

Abstract
We report the synthesis and analytical application of the first Cu2+‐selective synthetic ion channel based on peptide‐modified gold nanopores. A Cu2+‐binding peptide motif (Gly‐Gly‐His) along with two additional functional thiol derivatives inferring cation‐permselectivity and hydrophobicity was self‐assembled on the surface of gold nanoporous membranes comprising of about 5 nm diameter pores. These membranes were used to construct ion‐selective electrodes (ISEs) with extraordinary Cu2+ selectivities, approaching six orders of magnitude over certain ions. Since all constituents are immobilized to a supporting nanoporous membrane, their leaching, that is a ubiquitous problem of conventional ionophore‐based ISEs was effectively suppressed.
Keywords: copper-binding peptides, ion channels, ion-selective nanopores, self-assembled monolayers, sensors
Ion‐selective electrodes (ISEs) are one of the most established types of chemical sensors, commercialized for a wide variety of applications ranging from their use as indicator electrodes in titrations to the direct determination of ions in clinical analysis.1 This latter application has been largely aided by the implementation of ionophore‐based ISEs.2 Such electrodes are based on hydrophobic ion‐selective membranes (ISMs) most often comprising a plasticized polymer that forms an organic membrane phase immiscible with the aqueous sample. This membrane incorporates an ionophore and a lipophilic ion‐exchanger that confer ion selectivity and permselectivity, respectively, to the ISMs. The ion‐exchange equilibrium at the organic|aqueous phase boundaries generates a membrane potential that enables ultimately the potentiometric determination of the sample ion activity. All active constituents are dissolved in the hydrophobic membrane, and with a few exceptions,3 withhold solely by their lipophilicity. Therefore, using highly lipophilic constituents is essential to minimize their gradual leaching from the ISMs into the aqueous samples.4 The first ionophores matching this requirement were natural ionophores, such as valinomycin,5 the biological role of which is to facilitate ion transfer across cell membranes. The range of assessable ions were spectacularly enlarged by the implementation of synthetic ionophores with custom designed properties.2 By now the progress in ionophore development seems to have largely reached its limits while ISMs still suffers of leaching components and lack of selective lipophilic ionophores to certain ions. Therefore, here we explored the feasibility of using hydrophilic natural ionophores that could not be used previously in ISEs due to their incompatibility with the hydrophobic membrane matrices. Since the number of such ligands is very large6 this first study may open the way to enlarge the range of assessable ions by ISEs. To implement hydrophilic ligands a radically different ISM construction is needed. Inspired by biological ion channels that naturally integrate hydrophilic ligands we opted to mimic their selectivity filter by chemically modifying solid‐state nanopores.7 However, while cation and anion permselectivity can be achieved straightforwardly by inferring solid‐state nanopores with negative or positive surface charge, respectively,8 reaching single ion selectivity proved to be very challenging. A close mimicry of biological ion‐selective channels, for example, potassium ion channels,9 would imply to precisely distribute different functional groups to form selectivity filters in solid‐state nanopores. This approach is largely restricted to theoretical studies10 due to difficulties in the experimental realization, but even so the selectivities predicted are rather modest. From a fabrication perspective, the use of nanopores modified with functional molecules is clearly a less demanding approach. However, the reported solid‐state ion‐selective nanopores11 are in fact based on an ingeniously designed gating effect, that is, the ion to be determined modulates an ion current across the nanopores, rather than being selectively transported.12 We found earlier that ion selectivities exceeding that of biological ion channels can be obtained upon modifying nanopores with the same functional molecules as used in conventional ISMs.7 Thus, gold nanopores were modified with a mixed self‐assembled monolayer of a lipophilic silver ionophore, a cation exchanger and a lipophilic perfluorocarbon compound, all of them synthesized to have terminal thiol (or disulfide) groups for attachment. The role of the latter two components was to infer permselectivity and to restrict water from the pores, respectively. Upon reducing the nanopore diameters to have their surface modification controlling the ion transport properties, the respective ISMs exhibited excellent Ag+ selectivities.7 However, while this approach can integrate in principle functional components with widely different polarities still conventional lipophilic ionophores were used. Here we set to explore whether the concept can be extended to hydrophilic ionophores. For proof of principle we selected a hydrophilic Cu2+ complexing tripeptide (Gly‐Gly‐His) that was made amenable for attachment to gold nanopores by modifying it with a terminal cysteine and a glycine spacer (Scheme 1 B). This peptide was reported to fold into a stable tetradentate complex around the Cu2+ with a binding constant of (8.1±0.4)×1010 m −1.13
Scheme 1.
