

Summary
GPRC5B recruitment of Src family kinases has been implicated in diet-induced insulin resistance. However, the mechanism of this action is not fully understood. Here, we report that GPRC5B-mediated phosphorylation of sphingomyelin synthase 2 (SMS2) by Fyn is a crucial step in the development of insulin resistance. Lipid-induced metabolic stress augments SMS2 phosphorylation by facilitating the interaction of GPRC5B and SMS2. SMS2 phosphorylation reduces its ubiquitination, and consequently increases SMS2 protein abundance. Although ceramide and diacylglycerol (DAG) have been known to be central mediators of lipid-induced insulin resistance, the accumulation of these lipids fails to impair insulin signaling in SMS2 knockout mouse embryonic fibroblasts (MEFs). Conversely, exogenous expression of a phosphomimetic SMS2 impairs insulin action in SMS2 knockout MEFs under metabolic stress conditions. We demonstrate that SMS2-generated DAG in sphingomyelin synthesis inhibits insulin signaling through JNK activation. Thus, GPRC5B links sphingolipid metabolism to diet-induced insulin resistance via SMS2-dependent DAG production.
Subject Areas: Biochemical Mechanism, Molecular Mechanism of Behavior, Diabetology
Graphical Abstract
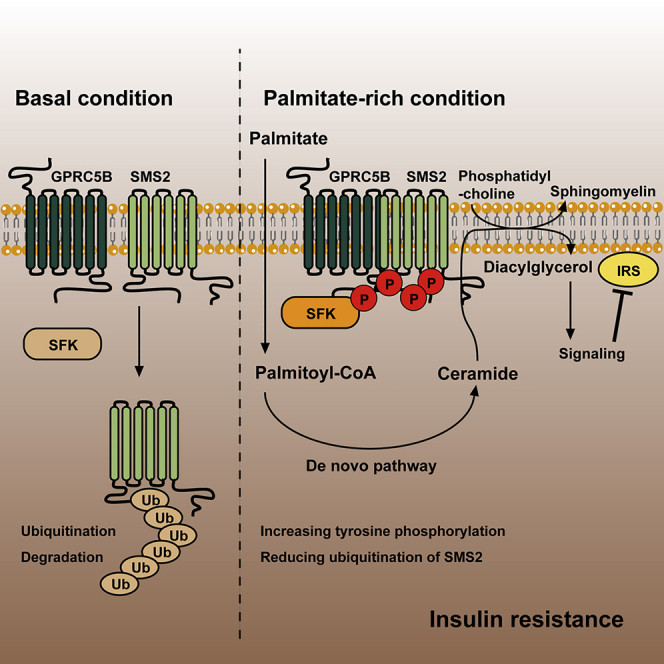
Highlights
-
•
Saturated fatty acids enhance interaction between GPRC5B and SMS2
-
•
GPRC5B facilitates tyrosine phosphorylation of SMS2 by recruiting Fyn
-
•
Tyrosine phosphorylation of SMS2 increases its protein abundance
-
•
SMS2-generated DAG is critical for the development of insulin resistance
Biochemical Mechanism; Molecular Mechanism of Behavior; Diabetology
Introduction
Diet-induced obesity is often associated with increased free fatty acid (FFA) levels in circulating blood; these are considered to be a major risk factor for the development of type 2 diabetes and cardiovascular disease (Boden, 1999, Groop et al., 1991). On a cellular level, an excess of unsaturated FFAs increases the triacylglycerol (TAG) pool in lipid droplets (Rosenthal, 1981). Excess saturated FFAs increase the diacylglycerol (DAG) pool in the endoplasmic reticulum (ER) (Blachnio-Zabielska et al., 2013, Montell et al., 2001), and the accumulation of ceramide (Cer) (Chavez and Summers, 2003). The accumulation of intracellular Cer and DAG is thought to modulate intracellular signaling cascades, leading to impaired insulin signaling in vitro and in vivo (Chavez and Summers, 2012, Erion and Shulman, 2010).
Several lines of evidence link sphingolipid metabolism, in particular Cer processing, to the development of diabetes, liver steatosis, and cardiovascular diseases (Chavez and Summers, 2012, Holland et al., 2007). The first step in sphingolipid biosynthesis toward the sphingoid base is catalyzed by serine-palmitoyl CoA transferase (SPT), facilitating the condensation of L-serine and palmitoyl CoA. Pharmacological inhibition of SPT activity led to the retention of insulin sensitivity in muscle cells with saturated FFA-induced stress (Watson et al., 2009). Similarly, SPT2 heterozygous deficiency protected mice from diet-induced insulin resistance (Li et al., 2011). Condensation of sphingoid base with acyl-CoA is facilitated by a family of six ceramide synthases (CerSs). Each CerS exhibited a distinct tissue distribution and preferred acyl chain length (Mullen et al., 2012). Human CerS6 mRNA expression in adipose tissues is positively correlated with body mass index (Turpin et al., 2014), and ablation of CerS5 or CerS6 genes in mice prevents high-fat-diet-induced obesity and insulin resistance (Gosejacob et al., 2016, Turpin et al., 2014). By contrast, CerS2 haploinsufficiency led to a compensatory increase in C16-Cer and raised the susceptibility to diet-induced hepatosteatosis and insulin resistance (Raichur et al., 2014).
Cer is a key intermediate of the sphingolipid metabolism and the last common precursor of sphingomyelin (SM) and glucosylceramide biosyntheses. SM biosynthesis is catalyzed by two key enzymes, sphingomyelin synthase 1 (SMS1) and SMS2. Both enzymes act as phosphatidylcholine:ceramide phosphocholine transferase, by facilitating transfer of the phosphorylcholine head group from the phosphatidylcholine (PC) to Cer, generating SM and DAG (Luberto and Hannun, 1998). SMS1 resides in the Golgi apparatus, and its deficiency leads to mitochondrial dysfunction and a defect in insulin secretion (Yano et al., 2011, Yano et al., 2013). SMS2 is localized in both the Golgi apparatus and the plasma membrane (PM), in particular in lipid rafts. Its deficiency ameliorates diet-induced obesity and insulin resistance (Li et al., 2011, Mitsutake et al., 2011, Sugimoto et al., 2016). Although SMS1 and SMS2 deficiencies produce only a marginal effect for Cer, DAG, and SM content in tissues or cells, the molecular mechanism underlying the related phenotypes in knockout mice remains elusive.
We previously reported that GPRC5B deficiency in mice protects from diet-induced obesity and insulin resistance. GPRC5B recruitment of Src-family kinase (SFK) Fyn was critical for chronic inflammation and insulin resistance in adipose tissues (Kim et al., 2012). This suggests that GPRC5B plays a significant role in the metabolic regulation of insulin-sensitive organs, including the CNS, muscles, and adipose tissues. Interestingly, under nutrient excess condition, GPRC5B as well as SMS2 knockout mice produced highly similar metabolic phenotypes, especially in regard to insulin resistance, raising the possibility of affecting a similar influence on lipid metabolism. Furthermore, both proteins are similarly distributed in the PM and Golgi apparatus, and they co-fractionate into detergent-resistant membranes. Thus, we hypothesized that GPRC5B and SMS2 might share a functional connection to mediate lipid metabolism under metabolic stress conditions. In this study, we determined whether there is a functional connection between GPRC5B and SMS2 concerning the regulation of sphingolipid metabolism and insulin signaling under FFA-induced metabolic stress conditions.
