

Abstract
Every single nucleotide change compatible with life is present in the human population today. Understanding these rare human variants defines an extraordinary challenge for genetics and medicine. The new clinical practice of sequencing many genes for hereditary cancer risk has illustrated the utility of clinical next-generation sequencing in adults, identifying more medically actionable variants than single gene testing. However, it has also revealed a linear relationship between length of DNA evaluated and number of rare “variants of uncertain significance” reported. We propose that careful approaches to phenotype-genotype inference, distinguishing between diagnostic and screening intent, and expanded use of family-scale genetics studies as a source of information on family-specific variants will reduce variants of uncertain significance reported to patients.
The Ubiquity of Rare, Family-Specific Genetic Variants
All genomic single nucleotide changes compatible with life are present in the human population today. With a point mutation rate about 1.6×10−8 per nucleotide per individual (Palamara et al., 2015) and a human population of 7.3×109, it is clear that each of the possible 9.7×109 nucleotide changes that is compatible with life exists somewhere in the human population (Figure 1). Indeed, the population is large enough that all single nucleotide changes compatible with reproduction have been inherited by someone alive today. In addition to single nucleotide variants, there are an unlimited number of possible insertions, deletions, and genomic rearrangements. More than 10 billion genetic variants present in living individuals today define an extraordinary challenge intrinsic to comprehensive medical interpretation of the human genome. Although the worldwide populations of other species such as cattle, mice, flies, and many bacteria are large enough to have their genomes similarly saturated, classifying rare human variation presents unique medical challenges and opportunities.
Figure 1. The Proportion of all Possible Single Nucleotide Genetic Variants Present in the Human Population.
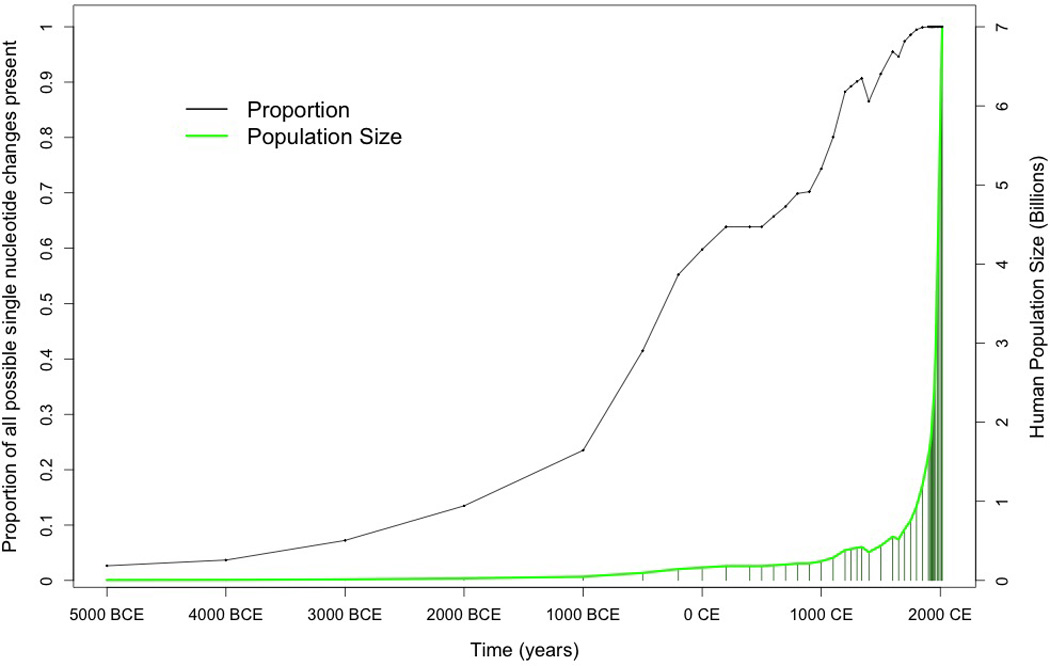
This proportion is related to the population size in relation to the size of the genome. Over 99% of all single nucleotide substitutions compatible with life have been present in at least one living human for about 200 years.
New variants arise with each birth and, if not passed on, disappear with each death. So, the growth in genetic variation parallels the growth of the human population. Most variants arose recently in our evolutionary history and are extremely rare (Fu et al., 2014, Lek et al., 2015). Although individually rare, collectively these one-in-amillion and one-in-a-billion variants are ubiquitous. Each of us has 50 to 100 de novo variants that were not in the genomes of our parents. On average, less than one of these per person will be in coding sequence (Veltman and Brunner, 2012). However, variants are passed from generation to generation, so each of us has several extremely rare, family-specific exonic variants that have arisen over the last few generations (Figure 1). Most recent variants are shared by only a few close relatives. Older variants may be shared by a few thousand descendants of a common ancestor. These variants are not distributed evenly in populations. We label them family-specific variants (see Glossary) because they cluster in the human population based on recent familial reproduction and migration patterns. Population based frequencies may present an inaccurate oversimplification of family-specific variant distribution and can lead to population stratification when used in case-control analysis (Jiang et al., 2013, Liu et al., 2013). Lineage-specific variants can be selected for and bred into strains of model organisms, but other methods are necessary to determine the functional effects of family-specific variants in humans (Box 1).
Box 1. The Clinician’s Corner.
Family-specific variants are collectively medically important. Large panel tests identify more actionable pathogenic variants than single gene tests in hereditary cancer and other diseases where panel testing is available.
A variant of uncertain significance should not alter medical management. Treatment decisions should be made based on other personal and family medical information. The variant may be revisited to determine if any additional information on the variant is available. New data may come from functional studies, which are developing rapidly, from updates on variant frequency or segregation data in variant databases, and from newly published research.
Family-based segregation studies are a way to classify patient variants. Most clinical laboratories offer family studies, but eligibility criteria vary widely. If a patient is interested in finding out more about his or her variant these may be a useful option.
Large-scale sequencing tests (gene panel, exome, and genome tests) often have diagnostic testing and screening components. Differences in pre-test probability for diagnostic and screening indications change the positive predictive value of the test for different indications. In the future, it may be possible to separate diagnostic and screening components of genomic testing.
There has been a push in research and clinical practice to perform more human genetic sequencing, which has led to the identification of many family-specific variants. Associated genomic discovery has led to clinical diagnosis and treatment in many clinical cases (Parsons et al., 2016, Yang et al., 2014). In this opinion article we discuss how advances in genetic sequencing have identified many medically actionable family-specific variants, and identified more variants classified as “variants of uncertain significance”, in the context of hereditary cancer testing. We describe overarching principles of rare variant classification, and propose several strategies to better classify family-specific variants so as to minimize the frequency of reporting variants of uncertain significance to patients.
Are Rare Genetic Variants Medically Important?
How should scientists and medical professionals interpret and report the clinical implications of these rare, family-specific genetic variants? Will knowing about them improve medical care, or just make it more confusing? How much effort should society spend on them, since the vast majority of family-specific variants are completely benign and the population-attributable risk of even the most medically actionable single family-specific variant is miniscule?
On the one hand, recent manuscripts reporting the findings of clinical panel testing for genes involved in hereditary cancer risk suggests some answers (Chong et al., 2014, Dewey et al., 2014, Judkins et al., 2015, Kurian et al., 2014, Kwong et al., 2016, Laduca et al., 2014, Lincoln et al., 2015, Mannan et al., 2016, Maxwell et al., 2014, Shirts et al., 2016, Susswein et al., 2015, Tung et al., 2014, Yorczyk et al., 2014). The main conclusion of each of these manuscripts is that panel testing for multiple known cancer risk genes increases diagnostic success rate by identifying more medically actionable family-specific genetic findings than testing one or a few genes. These articles illustrate that, when considered collectively, rare variants have a substantial impact on human health. In one clinic-based study of multi-gene testing in women with personal or family history of breast and/or ovarian cancer, genetic findings in seven different genes led to changes in medical management in seven of twelve patients (Bunnell et al., 2016). Gene panel testing has further been shown to have medical validity and to be cost effective in the context of colon cancer risk screening (Desmond et al., 2015, Gallego et al., 2015).
On the other hand, a finding that is usually downplayed or even excluded in reports of hereditary cancer risk testing (Chong et al., 2014, Judkins et al., 2015) is that there is a roughly linear relationship between the amount of DNA evaluated and the number of ultra-rare variants identified (Figure 2, Key Figure). Many ultra-rare variants are clinically classified as variants of uncertain clinical significance (VUS). Currently, at most clinical laboratories, for every diagnostic success there are approximately two patients that receive a report that may lead to more questions than answers (see below). We present results for cancer diagnostic testing, but the relationship between the amount of DNA sequenced and the number of family-specific VUS identified is likely similar in other areas of clinical genetic testing and bears broad implications.
Figure 2. Key Figure. Average Number of Variants of Uncertain Significance Relative to Number of Genes Sequenced.
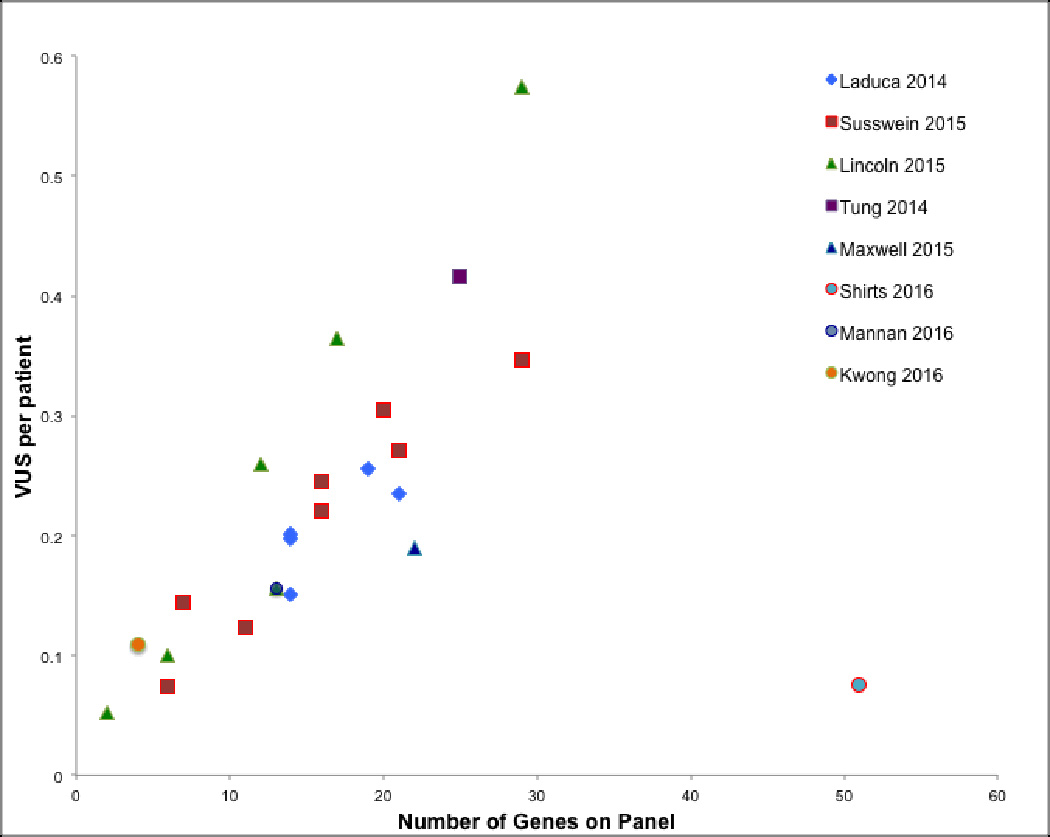
The graph presents the linear relationship between the average number of variants of uncertain significance reported per patient tested for hereditary cancer risk, and the number of genes sequenced at most commercial laboratories. Data were collected from published manuscripts by clinical laboratories performing sequence interpretation. (Kwong et al., 2016, Laduca et al., 2014, Lincoln et al., 2015, Mannan et al., 2016, Maxwell et al., 2014, Shirts et al., 2016, Susswein et al., 2015, Tung et al., 2014)
The Rise of “Variants of Uncertain Significance”
The term “variant of uncertain clinical significance” (VUS) was initially introduced in the context of early commercial clinical testing of BRCA1 and BRCA2 (Petrucelli et al., 2002). The term was intended to indicate that there was not enough evidence to determine with confidence if a variant was or was not associated with disease, or that relevant evidence was inconsistent. The term has now entered into the clinical genetics lexicon and professional recommendations for variant classification (Plon et al., 2008, Richards et al., 2015). Most VUS reported to patients for cancer risk are in genes that unquestionably harbor pathogenic high-risk pathogenic variants in some patients, although some VUS may be in newly identified genes for which the exact level of risk is not well defined.
How should a patient or physician interpret a VUS? As a construct designed by and for laboratories, VUS reports may not be helpful to patients. Official consensus is that medical management in the context of a VUS should be based only on other personal and family medical information, so a VUS should not alter clinical management (Lindor et al., 2013, Richards et al., 2015, Riley et al., 2012). However, in practice, VUS lead to patient and provider confusion, are often misinterpreted as significant positive findings, and do alter clinical management especially when non-specialists guide patients regarding genetic information (Culver et al., 2013, Murray et al., 2011, Richter et al., 2013). Patients with VUS findings are encouraged to periodically revisit their VUS and query about changes in variant classification (Lindor et al., 2013). If specific VUS are eventually reclassified as clinically important, patients may benefit.
Some have proposed not to report VUS identified in clinical testing to patients or clinicians (Culver et al., 2013, Richter et al., 2013)(Outstanding Questions Box). Although there are genetic counseling challenges to explaining the lack of clinical implications of a VUS, our opinion is that there will be societal and familial difficulties with laboratories not reporting rare variants. Not reporting VUS may dramatically reduce the rate of growth of societal genetic understanding (ACMG, 2015). In addition, in the context of medical care, a patient who has had VUS results withheld may spread incorrect information to relatives, communicating that a comprehensive sequencing test yielded negative results, which would suggest that other risk factors could explain family clusters, and that genetic causes might have been ruled out. Not reporting VUS results would be particularly problematic in a situation where other relatives receive results about the same variant, as this could cause medical confusion, potentially bringing biological relationships into question. Open communication is critical to increasing an understanding of family-specific variants whether the communication is between relatives or within the scientific and medical genetics communities.
Outstanding Questions Box.
Should clinical laboratories report rare variants not known to alter protein function as variants of uncertain significance when most of these variants are benign? There is currently a debate on the value of reporting VUS to patients and on the uncertainty of the possible consequences of not reporting variants. If laboratories do not report these variants will it decrease medical errors? Will it prevent patients from correctly identifying causes of hereditary risk or slow the growth of genetic knowledge?
How often are variants classified when strategies to “bend” the linear relationship between clinical sequence generated and reported variants of uncertain significance reported are used?
What clinical mechanisms will be necessary to decouple targeted genetic testing from genomic screening?
Crowd-sourced research and small-scale family studies will be necessary to classify all potentially meaningful variants; how can research and clinical resources be economically brought to the table for the purpose of classifying variants only seen in individual families?
What are the best ways to address real and perceived barriers to family centered care while addressing costs of care and concerns about patient autonomy and confidentiality?
Family-specific Variants and the Charge Towards Precision Medicine
Different databases may classify variants in different ways (Vail et al., 2015), and clinical laboratories report VUS at different rates (Figure 2, Key Figure). We were surprised that our laboratory was an outlier with far fewer VUS reports per case than other laboratories doing similar testing (Figure 2, Key Figure). We believe that a clear understanding of both population genetics and medical diagnostics principles can lead to variant interpretation that may mitigate the proliferation of VUS while facilitating open communication. Bending the linear relationship between DNA sequenced and VUS reported to patients by appropriately classifying a higher proportion of rare variants and reducing the number of VUS reported per gene, might help address concerns about VUS (Blazer et al., 2015, Culver et al., 2013, Murray et al., 2011, Richter et al., 2013, Shashi et al., 2016) and aid in fostering societal acceptance of clinical exome and genome scale sequencing. If this does not happen, then patients reading genomic sequencing reports risk missing critical information because of long lists of VUS (Blazer et al., 2015, Culver et al., 2013, Murray et al., 2011, Richter et al., 2013, Shashi et al., 2016).
We would like to highlight several general principles that, in our opinion, should guide variant interpretations. These illustrate how fields as diverse as population genetics, functional analysis in model organisms, and Bayesian statistics will be increasingly relevant to the entire medical field as the number of family-specific variants identified by broad genetic sequencing grows.
Precision Medicine and Evidence-Based Medicine
Almost all clinical assertions about rare genetic variants are based on comparing the predicted properties of novel variants to those of known variants (Richards et al., 2015). For example: we know that truncation variants in the first 90% of BRCA2 sequence cause increased breast and ovarian cancer risk, so we can confidently assert that never-before-seen early truncation in BRCA2 will be associated with increased cancer risk. Other classes of variants are more difficult to classify: Very late truncation variants in BRCA2, such as the p.Lys3326* variant (rs11571833), may not alter protein function, for example. Some missense variants in highly conserved functional domains alter protein function, and others do not (Jeyasekharan et al., 2013, Starita et al., 2015). Some gene domains tolerate variants and others do not (Dimitrova et al., 2016, Wang et al., 2016). Some splice-site variants cause exon skipping, but others do not or cause alternate splicing that does not substantially change protein function (de la Hoya et al., 2016). Sometimes, a variant alters gene expression or function in a way that causes an attenuated phenotype or a completely different phenotype (Hesson et al., 2015, Li et al., 2016). Consequently, the potential clinical effects of many classes of variants are still not well understood for most disease-associated genes.
The practice of comparing new variants to known variants is markedly different than relying on evidence from randomized trials; it is comparable to assessment of a new physical finding based entirely on assumptions from physiology principles. In the early 20th century, dramatic advances in medicine were made based on improvements in the understanding of physiology. Advances in knowledge of renal and cardiac physiology were sometimes immediately translated to clinical care when new understanding illuminated clear interventions (Granger, 1998, Pitts, 1946). We believe that society is in a similar translational era for genetics. The pace of genetic discovery is rapid with advances coming from single families that may lead directly to interventions (Chong et al., 2015, Enns et al., 2014, Parsons et al., 2016, Petrovski et al., 2015, Yang et al., 2014). However, the movement towards “precision medicine” using genetic information from rare variants may clash with the philosophy of “evidence-based” medicine where action that is based only on physiological understanding, is avoided pending evidence from randomized trials.
Unavoidable Statistical Uncertainty for Family-specific Risk Variants
Without large cohorts it is impossible to separate potential low risk variants from benign variants. Meta-analyses of thousands of cases and controls have been necessary to confidently estimate the 1.4 to 3 fold colon cancer risk associated with several relatively common APC and CHEK2 variants (Liang et al., 2013, Xiang et al., 2011). In genome-wide association studies (GWAS), tens of thousands of individuals have been tested to find common genetic variants responsible for 1.5 fold risk while leaving substantial uncertainty about lower levels of risk despite the enormity of these studies (Kar et al., 2016, Zeng et al., 2016). The scale of these studies illustrates the statistical impossibility of independently distinguishing low risk from no risk with any confidence for family-specific variants (Shirts et al., 2013). In these situations, once high-level risk is ruled out, all remaining family-specific variants must be either reported as VUS or treated as likely benign. Although we advocate for reporting VUS that may be clinically actionable to patients, we believe it is absurd and likely harmful to classify these likely benign variants that may hypothetically cause low risk as VUS.
The Promise and Peril of High-Throughput Analyses
A recent study generated high-throughput functional data on all possible single nucleotide changes the BRCA1 RING domain (Starita et al., 2015). Innovative applications of next-generation sequencing and CRISPR/Cas9 genome editing are enabling high-throughput evaluation of variants in many genes (Hart et al., 2015, Kretz et al., 2015, Sahni et al., 2015). Additional high-throughput functional data are fantastic, and will certainly improve variant prediction, but will not eliminate all uncertainty for several reasons. First, functional analyses favor specific mechanisms. For example, authors of the BRCA1 RING domain study noted above highlighted that pathogenic splice disrupting variants were not flagged by their analysis (Starita et al., 2015). In addition, with a few dozen known disease-causing variants driving clinical correlations for thousands of predictions, extrapolation beyond clinical evidence becomes a major concern. Lastly, current methods for evaluating the utility of high-throughput analyses are based on diagnostic testing, not population screening, so scores or likelihood ratios generated by functional studies may not be applicable to genomic screening. Functional studies will undoubtedly be increasingly useful, but will always have caveats.
Experience with computational ‘in silico’ predictions of variant pathogenicity is a cautionary example that illustrates these concerns. As it became clear that early amino acid similarity and evolutionary conservation-based algorithms were not uniformly useful, new tools have resulted in dozens of competing in silico predictions (Peterson et al., 2013). Algorithmic predictions uniformly present good performance characteristics for selected variants, but fail to perform as well in real-world testing. Just as functional tests often validate on small sets of variants, in silico scores are often validated on highly curated variant sets. Finally, computational algorithms, like variant classification guidelines and functional studies, were designed under the assumption of targeted diagnostic testing. Because of targeted validation and diagnostic testing assumptions in the genomic context, the best in silico tools have unacceptably low positive predictive value (Mather et al., 2016). This is because positive predictive value for targeted diagnostic testing is much higher than positive predictive value for test screening bearing a lower prior probability for screening tests. We illustrate this principle in Table 1. We assumed 5 to 20 clinically meaningful rare variants per genome. Even a predictive model with high sensitivity and specificity has low positive predictive value if thousands or millions of rare variants in a population are screened because of the relatively low pre-test probability that any specific variant is meaningful. Genomic and gene panel tests are usually initiated with a specific diagnostic objective with a small set of target genes, but these may subtly incorporate both diagnostic and screening components. In the context of panel testing, most tested genes may fit the clinical phenotype, but genes that do not fit the phenotype could be considered screening testing. In hereditary cancer panels, screening genes are positive in approximately 1% of cases (Shirts 2016). Genome testing for novel pediatric phenotypes relies on narrowing the search space to a set of genes in pathways related to the phenotype, but genomic tests may be easily expanded to screen for other known traits. We propose that variant analyses incorporate underlying testing assumptions and lower screening pre-test probabilities such that there may be many fewer VUS reported. Bayesian reasoning using underlying pretest probabilities is common in medical care, but current guidelines do not acknowledge that considering patient specific priors may be important in variant classification, reporting, and patient treatment decisions (Plon et al., 2008, Richards et al., 2015).
Table 1. Predicted Performance of High-Throughput Variant Classification.
The classification is performed with sensitivity of 0.95 and specificity of 0.95 using different sizes of sequencing tests. Sensitivity and specificity of 0.95 would define a very good test in a diagnostic setting with approximately 95% chance of correctly confirming a diagnosis (positive predictive value) and approximately 95% chance of correctly ruling out a diagnosis (negative predictive value). In clinical sequencing, as more sequence is evaluated, the genetic tests become less diagnostic, and more like screening tests as positive predictive value decreases.
| Size of sequencing test |
Example Test |
Estimated prior probability* |
Positive Predictive Value |
Negative Predictive Vale |
|---|---|---|---|---|
| Single gene | MLH1 | 0.5 | 0.95 | 0.95 |
| 4 to 5 genes |
MLH1, MSH2, MSH6, PMS2 |
0.1 | 0.679 | 0.994 |
| 20–25 genes | ColoNext, Coloseq, etc. | 0.02 | 0.279 | 0.9989 |
| 100 genes | TruSight | 0.005 | 0.087 | 0.9997 |
| 500 genes | - | 0.001 | 0.019 | 0.99995 |
| 20,000 genes | Exome | 0.00002** | 0.0004 | 0.999999 |
| 20,000 genes+ | Genome | 0.00000002** | 0.0000004 | 0.999999999 |
Prior probability that a variant causes a given clinical phenotype estimated as expected ratio of clinically important variants observed to total rare variants observed. Exact priors will vary depending on population and number of individuals tested.
Priors are generated from testing applied to a hypothetical population of individuals. Although each individual genome has only 40,000 to 200,000 rare variants(Genomes Project et al., 2015) more variants are present in the population and the performance. Performance is based on the entire population tested.
Returning to the Family
Family co-segregation studies may be the optimal way to generate evidence about family-specific variants (see Figure 3)(Mohammadi et al., 2009, Thompson et al., 2003). However, these require substantial effort to identify and test relatives. New paradigms are emerging that move components of this translational efforts closer to the patient, facilitating crowd-sourced genetics research (Garrett et al., 2016). Almost every clinical laboratory that performs panel genetic testing offers free testing of family members for VUS classification, although availability and inclusion criteria vary from laboratory to laboratory (Garrett et al., 2016). Despite opportunities, there are multiple barriers to family-centered translational genetics. The current medical paradigm and regulatory framework focuses on the relationship of the individual to health care providers and insurers, creating both real and perceived barriers to family-centered care that arise from patient payment mechanisms and confidentiality regulations (George et al., 2015, Weaver, 2016). These impede family-centered follow-up and efficient communication among family members for both pathogenic and uncertain variants.
Figure 3. Co-segregation Analysis in Breast Cancer.


Examples of co-segregation analysis in two example families with a history of breast cancer are shown. Individuals analyzed are in colored boxes and the likelihood ratios (LR) corresponding to co-segregation analysis of just those individuals are in colored text. Circles represent women, squares men, and lines indicate relationships. Filled circles indicate breast cancer diagnosis with the age at diagnosis listed below (BrCa ‘age’). “+” indicates a tested individual with the VUS, “-” indicates a tested individual without the VUS. (a) Example family with a hypothetical pathogenic BRCA1 variant. LR >20 is usually sufficient to classify a variant as likely pathogenic and LR >100 is usually classifies a variant as pathogenic. b) Example family with a hypothetical benign BRCA1 variant. LR <0.05 is usually sufficient to classify a variant as likely benign and LR < 0.01 strongly suggests a variant as benign. Including more relatives with a variant increases the probability that a variant will be classified. Figure modified with permission from findmyvariant.org.
Genetic Uncertainty and Clinical Practice
With family-specific variants, some uncertainty is unavoidable, but uncertainty managed by skilled practitioners has always been a hallmark of medical practice (Han et al., 2011). Professional guidelines suggest that 90% to 95% probability of variant pathogenicity is acceptable for medical decisions, and that 5% to 10% probability is low enough to suggest a variant is likely benign; everything else is classified a VUS (Plon et al., 2008, Richards et al., 2015). In informal clinician surveys, professional organizations that designed variant classification guidelines found that this level of genetic uncertainty is consistent with the uncertainty encountered in the context of other clinical decision-making; the best current evidence is used in tailored conversations about situation-specific options for optimal personal health (Richards et al., 2015). As outlined above, methods used to derive variant classifications are complicated and require the judgment of experts skilled in the art and science of genetic medicine. Genetic reports that provide information about rare, family-specific variants contain interpretations created by medical professionals that are, in our opinion, a consultative component of their clinical practice. These interpretations present a knowledgeable explanation of the best current evidence to be interpreted in the context of a specific patients’ clinical care.
Concluding Remarks
Novel family-specific variants of uncertain significance present a challenge and an opportunity. Clinical interpretation of these variants must rely on expert integration of multifactorial evidence. Our opinion is that for family-specific variants once high-penetrance has been ruled out the most appropriate classification is likely-benign, not VUS, because it is statistically impossible to prove or disprove low risk. We propose that considering the diagnostic or screening context of genomic testing and evaluating variants in different contexts using different pre-test probabilities will bend the linear relationship between amount of DNA sequenced and number of variants reported to patients (Outstanding Questions Box). The combinations of relatively rapid functional assays, such as assays for potential splice altering variants with family follow-up, are powerful, and when used well have allowed identification of new types of disease causing variants (Pritchard et al., 2013, Shirts et al., 2014). Given the ubiquity of family-specific variants, personalized medicine will necessarily lead to personalized research. New clinical paradigms of broad access to crowd-sourced, small-scale translational research should be welcomed (Outstanding Questions Box). Efforts such as ClinVar, Matchmaker Exchange, FindMyVariant, and PROMPT are facilitating this paradigm change (see Resources list). Just as many medical advances of the past century came from empowered physicians striving to understand the mechanisms underlying the physiology of a unique patient or family, we are entering an era where genomic tools may enable individual families to collaborate with physicians and researchers by exploring their unique family-specific variants.
Trends Box.
Panel testing of many genes associated with several clinical syndromes, such as hereditary cancer risk, was recently introduced and is becoming common.
Family-specific variants of uncertain significance are reported at a rate correlated with the amount of sequence tested in next-generation clinical sequencing tests.
With more inexpensive genetic sequencing, screening analysis is now increasingly being combined with diagnostic analysis. We propose that a clear distinction between these two objectives with different analyses based on pre-test probability will reduce the number of variants of uncertain significance reported.
New technology has facilitated high throughput functional studies, which will be useful, yet bearing caveats with regard to extrapolation beyond clinical evidence from a few well-understood variants.
Family segregation studies that have been mostly used for gene discovery in the past are re-emerging as an optimal way to classify extremely rare variants. Many clinical laboratories offer family studies but eligibility criteria vary widely.
Questions: * indicates correct answer
- Consensus is, that in the context of medical management, a VUS should:
- Be integrated with other risk information similarly to a pathogenic variant.
- Influence treatment in the same way a benign variant would.
- Not influence treatment decisions; treatment should be based on other personal and family information.*
- Prompt the medical provider to seek guidance from a genetics expert.
- Positive predictive value for in silico scores that predict variant pathogenicity
- Is always low in the context of very rare variants
- Is always high if the scores have very high sensitivity and high specificity
- Is extremely low in the context of diagnostic testing
- Is extremely low in the context of genomic screening*
- Which of the following is a good way to determine the clinical significance of a family-specific variant of uncertain significance:
- Co-segregation studies*
- Large case-control studies
- Population stratification
- Randomized controlled trials
- Which of following is true of both “precision medicine” and “evidence-based medicine”:
- Both are focused on classifying the strength of evidence and using high-quality evidence for medical decisions.
- Both are focused on making tailored medical decisions based on genetic understanding and scientific principles.
- Both seek evidence from medical genetics research that may improve medical care.*
- There is nothing true for both “precision medicine” and “evidence-based medicine.” These are opposing beliefs.
Acknowledgments
The authors would like to thank Mary-Claire King for critically reviewing this manuscript. Brian Shirts is a Damon Runyon-Rachleff Innovator supported in part by the Damon Runyon Cancer Research Foundation (DRR-33-15), by the NHGRI (R21HG008513), and by development funds from the Fred Hutch/University of Washington Cancer Consortium (NCI 5P30 CA015704-39). Colin Pritchard is supported by CDMRP award PC131820, and a 2013 Young Investigator Award from the Prostate Cancer Foundation. Tom Walsh is supported by the NCI (R01CA175716-04).
Glossary
- Family-specific variants
are extremely rare genetic variants that probably arose in the last few hundred years and cluster in the human population based on recent familial reproduction and migration patterns.
- Variant of uncertain significance or Variant of uncertain clinical significance
is a genetic variant, usually in a gene that has been associated with a disease, where the pathogenicity of the variant in relation to the disease can neither be confirmed nor ruled out because of lack of adequate information or conflicting data.
- Family co-segregation studies
evaluate the allelic segregation of a variant with a disease. Given a known model of disease penetrance these can generate likelihood ratios that a variant fits pathogenic or benign patterns.
- Bayesian Statistics
statistical methods that incorporate pre-test probabilities and testing likelihood ratios to generate post-test probabilities.
- Precision Medicine
refers to a medical model of prevention and treatment decisions tailored to patients based on individual variability in genes, environment, and lifestyle. Although not necessarily related to genetics, this term has increasingly been used to medicine tailored based on genetic information.
- in silico’ predictions
are a collection of computational tools designed to generate scores that correlate with biological consequences of genetic variants.
- Crowd-sourced genetics
is the strategy of using the community of healthcare providers and patients, linked through online resources, to advance genetic understanding and clinical genetic care.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
All authors declare they have no financial conflicts of interest
Resources
ClinVar http://www.ncbi.nlm.nih.gov/clinvar/ - ClinVar is a repository for variant interpretations submitted mostly by laboratories that perform clinical genetics sequencing.
Matchmaker Exchange http://www.matchmakerexchange.org/ - Matchmaker Exchange is a collection of multiple resources that seek to connect patients, clinicians, and researchers seeking to share rare phenotypes or genetic findings.
FindMyVariant http://findmyvariant.org/ - FindMyVariant is a study that teaches patients about family studies of VUS with information on how to identify and facilitate genetic testing of relatives that might yield more information their VUS.
PROMPT http://promptstudy.info/ - PROMPT is an online patient registry for people with positive or VUS findings from multi-gene testing.
References
- 1.ACMG. Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2015;17:505–507. doi: 10.1038/gim.2015.41. [DOI] [PubMed] [Google Scholar]
- 2.Blazer KR, Nehoray B, Solomon I, Niell-Swiller M, Culver JO, Uman GC, Weitzel JN. Next-Generation Testing for Cancer Risk: Perceptions, Experiences, and Needs Among Early Adopters in Community Healthcare Settings. Genet Test Mol Biomarkers. 2015;19:657–665. doi: 10.1089/gtmb.2015.0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bunnell AE, Garby CA, Pearson EJ, Walker SA, Panos LE, Blum JL. The Clinical Utility of Next Generation Sequencing Results in a Community-Based Hereditary Cancer Risk Program. J Genet Couns. 2016 doi: 10.1007/s10897-016-9985-2. [DOI] [PubMed] [Google Scholar]
- 4.Chong HK, Wang T, Lu HM, Seidler S, Lu H, Keiles S, Elliott AM. The validation and clinical implementation of BRCAplus: a comprehensive high-risk breast cancer diagnostic assay. PLoS One. 2014;9:e97408. doi: 10.1371/journal.pone.0097408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, Bamshad MJ. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet. 2015;97:199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Culver J, Brinkerhoff C, Clague J, Yang K, Singh K, Sand S, Weitzel J. Variants of uncertain significance in BRCA testing: evaluation of surgical decisions, risk perception, and cancer distress. Clin Genet. 2013;17:12097. doi: 10.1111/cge.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Hoya M, Soukarieh O, Lopez-Perolio I, Vega A, Walker LC, van Ierland Y, Spurdle AB. Combined genetic and splicing analysis of BRCA1 c.[594-2A>C; 641A>G] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desmond A, Kurian AW, Gabree M, Mills MA, Anderson MJ, Kobayashi Y, Ellisen LW. Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol. 2015;1:943–951. doi: 10.1001/jamaoncol.2015.2690. [DOI] [PubMed] [Google Scholar]
- 9.Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, Quertermous T. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311:1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitrova D, Ruscito I, Olek S, Richter R, Hellwag A, Turbachova I, Sehouli J. Germline mutations of BRCA1 gene exon 11 are not associated with platinum response neither with survival advantage in patients with primary ovarian cancer: understanding the clinical importance of one of the biggest human exons. A study of the Tumor Bank Ovarian Cancer (TOC) Consortium. Tumour Biol. 2016 doi: 10.1007/s13277-016-5109-8. [DOI] [PubMed] [Google Scholar]
- 11.Enns GM, Shashi V, Bainbridge M, Gambello MJ, Zahir FR, Bast T, Goldstein DB. Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet Med. 2014;16:751–758. doi: 10.1038/gim.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu W, Gittelman RM, Bamshad MJ, Akey JM. Characteristics of neutral and deleterious protein-coding variation among individuals and populations. Am J Hum Genet. 2014;95:421–436. doi: 10.1016/j.ajhg.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallego CJ, Shirts BH, Bennette CS, Guzauskas G, Amendola LM, Horike-Pyne M, Veenstra DL. Next-Generation Sequencing Panels for the Diagnosis of Colorectal Cancer and Polyposis Syndromes: A Cost-Effectiveness Analysis. J Clin Oncol. 2015;33:2084–2091. doi: 10.1200/JCO.2014.59.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garrett LT, Hickman N, Jacobson A, Bennett RL, Amendola LM, Rosenthal EA, Shirts BH. Family Studies for Classification of Variants of Uncertain Classification: Current Laboratory Clinical Practice and a New Web-Based Educational Tool. J Genet Couns. 2016 doi: 10.1007/s10897-016-9993-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genomes Project, C. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.George R, Kovak K, Cox SL. Aligning policy to promote cascade genetic screening for prevention and early diagnosis of heritable diseases. J Genet Couns. 2015;24:388–399. doi: 10.1007/s10897-014-9805-5. [DOI] [PubMed] [Google Scholar]
- 17.Granger HJ. Cardiovascular physiology in the twentieth century: great strides and missed opportunities. Am J Physiol. 1998;275:H1925–H1936. doi: 10.1152/ajpheart.1998.275.6.H1925. [DOI] [PubMed] [Google Scholar]
- 18.Han PK, Klein WM, Arora NK. Varieties of uncertainty in health care: a conceptual taxonomy. Med Decis Making. 2011;31:828–838. doi: 10.1177/0272989X11393976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Moffat J. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 20.Hesson LB, Packham D, Kwok CT, Nunez AC, Ng B, Schmidt C, Ward RL. Lynch syndrome associated with two MLH1 promoter variants and allelic imbalance of MLH1 expression. Hum Mutat. 2015;36:622–630. doi: 10.1002/humu.22785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeyasekharan AD, Liu Y, Hattori H, Pisupati V, Jonsdottir AB, Rajendra E, Venkitaraman AR. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat Struct Mol Biol. 2013;20:1191–1198. doi: 10.1038/nsmb.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang Y, Epstein MP, Conneely KN. Assessing the impact of population stratification on association studies of rare variation. Hum Hered. 2013;76:28–35. doi: 10.1159/000353270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Judkins T, Leclair B, Bowles K, Gutin N, Trost J, McCulloch J, Timms K. Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer. 2015;15:215. doi: 10.1186/s12885-015-1224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kar SP, Beesley J, Amin Al Olama A, Michailidou K, Tyrer J, Kote-Jarai Z, Lambrechts D. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-15-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kretz CA, Dai M, Soylemez O, Yee A, Desch KC, Siemieniak D, Ginsburg D. Massively parallel enzyme kinetics reveals the substrate recognition landscape of the metalloprotease ADAMTS13. Proc Natl Acad Sci U S A. 2015;112:9328–9333. doi: 10.1073/pnas.1511328112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, Ford JM. Clinical Evaluation of a Multiple-Gene Sequencing Panel for Hereditary Cancer Risk Assessment. J Clin Oncol. 2014;14:14. doi: 10.1200/JCO.2013.53.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwong A, Shin VY, Au CH, Law FB, Ho DN, Ip BK, Chan TL. Detection of Germline Mutation in Hereditary Breast and/or Ovarian Cancers by Next-Generation Sequencing on a Four-Gene Panel. J Mol Diagn. 2016;18:580–594. doi: 10.1016/j.jmoldx.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Laduca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, Chao E. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med. 2014;24:40. doi: 10.1038/gim.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, MacArthur D. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv. 2015 doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Woods SL, Healey S, Beesley J, Chen X, Lee JS, Chenevix-Trench G. Point Mutations in Exon 1B of APC Reveal Gastric Adenocarcinoma and Proximal Polyposis of the Stomach as a Familial Adenomatous Polyposis Variant. Am J Hum Genet. 2016;98:830–842. doi: 10.1016/j.ajhg.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang J, Lin C, Hu F, Wang F, Zhu L, Yao X, Zhao Y. APC polymorphisms and the risk of colorectal neoplasia: a HuGE review and meta-analysis. Am J Epidemiol. 2013;177:1169–1179. doi: 10.1093/aje/kws382. [DOI] [PubMed] [Google Scholar]
- 32.Lincoln SE, Kobayashi Y, Anderson MJ, Yang S, Desmond AJ, Mills MA, Ellisen LW. A Systematic Comparison of Traditional and Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Genes in More Than 1000 Patients. J Mol Diagn. 2015;17:533–544. doi: 10.1016/j.jmoldx.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Lindor NM, Goldgar DE, Tavtigian SV, Plon SE, Couch FJ. BRCA1/2 sequence variants of uncertain significance: a primer for providers to assist in discussions and in medical management. Oncologist. 2013;18:518–524. doi: 10.1634/theoncologist.2012-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Nicolae DL, Chen LS. Marbled inflation from population structure in gene-based association studies with rare variants. Genet Epidemiol. 2013;37:286–292. doi: 10.1002/gepi.21714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mannan AU, Singh J, Lakshmikeshava R, Thota N, Singh S, Sowmya TS, Subramanian K. Detection of high frequency of mutations in a breast and/or ovarian cancer cohort: implications of embracing a multi-gene panel in molecular diagnosis in India. J Hum Genet. 2016;61:515–522. doi: 10.1038/jhg.2016.4. [DOI] [PubMed] [Google Scholar]
- 36.Mather CA, Mooney SD, Salipante SJ, Scroggins S, Wu D, Pritchard CC, Shirts BH. CADD score has limited clinical validity for the identification of pathogenic variants in non-coding regions in a hereditary cancer panel. Genet Med. 2016 doi: 10.1038/gim.2016.44. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maxwell KN, Wubbenhorst B, D'Andrea K, Garman B, Long JM, Powers J, Nathanson KL. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med. 2014;11:176. doi: 10.1038/gim.2014.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohammadi L, Vreeswijk MP, Oldenburg R, van den Ouweland A, Oosterwijk JC, van der Hout AH, van Houwelingen HC. A simple method for co-segregation analysis to evaluate the pathogenicity of unclassified variants; BRCA1 and BRCA2 as an example. BMC Cancer. 2009;9 doi: 10.1186/1471-2407-9-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murray ML, Cerrato F, Bennett RL, Jarvik GP. Follow-up of carriers of BRCA1 and BRCA2 variants of unknown significance: variant reclassification and surgical decisions. Genet Med. 2011;13:998–1005. doi: 10.1097/GIM.0b013e318226fc15. [DOI] [PubMed] [Google Scholar]
- 40.Palamara PF, Francioli LC, Wilton PR, Genovese G, Gusev A, Finucane HK, Price AL. Leveraging Distant Relatedness to Quantify Human Mutation and Gene-Conversion Rates. Am J Hum Genet. 2015;97:775–789. doi: 10.1016/j.ajhg.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, Plon SE. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peterson TA, Doughty E, Kann MG. Towards precision medicine: advances in computational approaches for the analysis of human variants. J Mol Biol. 2013;425:4047–4063. doi: 10.1016/j.jmb.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrovski S, Shashi V, Petrou S, Schoch K, McSweeney KM, Dhindsa RS, Goldstein DB. Exome sequencing results in successful riboflavin treatment of a rapidly progressive neurological condition. Cold Spring Harb Mol Case Stud. 2015;1:a000257. doi: 10.1101/mcs.a000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petrucelli N, Lazebnik N, Huelsman KM, Lazebnik RS. Clinical interpretation and recommendations for patients with a variant of uncertain significance in BRCA1 or BRCA2: a survey of genetic counseling practice. Genet Test. 2002;6:107–113. doi: 10.1089/10906570260199357. [DOI] [PubMed] [Google Scholar]
- 45.Pitts RF. Kidney. Annu Rev Physiol. 1946;8:199–230. doi: 10.1146/annurev.ph.08.030146.001215. [DOI] [PubMed] [Google Scholar]
- 46.Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Tavtigian SV. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pritchard CC, Smith C, Marushchak T, Koehler K, Holmes H, Raskind W, Bennett RL. A mosaic PTEN mutation causing Cowden syndrome identified by deep sequencing. Genet Med. 2013;15:1004–1007. doi: 10.1038/gim.2013.51. [DOI] [PubMed] [Google Scholar]
- 48.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;5:30. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richter S, Haroun I, Graham TC, Eisen A, Kiss A, Warner E. Variants of unknown significance in BRCA testing: impact on risk perception, worry, prevention and counseling. Ann Oncol. 2013;24(Suppl 8):viii69–viii74. doi: 10.1093/annonc/mdt312. [DOI] [PubMed] [Google Scholar]
- 50.Riley BD, Culver JO, Skrzynia C, Senter LA, Peters JA, Costalas JW, Trepanier AM. Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns. 2012;21:151–161. doi: 10.1007/s10897-011-9462-x. [DOI] [PubMed] [Google Scholar]
- 51.Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Vidal M. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015;161:647–660. doi: 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shashi V, McConkie-Rosell A, Schoch K, Kasturi V, Rehder C, Jiang YH, McDonald MT. Practical considerations in the clinical application of whole-exome sequencing. Clin Genet. 2016;89:173–181. doi: 10.1111/cge.12569. [DOI] [PubMed] [Google Scholar]
- 53.Shirts BH, Casadei S, Jacobson AL, Lee MK, Gulsuner S, Bennett RL, Pritchard CC. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet Med. 2016:212. doi: 10.1038/gim.2015.212. [DOI] [PubMed] [Google Scholar]
- 54.Shirts BH, Jacobson A, Jarvik GP, Browning BL. Large numbers of individuals are required to classify and define risk for rare variants in known cancer risk genes. Genet Med. 2013;19:187. doi: 10.1038/gim.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shirts BH, Salipante SJ, Casadei S, Ryan S, Martin J, Jacobson A, Pritchard CC. Deep sequencing with intronic capture enables identification of an APC exon 10 inversion in a patient with polyposis. Genet Med. 2014;27:30. doi: 10.1038/gim.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fields S. Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics. 2015;200:413–422. doi: 10.1534/genetics.115.175802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, Chung WK. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2015 doi: 10.1038/gim.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thompson D, Easton DF, Goldgar DE. A full-likelihood method for the evaluation of causality of sequence variants from family data. Am J Hum Genet. 2003;73:652–655. doi: 10.1086/378100. Epub 2003 Jul 2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, Hartman AR. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2014;3:29010. doi: 10.1002/cncr.29010. [DOI] [PubMed] [Google Scholar]
- 60.Vail PJ, Morris B, van Kan A, Burdett BC, Moyes K, Theisen A, Eggington JM. Comparison of locus-specific databases for BRCA1 and BRCA2 variants reveals disparity in variant classification within and among databases. J Community Genet. 2015;6:351–359. doi: 10.1007/s12687-015-0220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, Johnson N. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J Clin Invest. 2016 doi: 10.1172/JCI87033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weaver M. The Double Helix: Applying an Ethic of Care to the Duty to Warn Genetic Relatives of Genetic Information. Bioethics. 2016;30:181–187. doi: 10.1111/bioe.12176. [DOI] [PubMed] [Google Scholar]
- 64.Xiang HP, Geng XP, Ge WW, Li H. Meta-analysis of CHEK2 1100delC variant and colorectal cancer susceptibility. Eur J Cancer. 2011;47:2546–2551. doi: 10.1016/j.ejca.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 65.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Eng CM. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yorczyk A, Robinson LS, Ross TS. Use of panel tests in place of single gene tests in the cancer genetics clinic. Clin Genet. 2014;16:12488. doi: 10.1111/cge.12488. [DOI] [PubMed] [Google Scholar]
- 67.Zeng C, Matsuda K, Jia WH, Chang J, Kweon SS, Xiang YB, Zheng W. Identification of Susceptibility Loci and Genes for Colorectal Cancer Risk. Gastroenterology. 2016;150:1633–1645. doi: 10.1053/j.gastro.2016.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]