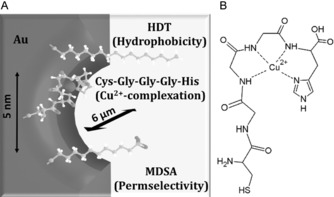
A) Schematic of the gold nanopore modification by self‐assembling a ternary mixture of thiol bearing functional compounds, i.e., a hydrophilic peptide ionophore, mercaptodecanesulfonate (MDSA) and hexadecanethiol (HDT), in gold nanopores of ca. 5 nm effective diameter and 6 μm length. B) The peptide ionophore—Cu2+ complex.
Gold nanoporous membranes were prepared by electroless plating of gold within the pores (Ø 30 nm) of track‐etched polycarbonate membranes.14 The gold plating was performed until the diameter of the nanopores was reduced to ca. 5 nm (360 min) as determined by N2 permeation experiments (Figure S1 in the Supporting Information). We found that such small diameter nanopores, in the range of the Debye length, owing to the surface adsorbed anions exhibit cation permselectivity even without functionalization that is, however, further improved by modification with mercaptodecanesulfonate (MDSA).8b For Cu2+ selectivity the chemical environment within the nanopore had to be adjusted, that is, the relative amounts of the three functional thiols in the self‐assembled layer formed within the nanopore had to be to finely tuned.
This was achieved by using a ternary mixture formulated in ethanol of the functional thiol derivatives at a fixed total concentration of 0.1 mm, reacted overnight with the gold nanoporous membrane (see experimental details in the Supporting Information). However, the composition of self‐assembled monolayers prepared with thiols of such widely different properties does not necessarily reflect the ratios established in the modification solutions. Contact angle measurements confirm the formation of a mixed self‐assembled layer with the dominance of linear chain thiols (e.g. perfluorocarbon thiols, Figure S2) over charged or branched thiol components in determining the surface polarity. Therefore, the inner pore modification proved to be a major challenge, especially due to the replacement of a lipophilic ionophore, as used in our earlier study, with a hydrophilic peptide. Using perfluorocarbon thiols (found optimal in our previous study)7 the resulted membranes even after lengthily optimizations preserved their Cu2+ selective response at most a few days. Since the surface rearrangement and gradual segregation of thiols having such widely different polarity featured as the most likely explanation for the loss of the ion‐selective response we changed from perfluorocarbon thiols to alkanethiols. To this end the nanopores were systematically modified with ternary mixtures of alkanethiols having different hydrocarbon chain lengths (C3, C8, C12 and C16), Cu2+‐selective peptide and MDSA in different ratios. Plotting the best potentiometric selectivity coefficients achieved with each alkanethiol against Mg2+( ) revealed a remarkable selectivity improvement upon increasing the chain length (Figure 1, inset). Selecting for further experiments hexadecanethiol and modifying the nanopores with various composition ternary mixtures revealed that the proper hydrophobic environment within the nanopore is critical for ion‐selectivity. Figure 1 shows the selectivity coefficients obtained with different ternary mixtures as a function of the membrane resistance determined by electrochemical impedance spectroscopy (EIS). Given the similarity of the nanopore diameters the resistance increase can be attributed mainly to the increased hydrophobicity of the nanopores. The optimal Cu2+ ‐selectivity was obtained for a modifying solution with 6:3:1 molar ratio of Cys‐Gly‐Gly‐Gly‐His peptide, HDT and MDSA. A water contact angle of 98° (±4°) was found for this layer (Figure S3) that is much higher than either solely the peptide (25±2°) or MDSA covered gold surfaces (23±1°). This indicates the formation of a hydrophobic layer and confirms again the dominance of linear chain alkanethiols in establishing the surface polarity.
Figure 1.
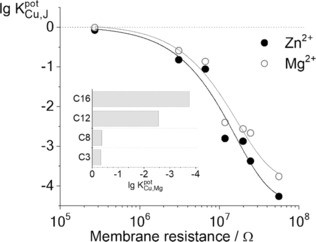
Logarithmic selectivity coefficients of gold nanoporous membrane‐based Cu2+‐selective electrodes for Zn2+ and Mg2+ as a function of the resistance of the corresponding membranes determined by EIS in a symmetrical cell setup with the peptide‐modified nanoporous membrane separating two half cells filled with 10 mm KCl. The nanoporous membrane was modified by various composition ternary thiol solutions using HDT to provide hydrophobicity. The inset shows the best selectivities over Mg2+ obtained with alkanethiols of various chain lengths (C3, C8, C12, C16).
For ion selectivity, the ligand needs to be in molar excess with respect of the cation exchanger. The obtained optimal composition is in line with this expectation, but the exact composition could not be foreseen. It is likely that, while the concept is general, its extrapolation to other hydrophilic ligands may need empiric optimization. However, the finding that the hydrophibicity of the nanopores in case of hydrophilic ligands is essential for proper potentiometric ion‐selectivity is expected to alleviate this process.
The potentiometric Cu2+ calibration curve of ISEs made by integrating nanoporous peptide‐modified membrane disks of 6 mm diameter into conventional electrode bodies1c, 7 is shown in Figure 2.
Figure 2.
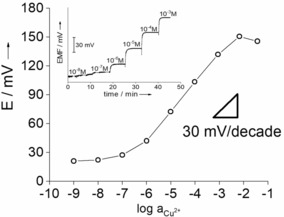
Typical potentiometric response of the optimized peptide‐modified gold nanoporous Cu2+‐selective electrodes (CGGGH peptide, HDT, MDSA 6:3:1 molar ratio). Inset: the corresponding time‐dependent potential traces.
Close to Nernstian behavior for a divalent ion (≈30 mV/decade) in the 10−3–10−6 m range and sub‐micromolar limit of detection were found along with a fast, drift‐free response (Figure 2, inset). The deviation from linearity at higher concentrations is due to partial loss of the cation permselectivity at high ionic strengths.8b
With a few exceptions the selectivity of the peptide‐based ISEs proved to be better than that of state of the art Cu2+ ‐ionophores in conventional plasticized PVC membranes (Table 1). Especially in case of alkali metal ions, which are ubiquitous in any biological or environmental sample, the selectivities approaching six orders of magnitude are spectacularly better for the peptide‐modified nanoporous membranes. To confirm that the Cu2+‐selective response is due to the hydrophilic peptide ionophore gold nanoporous membranes modified with an ethanolic solution of MDSA and HDT (in the same ratio as in the optimized composition), but without the peptide were also prepared. Practically no Cu2+ selectivity was observed for ISEs prepared with such membranes that confirms that the ion‐selectivity is generated by the Cu2+ complexing peptide.
Table 1.
Logarithmic selectivity coefficients of gold nanoporous membrane based Cu2+‐selective electrodes.
| Ion [J] |
|
|||
|---|---|---|---|---|
| Cu2+‐selective nanopore | peptide‐free nanopore | conventional ionophore‐based ISEs | ||
| Na+ | −5.56 | −0.71 | −3.7,[b] −4.61[d] | |
| K+ | −5.71 | −0.30 | −2.4,[a] −3.7,[b] −4.74[d] | |
| Li+ | −5.70 | −0.84 | ||
| Cs+ | −5.76 | 0.16 | ||
| H+ | −2.07 | −0.08 | −0.44[d] | |
| Ca2+ | −3.63 | −0.27 | −4.3,[b] −3.3,[c] −4.64[d] | |
| Mg2+ | −3.76 | 0.11 | −4.0,[b] −3.3,[c] −6.26[d] | |
| Zn2+ | −4.26 | 0.04 | −2.3,[a] −3.9,[b] −3.0,[c] −2.67[d] | |
| Cd2+ | −3.19 | 0.07 | −4.4,[a] −4.4,[b] −2.7,[c] −3.34[d] | |
| Ni2+ | −2.97 | −0.10 | −3.3,[a] −3.9,[b] −3.0[c] | |
| Pb2+ | −3.08 | 0.02 | −0.8,[a] −1.8,[b] −2.7,[c] −1.82[d] | |
| Cr3+ | −4.37 | −0.50 | −3.0[c] | |
Since all active components are immobilized to the nanoporous support, the peptide ligand‐based ion‐selective nanopores should be beneficial also in terms of suppressing the gradual loss of active components to aqueous samples. To accelerate the leaching process, the Cu2+‐selective nanopores‐based ISEs as well conventional plasticized PVC membranes based on o‐xylylene bis(N,N‐diisobutyldithiocarbamate) as Cu2+‐ionophore were stored in EtOH. For both types of electrodes, the original calibration curves were compared with those measured after storage in ETOH. Figure 3 shows that after 48 h in EtOH the calibration curves of the peptide‐modified nanopore‐based Cu2+‐selective electrodes are practically unchanged. However, the conventional PVC based membranes suffered a complete loss of their Cu2+‐selective response already after 8 h of storage in EtOH.
Figure 3.
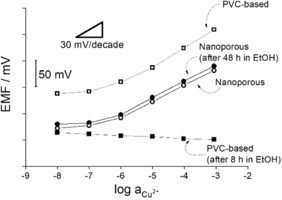
Calibration curves of conventional and nanoporous membrane‐based Cu2+‐selective electrodes, before and after their storage in ethanol. Conventional plasticised PVC membranes were based on o‐xylylene bis(N,N‐diisobutyldithiocarbamate) ionophore.
In conclusion, in this report we show that the concept of using a nanoporous membrane as solid support with all active membrane components immobilized on its surface enables the use of hydrophilic ligands such as metal binding peptides to construct ISEs. The sophisticated modification of gold nanopores by self‐assembling a ternary mixture of functional thiols proved to result in superior potentiometric ion‐selectivity, the prerequisite of which is the establishment of a properly hydrophobic environment within the nanopores. Thus the use of hydrophilic ionophores, but integrated in a hydrophobic environment, which is in contrast to the general trend of designing “aqueous” ion‐channel mimetic ion‐selective nanopores seems to offer clear benefits in terms of selectivity. The concept seems widely applicable except of ligands comprising thiols or disulfides in their binding sites (in case of using gold nanopores), that however can be addressed by using other nanoporous materials and surface chemistries. The significance of these findings may point well beyond the chemical sensing applications given the potential use of ion‐selective nanopores for desalination19 and energy harvesting devices.20
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Lendület program of the Hungarian Academy of Sciences (LP2013‐63) and ERA‐Chemistry (OTKA NN117637).
S. Papp, G. Jágerszki, R. E. Gyurcsányi, Angew. Chem. Int. Ed. 2018, 57, 4752.
References
- 1.
- 1a. Bakker E., Bühlmann P., Pretsch E., Chem. Rev. 1997, 97, 3083–3132; [DOI] [PubMed] [Google Scholar]
- 1b. Gyurcsányi R. E., Pretsch E. in Nanoelectrochemistry (Eds.: M. V. Mirkin, S. Amemiya), CRC Press Taylor & Francis Group, Boca Raton, 2015, pp. xiii, 849; [Google Scholar]
- 1c. Lindner E., Gyurcsányi R. E., Pretsch E., in Applications of supramolecular chemistry for 21st century technology (Ed.: H.-J. Schneider), Taylor & Francis, Boca Raton, 2012, pp. xi, 441. [Google Scholar]
- 2. Bühlmann P., Pretsch E., Bakker E., Chem. Rev. 1998, 98, 1593–1687. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Daunert S., Bachas L. G., Anal. Chem. 1990, 62, 1428–1431; [DOI] [PubMed] [Google Scholar]
- 3b. Reinhoudt D. N., Engbersen J. F. J., Brzozka Z., van der Vlekkert H. H., Honig G. W. N., Holterman H. A. J., Verkerk U. H., Anal. Chem. 1994, 66, 3618–3623; [Google Scholar]
- 3c. Püntener M., Vigassy T., Baier E., Ceresa A., Pretsch E., Anal. Chim. Acta 2004, 503, 187–194; [Google Scholar]
- 3d. Bereczki R., Gyurcsányi R. E., Agai B., Toth K., Analyst 2005, 130, 63–70; [DOI] [PubMed] [Google Scholar]
- 3e. Qin Y., Bakker E., Anal. Chem. 2003, 75, 6002–6010. [DOI] [PubMed] [Google Scholar]
- 4. Dinten O., Spichiger U. E., Chaniotakis N., Gehrig P., Rusterholz B., Morf W. E., Simon W., Anal. Chem. 1991, 63, 596–603. [DOI] [PubMed] [Google Scholar]
- 5. Pioda L. A. R., Stankova V., Simon W., Anal. Lett. 1969, 2, 665–674. [Google Scholar]
- 6. Sigel H., Martin R. B., Chem. Rev. 1982, 82, 385–426. [Google Scholar]
- 7. Jágerszki G., Takacs A., Bitter I., Gyurcsányi R. E., Angew. Chem. Int. Ed. 2011, 50, 1656–1659; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1694–1697. [Google Scholar]
- 8.
- 8a. Nishizawa M., Menon V. P., Martin C. R., Science 1995, 268, 700–702; [DOI] [PubMed] [Google Scholar]
- 8b. Makra I., Jágerszki G., Bitter I., Gyurcsányi R. E., Electrochim. Acta 2012, 73, 70–77. [Google Scholar]
- 9. Doyle D. A., Cabral J. M., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., Chait B. T., MacKinnon R., Science 1998, 280, 69–77. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Gong X., Li J., Xu K., Wang J., Yang H., J. Am. Chem. Soc. 2010, 132, 1873–1877; [DOI] [PubMed] [Google Scholar]
- 10b. Sint K., Wang B., Král P., J. Am. Chem. Soc. 2008, 130, 16448–16449. [DOI] [PubMed] [Google Scholar]
- 11. Lepoitevin M., Ma T., Bechelany M., Janot J.-M., Balme S., Adv. Colloid Interface Sci. 2017, 250, 195–213. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Siwy Z. S., Powell M. R., Petrov A., Kalman E., Trautmann C., Eisenberg R. S., Nano Lett. 2006, 6, 1729–1734; [DOI] [PubMed] [Google Scholar]
- 12b. Tian Y., Hou X., Wen L., Guo W., Song Y., Sun H., Wang Y., Jiang L., Zhu D., Chem. Commun. 2010, 46, 1682–1684; [DOI] [PubMed] [Google Scholar]
- 12c. Ali M., Nasir S., Ramirez P., Cervera J., Mafe S., Ensinger W., ACS Nano 2012, 6, 9247–9257. [DOI] [PubMed] [Google Scholar]
- 13. Yang W., Chow E., Willett G. D., Hibbert D. B., Gooding J. J., Analyst 2003, 128, 712–718. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Jirage K. B., Hulteen J. C., Martin C. R., Anal. Chem. 1999, 71, 4913–4918; [DOI] [PubMed] [Google Scholar]
- 14b. Jágerszki G., Gyurcsányi R. E., Höfler L., Pretsch E., Nano Lett. 2007, 7, 1609–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kamata S., Murata H., Kubo Y., Bhale A., Analyst 1989, 114, 1029–1031. [Google Scholar]
- 16. Kamata S., Bhale A., Fukunaga Y., Murata H., Anal. Chem. 1988, 60, 2464–2467. [Google Scholar]
- 17. Ren K., Talanta 1989, 36, 767–771. [DOI] [PubMed] [Google Scholar]
- 18. Szigeti Z., Bitter I., Toth K., Latkoczy C., Fliegel D. J., Gunther D., Pretsch E., Anal. Chim. Acta 2005, 532, 129–136. [Google Scholar]
- 19. Surwade S. P., Smirnov S. N., Vlassiouk I. V., Unocic R. R., Veith G. M., Dai S., Mahurin S. M., Nat. Nanotechnol. 2015, 10, 459. [DOI] [PubMed] [Google Scholar]
- 20. Guo W., Cao L., Xia J., Nie F.-Q., Ma W., Xue J., Song Y., Zhu D., Wang Y., Jiang L., Adv. Funct. Mater. 2010, 20, 1339–1344. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary