

Abstract
Background
Celastrol is an active ingredient of Chinese medicine Tripterygium wilfordii which is clinically used to treat the immune diseases. Currently, celastrol is documented as a potent agent for treating cancer and inflammatory disorders. This study was to investigate the effect of celastrol on cisplatin nephrotoxicity and the underlying mechanism.
Methods
Male C57BL/6 mice were treated with cisplatin (20 mg/kg) with or without celastrol treatment (1 and 2 mg/kg/day). In vitro, human proximal tubule epithelial cell line (HK−2) and mouse renal tubule epithelial cells (RTECs) were treated with cisplatin (5 μg/mL) with or without celastrol administration. Then renal injury and cell damage were evaluated.
Findings
In vivo, after celastrol treatment, cisplatin-induced kidney injury was significantly ameliorated as shown by the improvement of renal function (BUN, serum creatinine, and cystatin C), kidney morphology (PAS staining) and oxidative stress (MDA) and the suppression of renal tubular injury markers of KIM-1 and NGAL. Meanwhile, the renal apoptosis and inflammation induced by cisplatin were also strikingly attenuated in celastrol-treated mice. In vitro, celastrol treatment markedly inhibited cisplatin-induced renal tubular cell apoptosis, suppressed NF-κB activation, and improved mitochondrial function evidenced by the restored mtDNA copy number, mitochondrial membrane potential, and OXPHOS activity in cisplatin-treated renal tubular epithelial cells.
Interpretation
This work suggested that celastrol could protect against cisplatin-induced acute kidney injury possibly through suppressing NF-κB and improving mitochondrial function.
Fund
The National Natural Science Foundation of China, National Key Research and Development Program, and Natural Science Foundation of Jiangsu Province.
Keywords: Celastrol, AKI, Cisplatin, NF-κB, Mitochondrial Dysfunction
Research in context.
Evidence before this study
Celastrol is an active ingredient of Chinese medicine Tripterygium wilfordii and is used for the treatment of immune diseases. As one of the five traditional medicinal compounds listed by “Cell” journal, celastrol is becoming a potent agent for treating inflammatory disorders including arthritis, Crohn's disease, and Parkinson's disease because of its anti-inflammatory and anti-oxidative activity. However, its role in treating cisplatin nephrotoxicity, a common clinical complication, remains unknown.
Added value of this study
The data from current study demonstrated that celastrol could attenuate cisplatin nephrotoxicity possibly through inhibiting NF-κB activation and improving mitochondrial function.
Implications of all the available evidence
Besides the protective effect against cisplatin nephrotoxicity, celastrol was also documented as an effective drug for treating cancer. Thus, celastrol could play dual roles in treating cancer and preventing cisplatin nephrotoxicity.
Alt-text: Unlabelled Box
1. Introduction
Cis-diamminedichloroplatinum (cisplatin) is a potent chemotherapeutic agent used for treating various types of solid organ tumors [[1], [2], [3], [4]]. Unfortunately, its clinical use is limited by its side effects in normal tissues. Due to preferential accumulation of cisplatin in renal tubules, acute kidney injury (AKI) is a serious and frequent complication in cancer patients undergoing cisplatin chemotherapy [5]. In clinical practice, approximately 25–40% patients experience renal dysfunction after treatment with cisplatin [6]. Thus, a challenge on the prevention of cisplatin nephrotoxicity still lies ahead.
The proposed mechanisms of cisplatin-induced AKI include the generation of reactive oxygen species (ROS), mitochondrial dysfunction, caspase activation, DNA damage, and apoptotic cell death [[7], [8], [9], [10]]. Entry of cisplatin into tubular cells provokes oxidative stress followed by inflammation and subsequent cell apoptosis and necrosis, leading to cisplatin-induced renal dysfunction [[11], [12], [13]]. The overproduction of ROS due to the damage to DNA and mitochondria also inhibits anti-oxidant enzymes such as superoxide dismutase (SOD), catalase (CAT) and glutathione (GSH) and activates apoptosis in renal tubules [[14], [15], [16]]. Moreover, ROS are potent in triggering inflammation through the activation of NF-κB signaling pathway [17].
Celastrol, also known as tripterine, is an active ingredient of Chinese medicine Tripterygium wilfordii and is documented as a potent agent for treating inflammatory disorders including arthritis, Crohn's disease, and Parkinson's disease [18]. Accompanied by artemisinin, celastrol has been listed by “Cell” journal as one of the five traditional medicinal compounds most likely to be developed as a modern drug [19]. Research has found that celastrol can protect acute ischemic stroke-associated brain injury [20]. Moreover, it also can improve mitochondrial function via activation of PI3K-Akt signaling pathway in C2C12 myotubes [21]. In addition, celastrol promotes Nur77 translocation from the nucleus to mitochondria to remove the damaged mitochondria to alleviate oxidative stress and inflammation [22]. Interestingly, targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis [23]. Recent research also reported that celastrol ameliorated acute kidney injury caused by IR, which was associated with the inhibition of NF-κB activation and inflammation [24]. However, the effect of celastrol on cisplatin nephrotoxicity has not been investigated.
In this study, we investigated the effect of celastrol on cisplatin nephrotoxicity in both mice and cell models and the underlying mechanisms. The results suggested that celastrol could be used for preventing cisplatin-induced renal injury by inhibiting inflammation and protecting mitochondrial function.
2. Materials and methods
2.1. Reagents
Cisplatin and Celastrol (>98% pure) were purchased from Sigma-Aldrich (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and trypsin solution (EDTA) were bought from Gibco (Invitrogen, Grand Island, NY). Antibodies against NF-κB p65 (rabbit, monoclonal), phospho-NF-κB p65 (rabbit, monoclonal), TAK1 (rabbit, monoclonal), phospho-TAK1 (rabbit, monoclonal), phospho-IKKα/β (rabbit, monoclonal), caspase-3 (rabbit, monoclonal), cleaved caspase-3 (rabbit, monoclonal), Bax (rabbit, monoclonal), Bcl-2 (rabbit, monoclonal), and GAPDH were all provided by Cell Signaling Technology (Danvers, MA, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody was also ordered from Cell Signaling Technology (Danvers, MA, USA). Alexa Fluor® 488 dye was provided by Invitrogen.
2.2. Animals
Adult male C57BL/6 J mice (20–22 g) were provided by Laboratory Animal Center of Nanjing Medical University and were caged under a 12-h light-dark cycle in a temperature- and humidity-controlled (19–21 °C, 50–60%) room, fed a standard rodent diet and allowed free access to drinking water. All animal procedures were approved by the Nanjing Medical University Institutional Animal Care and Use Committee (registration number: IACUCIACUC 14030112–1).
2.3. Establishment of cisplatin-induced AKI model and celastrol treatment
For the test of celastrol effect on cisplatin-induced AKI, adult male mice were randomly divided into following groups (N = 8 per group): control group, cisplatin group, and cisplatin + celastrol group. The control mice and cisplatin group received single intraperitoneal (i.p.) injection of saline or cisplatin (20 mg/kg), while cisplatin + celastrol group was pretreated intraperitoneally with celastrol (1 mg/kg or 2 mg/kg) 24 h before the challenge with cisplatin (20 mg/kg). In a separate experiment, 1 mg/kg celastrol was given to the mice with cisplatin simultaneously. After 72 h cisplatin treatment, mice of all groups were sacrificed. The blood was collected, and the isolated serum was stored at −80 °C. Kidney tissues for histological analysis were fixed in 4% paraformaldehyde (PFA). The remaining kidney tissues were stored at −80 °C for mRNA and protein analysis [25].
2.4. Histological analysis
Kidneys from mice of all the groups were fixed in 4% paraformaldehyde (PFA) for 24 h at room temperature and embedded in paraffin. Sections (3 μm) were stained with PAS and analyzed by a pathologist in a blind procedure. A minimum of 10 fields for each kidney slide were examined and scored for pathological injury. A score from 0 to 4 was given for pathological assessment: 0, normal histology; 1, mild injury, 5% to 25% of tubules showed pathological damage; 2, moderate injury, 25% to 50% of tubules showed pathological damage; 3, severe injury, 50% to 75% showed pathological damage; and 4, almost all tubules in field of view were damaged. The average histological score for each sample was calculated [26]. The images were captured with an Olympus BX51 microscopy (Olympus, Center Valley, PA).
2.5. Serum biochemistry
Blood was collected from inferior caval vein after anesthesia and serum samples were collected by centrifugation at 1500 rpm for 20 min. The levels of Serum creatinine (SCr) and blood urea nitrogen (BUN) were evaluated using a serum biochemical autoanalyzer (Hitachi 7600 modular chemistry analyzer, Hitachi Ltd., USA).
2.6. ELISA
Serum levels of IL-6 and TNF-α were measured by ELISA kits (Dakewe Biotech, Beijing, China) in accordance with the manufacturer's instructions. The levels of Cystatin C in the serum were also determined by a mouse Cystatin C ELISA kit (E-EL-M0389C, Elascience, China) [27].
2.7. Kidney MDA assay
The MDA concentration in kidney tissues was detected with a commercial MDA assay kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer's instructions [28]. MDA values were normalized to the protein concentrations and expressed as nmoL/mg protein.
2.8. Cell culture
Human proximal tubule epithelial cell line (HK-2) and mouse renal tubule epithelial cells (RTECs) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured in DMEM/F-12 medium (Wisent, Canada) supplemented with 10% fetal bovine serum (GIBCO), penicillin (100 U/mL) and streptomycin (100 μg/mL) and maintained at 37 °C in 5% CO2 in a humidified incubator. Cells were grown to 80% confluence and pretreated with celastrol for 2 h. Then cisplatin (5 μg/mL) was added to the serum-free medium to stimulate RTECs or HK2 cells for 24 h.
2.9. Cell counting Kit-8 (CCK-8) assay
Cell viability was determined by CCK-8 assay kit (KGA317, KeyGen Biotech, China) [29]. Briefly, RTECs or HK2 cells were treated with celastrol (10 μM to 100 μM) for 24 h, and then 10 μL CCK-8 reagent was added to medium and incubated for 2 h. The absorbance was detected at 450 nm.
2.10. Quantitative reverse transcriptase PCR
Total tissue RNA extraction was performed using the RNAiso Plus reagent (TaKaRa Biotechnology Co., Ltd., Dalian, China) according to the manufacturer's protocol [26]. cDNA was generated from 1 μg total RNA using PrimeScript™ Reverse Transcriptase (TaKaRa Biotechnology Co., Ltd., Dalian, China). Quantitative PCR was subsequently carried out using SYBR Green Master Mix (Vazyme, Nanjing, China) on a QuantStudio 3 Real-time PCR System (Applied Biosystems, Foster City, CA, USA). The primers used for PCR amplification were listed in Table 1. Cycling conditions were 95 °C for 10 min, followed by 40 repeats of 95 °C for 15 s and 60 °C for 1 min. The mRNA was normalized to GAPDH and calculated using delta method from threshold cycle numbers. The relative mRNA expression levels were analyzed and expressed relative to threshold cycle values (ΔCt), then converted to fold changes using the 2 − ΔΔCt method as described previously [30].
Table 1.
Sequences of the primers for qRT-PCR.
| Gene | Primer Sequence (5′-3′) |
|---|---|
| Mouse MCP-1 | F: GCTCTCTCTTCCTCCACCAC R: ACAGCTTCTTTGGGACACCT |
| Mouse IL-1β | F: ACTGTGAAATGCCACCTTTTG R: TGTTGATGTGCTGCTGTGAG |
| Mouse IL-6 | F: ACAAAGCCAGAGTCCTTCAGAGAG R: TTGGATGGTCTTGGTCCTTAGCCA |
| Mouse kim-1 | F: ACATATCGTGGAATCACAACGAC R: ACTGCTCTTCTGATAGGTGACA |
| Mouse NGAL | F: GCAGGTGGTACGTTGTGGG R: CTCTTGTAGCTCATAGATGGTGC |
| Mouse Bax | F: TGGAGATGAACTGGACAGCAATAT R: GCAAAGTAGAAGAGGGCAACCAC |
| Mouse Bcl-2 | F: CTCAGGCTGGAAGGAGAAGAT R: AAGCTGTCACAGAGGGGCTAC |
| Mouse mtND1 | F:ATCCTCCCAGGATTTGGAAT R:ACCGGTAGGAATTGCGATAA |
| Mouse 18S | F:TTCGGAACTGAGGCCATGATT R:TTTCGCTCTGGTCCGTCTTG |
| Mouse COX-2 | F: AGGACTCTGCTCACGAAGGA R: TGACATGGATTGGAACAGCA |
| Mouse GAPDH | F: GTCTTCACTACCATGGAGAAGG R: TCATGGATGACCTTGGCCAG |
2.11. Western blotting
Twenty milligram frozen kidney cortex tissue was grinded in liquid nitrogen. Cells were seeded onto six well plates. Two hundred milliliters of lysis buffer (50 mM Tris, 150 mM NaCl, 10 mM EDTA, 1% Triton X-100, 200 mM sodium fluoride, and supplemented with 1 × protease inhibitor cocktail (Roche, 04693132001)) was added to grinded tissue or cells and incubated on ice for 30 min. Then samples were centrifuged for 15 min at 12,000 rpm [31]. Protein concentration was measured using the Bradford method,and 50 μg total protein was used for Western blotting analysis following standard methods with primary antibodies against Bax (1:1000), Bcl-2 (1:1000), Caspase-3 (1:1000), Cleaved caspase-3 (1:1000), NF-κB p65 (1:1000), phospho-NF-κB p65 (1:1000), TAK1 (1:1000), phospho-TAK1 (1:1000), and phospho-IKKα/β (1:1000), followed by addition of HRP-labeled secondary antibodies (1:2500). The blots were visualized by reaction with chemiluminescence (ECL) Plus Western blotting detection reagents (Millipore, Bedford, MA, USA). Densitometric analysis was performed by measuring the intensity of the band using Image J software (NIH, Bethesda, MD, USA).
2.12. Oxygen consumption rate
Cellular mitochondrial function was measured by using a Seahorse XF96 Extracellular Flux Analyzer and a Seahorse XF Cell Mito Stress Test Kit (Agilent Technologies, CA, USA). The mitochondrial function was examined by direct measurement of the oxygen consumption rate (OCR) of cells according to the manufacturer's instructions [32]. Briefly, the cells were seeded onto 96-well plates and treated with cisplatin and celastrol for 24 h. After replacing the culture medium to seahorse buffer, oligomycin (1 mol/L), FCCP (0.5 mol/L), and rotenone/antimycin A (0.5 mol/L) was automatically injected into the RTECs or HK2 cells. After recording, OCR values were calculated after normalizing with total protein amounts.