Results
Metabolic Stress Facilitates the Interaction of GPRC5B and SMS2, and SMS2 Phosphorylation
Our initial study focused on identifying potential GPRC5B-interacting partners and the consequences of the resulting GPRC5B-mediated phosphorylation. Because SMS2 is considered to be a candidate interacting partner of GPRC5B, we tested whether their association occurs in cells. We co-transfected cells with expression plasmids encoding SMS2 and either wild-type (5b-WT) or mutant (5b-YF) GPRC5B. This mutant has a defect in Fyn-recruiting activity (Kim et al., 2012). SMS2 specifically interacted with both wild-type (WT) and mutant GPRC5B (Figure 1A). There were no obvious changes in this interaction, suggesting that Fyn-recruiting activity does not influence the association of GPRC5B and SMS2.
Figure 1.

Metabolic Stress Facilitates the Interaction of GPRC5B and SMS2 and SMS2 Phosphorylation
(A) Interaction between GPRC5B and SMS2 was measured using co-immunoprecipitation (IP). COS7 cells were transfected with expression plasmids for SMS2-Flag and either 5b-WT or mutant 5b-YF. Empty vector (EV) was used as a negative control. SMS2-Flag was immunoprecipitated using Flag beads. GPRC5B in the immunoprecipitates was detected using SDS-PAGE and western blotting.
(B and C) Palmitate exposure enhances GPRC5B-SMS2 interaction. (B) COS7 cells were transfected with the indicated expression plasmids. After 24 hr of transfection, culture medium was replaced with serum-free medium containing palmitate-BSA complex for the indicated times. GPRC5B-Flag was immunoprecipitated using Flag beads. Immunoprecipitates were digested with PNGase F to remove N-linked glycans from GPRC5B and then subjected to SDS-PAGE. Data are means ± SEM (n = 3; ANOVA, **p < 0.01). (C) COS7 cells were co-transfected with expression plasmids for SMS2-AcGFP and GPRC5B-Flag. After 8 hr of palmitate treatment, cells were fixed and labeled with anti-Flag antibody. Alexa 546-labeled secondary antibody was used to visualize GPRC5B. Fluorescence images were obtained with an FV1000 confocal microscope (Olympus). Sensitized emission fluorescence resonance energy transfer (FRET) was measured. FRET values for individual cells are plotted, and means ± SEM superimposed (n = 52; Student's t test). Palm, palmitate; BSA, vehicle control. Scale bar, 10 μm.
(D) SFK inhibitor, Su6656 blocked palmitate-induced SMS2 phosphorylation. Transfected COS7 cells with the indicated expression plasmids were pretreated with 10 μM of Su6656 and then stimulated with 0.5 mM palmitate for 8 hr. After stimulation, SMS2-Flag was immunoprecipitated, and then tyrosine phosphorylation was detected using anti-phosphotyrosine antibody. Data are means ± SEM (n = 3; ANOVA, ***p < 0.001).
(E) GPRC5B-SMS2 interaction enhances SMS2 tyrosine phosphorylation by recruiting SFK under metabolic stress. After 8 hr of palmitate treatment, SMS was immunoprecipitated.
See also Figures S1 and S2.
Metabolic stress can result from chronic nutritional changes, such as that caused by starvation or by induced cellular stress related to nutrient excess (Wellen and Thompson, 2010). Cell culture in the medium supplemented with palmitate is a representative model system for lipid-induced insulin resistance. We next examined whether this interaction is affected by palmitate-induced metabolic stress. The association of GPRC5B and SMS2 significantly increased by palmitate exposure (Figure 1B). By contrast, GPRC5B's association with SMS1 did not increase. The enhanced interaction between GPRC5B and SMS2 was also demonstrated using fluorescence resonance energy transfer microscopy, showing that metabolic stress facilitates the association of these two proteins (Figure 1C).
Phorbol 12-myristate 13-acetate (PMA) is a potent activator for protein kinase C (PKC), and generates reactive oxygen species. This triggers subsequent signaling cascades leading to activate SFK. PMA treatment also enhanced the interaction between GPRC5B and SMS2 (Figure S1). Interestingly, SMS2-associated GPRC5B showed altered glycosylation patterns (Figures 1A and S1A). We confirmed N-linked glycosylation of GPRC5B by PNGase F digestion (Figure S1B). Differential glycosylation of GPRC5B-SMS2 complex suggests that their interaction occurs in the ER and Golgi apparatus to influence the glycosylation process.
Next, we determined whether SMS2 phosphorylation depends on the SFK-recruiting activity of GPRC5B. Pretreatment with Su6656, an SFK inhibitor, completely blocked palmitate-induced SMS2 phosphorylation (Figure 1D), indicating that SFK is involved in this process. In addition, palmitate-induced tyrosine phosphorylation of SMS2 was apparently increased in cells co-transfected with WT GPRC5B but not with mutant GPRC5B (GPRC5B-YF) (Figure 1E), suggesting that the SFK-recruiting activity of GPRC5B is critical for SMS2 phosphorylation under metabolic stress. Palmitate exposure did not affect SMS1 phosphorylation (Figure 1E). The SFK-recruiting activity of GPRC5B also enhances tyrosine phosphorylation of SMS2 after PMA treatment (Figure S1C). Pretreatment with Su6656 also reduced SMS2 phosphorylation by PMA exposure (Figure S1D).
Phosphorylation of Cytoplasmic Tyrosine Residues Increases SMS2 Protein Abundance
PMA treatment gradually elevated SMS2 abundance (Figure S1A), and SMS2 level was positively correlated with its tyrosine phosphorylation (Figures S1C and S1D). To examine how phosphorylation of SMS2 regulates its protein abundance, we assessed Fyn-mediated phosphorylation of SMS2. We observed significant elevation of SMS2 protein level accompanied by robust tyrosine phosphorylation in cells co-transfected with constitutively active Fyn (Fyn-CA) (Figure 2A). Although WT Fyn (Fyn-WT) increases SMS2 phosphorylation, SMS2 protein level does not change (Figure 2A). This suggests that persistent phosphorylation is required to increase SMS2 protein level. We also compared Fyn-mediated phosphorylation of SMS1 with that of SMS2 and observed that Fyn phosphorylated SMS1 to a lesser extent than it did SMS2 (Figure S2A).
Figure 2.

Tyrosine Phosphorylation of SMS2 Increases its Protein Abundance
(A) Tyrosine phosphorylation increases the expression level of SMS2. Co-transfection with constitutively active Fyn (Fyn-CA, Y528F) and SMS2 in COS7 cells increased the expression of SMS2. Kinase-dead mutant Fyn (Fyn-KD, K299M) did not affect SMS2 phosphorylation. To quantify tyrosine phosphorylation level precisely, we adjusted cell lysates to have similar amounts of SMS2 in the immunoprecipitate (see Transparent Methods). An EGFP expression plasmid was used as an internal standard to control transfection efficiency.
(B) Tyrosine phosphorylation increases cell surface expression of SMS2 in COS7 cells. Cell surface proteins were labeled with sulfo-NHS-biotin and purified using streptavidin beads.
(C) GPRC5B increases SMS2 protein abundance under metabolic stress. COS7 cells were co-transfected with SMS expression plasmids and either wild-type (5b-WT) or mutant GPRC5B (5b-YF) plasmids. After 6 hr of transfection, culture medium was replaced with serum-free medium containing BSA or palmitate-BSA complex (Palm) for 16 hr. An AcGFP expression plasmid was used as an internal standard to control transfection efficiency.
