed list of the enriched GO terms is given in Figure 5.
Figure 5.

Enriched GO terms of drought-responsive miRNA targets.
KEGG pathway analysis unraveled significant activation of hormone signaling, lipid and carbohydrate metabolism, and antioxidant defense under drought. The full list of significant pathways (adjusted P value < 0.05) and the number of differentially expressed genes enriched in each pathway are represented in Supplementary Table S4.
3.8.1. miRNA-Regulated Phytohormone Signaling
Plant hormone signal transduction represented the most significantly enriched pathway with 52 genes differentially expressed under drought. A number of miRNA-regulated phytohormones are known to participate in plant responses to abiotic stress, e.g., auxin, salicylic acid, ethylene, GAs, jasmonate acid, and abscisic acid.
Here we found that the expression of key genes in auxin synthesis and the ABA signaling pathway was induced under drought stress. For example, IAR1 transcripts are downregulated by increased miR528 under drought. IAR1 regulates auxin homeostasis by hydrolyzing an inactive form of auxin to release bioactive auxin [53]. The decreased IAR1 expression and hence decreased auxin level leads to the inhibition of adventitious rooting under drought. Several genes in the ABA signaling pathway were also activated under drought. ABA is a plant stress hormone produced under environmental stresses [54]. It is an apocarotenoid derivative produced via oxidative cleavage of carotenoids. Also, we found drought stress increases carotenoid synthesis, which suggests that carotenoid synthesis and the ABA signaling were cooperatively involved in the drought stress response.
3.8.2. Lipid and Carbohydrate Metabolism
Pathway analysis unraveled significant reduction of starch-sucrose metabolism. This includes a variety of sucrose synthases, amylase, and starch branching enzymes. Decreased starch and sucrose concentration is an adaptive feature under drought stress [55], since it allows plants to divert assimilates and energy intended for growth into protective responses for survival [54].
In the context of fatty acid metabolism, α-linolenic acid metabolism was significantly induced under drought. α-linolenic acid is an unsaturated fatty acid in membrane lipids [56]. It enhances plant tolerance by maintaining membrane fluidity during stress [57].
3.8.3. miRNA-Mediated Antioxidant Defense
The immediate response of plants to stress is accumulation of reactive oxygen species (ROS). To remove the excessive ROS, the cell produces an antioxidative enzyme system, e.g., POD, AO, and SOD that serve as ROS scavengers.
In this study, we found that the production of ROS scavengers is regulated by miRNAs, as illustrated in Figure 6. For example, miR528 targets genes coding ascorbate oxidase, a central component of the hydrogen peroxide scavenging network. Ascorbate metabolism reduces oxidative damage by maintaining the ascorbate redox state in the form of apoplastic dehydroascorbate [58]. In addition, miR528 targets SOD transcripts, which catalyzes the dismutation of O2 − and alleviates the damage from ROS. Drought also induces key enzyme genes involved in biosynthesis of several secondary metabolites, such as isoflavonoids and carotenoids which mitigate diverse stress in plants.
Figure 6.

Schematic diagram of miRNA-regulated metabolic processes in rice under drought stress. Under drought stress, rice responds with the following physiological changes: (1) activating hormonal signaling pathways, (2) immobilizing fatty acid and downregulating starch and sucrose metabolism, (3) producing antioxidant compounds for detoxification, and (4) inhibiting miRNA biogenesis.
A simple model was constructed according to the function of miRNA and target genes under drought stress (Figure 6).
4. Conclusion
In conclusion, this study employed a multiple regression model for integrating paired miRNA and mRNA expression profiles. The integrated approach uncovered high-confidence reciprocally expressed miRNA target pairs, supporting the existence of a coregulatory network triggered by drought stress. Further validation of these hub miRNAs as coregulatory nodes may facilitate the development of novel molecular markers for breeding of rice cultivars with enhanced drought tolerance. In-depth analysis of coregulatory networks will reveal the regulatory role of miRNAs in a broader range of a posttranscriptional network under drought stress.
Acknowledgments
This work was supported by the National Natural Science Foundation of China [grant numbers 31770903, 31400712, and 21676173] and Qinglan Project of Jiangsu Province.
Abbreviations
- ABA:
Abscisic acid
- AUC:
Areas under the ROC curve
- CIPK:
Calcineurin B-like protein interacting protein kinase
- ERF:
Ethylene-responsive element-binding factor
- GO:
Gene Ontology
- GT:
Glutathione transferase
- KEGG:
Kyoto Encyclopedia of Genes and Genomes
- NFYA:
Alpha subunit of CCAAT binding NFY transcription factors
- POD:
Peroxidase
- RH:
Relative humidity
- RLM-5′ RACE:
RNA ligase-mediated 5′ rapid amplification of cDNA ends
- ROC:
Receiver operating characteristic
- SOD:
Superoxide dismutase
- SPL:
Squamosa promoter binding protein-like.
Data Availability
The microarray data used to support the findings of this study have been deposited in the GEO repository (GSE99083).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Supplementary Materials
Table S1: oligonucleotide primers used in RT-qPCR.
Table S2: the specific primer sequences for RLM-5′ RACE of target genes.
Table S3: differentially expressed miRNAs under drought stress.
Table S4: enriched KEGG pathways of drought-responsive miRNA targets.
Figure S1: ROC curves of 13 drought-responsive miRNAs.
Figure S2: validation of drought-responsive miRNAs and coregulated target genes by RT-qPCR. The expression level is expressed as the mean fold changes of 3 replicates. Error bars depict the standard error (n = 3).
References
- 1.Ala U., Karreth F. A., Bosia C., et al. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(18):7154–7159. doi: 10.1073/pnas.1222509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lafitte H. R., Yongsheng G., Yan S., Li Z. K. Whole plant responses, key processes, and adaptation to drought stress: the case of rice. Journal of Experimental Botany. 2007;58(2):169–175. doi: 10.1093/jxb/erl101. [DOI] [PubMed] [Google Scholar]
- 3.Barnabas B., Jager K., Feher A. The effect of drought and heat stress on reproductive processes in cereals. Plant, Cell & Environment. 2007;31(1):11–38. doi: 10.1111/j.1365-3040.2007.01727.x. [DOI] [PubMed] [Google Scholar]
- 4.Unver T., Namuth-Covert D. M., Budak H. Review of current methodological approaches for characterizing microRNAs in plants. International Journal of Plant Genomics. 2009;2009:11. doi: 10.1155/2009/262463.262463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Research. 2013;42(D1):D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akdogan G., Tufekci E. D., Uranbey S., Unver T. miRNA-based drought regulation in wheat. Functional & Integrative Genomics. 2016;16(3):221–233. doi: 10.1007/s10142-015-0452-1. [DOI] [PubMed] [Google Scholar]
- 7.Covarrubias A. A., Reyes J. L. Post-transcriptional gene regulation of salinity and drought responses by plant microRNAs. Plant, Cell & Environment. 2010;33(4):481–489. doi: 10.1111/j.1365-3040.2009.02048.x. [DOI] [PubMed] [Google Scholar]
- 8.Shuai P., Liang D., Zhang Z., Yin W., Xia X. Identification of drought-responsive and novel Populus trichocarpa microRNAs by high-throughput sequencing and their targets using degradome analysis. BMC Genomics. 2013;14(1):p. 233. doi: 10.1186/1471-2164-14-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Q., Zhang Y. C., Wang C. Y., et al. Expression analysis of phytohormone-regulated microRNAs in rice, implying their regulation roles in plant hormone signaling. FEBS Letters. 2009;583(4):723–728. doi: 10.1016/j.febslet.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 10.Ding Y., Chen Z., Zhu C. Microarray-based analysis of cadmium-responsive microRNAs in rice (Oryza sativa) Journal of Experimental Botany. 2011;62(10):3563–3573. doi: 10.1093/jxb/err046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajyzadeh M., Turktas M., Khawar K. M., Unver T. miR408 overexpression causes increased drought tolerance in chickpea. Gene. 2015;555(2):186–193. doi: 10.1016/j.gene.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Li B., Qin Y., Duan H., Yin W., Xia X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. Journal of Experimental Botany. 2011;62(11):3765–3779. doi: 10.1093/jxb/err051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cagirici H. B., Alptekin B., Budak H. RNA sequencing and co-expressed long non-coding RNA in modern and wild wheats. Scientific Reports. 2017;7(1, article 10670) doi: 10.1038/s41598-017-11170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akpinar B. A., Budak H. Dissecting miRNAs in wheat D genome progenitor, Aegilops tauschii . Frontiers in Plant Science. 2016;7:p. 606. doi: 10.3389/fpls.2016.00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alptekin B., Langridge P., Budak H. Abiotic stress miRNomes in the Triticeae. Functional & Integrative Genomics. 2017;17(2-3):145–170. doi: 10.1007/s10142-016-0525-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akpinar B. A., Kantar M., Budak H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Functional & Integrative Genomics. 2015;15(5):587–598. doi: 10.1007/s10142-015-0453-0. [DOI] [PubMed] [Google Scholar]
- 17.Fard E. M., Bakhshi B., Keshavarznia R., Nikpay N., Shahbazi M., Salekdeh G. H. Drought responsive microRNAs in two barley cultivars differing in their level of sensitivity to drought stress. Plant Physiology and Biochemistry. 2017;118:121–129. doi: 10.1016/j.plaphy.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Aravind J., Rinku S., Pooja B., et al. Identification, characterization, and functional validation of drought-responsive microRNAs in subtropical maize inbreds. Frontiers in Plant Science. 2017;8:p. 941. doi: 10.3389/fpls.2017.00941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budak H., Akpinar A. Dehydration stress-responsive miRNA in Brachypodium distachyon: evident by genome-wide screening of microRNAs expression. OMICS: A Journal of Integrative Biology. 2011;15(11):791–799. doi: 10.1089/omi.2011.0073. [DOI] [PubMed] [Google Scholar]
- 20.Sunkar R., Zhou X., Zheng Y., Zhang W., Zhu J. K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biology. 2008;8(1):p. 25. doi: 10.1186/1471-2229-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou L., Liu Y., Liu Z., Kong D., Duan M., Luo L. Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. Journal of Experimental Botany. 2010;61(15):4157–4168. doi: 10.1093/jxb/erq237. [DOI] [PubMed] [Google Scholar]
- 22.Balyan S., Kumar M., Mutum R. D., et al. Identification of miRNA-mediated drought responsive multi-tiered regulatory network in drought tolerant rice, Nagina 22. Scientific Reports. 2017;7(1):p. 15446. doi: 10.1038/s41598-017-15450-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheah B. H., Jadhao S., Vasudevan M., Wickneswari R., Nadarajah K. Identification of functionally important microRNAs from rice inflorescence at heading stage of a qDTY4.1-QTL bearing near isogenic line under drought conditions. PloS One. 2017;12(10, article e0186382) doi: 10.1371/journal.pone.0186382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bakhshi B., Mohseni Fard E., Nikpay N., et al. MicroRNA signatures of drought signaling in rice root. PloS One. 2016;11(6, article e0156814) doi: 10.1371/journal.pone.0156814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giusti L., Mica E., Bertolini E., et al. microRNAs differentially modulated in response to heat and drought stress in durum wheat cultivars with contrasting water use efficiency. Functional & Integrative Genomics. 2017;17(2-3):293–309. doi: 10.1007/s10142-016-0527-7. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L., Xuan H., Zuo Y., et al. Topological characteristics of target genes regulated by abiotic-stress-responsible miRNAs in a rice interactome network. Functional & Integrative Genomics. 2016;16(3):243–251. doi: 10.1007/s10142-016-0481-4. [DOI] [PubMed] [Google Scholar]
- 27.Michel B. E., Kaufmann M. R. The osmotic potential of polyethylene glycol 6000. Plant Physiology. 1973;51(5):914–916. doi: 10.1104/pp.51.5.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pihur V., Datta S., Datta S. Reconstruction of genetic association networks from microarray data: a partial least squares approach. Bioinformatics. 2008;24(4):561–568. doi: 10.1093/bioinformatics/btm640. [DOI] [PubMed] [Google Scholar]
- 29.Li X., Gill R., Cooper N. G. F., Yoo J. K., Datta S. Modeling microRNA-mRNA interactions using PLS regression in human colon cancer. BMC Medical Genomics. 2011;4(1):p. 44. doi: 10.1186/1755-8794-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang D. W., Sherman B. T., Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 31.Budak H., Kantar M., Bulut R., Akpinar B. A. Stress responsive miRNAs and isomiRs in cereals. Plant Science. 2015;235:1–13. doi: 10.1016/j.plantsci.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 32.Zhao B., Liang R., Ge L., et al. Identification of drought-induced microRNAs in rice. Biochemical and Biophysical Research Communications. 2007;354(2):585–590. doi: 10.1016/j.bbrc.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z., Yu J., Li D., et al. PMRD: plant microRNA database. Nucleic Acids Research. 2010;38(Supplement 1):D806–D813. doi: 10.1093/nar/gkp818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai X., Zhao P. X. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Research. 2011;39(Supplement 2):W155–W159. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang Y., Xie K., Xiong L. Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. Journal of Experimental Botany. 2014;65(8):2119–2135. doi: 10.1093/jxb/eru072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang F., Luo X., Zhou Y., Xie J. Genome-wide identification of conserved microRNA and their response to drought stress in Dongxiang wild rice (Oryza rufipogon Griff.) Biotechnology Letters. 2016;38(4):711–721. doi: 10.1007/s10529-015-2012-0. [DOI] [PubMed] [Google Scholar]
- 37.Lozano R., Angosto T., Gomez P., et al. Tomato flower abnormalities induced by low temperatures are associated with changes of expression of MADS-box genes. Plant Physiology. 1998;117(1):91–100. doi: 10.1104/pp.117.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Usami T., Horiguchi G., Yano S., Tsukaya H. The more and smaller cells mutants of Arabidopsis thaliana identify novel roles for squamosa promoter binding protein-like genes in the control of heteroblasty. Development. 2009;136(6):955–964. doi: 10.1242/dev.028613. [DOI] [PubMed] [Google Scholar]
- 39.Sunkar R., Li Y. F., Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends in Plant Science. 2012;17(4):196–203. doi: 10.1016/j.tplants.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 40.Hasson A., Plessis A., Blein T., et al. Evolution and diverse roles of the cup-shaped cotyledon genes in Arabidopsis leaf development. The Plant Cell. 2011;23(1):54–68. doi: 10.1105/tpc.110.081448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiang Y., Huang Y., Xiong L. Characterization of stress-responsive CIPK genes in rice for stress tolerance improvement. Plant Physiology. 2007;144(3):1416–1428. doi: 10.1104/pp.107.101295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S. T., Sun X. L., Hoshino Y., et al. MicroRNA319 positively regulates cold tolerance by targeting OsPCF6 and OsTCP21 in rice (Oryza sativa L.) PloS One. 2014;9(3, article e91357) doi: 10.1371/journal.pone.0091357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morea E. G. O., da Silva E. M., e Silva G. F. F., et al. Functional and evolutionary analyses of the miR156 and miR529 families in land plants. BMC Plant Biology. 2016;16(1):p. 40. doi: 10.1186/s12870-016-0716-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao B., Ge L., Liang R., et al. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Molecular Biology. 2009;10(1):p. 29. doi: 10.1186/1471-2199-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Enright A. J., John B., Gaul U., Tuschl T., Sander C., Marks D. S. MicroRNA targets in Drosophila . Genome Biology. 2003;5(1):p. R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krek A., Grün D., Poy M. N., et al. Combinatorial microRNA target predictions. Nature Genetics. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 47.Pan C., Ye L., Zheng Y., et al. Identification and expression profiling of microRNAs involved in the stigma exsertion under high-temperature stress in tomato. BMC Genomics. 2017;18(1):p. 843. doi: 10.1186/s12864-017-4238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bian H., Xie Y., Guo F., et al. Distinctive expression patterns and roles of the miRNA393/TIR1 homolog module in regulating flag leaf inclination and primary and crown root growth in rice (Oryza sativa) The New Phytologist. 2012;196(1):149–161. doi: 10.1111/j.1469-8137.2012.04248.x. [DOI] [PubMed] [Google Scholar]
- 49.Zhao L. H., Zhou X. E., Yi W., et al. Destabilization of strigolactone receptor DWARF14 by binding of ligand and E3-ligase signaling effector DWARF3. Cell Research. 2015;25(11):1219–1236. doi: 10.1038/cr.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen H., Li Z., Xiong L. A plant microRNA regulates the adaptation of roots to drought stress. FEBS Letters. 2012;586(12):1742–1747. doi: 10.1016/j.febslet.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 51.Dong S., Zhang J., Sun D., et al. Identification of Magnaporthe oryzae-elicited rice novel miRNAs and their targets by miRNA and degradome sequencing. European Journal of Plant Pathology. 2018;151(3):629–647. doi: 10.1007/s10658-017-1399-y. [DOI] [Google Scholar]
- 52.Zhou M., Gu L., Li P., et al. Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica) Frontiers in Biology. 2010;5(1):67–90. doi: 10.1007/s11515-010-0007-8. [DOI] [Google Scholar]
- 53.Li T., Li H., Zhang Y. X., Liu J. Y. Identification and analysis of seven H2O2-responsive miRNAs and 32 new miRNAs in the seedlings of rice (Oryza sativa L. ssp. indica) Nucleic Acids Research. 2011;39(7):2821–2833. doi: 10.1093/nar/gkq1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu J. K. Salt and drought stress signal transduction in plants. Annual Review of Plant Biology. 2002;53(1):247–273. doi: 10.1146/annurev.arplant.53.091401.143329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Padmalatha K. V., Dhandapani G., Kanakachari M., et al. Genome-wide transcriptomic analysis of cotton under drought stress reveal significant down-regulation of genes and pathways involved in fibre elongation and up-regulation of defense responsive genes. Plant Molecular Biology. 2012;78(3):223–246. doi: 10.1007/s11103-011-9857-y. [DOI] [PubMed] [Google Scholar]
- 56.Lenka S. K., Katiyar A., Chinnusamy V., Bansal K. C. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnology Journal. 2011;9(3):315–327. doi: 10.1111/j.1467-7652.2010.00560.x. [DOI] [PubMed] [Google Scholar]
- 57.Prince S. J., Joshi T., Mutava R. N., et al. Comparative analysis of the drought-responsive transcriptome in soybean lines contrasting for canopy wilting. Plant Science. 2015;240:65–78. doi: 10.1016/j.plantsci.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 58.Wu L., Zhang Q., Zhou H., Ni F., Wu X., Qi Y. Rice microRNA effector complexes and targets. The Plant Cell. 2009;21(11):3421–3435. doi: 10.1105/tpc.109.070938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: oligonucleotide primers used in RT-qPCR.
Table S2: the specific primer sequences for RLM-5′ RACE of target genes.
Table S3: differentially expressed miRNAs under drought stress.
Table S4: enriched KEGG pathways of drought-responsive miRNA targets.
Figure S1: ROC curves of 13 drought-responsive miRNAs.
Figure S2: validation of drought-responsive miRNAs and coregulated target genes by RT-qPCR. The expression level is expressed as the mean fold changes of 3 replicates. Error bars depict the standard error (n = 3).
Data Availability Statement
The microarray data used to support the findings of this study have been deposited in the GEO repository (GSE99083).