2.13. TUNEL assay
In situ cell death was measured by TdT–mediated dUTP Nick-End Labeling (TUNEL) in situ cell death detection kit (Roche) according to the manufacturer's instructions [32]. Detection of the apoptotic cells showing green fluorescence was performed by fluorescence microscopy.
2.14. Annexin V/PI double staining
After treatment, cells were washed three times with PBS, trypsinized, centrifuged (1500 rpm at room temperature) for 5 min, adjusted to 5 × 104/ml and double-stained with annexin V-FITC and PI (Annexin V-FITC Apoptosis Detection Kit, BD Biosciences, San Diego, CA) according to the manufacturer's instructions [27]. After incubation for 15 min at room temperature in the dark, the fluorescent intensity was measured using a flow cytometer (BD Biosciences, San Diego, CA).
2.15. Determination of mitochondrial ROS and mitochondrial membrane potential (MMP)
Measurement of cell mitochondrial reactive oxygen species (ROS) production was measured using MitoSOX™ Red mitochondrial superoxide indicator (Life Technologies) [33]. Briefly, 50 μg MitoSOX™ mitochondrial superoxide indicator was dissolved in 13 μL of dimethylsulfoxide (DMSO) to make 5 mM MitoSOX™ reagent stock solution. Then the reagent stock solution was diluted using buffer to make 5 μM MitoSOX™ reagent working solution. Then 1.0 mL of 5 μM MitoSOX™ reagent working solution was applied to cover cells adhering to coverslip(s) to incubate for 10 min at 37 °C in dark. After incubation, mitochondrial superoxide anion in RTEC or HK2 cells was analyzed by flow cytometry. The MMP △Ψm of cells was determined using the mitochondrial membrane potential assay kit (Beyotime Biotechnology) with JC-1 and the quantification of JC-1 fluorescence were detected with flow cytometry as described previously [34].
2.16. Immunofluorescence staining
To visualize the expression and localization of NF-κB in cultured renal tubular cells, cultured cells were incubated with a rabbit phospho-NF-κB p65 antibody (1:200; Cell Signaling Technology) as a primary antibody at 4 °C overnight. Then cells were incubated in Alexa Fluor® 488 dye (1:400, Invitrogen) for 60 min. Finally, cells were visualized using Carl Zeiss LSM 5 PASCAL laser scanning confocal microscopy. Negative controls were performed using murine IgG instead of primary antibodies. An average score of the immunofluorescence was calculated as described previously [35].
2.17. Statistical analysis
All data were presented as the mean ± SD from triplicate experiments performed in a parallel manner unless otherwise indicated. Statistically significant differences were determined by ANOVAs followed by Bonferroni's Multiple Comparison Test or Student's t-test using GraphPad Prism 6 software. A value of P < 0.05 was considered significant.
3. Results
3.1. Celastrol attenuated cisplatin-induced acute kidney injury in mice
To investigate the effect of celastrol on renal dysfunction caused by cisplatin, kidney tissues collected at 72 h after cisplatin treatment were stained with PAS and scored. The kidneys obtained from cisplatin group demonstrated obvious features of tubular injury, including tubular dilatation, loss of brush border, cytoplasmic vacuoles, cast formation, and inflammatory cell infiltration compared with the control group. Strikingly, these histological lesions were significantly attenuated in mice treated with celastrol (Fig. 1a&b). In agreement with the improved renal morphology, enhanced BUN, serum Cr, and serum cystatin C levels after 72 h cisplatin treatment were significantly reversed by celastrol (cisplatin vs. cisplatin + celastrol: BUN, 75.22 ± 10.66 vs. 31.05 ± 10.88, p < 0.001, one-way ANOVA; serum creatinine, 1.178 ± 0.3083 vs. 0.3732 ± 0.1838, p < 0.001; and cystatin C: 16.00 ± 1.68 vs. 11.27 ± 1.891, p < 0.001, one-way ANOVA) (Fig. 1c-e).
Fig. 1.

Celastrol attenuated cisplatin-induced acute kidney injury in mice.
(a) Representative images of periodic acid-Schiff staining (×200 and ×400) of kidneys. (b) Tubular injury score in mice. Data were presented as means ± SD of 10 random fields from each kidney. Blood urea nitrogen (c), serum creatinine (d), serum cystatin C (e), and MDA (f) levels were measured. (g) qRT-PCR analyses of renal KIM-1 and NGAL mRNA expression. GAPDH was used as internal control. Data were presented as means ± SD (n = 8 in each group). All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ## P < 0.01, ### P < 0.001, * P < 0.05, *** P < 0.001. CP: cisplatin.
MDA level in kidney was also measured to evaluate the oxidative stress caused by cisplatin. As shown by the data, cisplatin increased MDA concentration by 1.69 folds, which was blocked by 57.84% (p < 0.001, one-way ANOVA) (Fig. 1f). Consistently, we also observed that celastrol decreased the tubular injury markers of kidney injury molecule 1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) by 87.28% and 79.42% (p < 0.001, one-way ANOVA), respectively (Fig. 1g). These findings demonstrated that celastrol attenuated cisplatin-induced acute kidney injury in mice. It must be noted that we did not find obvious toxic effects of celastrol on kidney morphology and the functions of kidney, liver and heart (Fig. 6a-c).
Fig. 6.

Evaluation of toxic effects of celastrol in mice.
(a) Blood urea nitrogen and serum creatinine levels were measured. Data were presented as means ± SD, n = 6 in each group. (b) Representative images of periodic acid-Schiff staining (×200 and ×400) of kidneys. (c) ALT, AST, and LDH levels were measured. Data were presented as means ± SD, n = 6 in each group. All experiments were duplicated for three times. Statistically significant differences were determined by Student's t-test.
3.2. Celastrol inhibited inflammatory response in the kidneys of mice treated with cisplatin
Pro-inflammatory cytokines and mediators such as IL-1β, TNF-a, IL-6, MCP-1, and COX-2 were reported to play important roles in renal injury induced by cisplatin. Therefore, we investigated celastrol effect on inflammation in the present experimental setting (Fig. 2a&b). qRT-PCR results showed that cisplatin-induced upregulation of IL-1β, IL-6, MCP-1, and COX-2 was strikingly reduced by 94.20%, 89.27%, 81.59%, 75.70%, respectively, after celastrol treatment. By ELISA, we confirmed that enhanced protein levels of IL-6 and TNF-α in circulation were decreased by 57.14% and 78.57%, respectively (p < 0.01, one-way ANOVA). These results indicated an anti-inflammatory effect of celastrol in cisplatin-induced AKI.
Fig. 2.
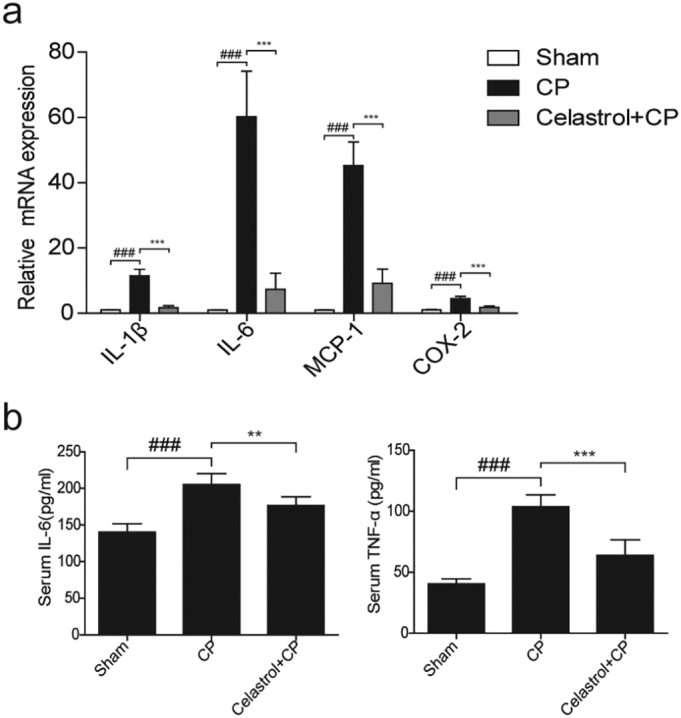
Celastrol inhibited inflammatory response in the kidney during cisplatin treatment.
(a) qRT-PCR analyses of renal IL-1β, IL-6, MCP-1, and COX-2 mRNA levels in kidneys of mice. (b) Enzyme linked immunosorbent assay analyses of IL-6 and TNF-α in kidney tissues. Data were expressed as means ± SD (n = 8 in each group). All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ### P < 0.001, ** P < 0.01, *** P < 0.001.
3.3. Celastrol ameliorated cisplatin-induced apoptosis in the kidney
By TUNEL staining, we evaluated apoptotic response in the kidneys of AKI mice (Fig. 3a). There were rare TUNEL-positive cells in the kidneys of control mice. However, the number of TUNEL-positive cells was significantly increased in the kidneys of cisplatin-treated mice, which was strikingly blocked by 67.67% after celastrol administration. Consistently, the dysregulation of apoptosis-associated proteins of Bax, Bcl-2 and cleaved-caspase-3 was significantly reversed by celastrol treatment compared to cisplatin group (Fig. 3b). Meanwhile, the changes of Bax and Bcl-2 mRNA levels analyzed by qRT-PCR showed a similar pattern as their protein levels (Fig. 3c). These findings indicated that celastrol ameliorated cisplatin-induced apoptosis in vivo.
Fig. 3.

Celastrol ameliorated cisplatin-induced apoptosis in kidney.
(a) Transferase dUTP nick-end labeling (TUNEL) staining (original magnification ×630; green: TUNEL; blue: DAPI; red arrow: indicating TUNEL positive cells). (b) Western blotting analyses of Bax, Bcl-2 and cleaved caspase-3 levels in the kidneys of cisplatin-treated mice with or without celastrol administration. GAPDH was used as loading control. (c) qRT-PCR analyses of renal Bax, Bcl-2 mRNA expressions. Data were expressed as means ± SD (n = 8 in each group). All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, # P < 0.05, ### P < 0.001, ** P < 0.01, *** P < 0.001.
3.4. Evaluation of the dose and time effects of celastrol in protecting against cisplatin-induced acute kidney injury
Furthermore, we tested the time and dose effects of celastrol on protecting against cisplatin nephrotoxicity. As shown in Fig. 4a–f, 2 mg/kg celastrol did not further potentiate the protective effect on kidney compared to 1 mg/kg celastrol treatment, indicating that 1 mg/kg celastrol in mice already reached its maximum in benefiting the kidney in this disease model. Moreover, we observed that simultaneous treatment of 1 mg/kg celastrol with cisplatin showed similar effect as 24 h pretreatment of celastrol on protecting renal function (Fig. 5a–c). Meanwhile, the increment of NF-κB p65 and p-NF-κB p65 induced by cisplatin was similarly blocked by before and simultaneous treatments of celastrol (Fig. 5d), indicating that celastrol might ameliorate cisplatin-induced renal inflammation via inhibiting NF-κB signaling.
Fig. 4.

Evaluation of the dose effect of celastrol on cisplatin-induced acute kidney injury in mice.
(a) Representative images of periodic acid-Schiff staining (×200 and ×400) of kidneys. (b) Tubular injury score in mice. Data were presented as means ± SD of 10 random fields from each kidney. Blood urea nitrogen (c), serum creatinine (d), serum cystatin C (e) and MDA (f) levels were measured. Data were presented as means ± SD (n = 8 in each group). All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ### P < 0.001, ** P < 0.01, *** P < 0.001.
Fig. 5.

Evaluation of the time effect of celastrol treatment on cisplatin-induced acute kidney injury in mice.
(a) Representative images of periodic acid-Schiff staining (×200 and ×400) of kidneys. (b) Tubular injury score in mice. Data were presented as means ± SD of 10 random fields from each kidney. Blood urea nitrogen, serum creatinine (c) levels were measured. Data were presented as means ± SD (n = 6 in each group). (d) Western blots of NF-κB p65 and phospho-NF-κB p65 protein levels in kidney tissues. All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ### P < 0.001, *** P < 0.001. Pre: pretreatment; Sim: simultaneous treatment.
3.5. Celastrol protected against cisplatin-induced proximal tubular cell apoptosis in vitro
To examine the cytotoxicity of celastrol, cell viability assay was performed in cultured RTECs using a CCK8 assay kit. With celastrol treatment at increasing concentrations from 10 nM to 100 nM for 24 h, we found the concentrations of celastrol within 50 nM were safe for cells (Fig. 7a). Then RTECs were pretreated with celastrol (50 nM) for 2 h before cisplatin treatment. We found that the apoptosis induced by cisplatin was significantly inhibited by 36.31% after celastrol treatment (Fig. 7b&c). In parallel to ameliorated cell apoptosis, enhanced Bax and cleaved-caspase-3 was significantly decreased by celastrol treatment in cisplatin-treated RTECs (Fig. 7d&e). Meanwhile, the reduction of protein and mRNA expressions of Bcl-2 could be significantly alleviated by celastrol (Fig. 7d&e). Moreover, elevated mRNA expressions of KIM-1 and NGAL induced by cisplatin were markedly blunted by 69.80% and 44.84%, respectively, after celastrol treatment in RTECs (Fig. 7f). In a dose-dependent study, we observed less effect of 25 nM celastrol in antagonizing cisplatin-induced cell apoptosis (Fig. 8a–c). These data showed that celastrol protected against cisplatin-induced renal tubular cell apoptosis in vitro.
Fig. 7.

Celastrol protected against cisplatin-induced renal tubular cell apoptosis in vitro.
(a) Cell viability was analyzed by CCK8 assay after treatment with celastrol for 24 h at increasing concentrations ranging from 10 μM to 100 μM. Data were presented as means ± SD. ***P < 0.001 vs. vehicle group, n = 3 in each group. (b) Representative images of FACS analysis of Annexin V and PI staining. RTECs were pretreated with celastrol, and then incubated with cisplatin (5 μg/mL) for 24 h. (c) Percentage of apoptotic cells was determined by FACS. Data were presented as means ± SD. n = 3 in each group. (d) Western blots of Bax, Bcl-2 and cleaved caspase-3. (e) qRT-PCR analyses of renal Bax and Bcl-2 mRNA expressions. (f) qRT-PCR analyses of renal KIM-1 and NGAL mRNA expressions. GAPDH was used as internal control. All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ## P < 0.01, * P < 0.05, ** P < 0.01, ***P < 0.001.
Fig. 8.

Evaluation of the dose effect of celastrol on cisplatin-induced RTEC apoptosis.
(a) Representative images of FACS analysis of Annexin V and PI staining. RTECs were pretreated with celastrol (25 and 50 nM), then incubated with cisplatin (5 μg/mL) for 24 h. (b) Percentage of apoptotic cells was determined by FACS. (c) qRT-PCR analyses of renal Bax, Bcl-2 mRNA expressions. GAPDH was used as internal control. Data were presented as means ± SD. n = 3 in each group. All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ## P < 0.01, ### P < 0.001, * P < 0.05, ** P < 0.01, ***P < 0.001.
3.6. Celastrol suppressed the activation of NF-κB signaling in RTECs treated with cisplatin
Celastrol, as an NF-κB inhibitor, had been proved to protect kidney against ischemia-reperfusion-induced injury in rats [24]. To explore the mechanism of celastrol in protecting against cisplatin-induced tubular injury, we checked the nuclear translocation of NF-κB p65 in RTECs pretreated with celastrol. The number of cells with nuclear translocation of NF-κB p65 was examined by immunofluorescence. As shown by the data, the cells with NF-κB p65 nuclear translocation were increased after cisplatin treatment, suggesting the activation of NF-κB. Strikingly, celastrol treatment markedly blocked NF-κB p65 nuclear translocation induced by cisplatin (Fig. 9a). Furthermore, the expressions of NF-κB pathway-related proteins including NF-κB p65, phospho-NF-κB p65, TAK1, phospho-TAK1, and phospho-IKKα/β in RTECs were detected by Western blotting (Fig. 9b). Similarly, these proteins in RTECs were higher in cisplatin-treated cells as compared with the control group, which was significantly blocked by celastrol treatment. Then we examined the mRNA levels of pro-inflammatory cytokines of IL-1β, IL-6, and COX-2 by qRT-PCR analysis and found that these enhanced pro-inflammatory factors were all blunted by celastrol (Fig. 9c). These results indicated that celastrol ameliorated cisplatin-induced inflammation possibly by blocking NF-κB activation.
Fig. 9.
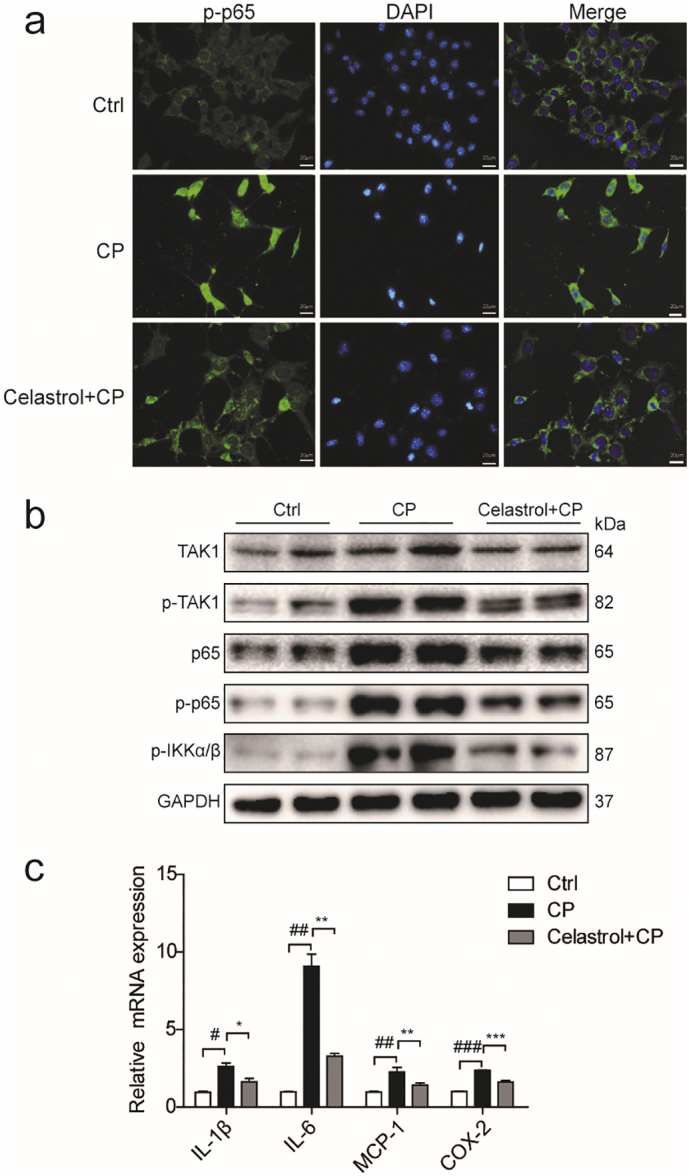
Celastrol suppressed the activation of NF-κB signaling in RTECs treated with cisplatin.
(a) Representative immunofluorescence staining of NF-κB p65 in control (saline), cisplatin, and cisplatin + celastrol groups (Original magnification×400; green: NF-κB p65; blue: DAPI). RTECs were pretreated with celastrol, then incubated with cisplatin (5 μg/mL) for 24 h. (b) Western blots of NF-κB p65, phospho-NF-κB p65, TAK1, phospho-TAK1, and phospho-IKKα/β protein levels in RTECs. (c) qRT-PCR analyses of IL-1β, IL-6, and COX-2 mRNA expressions in RTECs. GAPDH was used as internal control. Data were presented as means ± SD. n = 3 in each group. All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, # P < 0.05, ## P < 0.01, ### P < 0.001, * P < 0.05, ** P < 0.01, *** P < 0.001.
3.7. Celastrol ameliorated cisplatin-induced mitochondrial dysfunction in vitro
Mitochondria play an integral role in cell death, autophagy, immunity, and inflammation. Recent research found that celastrol could promote Nur77 translocation from the nucleus to mitochondria to remove the damaged mitochondria to alleviate inflammation [22]. Thus, we hypothesized that celastrol may also improve mitochondrial function in this experimental setting. To test this hypothesis, we examined the effect of celastrol on mitochondrial function in cisplatin-treated RTECs. First, we examined the effect of celastrol on mitochondrial ROS production in cisplatin-treated RTECs using MitoSOX™ Red Mitochondrial Superoxide Indicator. As shown in Fig. 10a, celastrol treatment obviously reduced cisplatin-induced mitochondrial ROS accumulation by 57.28% (p < 0.001, one-way ANOVA). Next, we determined the copy number of mitochondrial DNA (mtDNA) by qRT-PCR. As shown in Fig. 10b, cisplatin treatment reduced the number of mtDNA, which was significantly ameliorated by celastrol (+54.01%, p < 0.05, one-way ANOVA). Then we checked the mitochondrial membrane potential (MMP) using JC-1 fluorescent probe and found that celastrol treatment obviously alleviated cisplatin-induced MMP decline (+51.60%, p < 0.01, one-way ANOVA) (Fig. 10c). Finally, we detected the mitochondrial OXPHOS activity and found that cisplatin diminished basal oxygen consumption rate (OCR, representative of the basal mitochondrial OXPHOS activity) and significantly reduced spare respiratory capacity by using the seahorse 96xf. Strikingly, celastrol treatment alleviated OCR and maximal respiration (+74.09%, p < 0.001, one-way ANOVA) in RTECs treated with cisplatin (Fig. 10d&e). These findings indicated that celastrol ameliorated the mitochondrial dysfunction of renal tubular cells, which may contribute to the protective effect of celastrol against cisplatin nephrotoxicity to some degree.
Fig. 10.

Celastrol ameliorated cisplatin-induced mitochondrial dysfunction in vitro.
(a) FACS analysis of mitochondrial ROS accumulation using MitoSOX™ Red Mitochondrial Superoxide Indicator in RTECs. RTECs were pretreated with celastrol, and then cultured with cisplatin (5 μg/mL) for 24 h. (b) qRT-PCR analysis of mtDNA in RTECs. 18 s was used as internal control. (c) Analysis of mitochondrial membrane potential (MMP) using JC-1 fluorescent probe in RTECs. (d-e) Seahorse 96xf detected basal oxygen consumption rate (OCR, representative of the basal mitochondrial OXPHOS activity) and spare respiratory capacity. Data were presented as means ± SD. n = 3 in each group. All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ## P < 0.01, ### P < 0.001, * P < 0.05, ** P < 0.01, *** P < 0.001.
3.8. Celastrol ameliorated cisplatin-induced HK2 cell injury in vitro
Furthermore, we confirmed the celastrol effect on cisplatin nephrotoxicity using human renal epithelial cells (HK2). Cell viability assay also showed celastrol at the concentration within 50 nM are safe in HK2 cells (Fig. 11a). Similar as the results shown in RTEC studies, celastrol strikingly inhibited cisplatin-induced apoptosis, inflammation, and mitochondrial dysfunction in HK2 cells (Fig. 11b–h).
Fig. 11.

Celastrol ameliorated cisplatin-induced HK2 cell injury in vitro.
(a) Cell viability was analyzed by CCK8 assay after treatment with celastrol for 24 h at increasing concentrations ranging from 10 μM to 100 μM. Data were presented as means ± SD, n = 3 in each group. (b) Representative images of FACS analysis of Annexin V and PI staining. HK2 were pretreated with celastrol, and then incubated with cisplatin (5 μg/mL) for 24 h. (c) Western blots of Bax, Bcl-2, and cleaved caspase-3. (d) Western blots of TAK1, phospho-TAK1, NF-κB p65, phospho-NF-κB p65, and phospho-IKKα/β. (e) Analysis of mitochondrial membrane potential (MMP) using JC-1 fluorescent probe in HK2. (f) FACS analysis of mitochondrial ROS accumulation using MitoSOX™ Red Mitochondrial Superoxide Indicator in HK2 cells. (g-h) Seahorse 96xf detected basal oxygen consumption rate (OCR, representative of the basal mitochondrial OXPHOS activity) and spare respiratory capacity. GAPDH was used as internal control. Data were presented as means ± SD. n = 3 in each group. All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ## P < 0.01, ### P < 0.001, ** P < 0.01, *** P < 0.001.
3.9. Role of NF-κB signaling in mediating celastrol effect against cisplatin nephrotoxicity in vitro
Finally, we used QNZ, a NF-κB inhibitor, to explore whether celastrol protected renal tubular cells by inhibiting NF-κB pathway in this study. As shown in Fig. 12a&b, when RTECs and HK2 cells were pretreated with QNZ (100 nM), celastrol failed to further ameliorate cisplatin-induced cell death. These results indicated that the protective effect of celastrol on cisplatin nephrotoxicity could be dependent on the inhibition of NF-κB to some extent.
Fig. 12.

Evaluation of NF-κB signaling in mediating celastrol effect against cisplatin nephrotoxicity in vitro.
(a-b) Representative images of FACS analysis of Annexin V and PI staining. RTECs or HK2 cells were pretreated with QNZ and celastrol, and then incubated with cisplatin (5 μg/mL) for 24 h. Data were presented as means ± SD. n = 3 in each group. All experiments were duplicated for three times. Statistically significant differences were determined by one-way ANOVA, ### P < 0.001, *** P < 0.001.
4. Discussion
In kidney diseases, celastrol had been found to inhibit renal oxidative stress and inflammation, decrease the urinary excretion of albumin, and improve renal function in diabetic mice [36]. Moreover, celastrol also could protect kidneys against ischemia-reperfusion-induced injury in rats [24]. Thus, we hypothesize that celastrol might have the potential as an effective drug for treating cisplatin nephrotoxicity. To our knowledge, the present study was the first one to investigate the effect of celastrol on cisplatin-induced acute kidney injury. Treatment with celastrol improved renal dysfunction, renal tubular damage, oxidative stress (MDA) in line with the inactivation of NF-κB signaling and attenuated mitochondrial dysfunction in response to cisplatin challenge, suggesting that celastrol was able to protect against cisplatin nephropathy possibly through inhibiting NF-κB-mediated inflammation and improving mitochondrial function.
Acute kidney injury (AKI) caused by sepsis, ischemia or nephrotoxic agents is characterized by abrupt and reversible kidney dysfunction [37]. The pathogenesis of AKI involves complex pathway crosstalk associated with oxidative stress, inflammation, and apoptosis. Moreover, AKI is also an important pathogenic factor in the development and progression of chronic kidney disease [11,35]. Nephrotoxicity is a major side effect in cisplatin chemotherapy that limits the use of cisplatin in cancer patients. Nowadays attention has been focused on discovering compounds of natural origin in preventing renal tubular cell injury and accelerating tubular cell regeneration in AKI [13,38]. Celastrol is one of the bioactive components derived from the Chinese herb Tripterygium wilfordii, which has been used for the treatment of inflammatory and autoimmune diseases such as primary glomerular diseases in clinic for decades [39]. Pharmacologically, celastrol shows powerful antioxidant, anti-inflammatory, and anticancer properties [18]. However, potential toxicity restricts the further application of celastrol in clinic. It has been reported that celastrol could lead cardiotoxicity [40]. In our prior research, we also found that celastrol aggravated LPS-induced inflammation in mice [41]. It was reported that the IC50 values of celastrol on murine mesangial HBZY-1 cells and RAW264.7 cells were 3715 ± 342 nmol/L and 360 ± 41 nmol/L [42], respectively. The dose we used in cells in the present study was much less than above values.
It was well established that a single injection of cisplatin at a dose of 20 mg/kg body weight in mouse could cause obvious renal injury shown by tubular cell apoptosis, necrosis, and cast formation, leading to the renal dysfunction [43]. In the present study, we observed that cisplatin-induced morphological changes of kidney tubules and renal dysfunction were markedly attenuated by celastrol in line with improved oxidative stress and cellular apoptosis. These in vivo evidences strongly suggested a potential of celastrol in treating cisplatin nephrotoxicity in clinic. Furthermore, the direct effect of celastrol on cisplatin-induced renal tubular cell injury and the underlying mechanisms need to be investigated. As shown by the in vitro data, cisplatin-induced apoptotic response in renal tubular cells was remarkably ameliorated by celastrol administration, indicating a direct effect of celastrol on protecting against tubular cell injury in the present experimental setting.
It is known that NF-κB signaling pathway controls the expressions of a number of genes including inflammatory cytokines and chemokines, all of which play pivotal roles in controlling inflammation [44]. NF-κB activation is also associated with increased ROS generation and regulates various cellular responses including apoptosis. Due to these pathogenic characteristics, targeting NF-κB pathway has been of interest for the treatment of many inflammatory diseases including AKI. Here we examined the activation of NF-κB and found that celastrol significantly inhibited NF-κB activation induced by cisplatin in RTECs and HK2 cells. Notably, we observed that celestrol failed to further attenuate cell death when NF-κB was inactivated in cisplatin-treated renal tubular cells by a specific NF-κB inhibitor,suggesting that celastrol protected against cisplatin nephrotoxicity by inhibiting NF-κB signaling. This is in agreement with current concept that celastrol may serve as a novel NF-κB inhibitor [24].
Mitochondrial dysfunction also plays a central role in tissue damage caused by cisplatin [15]. The alterations of mtDNA copy number, mitochondrial ROS, and MMP were usually used as the markers of mitochondrial dysfunction [45]. Mitochondria are known as the main intracellular sites for ROS production. Under the pathological condition, mitochondrial ROS could promote pro-inflammatory and apoptotic responses, which ultimately results in tubular injury. In renal tubular cells, celastrol significantly improved cisplatin-induced mitochondrial abnormality as shown by the restoration of mtDNA copy number, MMP, and mitochondrial OXPHOS activity, together with the blockade of mitochondrial ROS production. These results highly suggested that the celastrol may protect mitochondria to ameliorate cisplatin nephrotoxicity to some extent. However, the relationship between the inflammation and oxidative stress in this experimental setting was still unclear and need future studies.
In summary, our study demonstrated that celastrol could ameliorate cisplatin-induced nephrotoxicity by antagonizing NF-κB-mediated inflammation and protecting mitochondrial function. The findings highly suggested a translational potential of celastrol as a natural drug for treating cisplatin nephrotoxicity in clinic.
Funding sources
This work was supported by Grants from the National Natural Science Foundation of China (nos. 81600557, 81600532, 81600352, 81873599, 81670647, 81700604 and 81570616), the National Key Research and Development Program (no. 2016YFC0906103), the Natural Science Foundation of Jiangsu Province (no. BK20170148), and the Nanjing Medical University (2016NJMUZD051).
Conflicts of interest
The authors declare no competing financial interest.
Author contributions
X.Y., Z.J., and A.Z. designed the study. X.Y., X.M., M.X., X.Z., and Y.Z. performed experiments. X.Y., X.M., M.X., and G.D. analyzed the data. X.Y., S.H., Z.J., and A.Z. interpreted the results. X.Y., Z.J., and A.Z. wrote the manuscript.
Contributor Information
Aihua Zhang, Email: zhaihua@njmu.edu.cn.
Zhanjun Jia, Email: zhanjun.jia@hsc.utah.edu.
References
- 1.Wang D., Lippard S.J. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 2.Cohen S.M., Lippard S.J. Vol. 67. 2001. Cisplatin: from DNA damage to cancer chemotherapy. Progress in nucleic acid research and molecular biology; pp. 93–130. [DOI] [PubMed] [Google Scholar]
- 3.Arany I., Safirstein R.L. Cisplatin nephrotoxicity. Semin Nephrol. 2003;23(5):460–464. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 4.Hoek J., Bloemendal K.M., van der Velden L.A., van Diessen J.N., van Werkhoven E., Klop W.M. Nephrotoxicity as a Dose-Limiting Factor in a High-Dose Cisplatin-Based Chemoradiotherapy Regimen for Head and Neck Carcinomas. Cancer. 2016;8(2) doi: 10.3390/cancers8020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maimaitiyiming H., Li Y., Cui W., Tong X., Norman H., Qi X. Increasing cGMP-dependent protein kinase I activity attenuates cisplatin-induced kidney injury through protection of mitochondria function. Am J Physiol Renal Physiol. 2013;305(6):F881–F890. doi: 10.1152/ajprenal.00192.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan M.A., Liu J., Kumar G., Skapek S.X., Falck J.R., Imig J.D. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013;27(8):2946–2956. doi: 10.1096/fj.12-218040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakraborty P., Roy S.S., Sk U.H., Bhattacharya S. Amelioration of cisplatin-induced nephrotoxicity in mice by oral administration of diphenylmethyl selenocyanate. Free Radic Res. 2011;45(2):177–187. doi: 10.3109/10715762.2010.521155. [DOI] [PubMed] [Google Scholar]
- 8.Pan H., Shen Z., Mukhopadhyay P., Wang H., Pacher P., Qin X. Anaphylatoxin C5a contributes to the pathogenesis of cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol. 2009;296(3):F496–F504. doi: 10.1152/ajprenal.90443.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nozaki Y., Nikolic-Paterson D.J., Yagita H., Akiba H., Holdsworth S.R., Kitching A.R. Tim-1 promotes cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2011;301(5):F1098–F1104. doi: 10.1152/ajprenal.00193.2011. [DOI] [PubMed] [Google Scholar]
- 10.Saad A.A., Youssef M.I., El-Shennawy L.K. Cisplatin induced damage in kidney genomic DNA and nephrotoxicity in male rats: the protective effect of grape seed proanthocyanidin extract. Food and chemical toxicology : an international journal published for the British Industrial. Biol Res Assoc. 2009;47(7):1499–1506. doi: 10.1016/j.fct.2009.03.043. [DOI] [PubMed] [Google Scholar]
- 11.Ozkok A., Edelstein C.L. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014;2014 doi: 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Domitrovic R., Cvijanovic O., Pugel E.P., Zagorac G.B., Mahmutefendic H., Skoda M. Luteolin ameliorates cisplatin-induced nephrotoxicity in mice through inhibition of platinum accumulation, inflammation and apoptosis in the kidney. Toxicology. 2013;310:115–123. doi: 10.1016/j.tox.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Ma P., Zhang S., Su X., Qiu G., Wu Z. Protective effects of icariin on cisplatin-induced acute renal injury in mice. Am J Transl Res. 2015;7(10):2105–2114. [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava S., Sinha D., Saha P.P., Marthala H., D'Silva P. Magmas functions as a ROS regulator and provides cytoprotection against oxidative stress-mediated damages. Cell Death Dis. 2014;5:e1394. doi: 10.1038/cddis.2014.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang C., Dong Z. Mitochondria in Kidney Injury: When the Power Plant Fails. J Am Soc Nephrol. 2016;27(7):1869–1872. doi: 10.1681/ASN.2015111277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall A.M., Schuh C.D. Mitochondria as therapeutic targets in acute kidney injury. Curr Opin Nephrol Hypertens. 2016;25(4):355–362. doi: 10.1097/MNH.0000000000000228. [DOI] [PubMed] [Google Scholar]
- 17.Oh G.S., Kim H.J., Choi J.H., Shen A., Choe S.K., Karna A. Pharmacological activation of NQO1 increases NAD(+) levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2014;85(3):547–560. doi: 10.1038/ki.2013.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kannaiyan R., Shanmugam M.K., Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303(1):9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 19.Liu J., Lee J., Salazar Hernandez M.A., Mazitschek R., Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161(5):999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang M., Liu X., Zhang D., Wang Y., Hu X., Xu F. Celastrol treatment protects against acute ischemic stroke-induced brain injury by promoting an IL-33/ST2 axis-mediated microglia/macrophage M2 polarization. J Neuroinflammation. 2018;15(1):78. doi: 10.1186/s12974-018-1124-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abu Bakar M.H., Tan J.S. Improvement of mitochondrial function by celastrol in palmitate-treated C2C12 myotubes via activation of PI3K-Akt signaling pathway. Biomed Pharmacother. 2017;93:903–912. doi: 10.1016/j.biopha.2017.07.021. [DOI] [PubMed] [Google Scholar]
- 22.Hu M., Luo Q., Alitongbieke G., Chong S., Xu C., Xie L. Celastrol-Induced Nur77 Interaction with TRAF2 Alleviates Inflammation by Promoting Mitochondrial Ubiquitination and Autophagy. Mol Cell. 2017;66(1) doi: 10.1016/j.molcel.2017.03.008. [141-53 e6] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo L., Luo S., Du Z., Zhou M., Li P., Fu Y. Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis. Nat Commun. 2017;8(1):878. doi: 10.1038/s41467-017-00834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu C., He W., Kuang Y., Ren K., Gou X. Celastrol protects kidney against ischemia-reperfusion-induced injury in rats. J Surg Res. 2014;186(1):398–407. doi: 10.1016/j.jss.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 25.Li Z., Xu K., Zhang N., Amador G., Wang Y., Zhao S. Overexpressed SIRT6 attenuates cisplatin-induced acute kidney injury by inhibiting ERK1/2 signaling. Kidney Int. 2018;93(4):881–892. doi: 10.1016/j.kint.2017.10.021. [DOI] [PubMed] [Google Scholar]
- 26.Weidemann A., Bernhardt W.M., Klanke B., Daniel C., Buchholz B., Campean V. HIF activation protects from acute kidney injury. J Am Soc Nephrol. 2008;19(3):486–494. doi: 10.1681/ASN.2007040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y., Yu X., Zhang Y., Ding G., Zhu C., Huang S. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin Sci. 2018;132(7):825–838. doi: 10.1042/CS20171625. [DOI] [PubMed] [Google Scholar]
- 28.Liang C., Wang X., Hu J., Lian X., Zhu T., Zhang H. PTPRO Promotes Oxidized Low-Density Lipoprotein Induced Oxidative Stress and Cell Apoptosis through Toll-Like Receptor 4/Nuclear Factor kappaB Pathway. Cell Physiol Biochem. 2017;42(2):495–505. doi: 10.1159/000477596. [DOI] [PubMed] [Google Scholar]
- 29.Teng J., Liu M., Su Y., Li K., Sui N., Wang S. Down-regulation of GRP78 alleviates lipopolysaccharide-induced acute kidney injury. Int Urol Nephrol. 2018 doi: 10.1007/s11255-018-1911-0. (in press) [DOI] [PubMed] [Google Scholar]
- 30.Rajeevan M.S., Ranamukhaarachchi D.G., Vernon S.D., Unger E.R. Use of real-time quantitative PCR to validate the results of cDNA array and differential display PCR technologies. Methods. 2001;25(4):443–451. doi: 10.1006/meth.2001.1266. [DOI] [PubMed] [Google Scholar]
- 31.Galgamuwa R., Hardy K., Dahlstrom J.E., Blackburn A.C., Wium E., Rooke M. Dichloroacetate Prevents Cisplatin-Induced Nephrotoxicity without Compromising Cisplatin Anticancer Properties. J Am Soc Nephrol. 2016;27(11):3331–3344. doi: 10.1681/ASN.2015070827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh C.J., Ha C.M., Choi Y.K., Park S., Choe M.S., Jeoung N.H. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 2017;91(4):880–895. doi: 10.1016/j.kint.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Kudin A.P., Bimpong-Buta N.Y., Vielhaber S., Elger C.E., Kunz W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2004;279(6):4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- 34.Wang S., Zhang C., Niyazi S., Zheng L., Li J., Zhang W. A novel cytoprotective peptide protects mesenchymal stem cells against mitochondrial dysfunction and apoptosis induced by starvation via Nrf2/Sirt3/FoxO3a pathway. J Transl Med. 2017;15(1):33. doi: 10.1186/s12967-017-1144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Y., Ni J., Chen S., Bai M., Lin J., Ding G. MicroRNA-709 Mediates Acute Tubular Injury through Effects on Mitochondrial Function. J Am Soc Nephrol. 2018;29(2):449–461. doi: 10.1681/ASN.2017040381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J.E., Lee M.H., Nam D.H., Song H.K., Kang Y.S., Lee J.E. Celastrol, an NF-kappaB inhibitor, improves insulin resistance and attenuates renal injury in db/db mice. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0062068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doi K., Rabb H. Impact of acute kidney injury on distant organ function: recent findings and potential therapeutic targets. Kidney Int. 2016;89(3):555–564. doi: 10.1016/j.kint.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Baradaran A., Nasri H., Rafieian-Kopaei M. Protection of renal tubular cells by antioxidants: current knowledge and new trends. Cell J. 2015;16(4):568–571. doi: 10.22074/cellj.2015.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang X., Xu F., Chen D.M., Zeng C.H., Liu Z.H. The clinical course and long-term outcome of primary focal segmental glomerulosclerosis in Chinese adults. Clin Nephrol. 2013;80(2):130–139. doi: 10.5414/CN107607. [DOI] [PubMed] [Google Scholar]
- 40.Sun H., Liu X., Xiong Q., Shikano S., Li M. Chronic inhibition of cardiac Kir2.1 and HERG potassium channels by celastrol with dual effects on both ion conductivity and protein trafficking. J Biol Chem. 2006;281(9):5877–5884. doi: 10.1074/jbc.M600072200. [DOI] [PubMed] [Google Scholar]
- 41.Wu M., Chen W., Yu X., Ding D., Zhang W., Hua H. Celastrol aggravates LPS-induced inflammation and injuries of liver and kidney in mice. Am J Transl Res. 2018;10(7):2078–2086. [PMC free article] [PubMed] [Google Scholar]
- 42.Fang W., Peng F., Yi T., Zhang C., Wan C., Xu H. Biological activity and safety of Tripterygium extract prepared by sodium carbonate extraction. Molecules. 2012;17(9):11113–11123. doi: 10.3390/molecules170911113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim Y.J., Lee M.Y., Son H.Y., Park B.K., Ryu S.Y., Jung J.Y. Red ginseng ameliorates acute cisplatin-induced nephropathy. Planta Med. 2014;80(8–9):645–654. doi: 10.1055/s-0034-1368571. [DOI] [PubMed] [Google Scholar]
- 44.Baker R.G., Hayden M.S., Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011;13(1):11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan Y., Huang S., Wang W., Wang Y., Zhang P., Zhu C. Activation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012;82(7):771–789. doi: 10.1038/ki.2012.188. [DOI] [PubMed] [Google Scholar]