(D) Tyrosine phosphorylation inhibits polyubiquitination of SMS2. HEK293 cells were co-transfected with the indicated expression plasmids. Polyubiquitination of SMS2 was detected from Flag immunoprecipitates by western blotting using anti-hemagglutinin (HA) antibody.
(E) Palmitate treatment reduces polyubiquitination of SMS2. COS7 cells were co-transfected with the indicated expression plasmids. After 8 hr of palmitate treatment, polyubiquitination of SMS2 was detected.
(A–C) Data are means ± SEM (n = 3; ANOVA, *p < 0.05, **p < 0.01, ****p < 0.0001). See also Figures S1 and S2.
As differential intracellular localization of SMS1 and SMS2 may reflect their distinct functions, we sought to determine whether phosphorylation of SMS2 affects its localization in the PM. Cell surface biotin labeling revealed that tyrosine phosphorylation also increased SMS2 levels in the PM (Figure 2B).
Because persistent phosphorylation is required to produce elevated SMS2 levels, we examined whether GPRC5B controlled SMS2 levels by treating cells with palmitate and then measuring SMS1 and SMS2 levels. Palmitate significantly increased SMS2 levels in cells co-transfected with 5b-WT but not in cells co-transfected with 5b-YF (Figure 2C, right). Moreover, palmitate treatment did not affect SMS1 levels (Figure 2C, left). These observations suggest that GPRC5B mediates the persistent phosphorylation of SMS2 and that this phosphorylation depends on GPRC5B interaction with SMS2.
According to the hidden Markov model, SMS2 possesses six transmembrane domains (Figure 3A). The number of amino acids in the N- and C-terminal tails of human SMS2 accounts for approximately 41% of the total amino acids in the protein (Figure 3A). Prediction of intrinsically disordered regions in SMS2 reveals that most are located in the cytoplasmic tails (Figures 3A and S3A). We speculate that SMS2 protein stability may be linked to a disorder-to-order transition induced by phosphorylation. Proteins containing long disordered segments tend to have a shorter half-life (van der Lee et al., 2014). Therefore, we tested whether phosphorylation affects the ubiquitination of SMS2, because ubiquitination is the typical process by which unfolded proteins are degraded in cells. As expected, tyrosine phosphorylation remarkably suppressed the polyubiquitination of SMS2 (Figure 2D). Furthermore, palmitate treatment also apparently reduced the polyubiquitination of SMS2 (Figure 2E). On the other hand, phosphorylation moderately affected SMS1 ubiquitination, which depended on the extent of its phosphorylation by Fyn (Figure S2B). These observations suggest that GPRC5B-mediated phosphorylation is linked to increased SMS2 levels under metabolic stress.
Figure 3.

Phosphomimetic Mutations Increase SMS2 Protein Abundance
(A) Illustration showing the putative transmembrane topology of SMS2. Identified phosphotyrosines (orange), cytoplasmic phosphotyrosine candidates (yellow), and active sites (green squares) in SMS2 are indicated. Intrinsically disordered region of SMS2 was predicted by Database of Disordered Protein Predictions (D2P2), http://d2p2.pro/.
(B–D) Phosphomimetic mutations enhance SMS2 protein abundance. (B) HEK293 cells were co-transfected with expression plasmids for AcGFP and either wild-type (WT) or phosphomimetic mutants of SMS2-Flag. The expression levels of the indicated mutants were compared with that of Y56E mutant (hatched bars). (C) The expression levels of the indicated phosphomimetics were compared with that of phosphorylated wild-type SMS2 (hatched bars). (D) Multiple tyrosine-to-glutamate substitutions increased SMS2 expression levels compared with WT.
(E) Phosphorylation or phosphomimetic mutation did not disrupt the enzymatic activity of SMS2. Super-SMS2 is a Y24/56/59/151/319/351E mutant. In vitro SM synthesis activity was analyzed by high-performance thin-layer chromatography (top). The phosphorylation status and the SMS amount in immunoprecipitates were confirmed by western blotting (bottom).
(F) Super-SMS2 is less polyubiquitinated than wild-type SMS2. HEK293 cells were co-transfected with expression plasmids for hemagglutinin (HA)-ubiquitin plasmid and either SMS2-Flag or Super-SMS2-Flag for 24 hr. SMS2-Flag and Super-SMS2-Flag were immunoprecipitated using Flag beads. Polyubiquitination was detected using anti-HA antibody.
(B–D) Data are means ± SEM (n = 3; ANOVA, **p < 0.01, ***p < 0.001). ns, Not significant; ud, undetectable. See also Figures S3 and S4.
Phosphomimetic Mutations Increase SMS2 Protein Abundance
Using mass spectrometry, we identified three N-terminal tyrosine residues (Y24, Y56, and Y59) that were phosphorylated by Fyn-CA (data not shown; UniProtKB: Q8NHU3, phosphorylation residues were deposited in the PhosphoSitePlus database, https://www.phosphosite.org/homeAction.action). Other potential phosphotyrosine residues (Y151, Y243, Y319, and Y351) are evolutionarily conserved (Figures 3A and S3B), indicating that they have a possible effect on SMS2 protein levels. However, the distribution of many basic amino acids around potential phosphotyrosine residues makes it difficult to determine whether these candidates are indeed phosphorylated. This is because the peptide fragments produced by protease digestion are too short to be analyzed using standard identification methods. To solve this problem, we used SMS2 mutants to determine whether site-specific mutations of SMS2 cytoplasmic tyrosine residues affect SMS2 protein levels.
Initially, we tested the effect of single point mutations (tyrosine to phenylalanine) or phosphomimetic mutations (tyrosine to glutamate) on SMS2 protein abundance. With the exception of cells transfected with Y59F and Y351F mutants, cells transfected with all the other tyrosine-to-phenylalanine mutants expressed decreased SMS2 levels compared with WT under basal conditions (Figure S4A). In the presence of Fyn-CA, Y59F, Y151F, and Y351F mutants failed to reduce SMS2 expression compared with WT (Figure S4B). On the other hand, all the single phosphomimetic mutations, except the Y351E mutant, increased SMS2 expression levels (Figure S4C). However, cells expressing Y56E, Y243E, and Y351E mutants still showed increased SMS2 expression compared with cells expressing phosphorylated WT in the presence of Fyn-CA (Figure S4B). We could not figure out site-specific mutations affecting SMS2 protein levels. For instance, the Y351F mutation had no effect on basal or phosphorylated expression levels (Figures S4A and S4B). In addition, Y351E did not affect basal expression levels. However, cells expressing both Y351E and Fyn-CA still expressed elevated SMS2 levels, suggesting that this mutation affected phosphorylation of other tyrosine residues (Figures S4C and S4D). These findings indicate that multiple redundant tyrosine phosphorylation of residues regulates SMS2 protein levels.
We prepared various combinations of phosphomimetic mutants from Y56E (Figures 3B and 3C). Interestingly, double mutations comprising Y56E and Y243E failed to enhance expression (Figures 3B–3D), even though single mutations of either Y56E or Y243E increased expression compared with WT (Figure S4C). This presumes that there is a plausible electrostatic interaction between the N terminus and the second cytoplasmic loop to maintain stability. In the analysis of various combinations of mutations at multiple tyrosine sites, the highest expression levels were observed in cells expressing mutations at Y24/56/59/151/319/351E (Figure 3D). This artificial construct, referred to as Super-SMS2, was used in further experiments. The enzymatic activity of SMS2 was not disrupted by tyrosine phosphorylation or phosphomimetic mutations (Figure 3E). Super-SMS2 showed reduced ubiquitination comparable to that of phosphorylated SMS2 (Figure 3F). Taken together, these observations suggest that redundant multiple phosphorylation of cytoplasmic tyrosine residues lead to reduced SMS2 ubiquitination and increased SMS2 protein abundance.
GPRC5B Deficiency Suppresses Palmitate-Induced Accumulation of Cer and DAG
Cer and DAG are important bioactive lipids that have been implicated in the regulation of cellular signaling pathways leading to insulin resistance under metabolic stress. Therefore, we determined whether GPRC5B affects the intracellular accumulation of Cer and DAG after palmitate exposure in mouse embryonic fibroblasts (MEFs). GPRC5B deficiency suppressed palmitate-induced accumulation of Cer and DAG (Figures 4A and 4B). We expected SM levels in WT cells to be elevated, because palmitate exposure increases SMS2 protein levels in a GPRC5B-dependent manner (cf. Figure 1E). However, palmitate exposure did not alter SM levels in either WT or GPRC5B knockout MEFs (Figures 4A and 4B), likely because of the tight regulation of SM homeostasis (Luberto and Hannun, 1998, Tafesse et al., 2007).
Figure 4.

GPRC5B Deficiency Suppresses Palmitate-Induced Accumulation of Cer and DAG
(A and B) Lipid compositions of MEFs in the indicated culture conditions were analyzed by high-performance thin-layer chromatography (HPTLC). Lipids were extracted from cultured cells in serum-free medium supplemented with BSA or palmitate-BSA complex for 16 hr. (A) Total lipid extracts (left) and saponified lipids (right) were developed on an HPTLC plate and visualized with copper sulfate/phosphoric acid. (B) Quantification of SM, Cer, and DAG contents in MEFs.
(C and D) Lipid compositions in MEFs analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). (C) LC-MS/MS analyses revealed that palmitate exposure induces de novo (sphinganine and dihydroceramides) and recycling (sphingosine and Cer) pathways for Cer synthesis in WT MEFs. (D) LC-MS/MS quantification of different molecular species of Cer and dihydroceramide.
(E and F) Lipid compositions of SMS2−/− MEFs transfected with an expression plasmid for Super-SMS2. After 24 hr of transfection, cells were cultured in serum-free DMEM containing BSA or palmitate-BSA complex for 16 hr. Image of HPTLC plate (E) and quantification of the indicated lipids (F) are shown.
(B–D) and (F) Data are means ± SEM (n = 3; ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). ns, Not significant.
Lipid analysis using liquid chromatography-tandem mass spectrometry revealed that sphinganine and dihydroceramides—products of de novo sphingolipid synthesis—were elevated in WT cells compared with GPRC5B knockout cells under metabolic stress (Figure 4C). Palmitate exposure increased Cer and sphingosine levels in WT compared with GPRC5B knockout MEFs (Figure 4C). In WT cells, Cer species containing a C16:0 acyl chain were predominantly elevated, increasing by 4-fold approximately. Very-long-chain Cers increased by about 1.5-fold following palmitate exposure (Figure 4D). By contrast in WT cells, C16:0 dihydroceramide predominantly decreased compared with very-long-chain dihydroceramides (Figure 4D). This molecular species difference of the way dihydroceramides and Cers are affected suggests that palmitate-dependent accumulation of C16:0 Cer is generated via both de novo and recycling pathways.
Since GPRC5B mediated the increase in SMS2 levels in palmitate-treated cells, we next determined whether SMS2 affects the accumulation of Cer. Transfecting SMS2 knockout cells with Super-SMS2 restored SM levels but did not affect palmitate-dependent Cer and DAG levels (Figures 4E and 4F). Further analysis of lipid content in SMS1, SMS2, and SMS1/2 knockout cells revealed no apparent palmitate-dependent changes in lipid levels, although Cer and DAG levels were moderately elevated in SMS1 knockout MEFs (Figures S5A and S5B). These observations suggest that palmitate-dependent accumulation of Cer is independent of SMS2 activity.
GPRC5B Enhances Metabolic Stress-Induced Sphingolipid Metabolism
We next investigated the change in Cer flux after palmitate exposure. After 2 hr of pulse labeling with NBD-C6-Cer (N-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-D-erythro-sphingosine), we quantified fluorescent Cer metabolites by high-performance thin-layer chromatography. Under basal conditions, total fluorescent lipid metabolites were comparable in WT and GPRC5B knockout MEFs (Figure 5A). Total fluorescent lipids were approximately 2-fold higher in WT than in GPRC5B knockout MEFs when cells were cultured in palmitate-containing medium (Figure 5A). NBD-derived SM, glucosylceramide (GlcCer), and Cer were elevated in WT cells compared with GPRC5B knockout MEFs under metabolic stress (Figure 5A). These observations suggest that GPRC5B increases the Cer pool in cellular membranes during metabolic stress. When cells were pulse labeled with NBD-SM for 2 hr, palmitate treatment slightly increased the total fluorescent lipid levels (Figure 5B). However, NBD-Cer and NBD-GlcCer showed an approximately 2-fold increase in WT compared with GPRC5B knockout MEFs under metabolic stress (Figure 5B). These results suggest that metabolic-stress-induced increase in sphingolipids is Cer dominant.
Figure 5.

GPRC5B Enhances Metabolic Stress-Induced Sphingolipid Metabolism
(A) Under metabolic stress, the level of Cer is greater in WT than in GPRC5B knockout MEFs. Cells were cultured in medium containing BSA or palmitate-BSA complex for 14 hr and then labeled with 5 μM NBD-ceramides for 2 hr at 37°C. Lipid extracts corresponding to the same amount of proteins were spotted onto high-performance thin-layer chromatography (HPTLC) plates and developed.
(B) Increased SM degradation contributes to expansion of the Cer pool in palmitate-treated WT MEFs. Cells were cultured in medium containing BSA or palmitate-BSA complex for 14 hr and then labeled with NBD-SM for 2 hr at 37°C. Lipid extracts corresponding to the same amount of proteins were spotted onto HPTLC plates and developed.
(C) NBD-SM flux in palmitate-treated cells. Cells were cultured in the presence or absence of palmitate-BSA complex for 14 hr and then labeled with 5 μM NBD-SM for 30 min on ice. Cells were washed three times and then chased in fresh medium and incubated at 37°C for the indicated times. Lipid extracts were spotted onto HPTLC plates and developed. Quantification of lipids is expressed as percentage of total fluorescence.
(D) Inhibition of neutral sphingomyelinase, not of acid sphingomyelinase, blocks NBD-Cer formation from NBD-SM. Cells were pretreated with 10 μM GW4869 or 50 μM amitriptyline for 24 hr and then exposed to palmitate. After culturing in palmitate-containing media for 14 hr, cells were labeled with 5 μM NBD-SM for 30 min on ice and then chased in fresh medium.
Data are means ± SEM (n = 3; ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). See also Figure S5.
As mentioned above, palmitate-induced accumulation of Cer may come from two biosynthetic pathways, de novo and recycling. In the recycling pathway, sphingomyelinase-mediated degradation of SM is a key step for generating Cer (Kitatani et al., 2008). In chase analysis, labeled NBD-SM was gradually degraded to NBD-Cer, which was used as a precursor for subsequent synthesis of NBD-GlcCer (Figure S6). Under basal conditions, the NBD-SM degradation rate in WT and GPRC5B knockout MEFs was similar (Figure 5C). However, under metabolic stress, NBD-SM degradation and subsequent NBD-GlcCer synthesis occurred more rapidly in WT than in GPRC5B knockout MEFs (Figures 5C and S6). Pretreating cells with GW4869, a selective inhibitor of neutral sphingomyelinase, inhibited NBD-Cer generation from NBD-SM, indicating that, during metabolic stress, the recycling pathway is enhanced (Figure 5D). Pretreating cells with amitriptyline, an inhibitor of acid sphingomyelinase, however, did not affect SM degradation (Figure 5D). Taken together, these observations are consistent with the results that GPCR5B mediates metabolic-stress-induced Cer accumulation through both de novo and recycling pathways.
Metabolic Stress Enhances SMS2-Mediated Generation of DAG
Labeling cells with NBD-Cer is the typical method used to measure the in situ activity of GlcCer synthase and SM synthases (Asano et al., 2012, Gupta et al., 2010). Palmitate exposure changes lipid composition in cellular membrane. The accumulated Cer and DAG in cellular membrane may impede precise measuring of SMS activity, because SMS can also catalyze the reverse reaction (Huitema et al., 2004, van Helvoort et al., 1994). Although pulse labeling with NBD-Cer showed that NBD-SM levels in WT MEFs are elevated under metabolic stress (Figure 5A), it was not obvious whether this was caused by increased SMS2 levels or by increased Cer levels. Therefore, we measure SMS2 activity by a modified method using NBD-PC (Villani et al., 2008). The SMS-mediated biochemical reaction yields SM and DAG. This assay taps into the Cer-dependent generation of DAG from PC. Since cellular membranes contain much more PC than Cer or DAG, this method may minimize interference derived from endogenous DAG or Cer to measure SMS activity in cells.
We confirmed that Super-SMS2-transfected cells had the highest phosphorylcholine transferase activity using the conventional in vitro SMS assay (Asano et al., 2012) (Figure 6A). In the assay of SMS activity using NBD-PC, a large increase in NBD-DAG was observed in cells co-labeled with NBD-PC and C6-Cer, indicating that NBD-DAG generation was apparently triggered by the addition of exogenous C6-Cer (Figure 6B). We then examined whether this reaction was catalyzed by SMS1 or SMS2. Transfection with expression constructs for SMS2 or Super-SMS2 significantly increased exogenous C6-Cer-dependent NBD-DAG production in SMS1/2 double-knockout MEFs (Figure 6C). By contrast, transfection with empty vector or expression construct for SMS1 failed to produce NBD-DAG (Figure 6C). Palmitate exposure increased PC-derived DAG production in WT cells compared with that in GPRC5B knockout MEFs (Figure 6D). We confirmed that transfection of GPRC5B in GPRC5B knockout MEFs restored PC-derived DAG formation under metabolic stress (Figure 6E). These observations suggest that the increase of PC-derived DAG production in WT cells under palmitate-induced metabolic stress is caused by increased SMS2 protein levels or by increased endogenous Cer levels.
Figure 6.

Metabolic Stress Enhances SMS2-Generated DAG
(A) In vitro SMS assay. SMS2 was derived from cell lysates from SMS1/2 double-knockout MEFs transfected with the indicated constructs. Equal protein amounts of cell lysate were mixed with liposomes prepared from egg PC and NBD-Cer and then incubated in 37°C for 1 hr. Lipids were extracted from the reaction mixture and then synthesized SM was measured by high-performance thin-layer chromatography (HPTLC).
(B) Time course analysis of PC-derived DAG formation induced by the addition of exogenous C6-Cer. Cells were labeled with NBD-C6-PC alone or NBD-C6-PC plus C6-Cer for 30 min on ice, washed, and chased at the indicated times. Extracted lipids were developed via HPTLC, and fluorescence images were captured with a LAS3000 imaging system. To visualize total lipids, the same HPTLC plate was treated with cupric acid. PC-derived DAG was quantified from the fluorescence image.
(C) SMS2 expression is required for Cer-Dependent DAG formation from PC. SMS1/2 double-knockout MEFs were transfected with the indicated expression plasmids. After 48 hr of transfection, cells were labeled with NBD-PC or NBD-PC plus 5 μM C6-Cer for 30 min on ice and then washed twice and incubated at 37°C for 10 min. Fluorescence intensity of the extracted lipids was measured using a microplate reader, and the amount of lipids was determined. Same amount of fluorescent lipids was spotted on an HPTLC plate and developed.
(D) GPRC5B deficiency reduces PC-derived DAG formation under metabolic stress. Cells were cultured in medium containing BSA or palmitate-BSA complex for 16 h and then labeled with NBD-PC for 30 min on ice. After NBD-PC labeling, cells were washed twice with fresh medium and incubated at 37°C for 15 min. The same amount of fluorescent lipids was spotted onto an HPTLC plate and developed.
(E) GPRC5B enhances PC-derived DAG production by palmitate exposure. GPRC5B knockout MEFs were transfected with expression plasmid for human GPRC5B. After exposure to palmitate for 16 hr, cells were labeled with NBD-PC for 30 min on ice and then washed twice and incubated in 37°C for 15 min. NBD-DAG formation from NBD-PC were measured.
(A–E) Data are means ± SEM (n = 3; ANOVA, **p < 0.01, ****p < 0.0001).
SMS2-Generated DAG Is Responsible for JNK-Dependent Impairment of Insulin Signaling
Because Cer and DAG are bioactive lipids that are thought to impair insulin signaling, we measured insulin-evoked AKT activation in SMS knockout MEFs. Surprisingly, palmitate exposure failed to impair insulin signaling in SMS2 knockout MEFs (Figure 7A), even though SMS2-null cells showed comparable accumulation of Cer and DAG under metabolic stress (Figure S6).
Figure 7.

SMS2-Generated DAG Is Responsible for JNK-Dependent Impairment of Insulin Signaling
(A) SMS2−/− MEFs are insulin sensitive. Cells were pretreated with BSA or palmitate-BSA complex in serum-free medium for 16 hr and then stimulated with 1 nM insulin for 5 min. Insulin-induced phosphorylation (S473) of AKT was measured by western blotting.
(B) Super-SMS2 expression inhibits insulin sensitivity. SMS2 or Super-SMS2 was expressed in SMS2 knockout MEFs by transfection for 24 hr. After exposure in the medium containing 0.5 mM palmitate-BSA for 16 hr, cells were stimulated with 2 nM insulin for 5 min. AKT phosphorylation (S473) was measured by western blotting.
(C) Super-SMS2 enhances palmitate-induced JNK activity. SMS2−/− MEFs were transfected with empty vector or expression plasmid for Super-SMS2. After 24 hr of transfection, cells were cultured in medium containing BSA or palmitate-BSA complex for 16 hr. Phosphorylation (T183/Y185) of active JNK and IRS1 (S1101) were measured by western blotting.
(D) Super-SMS2 expression enhances the formation of metabolic-stress-induced DAG from PC. Transfection and culture conditions were the same as described in (C). Cells were labeled with NBD-PC for 30 min on ice. Cells were washed twice with fresh medium and then incubated in 37°C for 15 min.
(E and F) GPRC5B deficiency lessens palmitate-induced insulin resistance (E) and JNK activity (F). Wild-type and GPRC5B−/− MEFs were cultured in serum-free medium supplemented with BSA or palmitate-BSA complex for 16 hr. Cells were stimulated with 1 nM insulin for 5 min.
(G) Expression of Super-SMS2 in GPRC5B−/− MEFs restores palmitate-induced insulin resistance and JNK activation. Experimental conditions were the same as described in (B).
(H) Schematic diagram illustrating SMS2-mediated insulin resistance under metabolic stress. Metabolic stress induces SMS2 phosphorylation, which results in SMS2 upregulation. In parallel, metabolic stress also induces the accumulation of Cer, which are readily converted to SM to produce DAG via SMS2 in the plasma membrane. SMS2-produced DAG is considered to be a second messenger that activates the PKC-JNK axis. This is the primary means of impairing insulin action.
(B–G) Data are means ± SEM (n = 3; ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). ns, Not significant. See also Figures S6 and S7.
It is well known that DAG is a potent mediator for the activation of PKC (Takai et al., 1979). Transfecting SMS2 knockout MEFs with either SMS2 or Super-SMS2 restored palmitate-induced insulin resistance (Figure 7B). Expression of Super-SMS2 in SMS2 knockout MEFs was sufficient to activate JNK under metabolic stress (Figure 7C). In addition, we observed increased phosphorylation of IRS1 (Ser1101) in Super-SMS2-transfected MEFs, suggesting that PKCθ was activated (Li et al., 2004) (Figure 7C). Importantly, we also observed a remarkable elevation of PC-derived DAG formation in Super-SMS2-transfected MEFs (Figure 7D) that was strongly correlated with JNK activation (Figure 7C). This suggests that SMS2-generated DAG is a critical mediator of palmitate-induced impairment of insulin signaling via PKC-JNK axis.
We next examined insulin signaling in GPRC5B knockout MEFs under metabolic stress. As expected, insulin-evoked AKT phosphorylation was increased in GPRC5B knockout MEFs compared with that in WT (Figure 7E). This indicated that GPRC5B deficiency protected the cells from lipid-induced insulin resistance. At the same time, palmitate-induced JNK activity was higher in WT than in GPRC5B knockout MEFs (Figure 7F). Since palmitate treatment increases SMS2-generated DAG in WT cells (Figures 6D and 6E), it suggests that GPRC5B mediates SMS2-generated DAG production leading to JNK-dependent insulin resistance. We demonstrated that transfection of Super-SMS2, but not of SMS2, into GPRC5B knockout MEFs impairs insulin signaling under metabolic stress (Figure 7G). Taken together, these observations provide evidence that GPRC5B plays a critical role in palmitate-induced insulin resistance by regulating sphingolipid metabolism at the cellular and molecular levels.
In summary, we demonstrated that palmitate exposure increases SMS2 levels through a GPRC5B-mediated tyrosine phosphorylation. With metabolic stress, GPRC5B recruitment of SFK plays an important role in the phosphorylation of SMS2. Concurrently, GPRC5B activates de novo Cer synthesis and Cer recycling pathways, causing Cer to accumulate, which is required for the generation of DAG by SMS2. Thus, under conditions of metabolic stress, SMS2-generated DAG plays a pivotal role in impairing insulin signaling by enhancing the PKC-JNK signaling axis. These events are summarized in Figure 7H.
Discussion
Understanding the physiological mechanism underlying saturated-FFA-induced insulin resistance has remained elusive for many years, because several complicated pathways including lipid metabolism and signal transduction are involved. In the present study, GPRC5B knockout MEFs limit the accumulation of Cer and DAG induced by palmitate exposure (Figures 4A–4D). This is accompanied by a reduction in SM degradation (Figures 5C and S5), suggesting that palmitate-induced Cer accumulation occurs not only through de novo Cer synthesis but also through Cer recycling pathways. Activated recycling pathways may also contribute to tight regulation of SM homeostasis. In our experiments, palmitate did not alter SM levels (Figures 4A and 4B), even though a GPRC5B-mediated event increased SMS2 levels in palmitate-treated cells (Figure 2C). This is consistent with modest changes in SM content reported for CHO cells overexpressing SMS1 and SMS2, and in cells in which SMS was knocked down using small interfering RNA(Ding et al., 2008).
To understand how Cers are related to the development of insulin resistance, it is important to consider their subcellular localization. Cers are synthesized in the ER and are rapidly converted to SM or glucosylceramides in the Golgi apparatus, whereby Cer transport protein (CERT) plays a key role in SM synthesis (Hanada et al., 2003). SMS1-induced DAG-dependent protein kinase D (PKD) activation at the Golgi apparatus negatively regulates CERT activity by controlling phosphoinositide 4-phosphate levels (Capasso et al., 2017). This negative feedback loop governs the central metabolic flow of sphingolipids. Consequently, this may restrict SM and DAG levels at the Golgi apparatus. By contrast, sphingolipid metabolism at the PM, in particular the recycling pathway, is different from what occurs at the Golgi apparatus. In the present study, we observed that PC-derived DAG production is completely dependent on SMS2, but not SMS1 activity, which reflects different DAG subcellular compartments (Figure 6C). Cer-dependent insulin resistance may occur by blocking the translocation of AKT to the PM (Stratford et al., 2001), or by activating protein phosphatase 2A-dephosphorylated AKT (Chavez et al., 2003, Teruel et al., 2001). Despite considerable Cer accumulation in palmitate-treated SMS2 knockout MEFs (Figure S6), we did not observe impaired insulin signaling in SMS2 knockout MEFs (Figures 7A and 7B).
Like Cers, DAG is also an important bioactive lipid related to insulin resistance. Increased intracellular DAG leads to activation of PKCθ and PKCɛ in skeletal muscle and liver, respectively, which, in turn, results in impaired IRS1-IRS2-dependent insulin signaling (Galbo et al., 2013, Jornayvaz and Shulman, 2012, Li et al., 2004, Szendroedi et al., 2014). In our model system, palmitate-induced DAG accumulation may be generally accomplished by de novo synthesis; DAG is synthesized by the subsequent esterification of fatty acyl-CoA to glycerol-3-phosphate and dephosphorylation in the ER. We demonstrated that there are no apparent changes in the extent of palmitate-induced DAG accumulation between WT and SMS2 knockout MEFs (Figure S6). Nevertheless, insulin sensitivity is not disrupted in SMS2 knockout MEFs (Figure 7A). Very importantly, palmitate-dependent impairment of insulin signaling is strongly correlated with SMS2-generated DAG (Figures 6B–6E, and 7D). A high expression level of SMS2 exacerbates palmitate-induced insulin resistance (Figures 7B and 7G). These results suggest that the DAG compartment in the PM strongly affects the triggering signaling cascades related to insulin resistance, rather than intracellular DAG in the ER.
Saturated fatty acids activate JNK signaling pathway. This activation is critical for impairing insulin action (Jaeschke and Davis, 2007). Tyrosine phosphorylation events mediated by c-Src or Fyn are required for palmitate-induced JNK activation in fibroblasts (Holzer et al., 2011, Kant et al., 2017). We confirmed that pretreatment of SFK inhibitor prevents palmitate-stimulated JNK activation (Figure S7A). PKCs are also critical for palmitate-induced JNK activation related to insulin resistance. Loss of PKCs prevents palmitate-stimulated JNK activation (Jaeschke and Davis, 2007). PMA-induced JNK activation is not altered in GPRC5B knockout MEFs, indicating that the PKC-JNK axis is intact in these cells (Figure 7B). We demonstrated that SMS2-produced DAG is sufficient for activating JNK (Figures 7C and 7G). This suggests that SMS2 expression in this pathway is functionally significant.
SMS1 and SMS2 are essential enzymes for SM synthesis. However, there is little evidence to show how they are regulated. In the present study, we demonstrated that GPRC5B and SMS2 show increased association, and subsequent tyrosine phosphorylation by recruiting an SFK occurs in palmitate-exposed cells (Figures 1B–1E). Multiple phosphorylation on cytoplasmic tyrosine residues plays an important role in regulating SMS2 protein abundance (Figures 2 and S1). Thus a transmembrane adapter-like function of GPRC5B is critical for the cellular response to palmitate excess. We conclude that GPRC5B plays a key role in the mechanism underlying the formation of SMS2-generated DAG during metabolic stress-associated sphingolipid metabolism, which is required for lipid-induced insulin resistance. We believe that our findings contribute to a deeper understanding of the role of sphingolipid metabolism in the maintenance of cellular homeostasis and controlling the cellular metabolic stress response.
Limitations of the Study
We emphasize that our results demonstrate GPRC5B-mediated phosphorylation and accumulation of SMS2 under metabolic stress conditions in transfected cell lines. Although we demonstrated the increased DAG generation mediated by SMS2 activity in palmitate-treated MEFs, we could not show a concomitant increase of endogenous SMS2 protein abundance in MEFs. The increased SMS2-mediated DAG generation in palmitate-treated MEFs likely represents a critical step for lipid-induced insulin resistance. Absence of validated SMS1 and SMS2 antibodies for use in cultured cells and mice limited further experimental insights in this study.
We note that our results highlight the biological significance of GPRC5B and SMS2 in cellular model systems under metabolic stress, but further investigations in vivo are required to clarify the pathophysiological role of GPRC5B in obesity and type 2 diabetes.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank professor T. Okazaki and Dr. M. Taniguchi for kindly providing SMS knockout MEFs and SMS-expression plasmids. We thank Ms. T. Shimizu for technical assistance. We are grateful to the Support Unit for Bio-Material Analysis, Research Resources Division, RIKEN Center for Brain Science, for help with the nucleotide sequencing analysis. This work was in part supported by Integrated Lipidology Program of RIKEN (Y.H.), and RIKEN Brain Science Institute (Y.H.).
Author Contributions
Conceptualization, Y.-J.K. and Y.H.; Methodology, Y.-J.K.; Investigation, Y.-J.K. and P.G.; Writing – Original Draft, Y.-J.K. and Y.H.; Funding Acquisition, Y.H.; Resources, Y.-J.K.; Supervision, Y.-J.K. and Y.H.
Declaration of Interests
The authors declare no competing interests.
Published: October 26, 2018
Footnotes
Supplemental Information includes Transparent Methods and seven figures and can be found with this article online at https://doi.org/10.1016/j.isci.2018.10.001.
Data and Software Availability
Intrinsically disordered regions in SMS1 and SMS2 were predicted by the Database of Disordered Protein Predictions (D2P2, http://d2p2.pro/)
Supplemental Information
References
- Asano S., Kitatani K., Taniguchi M., Hashimoto M., Zama K., Mitsutake S., Igarashi Y., Takeya H., Kigawa J., Hayashi A. Regulation of cell migration by sphingomyelin synthases: sphingomyelin in lipid rafts decreases responsiveness to signaling by the CXCL12/CXCR4 pathway. Mol. Cell Biol. 2012;32:3242–3252. doi: 10.1128/MCB.00121-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blachnio-Zabielska A.U., Zabielski P., Jensen M.D. Intramyocellular diacylglycerol concentrations and [U-(13)C]palmitate isotopic enrichment measured by LC/MS/MS. J. Lipid Res. 2013;54:1705–1711. doi: 10.1194/jlr.D035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G. Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc. Assoc. Am. Phys. 1999;111:241–248. doi: 10.1046/j.1525-1381.1999.99220.x. [DOI] [PubMed] [Google Scholar]
- Capasso S., Sticco L., Rizzo R., Pirozzi M., Russo D., Dathan N.A., Campelo F., van Galen J., Holtta-Vuori M., Turacchio G. Sphingolipid metabolic flow controls phosphoinositide turnover at the trans-Golgi network. EMBO J. 2017;36:1736–1754. doi: 10.15252/embj.201696048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez J.A., Knotts T.A., Wang L.P., Li G., Dobrowsky R.T., Florant G.L., Summers S.A. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem. 2003;278:10297–10303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- Chavez J.A., Summers S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003;419:101–109. doi: 10.1016/j.abb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- Chavez J.A., Summers S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15:585–594. doi: 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Ding T., Li Z., Hailemariam T., Mukherjee S., Maxfield F.R., Wu M.P., Jiang X.C. SMS overexpression and knockdown: impact on cellular sphingomyelin and diacylglycerol metabolism, and cell apoptosis. J. Lipid Res. 2008;49:376–385. doi: 10.1194/jlr.M700401-JLR200. [DOI] [PubMed] [Google Scholar]
- Erion D.M., Shulman G.I. Diacylglycerol-mediated insulin resistance. Nat. Med. 2010;16:400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbo T., Perry R.J., Jurczak M.J., Camporez J.P., Alves T.C., Kahn M., Guigni B.A., Serr J., Zhang D., Bhanot S. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc. Natl. Acad. Sci. U S A. 2013;110:12780–12785. doi: 10.1073/pnas.1311176110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosejacob D., Jager P.S., Vom Dorp K., Frejno M., Carstensen A.C., Kohnke M., Degen J., Dormann P., Hoch M. Ceramide synthase 5 Is essential to maintain c16:0-ceramide pools and contributes to the development of diet-induced obesity. J. Biol. Chem. 2016;291:6989–7003. doi: 10.1074/jbc.M115.691212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groop L.C., Saloranta C., Shank M., Bonadonna R.C., Ferrannini E., DeFronzo R.A. The role of free fatty acid metabolism in the pathogenesis of insulin resistance in obesity and noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1991;72:96–107. doi: 10.1210/jcem-72-1-96. [DOI] [PubMed] [Google Scholar]
- Gupta V., Patwardhan G.A., Zhang Q.-J., Cabot M.C., Jazwinski S.M., Liu Y.-Y. Direct quantitative determination of ceramide glycosylation in vivo: a new approach to evaluate cellular enzyme activity of glucosylceramide synthase. J. Lipid Res. 2010;51:866–874. doi: 10.1194/jlr.D002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., Nishijima M. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- Holland W.L., Brozinick J.T., Wang L.P., Hawkins E.D., Sargent K.M., Liu Y., Narra K., Hoehn K.L., Knotts T.A., Siesky A. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Holzer R.G., Park E.J., Li N., Tran H., Chen M., Choi C., Solinas G., Karin M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitema K., van den Dikkenberg J., Brouwers J.F.H.M., Holthuis J.C.M. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004;23:33. doi: 10.1038/sj.emboj.7600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke A., Davis R.J. Metabolic stress signaling mediated by mixed-lineage kinases. Mol. Cell. 2007;27:498–508. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jornayvaz F.R., Shulman G.I. Diacylglycerol activation of protein kinase cɛ and hepatic insulin resistance. Cell Metab. 2012;15:574–584. doi: 10.1016/j.cmet.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant S., Standen C.L., Morel C., Jung D.Y., Kim J.K., Swat W., Flavell R.A., Davis R.J. A protein scaffold coordinates SRC-mediated JNK activation in response to metabolic stress. Cell Rep. 2017;20:2775–2783. doi: 10.1016/j.celrep.2017.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.J., Sano T., Nabetani T., Asano Y., Hirabayashi Y. GPRC5B activates obesity-associated inflammatory signaling in adipocytes. Sci. Signal. 2012;5:ra85. doi: 10.1126/scisignal.2003149. [DOI] [PubMed] [Google Scholar]
- Kitatani K., Idkowiak-Baldys J., Hannun Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008;20:1010–1018. doi: 10.1016/j.cellsig.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Soos T.J., Li X., Wu J., Degennaro M., Sun X., Littman D.R., Birnbaum M.J., Polakiewicz R.D. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101) J. Biol. Chem. 2004;279:45304–45307. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
- Li Z., Zhang H., Liu J., Liang C.P., Li Y., Li Y., Teitelman G., Beyer T., Bui H.H., Peake D.A. Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol. Cell Biol. 2011;31:4205–4218. doi: 10.1128/MCB.05893-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luberto C., Hannun Y.A. Sphingomyelin synthase, a potential regulator of intracellular levels of ceramide and diacylglycerol during SV40 transformation. Does sphingomyelin synthase account for the putative phosphatidylcholine-specific phospholipase C? J. Biol. Chem. 1998;273:14550–14559. doi: 10.1074/jbc.273.23.14550. [DOI] [PubMed] [Google Scholar]
- Mitsutake S., Zama K., Yokota H., Yoshida T., Tanaka M., Mitsui M., Ikawa M., Okabe M., Tanaka Y., Yamashita T. Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J. Biol. Chem. 2011;286:28544–28555. doi: 10.1074/jbc.M111.255646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell E., Turini M., Marotta M., Roberts M., Noe V., Ciudad C.J., Mace K., Gomez-Foix A.M. DAG accumulation from saturated fatty acids desensitizes insulin stimulation of glucose uptake in muscle cells. Am. J. Physiol. Endocrinol. Metab. 2001;280:E229–E237. doi: 10.1152/ajpendo.2001.280.2.E229. [DOI] [PubMed] [Google Scholar]
- Mullen T.D., Hannun Y.A., Obeid L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012;441:789–802. doi: 10.1042/BJ20111626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichur S., Wang S.T., Chan P.W., Li Y., Ching J., Chaurasia B., Dogra S., Ohman M.K., Takeda K., Sugii S. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014;20:687–695. doi: 10.1016/j.cmet.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Rosenthal M.D. Accumulation of neutral lipids by human skin fibroblasts: differential effects of saturated and unsaturated fatty acids. Lipids. 1981;16:173–182. doi: 10.1007/BF02535435. [DOI] [PubMed] [Google Scholar]
- Stratford S., DeWald D.B., Summers S.A. Ceramide dissociates 3'-phosphoinositide production from pleckstrin homology domain translocation. Biochem. J. 2001;354:359–368. doi: 10.1042/0264-6021:3540359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M., Shimizu Y., Zhao S., Ukon N., Nishijima K., Wakabayashi M., Yoshioka T., Higashino K., Numata Y., Okuda T. Characterization of the role of sphingomyelin synthase 2 in glucose metabolism in whole-body and peripheral tissues in mice. Biochim. Biophys. Acta. 2016;1861:688–702. doi: 10.1016/j.bbalip.2016.04.019. [DOI] [PubMed] [Google Scholar]
- Szendroedi J., Yoshimura T., Phielix E., Koliaki C., Marcucci M., Zhang D., Jelenik T., Muller J., Herder C., Nowotny P. Role of diacylglycerol activation of PKCtheta in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. U S A. 2014;111:9597–9602. doi: 10.1073/pnas.1409229111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafesse F.G., Huitema K., Hermansson M., van der Poel S., van den Dikkenberg J., Uphoff A., Somerharju P., Holthuis J.C. Both sphingomyelin synthases SMS1 and SMS2 are required for sphingomyelin homeostasis and growth in human HeLa cells. J. Biol. Chem. 2007;282:17537–17547. doi: 10.1074/jbc.M702423200. [DOI] [PubMed] [Google Scholar]
- Takai Y., Kishimoto A., Kikkawa U., Mori T., Nishizuka Y. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochem. Biophys. Res. Commun. 1979;91:1218–1224. doi: 10.1016/0006-291x(79)91197-5. [DOI] [PubMed] [Google Scholar]
- Teruel T., Hernandez R., Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50:2563–2571. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- Turpin S.M., Nicholls H.T., Willmes D.M., Mourier A., Brodesser S., Wunderlich C.M., Mauer J., Xu E., Hammerschmidt P., Bronneke H.S. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014;20:678–686. doi: 10.1016/j.cmet.2014.08.002. [DOI] [PubMed] [Google Scholar]
- van Helvoort A., van't Hof W., Ritsema T., Sandra A., van Meer G. Conversion of diacylglycerol to phosphatidylcholine on the basolateral surface of epithelial (Madin-Darby canine kidney) cells. Evidence for the reverse action of a sphingomyelin synthase. J. Biol. Chem. 1994;269:1763–1769. [PubMed] [Google Scholar]
- van der Lee R., Lang B., Kruse K., Gsponer J., Sánchez de Groot N., Huynen Martijn A., Matouschek A., Fuxreiter M., Babu M.M. Intrinsically disordered segments affect protein half-life in the cell and during evolution. Cell Rep. 2014;8:1832–1844. doi: 10.1016/j.celrep.2014.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani M., Subathra M., Im Y.B., Choi Y., Signorelli P., Del Poeta M., Luberto C. Sphingomyelin synthases regulate production of diacylglycerol at the Golgi. Biochem. J. 2008;414:31–41. doi: 10.1042/BJ20071240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M.L., Coghlan M., Hundal H.S. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem. J. 2009;417:791–801. doi: 10.1042/BJ20081149. [DOI] [PubMed] [Google Scholar]
- Wellen K.E., Thompson C.B. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol. Cell. 2010;40:323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M., Watanabe K., Yamamoto T., Ikeda K., Senokuchi T., Lu M., Kadomatsu T., Tsukano H., Ikawa M., Okabe M. Mitochondrial dysfunction and increased reactive oxygen species impair insulin secretion in sphingomyelin synthase 1-null mice. J. Biol. Chem. 2011;286:3992–4002. doi: 10.1074/jbc.M110.179176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M., Yamamoto T., Nishimura N., Gotoh T., Watanabe K., Ikeda K., Garan Y., Taguchi R., Node K., Okazaki T. Increased oxidative stress impairs adipose tissue function in sphingomyelin synthase 1 null mice. PLoS One. 2013;8:e61380. doi: 10.1371/journal.pone.0061380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.