

Abstract
The photoenzymatic decarboxylation of fatty acids to alkanes is proposed as an alternative approach for the synthesis of biodiesel. By using a recently discovered photodecarboxylase from Chlorella variabilis NC64A (CvFAP) we demonstrate the irreversible preparation of alkanes from fatty acids and triglycerides. Several fatty acids and their triglycerides are converted by CvFAP in near‐quantitative yield and exclusive selectivity upon illumination with blue light. Very promising turnover numbers of up to 8000 were achieved in this proof‐of‐concept study.
Keywords: biocatalysis, biofuels, decarboxylation, fatty acids, photocatalysis
The conversion of (waste) fatty acids and oils into biofuels has been the focus of research for decades.1 The most widely used approach is to transform fatty acids (esters) into the corresponding methyl and ethyl esters (FAMEs and FAEEs, respectively). The hydrolase‐catalyzed transesterification of oils and fats has, in particular, been in focus because of the mild reaction conditions. Equilibrium issues, however, still challenge the practicability of this approach. The decarboxylation of fatty acids into the corresponding C1‐shortened alkanes may be an interesting alternative to the (trans)esterification strategy (Scheme 1).
Scheme 1.
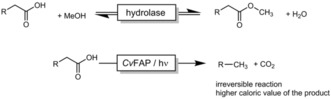
Enzymatic transesterification (top) and decarboxylation (bottom) reactions for the synthesis of biofuels.
On the one hand, the specific heat of combustion of alkanes is somewhat (ca. 9 %) higher than that of the corresponding FAMEs.1 On the other hand, the irreversible decarboxylation of fatty acids should lead to simpler reaction schemes compared to the reversible (trans)esterification procedure, where issues such as the water content (leading to saponification and catalyst inactivation) and equilibrium (generally significant molar surpluses of methanol or ethanol are required to achieve near‐full conversion) arise.
Established chemical methods for the decarboxylation of fatty acids require rather harsh reaction conditions and rare‐metal catalysts, which will challenge the overall eco‐efficiency of the proposed alkane synthesis.2 Recent advancements in photochemical decarboxylation make use of Pd‐doped TiO2 and much milder reaction conditions.3 However, one common issue with all the classical chemical processes for the decarboxylation of fatty acids is their rather poor selectivity, with complex product mixtures being obtained as a result of Kolbe‐ and Hofer–Moest‐type side reactions.
Enzymatic counterparts are so far limited to the oxidative decarboxylation of fatty acids to terminal alkenes4 or to activated carboxylic acids such as malonic acids5 or aromatic carboxylates.6
Very recently Beisson and co‐workers reported on an algal fatty acid photodecarboxylase from Chlorella variabilis NC64A (CvFAP).7 This flavoenzyme catalyzes the light‐dependent decarboxylation of some long‐chain fatty acids to the corresponding C1‐shortened alkanes. CvFAP requires photoactivation by blue light (450 nm), thus indicating that only the photoexcited FAD in the enzyme's active site is catalytically active. Examples of photoenzymes are quite rare: besides CvFAP, only flavin‐dependent DNA‐repair enzymes8 and protochlorophyllide oxidoreductases9 are known. We therefore set out to explore the preparative potential of CvFAP for the production of alkanes from biobased fatty acids and triglycerides.
Two variants of CvFAP were recombinantly produced in Escherichia coli: one variant comprises the complete sequence of CvFAP (residues 1–654, full‐length, Figure S1 in the Supporting Information), while the second variant lacks a predicted chloroplast‐targeting sequence and comprises residues 62–654 of CvFAP (short‐length, Figure S1). After production in Escherichia coli, the crude extract as well as the purified enzyme were used for catalytic experiments. While the short‐length CvFAP showed a good overproduction in E. coli and was purified in one step to a purity of about 40 %, full‐length CvFAP showed almost no overproduction in E. coli and there was no clear band by SDS‐PAGE after purification (Figure S2). Although both enzyme preparations showed significant decarboxylation activity (Figures S5 and S6), we continued our investigations using the short‐length CvFAP.
Interestingly, the CvFAP exhibited a higher activity and robustness in crude enzyme preparations than purified preparations. The activity of the purified enzyme was doubled when reconstituted with filtered E. coli cell extract. Pre‐incubating the enzyme under blue light causes a loss of activity, which is less pronounced when pre‐incubating the enzyme in filtered E. coli cell extract (Table S1). We currently have no satisfying explanation for the stabilization effect and further experiments will be necessary to shed more light on this. Crude cell‐free extracts were used in all further experiments.
It is worth mentioning that no background activity was found in the crude extracts prepared from E. coli cells containing an empty vector. Furthermore, performing experiments either in the absence of blue light or in the absence of CvFAP (or using thermally inactivated cell extract) gave no determinable conversion of, for example, palmitic acid. Performing the photoenzymatic reaction under different atmospheric conditions (air, Ar, N2, or H2) gave essentially the same result (i.e. full conversion of palmitic acid within 3 h, Table S2). DMSO was applied as a cosolvent to increase the solubility of the hydrophobic fatty acids, such as palmitic acid. CvFAP tolerates up to 50 vol % DMSO (Figure S7). In further studies, the reaction mixture contained 30 vol % DMSO.
Under these conditions, we were pleased to observe a smooth conversion of palmitic acid into pentadecane in the presence of CvFAP and blue light (Figure 1). The reaction was strictly light‐dependent. Further experiments showed that the rate of pentadecane production depended on the enzyme concentration, the intensity of the light source and, to some minor extent, the reaction temperature (Figures S5, S8, and S9).
Figure 1.

Photoenzymatic decarboxylation of palmitic acid into pentadecane. Conditions: [CvFAP]=6.0 μm, [palmitic acid]=13 mm, Tris‐HCl (pH 8.5, 100 mm), 30 % DMSO, illumination with blue light.
Next, we explored the scope of the photoenzymatic decarboxylation reaction further (Table 1). A broad range of different fatty acids was converted, with full conversion being observed for some of them and with quite favorable turnover numbers (TON, that is, concentration of product divided by concentration of the biocatalyst) for the enzyme.
Table 1.
Substrate scope of the photoenzymatic decarboxylation reaction.[a]

| Substrate | [Product] [mm] | Conversion [%][b] | TON (CvFAP)[c] |
|---|---|---|---|
| C12H24O2
(lauric acid) |
3.0 | 11 | 500 |
| C14H28O2
(myristic acid) |
6.9 | 25 | 1150 |
| C16H32O2
(palmitic acid) |
27.7 | 96 | 4610 |
| C17H34O2
(margaric acid) |
28.7 | 96 | 4780 |
| C18H36O2
(stearic acid) |
26.1 | 92 | 4350 |
| C18H34O2 (Δ9) (oleic acid) |
17.7 | 65[d] | 2950 |
| C18H32O2 (Δ9, 12) (linoleic acid) |
14.6[c] | 49[d] | 2600 |
| C20H40O2
(arachidic acid) |
25.7 | 90 | 4580 |
[a] Reaction conditions: [substrate]=30 mm, [CvFAP]=6.0 μm, Tris‐HCl buffer (pH 8.5, 100 mm), 30 % DMSO, illumination with blue light (intensity=13.7 μE L−1 s−1) for 14 h. [b] Conversion: [product]final×([product]final+[substrate]final)−1. [c] TON: [product]final×[CvFAP]−1. [d] Due to the lack of a standard reference, the conversion was calculated by using: conversion=([substrate]initial− [substrate]final)×[substrate]initial −1.
In accordance with previous observations,7a CvFAP showed the highest activity with long‐chain fatty acids (C>14). Interestingly, the conversions of oleic acid and linoleic acid were significantly lower than of the fully saturated counterpart (stearic acid). We therefore investigated the relative activity of CvFAP towards some oleic acid isomers (cis/trans and regioisomers, Table S3) and docked these isomers into the crystal structure of CvFAP (PDB: 5NCC; Figures S14–17).7a A good correlation was found between the initial decarboxylation rate and the distance of the substrate carboxylate group from the flavin cofactor (Table S3). Hence, the differences in the conversion and rate observed for the different substrates may be assigned to differences in the substrate binding in the CvFAP access channel and positioning of the reactive group towards the cofactor.
A preparative‐scale synthesis was performed under the optimal conditions (see the Supporting Information), with 155 mg of pentadecane being obtained. This corresponded to a conversion of 79 % (TON of CvFAP 7916) and a 61 % yield of the isolated product (Figures S11 and S12).
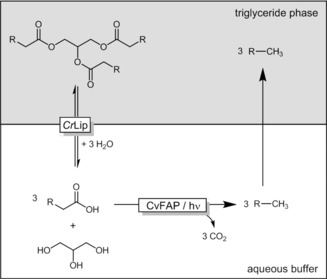
In view of the envisaged production of alkanes from (waste) oils and fats we also investigated a bienzymatic cascade comprising the lipase‐catalyzed hydrolysis of triolein to the free oleic acid and glycerol followed by the CvFAP‐catalyzed photodecarboxylation reaction (Table 2).
Table 2.
Photo‐biocatalytic cascade for the transformation of triolein into (Z)‐heptadec‐8‐ene.[a]
| Entry | Conditions | [CrLip] [U mL−1] |
[Product] [mm] | TON (CvFAP) |
|---|---|---|---|---|
| 1 | two‐step, pH 7.5 | 890 | 16.4 | 1366 |
| 2 | two‐step, pH 7.5 | 89 | 27.7 | 2308 |
| 3 | two‐step, adjustment of pH after step 1 | 890 | 34.5 | 2875 |
| 4 | one‐step, pH 7.5 | 890 | 19.8 | 1650 |
| 5 | homogeneous, two‐step, pH7.5 | 2500 | 49.7 (83 % conversion) | 8280 |
[a] General conditions for the two‐step procedure: triolein/Tris‐HCl buffer (pH 7.5, 100 mm) phase ratio=1:1 (v/v); CrLip (lipase from Candida rugosa); T=37 °C; step 1: reaction time=12 h; followed by step 2: [CvFAP]=6 μm; irradiation with blue light (450 nm; intensity = 13.7 μE L−1 s−1); reaction time=20 h. TON: [product]final×[CvFAP]−1.
The two‐step cascade using homogeneously dissolved triolein (20 mm) gave a satisfying overall yield of more than 80 % (Table 2, entry 5) and a respectable turnover number for the photodecarboxylase of 8280, which encouraged us to proceed to a cosolvent‐free system with two liquid phases. A first experiment using 890 U of lipase gave a relatively low product yield of 16 mm, which was attributed to a drop in the pH value of the aqueous phase to 5.2 and the decreased CvFAP activity (Table 2, entry 1). Indeed, either lowering the lipase activity or adjusting the pH value after the hydrolysis reaction led to product formation increasing significantly (Table 2, entries 2 and 3). Performing both steps simultaneously (one‐step; Table 2, entry 4) was shown to be practical, although the product yield and consequently the TON of the CvFAP were rather modest. Again, a drop in the pH value of the aqueous phase was observed. Optimization of the relative activities of the lipase and photodecarboxylase will circumvent this limitation and reveal the full potential of the proposed photobiocatalytic synthesis of alkanes from triglycerides.
Overall, we have demonstrated the synthetic potential of the novel photodecarboxylase from Chlorella variabilis NC64A. We are convinced that further optimizations of the reaction setup and of the biocatalyst(s) will yield a practical approach to valorize non‐edible triglycerides and acids into biofuels.
Experimental Section
Cloning of Cv FAP
For the production of the CvFAP in E. coli, two constructs were designed based on a previously reported construct.7a The constructs both consist of sequentially a 6× His‐tag, thioredoxin (TrxA) tag, tobacco etch virus (TEV) protease cleavage site, and the gene coding for CvFAP (GenBank: KY511411.1). The first construct comprises the full‐length sequence (residues 1–654) of CvFAP, while the second construct lacks the residues encoding a predicted chloroplast‐targeting sequence and thereby comprises residues 62–654 of CvFAP (Figure S1). The sequence coding for CvFAP was codon‐optimized for expression in E. coli. The construct was synthesized by Baseclear (Leiden, the Netherlands) and cloned into a pET28a vector using NdeI and HindIII restriction sites. Competent E. coli BL21 (DE3) cells (NEB) were transformed with the plasmid for recombinant enzyme production.
Preparation of the cell‐free extract containing Cv FAP (full‐length/short‐length)
10 mL precultures (terrific broth (TB) medium, containing 50 μg mL−1 kanamycin) were inoculated with E. coli BL21 (DE3) cells harboring the designed pET28a‐His‐TrxA‐CvFAP plasmid and grown overnight. The precultures were used to inoculate large cultures (500 mL TB + 50 μg mL−1 kanamycin in 2 L shake flasks). Cells were grown at 37 °C and 180 rpm until an optical density (OD600) between 0.7–0.8 was reached. Protein production was induced by the addition of 0.5 mm IPTG and the cells were left at 17 °C and 180 rpm for about 20 h. Cells were harvested by centrifugation (11 000 g at 4 °C for 10 min), washed with Tris‐HCl buffer (50 mm, pH 8, containing 100 mm NaCl), and centrifuged again. The cell pellet was resuspended in the same buffer, and 1 mm PMSF added. Cells were lysed by passing them twice through a Multi Shot Cell Disruption System (Constant Systems Ltd, Daventry, UK) at 1.5 kbar, followed by centrifugation of the cell lysate (38 000 g at 4 °C for 1 h). After centrifugation, 5 % glycerol (w/v) was added to the soluble fraction, the cell extract was separated into aliquots, frozen in liquid nitrogen, and stored at −80 °C.
The total protein content of the cell extract was determined by a BCA Assay (Interchim), using BSA as a standard. The production of CvFAP was analyzed by SDS‐PAGE (Figure S2) using a Criterion Cell electrophoresis system (Bio‐Rad). Precision Plus Protein Standard (Bio‐Rad) was used as a molecular‐weight marker. The gel was analyzed using a gel imaging system (GBox, Syngene, Cambridge, UK) and the amount of CvFAP in the cell extract was estimated from the relative intensity of the bands on the gel.
As a control, a cell‐free extract of E. coli BL21 (DE3) cells harboring an empty pET28a vector was prepared according to the same procedure.
Photocatalytic reactions
The photoenzymatic decarboxylation reactions using CvFAP were performed at 37 °C in a total volume of 1.0 mL Tris‐HCl buffer (pH 8.5, 100 mm) containing 30 vol % DMSO as cosolvent. Unless mentioned otherwise, 200 μL DMSO containing 65.5 mm palmitic acid, 100 μL pure DMSO, 500 μL Tris‐HCl buffer (pH 8.5, 100 mm), and 200 μL CvFAP stock solution (30 μm cell extract in Tris‐HCl buffer) were added to a transparent glass vial (total volume 5.0 mL). The vial was sealed and exposed to blue LED light under gentle magnetic stirring. The homemade setup is shown in Figure S3. The final conditions of this reaction were: [palmitic acid]=13.1 mm and [CvFAP]=6 μm in Tris‐HCl pH (8.5, 100 mm) and 30 vol % DMSO. At intervals, aliquots were withdrawn and the substrates and products were extracted with twice the volume of ethyl acetate (containing 5 mm 1‐octanol as an internal reference). The remaining organic phase was analyzed by gas chromatography.
Enzymatic cascade reactions transforming triglycerides into alkanes
Two‐step approach: A certain amount of lipase from Candida rugose in 500 μL Tris‐HCl buffer (pH 8.5, 100 mm) and 500 μL triolein as an organic phase were added to a transparent glass vial (total volume 5.0 mL). The hydrolysis of triolein was then performed at 37 °C in a thermal shaker (700 rpm). After 12 h, 200 μL CvFAP stock solution (30 μm cell extract in Tris‐HCl buffer) was added and the mixture was exposed to blue LED light under gentle magnetic stirring at 37 °C. After 20 h, 10 μL of the organic phase was withdrawn and treated with 30 μL NaOH (12 m) at 70 °C for 1.5 h. Subsequently, the mixture was extracted with ethyl acetate (containing 5 mm 1‐octanol as an internal reference) and analyzed by gas chromatography.
One‐step approach: The same reaction conditions as described in the two‐step approach were used, except that after the addition of all the reaction components into the reaction vial, the mixture was illuminated by blue LED light under gentle magnetic stirring at 37 °C.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Netherlands Organisation for Scientific Research for financial support through a VICI grant (no. 724.014.003). We thank Dr. Jonathan Z. Bloh for the characterization of our homemade light setup.
M. M. E. Huijbers, W. Zhang, F. Tonin, F. Hollmann, Angew. Chem. Int. Ed. 2018, 57, 13648.
References
- 1. Aransiola E. F., Ojumu T. V., Oyekola O. O., Madzimbamuto T. F., Ikhu-Omoregbe D. I. O., Biomass Bioenergy 2014, 61, 276–297. [Google Scholar]
- 2.
- 2a. Witsuthammakul A., Sooknoi T., Catal. Sci. Technol. 2016, 6, 1737–1745; [Google Scholar]
- 2b. Sun K., Schulz T. C., Thompson S. T., Lamb H. H., Catal. Today 2016, 269, 93–102; [Google Scholar]
- 2c. Ford J. P., Thapaliya N., Kelly M. J., Roberts W. L., Lamb H. H., Energy Fuels 2013, 27, 7489–7496; [Google Scholar]
- 2d. Ford J. P., Immer J. G., Lamb H. H., Top. Catal. 2012, 55, 175–184. [Google Scholar]
- 3.
- 3a. Ngo S., Betts L. M., Dappozze F., Ponczek M., George C., Guillard C., J. Photochem. Photobiol. A 2017, 339, 80–88; [Google Scholar]
- 3b. Hamid S., Ivanova I., Jeon T. H., Dillert R., Choi W., Bahnemann D. W., J. Catal. 2017, 349, 128–135. [Google Scholar]
- 4.
- 4a. Grant J. L., Hsieh C. H., Makris T. M., J. Am. Chem. Soc. 2015, 137, 4940–4943; [DOI] [PubMed] [Google Scholar]
- 4b. Xu H. F., Ning L. L., Yang W. X., Fang B., Wang C., Wang Y., Xu J., Collin S., Laeuffer F., Fourage L., Li S. Y., Biotechnol. Biofuels 2017, 10, 15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Dennig A., Kurakin S., Kuhn M., Dordic A., Hall M., Faber K., Eur. J. Org. Chem. 2016, 3473–3477; [Google Scholar]
- 4d. Dennig A., Kuhn M., Tassoti S., Thiessenhusen A., Gilch S., Bülter T., Haas T., Hall M., Faber K., Angew. Chem. Int. Ed. 2015, 54, 8819–8822; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8943–8946; [Google Scholar]
- 4e. Bojarra S., Reichert D., Grote M., Baraibar Á. G., Dennig A., Nidetzky B., Mügge C., Kourist R., ChemCatChem 2018, 10, 1192–1201; [Google Scholar]
- 4f. Zachos I., Gassmeyer S., Bauer D., Sieber V., Hollmann F., Kourist R., Chem. Commun. 2015, 51, 1918–1921. [DOI] [PubMed] [Google Scholar]
- 5. Gómez Baraibar A., Reichert D., Mugge C., Seger S., Groger H., Kourist R., Angew. Chem. Int. Ed. 2016, 55, 14823–14827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15043–15047. [Google Scholar]
- 6. Wuensch C., Pavkov-Keller T., Steinkellner G., Gross J., Fuchs M., Hromic A., Lyskowski A., Fauland K., Gruber K., Glueck S. M., Faber K., Adv. Synth. Catal. 2015, 357, 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Sorigué D., Légeret B., Cuiné S., Blangy S., Moulin S., Billon E., Richaud P., Brugière S., Couté Y., Nurizzo D., Müller P., Brettel K., Pignol D., Arnoux P., Li-Beisson Y., Peltier G., Beisson F., Science 2017, 357, 903–907; [DOI] [PubMed] [Google Scholar]
- 7b. Sorigue D., Legeret B., Cuine S., Morales P., Mirabella B., Guedeney G., Li-Beisson Y., Jetter R., Peltier G., Beisson F., Plant Physiol. 2016, 171, 2393–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang M., Wang L., Shu S., Sancar A., Zhong D., Science 2016, 354, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heyes D. J., Hardman S. J. O., Hedison T. M., Hoeven R., Greetham G. M., Towrie M., Scrutton N. S., Angew. Chem. Int. Ed. 2015, 54, 1512–1515; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1532–1535. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary