

Abstract
Drug-drug interactions have been demonstrated to alter CYP2D6 enzyme phenotype due to inhibitor ingestion though it is unclear how substrate interactions affect phenotype. This was a pragmatic clinical trial examining the kinetics of a CYP2D6 enzyme probe drug with and without CYP2D6 dependent substrates. Patients were enrolled into an inpatient study unit, orally administered a 2 mg microdose of dextromethorphan to probe enzyme activity with and without CYP2D6 dependent drug-drug interactions. Thirty-nine subjects were enrolled in this trial. Twelve subjects were on no CYP2D6 dependent drugs and 27 were on one or more CYP2D6 dependent drugs. There were 1 poor metabolizer, 5 intermediate metabolizers, 31 normal metabolizers, and 2 ultra-rapid metabolizers. Those with co-ingestion of another CYP2D6 dependent drug were 9.49 (95% CI: 1.54, 186.41; p = 0.01) times more likely to have genotype-phenotype discordance based upon the 3 hours DX/DM ratio. CYP2D6 substrate co-ingestions can cause genotype-phenotype discordance.
Keywords: CYP2D6, discordance, enzyme activity, metoprolol, pharmacogenomics, phenotype
INTRODUCTION
Cytochrome 2D6 (CYP2D6) is one of the most important drug metabolizing enzymes for human elimination of xenobiotics.(1, 2) The enzyme metabolizes approximately 25% of all drugs and genetic polymorphism in the CYP2D6 gene affects the pharmacokinetics of approximately 50% of these drugs.(3) This polymorphism can lead to drug ineffectiveness,(4–6) adverse drug events (ADEs),(7–9) or supra-effective therapy,(5, 6) even at low doses.
With a few notable exceptions, such as codeine, genotyping of CYP2D6 has not resulted in better prediction of drug effectiveness or safety commensurate with the number of drugs the enzyme metabolizes.(10–12) A contributing factor to this may be due to discordance of the predicted phenotype from the underlying CYP2D6 genotype.(12) For instance, the strong CYP2D6 inhibitor, paroxetine, has been shown to alter the pharmacokinetics of metoprolol leading to CYP2D6 genotype-phenotype discordance.(13) This shift in enzyme phenotype may not be limited to enzyme inhibitors. CYP2D6 substrates may occupy the enzyme’s active site thereby interfering with the metabolism of additional drugs, resulting in genotype-phenotype discordance. For instance, the analgesic effectiveness of hydrocodone, which is dependent upon CYP2D6 for conversion to an active metabolite hydromorphone, is better predicted by the absence of a CYP2D6 drug-drug interaction than CYP2D6 genotype.(11) This phenotypic conversion may be variable between patients with different genotypes.
Polypharmacy is becoming increasingly common, and the effect of drug-drug interactions on drug effectiveness and safety is underappreciated. Forty-eight percent of people in the US take at least one prescription medication and more than 76% of people 60 years or older are on two or more.(14–16) The clinical effects of drug-drug interactions in people with genetic polymorphism adds additional complexity to this problem. Given the frequency of polypharmacy, we must strive to understand these drug-drug and drug-gene interactions in order to better predict the effectiveness and safety of prescribed drugs. Our primary objective was to determine if CYP2D6 drug substrates result in genotype-phenotype discordance of enzyme function and secondarily, whether this translates to discordance of predicted clinical effect.
RESULTS
Thirty-nine subjects were enrolled in the pragmatic phenotyping analysis. CYP2D6 genotypes were not known at the time of the kinetic analyses. The demographic distribution was similar between groups (Table 1). Subjects were taking between zero and twenty-four total drugs. Twelve subjects were taking zero CYP2D6 dependent drugs and constituted the no-interaction group. The number of CYP2D6 dependent drugs taken in the interaction group was one (n=16), 2 (n=8), and 3 (n=3). All the co-ingested drugs patients reported taking within 3 half lives were found and confirmed by the qualitative urine drug screen performed by Labcorp.(17) Twenty-five (62.5%) subjects were taking metoprolol succinate for management of their hypertension. As expected, the majority of subjects were normal metabolizers (n=31, 79%). The negative log of the 3 hour DX/DM ratio was predicted by the negative log of DM AUC (R square 0.85, p<0.0001) with this microdose protocol.
Table 1.
Demographics of study participants.
| Characteristic | All N=39 |
Not Taking a CYP2D6 Dependent Drug n=12 |
Taking CYP2D6 Dependent Drug n=27 |
|---|---|---|---|
| Age, median (IQR) | 58 (53, 64) | 60 (58, 65) | 57 (52, 63) |
| Male Gender, n (%) | 22 (56%) | 7 (58%) | 15 (56%) |
| Race, n (%) Black or African American White |
17 (44%) 22 (56%) |
2 (17%) 10 (83%) |
15 (56%) 12 (44%) |
| Hispanic Ethnicity, n (%) Hispanic or Latino |
4 (10%) |
2 (17%) |
2 (7%) |
| CYP2D6 Genotype, n (%) Poor Intermediate Normal Ultra rapid |
1 (3%) 5 (13%) 31 (79%) 2 (5%) |
0 (0%) 3 (25%) 9 (75%) 0 (0%) |
1 (4%) 2 (7%) 22 (81%) 2 (7%) |
| Number of CYP2D6 Dependent Drugs, including metoprolol succinate 0 1 2 3 |
12 (31%) 16 (41%) 8 (21%) 3 (8%) |
12 (100%) NA NA NA |
NA 16 (59%) 8 (30%) 3 (11%) |
| Taking a CYP2D6 inhibitor, n | 4 | NA | 4 |
| DM/DX Ratio, median (IQR) | 0.01 (0.01- 0.11) |
0.003 (0.003–0.005) | 0.045 (0.009–0.123) |
| MT/MT OH Ratio, median (IQR) |
1.80 (1.12- 4.51) |
NA | 1.80 (1.12–4.51) |
Genotype-Phenotype Discordance:
Genotype-phenotype discordance was observed in 14 (35%) of the subjects (Table 2). Those with co-ingestion of another CYP2D6 dependent drug were 9.49 (95% CI: 1.54, 186.41; p = 0.01) times more likely to have genotype-phenotype discordance based upon the 3-hour DX/DM ratio. There was one subject that was taking no CYP2D6 dependent co-ingestions that genotyping demonstrated to be a normal metabolizer but phenotyping showed them to be an intermediate metabolizer. Age, ethnicity, and co-ingestion of CYP2D6 dependent drugs were determined to meet covariate selection criteria and were included in the full model. There was collinearity between ethnicity and CYP2D6 co-ingestion, and therefore ethnicity was removed from the model. When included in the full model, age was found to be nonsignificant (p = 0.21) in the presence of all other variables. Lastly, genotype was found to be insignificant when stratified by co-ingestion and excluding the person with a PM genotype (p = 0.26). See Table 3 for model estimates.
Table 2.
Subjects with genotype-phenotype discordance stratified by the number of CYP2D6 dependent drugs co-ingested.
| Number of CYP2D6 dependent drugs |
0 n=12 |
1 n=16 |
2 n=8 |
3 n=3 |
|---|---|---|---|---|
|
Number (%) of subjects with discordance based on the 3-hour DX/DM ratio n=14 subjects |
1 (8%) | 7 (43%) | 4 (50%) | 2 (67%) |
Table 3.
Chi-Square of Model Variables Predicting Genotype-Phenotype Discordance, univariate and multivariate analyses.
| Univariate Analyses | |||
|---|---|---|---|
| Variable | Chi-Square | DF | p-value |
| Age | 2.94 | 1 | 0.09 |
| Race | 0.36 | 1 | 0.64 |
| Ethnicity | 2.98 | 1 | 0.08 |
| Gender | 0.49 | 1 | 0.48 |
| CYP2D6 Co-ingestion | 6.24 | 1 | 0.01 |
| CYP2D6 Genotype | 1.16 | 3 | 0.13 |
| Combined Genotype | 1.16 | 1 | 0.28 |
| Derived Ki term | 3.33 | 1 | 0.07 |
| Multivariate Analyses | |||
| Age | 1.59 | 1 | 0.21 |
| CYP2D6 Genotype | 1.013 | 3 | 0.26 |
| CYP2D6 Co-ingestion | 3.933 | 1 | 0.047 |
| Ranked Dose over Ki | 3.99 | 1 | 0.05 |
Drug-Drug Interaction Effect on AUC:
The derived drug-drug interaction term (derived Ki term) was insufficient to predict genotype-phenotype discordance in isolation (p = 0.07, Table 3). There was still no association when the model controlled for age, CYP2D6 genotype, and CYP2D6 co-ingestion (p = 0.05, Table 3). Overall, patients that were taking a CYP2D6 dependent drug had a larger DM AUC (Figure 1) suggesting decreased enzyme metabolic activity. Those who co-ingested CYP2D6 drugs had 21.93 times increase in their DM AUC (CI 95%: 6.72, 71.52). We were unable to stratify this drug-drug interaction effect by the number of co-ingestants beyond the primary covariate of CYP2D6 co-ingestion captured as a binary variable, likely due to smaller numbers in this stratified sample (Table 4). Four patients were taking CYP2D6 inhibitors. Co-ingestion of CYP2D6 dependent drugs was still significant when included in the multivariate model with CYP2D6 inhibitor for DM AUC (p=0.002).
Figure 1:
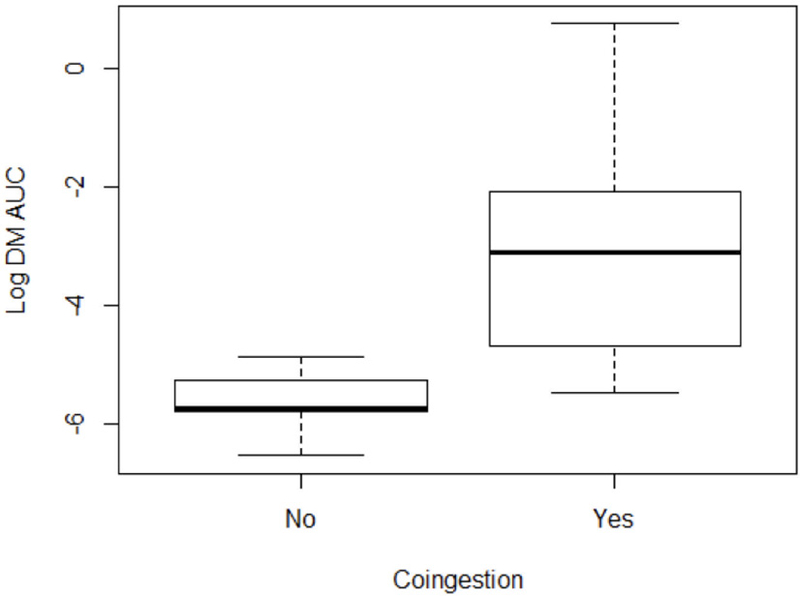
Distribution of the Log DM AUC values given whether or not the subject was co-ingesting any CYP2D6 dependent drugs
Table 4:
Univariate model predicting log DM AUC stratified by number of co-ingested CYP2D6 dependent drugs.
| Number of CYP2D6 drugs Co-ingested |
Estimate |
Standard Error |
P-value |
|---|---|---|---|
| Co-ingestion (yes/No) |
0.004* |
||
| Zero vs Two or more | 2.272 | 0.897 | 0.02* |
| Zero vs One | 2.963 | 0.795 | 0.001* |
| One vs Two or More | 0.691 | 0.833 | 0.417 |
When controlled for metoprolol dose, the median MT/MT-OH ratios at 3 hours significantly increased from phenotypically normal metabolizers (1.21 [IQR: 0.54, 1.73]), to intermediate metabolizers (4.20 [IQR: 3.22, 4.95]), to poor metabolizers (90.30 [IQR: 23.83, 489.09]) (p=0.0026).
Clinical Effects of CYP2D6 Phenotype:
In the subset of subjects taking metoprolol, there was no association between systolic blood pressure decline of 10% from baseline and phenotype (p=0.093, Table 5) or the derived Ki variable (p=0.19, Table 4). Similarly, there was no association between heart rate decline of 10% from baseline and phenotype (p=0.22, Table 5). When the phenotypes were dichotomized into PM/IM and NM/UM groups, blood pressure control was predicted by combined phenotype (p=0.03, Table 5), but heart rate decline was not predicted these combined phenotype groups (p=0.08, Table 5).
Table 5.
Clinical effects in those taking metoprolol predicted by phenotype, the Ki formula, and combined phenotype (PM/IM or NM/UM). Adjusted for metoprolol does.
| Univariate Analyses | ||||
|---|---|---|---|---|
|
Outcome |
Predictor |
LR Chisq |
Df |
Pr(>Chisq) |
| 10 % Blood pressure decline |
||||
| Phenotype | 4.754 | 2 | 0.090 | |
| Ki | 1.699 | 1 | 0.190 | |
| Combined Phenotype | 4.702 | 1 | 0.029* | |
| 10% Heart rate decline |
||||
| Phenotype | 3.016 | 2 | 0.175 | |
| Ki | 0.577 | 1 | 0.257 | |
| Combined Phenotype | 2.964 | 1 | 0.069 | |
| Composite of either 10% Blood pressure or HR decline |
||||
| Phenotype | 6.289 | 2 | 0.029* | |
| Ki | 0.790 | 1 | 0.273 | |
| Combined Phenotype | 5.190 | 1 | 0.020* | |
The composite secondary outcome of 10% decline in SBP or HR was predicted by the CYP2D6 phenotype. This composite outcome was more likely in patients that were phenotypically intermediate or poor metabolizers (p = 0.044, Table 5). The association was most profound in phenotypically intermediate metabolizers (p=0.021); the poor metabolizer phenotype did not predict the composite outcome, due to small numbers in that subset (p=0.361).
Adverse drug events (ADEs):
There were a total of eight reported events captured by the ADE screen. Four of these events were present prior to probe drug administration and thus were not considered associated with the probe drug. One patient described itching at the three-hour time point though no rash was present, and this symptom resolved prior to the final time point. One patient reported a subjective feeling of abdominal pain at the six-hour time point. The patient was observed, and the symptoms resolved after the subject ate some crackers. Two patients reported mild drowsiness at the four-hour time point, and this resolved in one subject, but remained present at discharge in the second subject.
LIMITATIONS
While appropriately powered for our stated primary outcome, the number of patients in this study are small. This means that we can not definitely state how CYP2D6 co-ingestion changes enzyme activity on the population level. The study only included 2 PMs, 4 UMs, and 10 IMs, by genotype, which limits this study’s ability to determine the interaction between the underlying genotype and the drug-gene interaction. However, we would not expect PMs or IMs to have increased enzyme activity when faced with an increased substrate load. The clinical effect data associated with metoprolol are also limited by the number of subjects in this subset. We will continue to enroll patients to further explore these trends.
This pragmatic trial allowed patients to take their own medications per their routine. This resulted in variability in timing of their ingestion. We opted for this approach because we desired to examine this interaction in a real world, pragmatic fashion. Examination of drug-drug interactions in this way is inherently less likely to identify clinical effects. Therefore, our study was biased toward the null hypothesis, that substrate ingestion does not cause genotype-phenotype discordance. Thus, our findings are suggestive that genotype-phenotype discordance may be more profound if all interacting drugs are taken at the same time.
The co-ingestion of CYP2D6 inhibitors, while not significant as a co-variate, may have contributed to the inability to stratify the co-ingestion variable by number of CYP2D6 drugs ingestions. There were only 4 subjects that co-ingested an inhibitor; 1 in which the inhibitor was the only CYP2D6 dependent drug taken, 1 was taking 2 CYP2D6 dependent drugs, and 2 were taking 3 CYP2D6 dependent drugs (one of which was the inhibitor). The ingestion of an inhibitor is likely to lead to more profound genotype-phenotype conversion, as demonstrated by Parker, et al.(13) Inclusion of inhibitor co-ingestant as a covariate demonstrated that the ingestion of CYP2D6 dependent substrates remained significant, which argues against inhibitor co-ingestion driving the reported association.
We utilized a microdose of DM as our probe drug. This lower dose was chosen to minimize ADEs and prevent competitive inhibition. The 3 hour DX/DM ratio was used by Frank, et al and this is the first report using the 3 hour/hour DX/DM ratio with microdose DM. We have provided the analysis that demonstrates the 3 hour microdose ratio predicts the phenotype calculated by the DM AUC, however, this was not the primary aim of this study. The association appears to hold, however a study specifically designed to validate this method may be warranted to confirm this finding. We hypothesized that the derived Ki term would allow for more specific stratification of the substrates taking into account the individual drug dose and the degree of enzyme inhibition. This approach may have been limited by pulling Kis from 2 separate in-vitro studies rather than a single unified source. Kis are inherently method dependent and vary between laboratories which may have limited the performance of the derived term. The Kis we incorporated may themselves not be accurate in individual patients with enzyme polymorphism. The more general binary co-ingestion term was more successful at predicting enzyme phenotype.
DISCUSSION
These data strongly suggest that co-ingestion of CYP2D6 substrates leads to genotype-phenotype discordance. This association with genotype-phenotype discordance and alteration in DM AUC remains present even without inhibitors which confirms an effect of substrates on enzyme phenotype. The one subject not taking any CYP2D6 co-ingestants that was genotyped as a CYP2D6 NM but was phenotyped as an IM was just barely in the IM ratio group. This patient had been taking zolpidem, which we do not consider a CYP2D6 dependent drug because less than 3% of the parent compound is metabolized via this pathway.(18) Zolpidem may have contributed to this phenotype shift or this patient may have a variant in the CYP2D6 gene that we did not capture with our assay. This genotype-phenotype discordance is associated with altered clinical effects in those taking metoprolol, as we’ve demonstrated with the composite blood pressure and heart rate outcome. Understanding shifts in phenotype due to interaction at the drug metabolizing enzyme level allows a more detailed understanding of the pathophysiology of drug-drug interactions. Other investigators have demonstrated alteration of pharmacokinetics and pharmacodynamics with CYP2D6 inhibitors though this work has never been extended to examine the effects of enzyme substrates on these parameters.(13) While co-ingestion may not always alter the clinical effects, enzyme substrates can have profound effects on drug metabolizing enzyme activity as well as the clinical effects of co-ingested drugs.
This trial attempted to account for variable enzyme effects caused by different drug substrates utilizing a derived term that accounted for all CYP2D6 dependent drugs and their doses. Other probe drugs, as well as other substrates, may yield different shifts in the enzyme phenotype. These shifts are ultimately dependent upon how tightly the enzyme binds each individual drug, and at what dose. For instance, a low dose drug that binds very tightly may still be out-competed by a drug at high dose even if the enzyme has lower affinity for the drug. It is likely that the resulting enzyme activity is altered differentially depending upon the specific substrate; the substrate affinity for the enzyme and the dose may be important factors that affect enzyme activity. We chose DM because of the high enzyme affinity for this drug but different probe drugs are likely to give different results. Whilst our pre-specified analysis did not identify a unifying term, we did observe a trend that suggests the need to enroll more subjects for future analyses. Ultimately, with no prior data to guide us, it was unclear how many subjects we would need to examine this derived term. Additionally, these data are likely stratified by underlying enzyme function and, perhaps, allelic variation. Thus, a larger study focused on this question is warranted.
We found no association with drug-drug interactions and ADEs. This was expected since the most common adverse drug events are off-target effects, such as nausea. ADEs that are exaggerations of the intended clinical pharmacology may be more common, may be predicted, and subsequently prevented by starting at lower doses in those with at risk genotypes. In this example, poor metabolizers were expected to tolerate lower doses of metoprolol, as demonstrated by other investigators.(19) Clinically, patients that have lower enzyme activity don’t tolerate significant up-titration of an enzyme dependent drug which minimizes the associations of ADEs in ecologic studies.(20, 21) Starting at a low dose and titrating upward is the safest method when no genotype information is available. It may be possible to start patients on higher doses in those with normal enzyme function and no drug-drug interaction or utilize a different drug entirely in patients not likely to meet therapeutic levels, such as those with ultra-rapid enzyme activity.
We were not able to predict phenotypes stratified by the number of CYP2D6 co-ingestions. This is likely due to the lower number of subjects ingesting more than two CYP2D6 dependent drugs and the presence of CYP2D6 inhibitors interspersed throughout the groups. Our results demonstrate that the composite clinical phenotype of BP and HR decline is affected by CYP2D6 dependent drug-drug interactions. A large trial that includes genotyping and drug-drug interactions must be undertaken to definitely determine the utility of genotyping in metoprolol treatment.
In conclusion, CYP2D6 substrate co-ingestion leads to genotype-phenotype discordance. Enzyme phenotype is associated with clinical effect in those taking metoprolol succinate. Larger studies that phenotype patients with broader CYP2D6 genotype representation and increased stratification of co-ingested CYP2D6 dependent drugs may allow for predictive modeling of clinical drug effects.
METHODS
Study Design/setting:
This was a prospective pharmacokinetic study in the University of Colorado Denver (UCD) Colorado Clinical Translational Science Institute (CCTSI) Clinical Translational Research Center (CTRC). The CTRC inpatient facilities include nursing staff, bionutritional support, and phlebotomy for pharmacokinetic analyses. The study was approved by the local institutional review board.
Patients:
A subset of patients from a parent clinical trial (NCT02293096) with uncontrolled HTN between 30 and 80 years of age were enrolled. Exclusion criteria included end-stage liver disease, glomerular filtration rate < 60 ml/min/1.73 m2, pregnancy, American Association of Anesthesiologists (ASA) classification of > 3, prisoners or wards of the state, decisionally challenged individuals, heart rate < 60 beats per minute, AV block > 240 msec, active reactive airway disease, illicit drug use in the preceding 30 days (excluding marijuana), allergy to metoprolol succinate, or severe peripheral arterial circulatory disorders.
CYP2D6 Genotyping and Phenotype Determination
Genomic DNA was extracted from whole blood via the Puregene® Blood Core kit B (Qiagen) according to the manufacturer’s instructions. CYP2D6 and ADRB1 were genotyped using the Multiplex SNaPshot technique previously described.(22) This assay detects 20 CYP2D6 clinically significant variants and identifies copy number variants. Predicted phenotypes were determined utilizing CYP2D6 activity score, as described by Gaedigk et al.(23) The CYP2D6 enzyme phenotype was determined based upon the 3 hour DM/DX ratio with ranges validated by Jurica et al.(24) Subjects were determined to have genotype-phenotype discordance if the phenotype predicted by the underlying genotype was different than the measured phenotype, determined by probe drug metabolism.
CYP2D6 probe and kinetic time points
An oral 2 mg microdose of dextromethorphan was administered as the phenotyping probe drug. Microdosing (2 mg versus the typical therapeutic dose of 20 mg) of dextromethorphan allows for subtherapeutic dosing, thus minimizing the risk of probe-drug interaction and adverse drug events(25). Measurement of serum dextromethorphan (DM) and its metabolite, dextrophan (DX), is considered the most reliable CYP2D6 phenotyping probe.(24, 26)
The parent compound and the metabolite were detected in EDTA plasma samples obtained at time 0, 1, 4, and 6 hours post dose. A reversed-phase ultra-performance liquid chromatographic tandem mass spectrometry (UPLC-MS/MS) assay for the determination of DM, its metabolite DX, metoprolol (MT), and its metabolite alphahydroxy metroprolol (MT-OH) in human EDTA plasma was validated.(24)
This method utilized stable labeled deuterated internal standards (DM-IS, DX-IS, MT-IS, MT-OH IS). The samples were treated with β-glucuronidase solution to convert dextrorphan–o-glucuronide to dextrorphan prior to extraction. A solid phase extraction procedure utilizing Strata-WCX cartridges was used to prepare the samples for analysis. The chromatographic separation was performed on a Waters 1.8 µm HSS T3, 100×2.1mm reverse-phase analytical column. Two different mobile phases were utilized for a gradient elution for the compounds. Mobile phase A consisted of 5:95 (acetonitrile:ultrapure water) containing 0.1 % formic acid (v:v). Mobile phase B consisted of 90:10 (acetonitrile:ultrapure water) containing 0.1% formic acid (v:v). The gradient separation changed linearly from 90% mobile phase A to 50% mobile phase A over 3 minutes at a flow rate of 0.50mL/min. The detection of DM, DX, MT, MT-OH, and their respective internal standards were achieved by protonated electrospray ionization on a TSQ Vantage detector. Precursor/product SRM transitions (m/z) in the positive ion mode were 284.4/116.2 and 289.4/121.2 for MT-OH and MT-OH IS, respectively, and 268.4/116.2 and 275.4/123.2 for MT and MT-IS, respectively. Precursor/product transitions (m/z) for positive ion mode were 258.4/157.2 and 261.4/157.2 for DX and DX-IS, respectively, 272.4/171.2 for DM and DM-IS, respectively, 434.402/258.402 for dextrorphan–o-glucuronide was also evaluated for completeness of enzymatic treatment. MT-OH, DM, MT utilize a linear (1/x weighted) regression while DX utilized a quadratic (1/x weighted) regression. MT, and MT-OH had a quantifiable range of 0.25ng/mL to 250ng/mL with a lower limit of quantitation (LLOQ) of 0.25ng/mL. DM and DX had a quantifiable range of 0.025ng/mL to 25.0ng/mL with a LLOQ of 0.025ng/ml. The assay used 0.2mL of human plasma.
The DM/DX 3-hour ratios were calculated to determine the CYP2D6 phenotype of each patient (based on cutoff values determined by Jurica J, et al(24)). Three of the DM 3-hour concentrations were below the limit of quantitation (BLQ), therefore half the LLOQ was imputed for these BLQ values based on the presence of DX . The MT:MT-OH 3-hour ratios were also calculated. One MT 3-hour concentration and one MT-OH 3-hour concentration were BLQ and half of the LLOQ was imputed for these values based on the presence of MT-OH and MT, respectively.
Drug Co-ingestion
A structured drug co-ingestion tool was utilized to captured prescriptions, over-the-counter medications, vitamins, and supplements. This tool captured the doses and times of the last dose of co-ingestants. Any drug taken within 3 of its half-lives was considered to have a potential CYP2D6 drug-drug interaction. We confirmed that stated drugs were taken using a comprehensive qualitative urine drug screen. This method did not confirm stated timing of ingestion, but demonstrated the patients were taking the drugs they claimed and no others. Subjects took all medications per their routine, with the exception of the dextromethorphan probe drug and the metoprolol succinate (when part of the subject’s medication list), which was taken after obtaining the baseline kinetic sample. When metoprolol was taken, it was scheduled by protocol to be at least 20 hours after the patient’s prior dose.
Area Under the Curve modeling
The area under the curve (AUC) was determined for DM, DX, MT, and MT-OH utilizing the trapezoidal rule,
The tail (+ Clast/ke) was not included in the AUC calculation for MT and MT-OH as these were at steady state concentrations. The DM/DX and MT/MT-OH AUC ratios were calculated.
Drug-drug interaction modeling
A unique CYP2D6 drug-drug interaction variable was derived in an attempt account for interaction as a single term. This term was derived from the sum of the doses of CYP2D6 dependent drugs divided by sum of each CYP2D6 substrate inhibition constant (Ki) for each drug, as determined by Shen et al, Vandenbrink et al Zhou et al, and Kerry et al.(2, 27–29) The Ki values are provided in Appendix 1.
[Dose1+Dose2+Dose3+Dosen…]/[Ki-1+Ki-2+Ki-3+Ki-n…]
Adverse drug events:
All adverse drug events during the trial were captured using a structured form. Abdominal pain, nausea, vomiting, diarrhea, shortness of breath, bradycardia, ventricular dysrhythmias, lightheadedness, syncope, rash/itching, and other adverse drug events, reported by the patient but not listed above, prior to probe administration and at every kinetic analysis time point were captured.
Metoprolol Clinical Effectiveness Outcomes:
A clinical effectiveness analysis was performed in the subset of subjects taking metoprolol. The primary outcome for this analysis was systolic blood pressure (SBP) decline of 10% from baseline, captured in the parent clinical trial over 4 weeks of therapy. Secondary outcomes were heart rate (HR) decline of 10% from baseline, and a composite of SBP and/or HR decline of 10%. Heart and blood pressure changes were not expected and were not observed during this trial since metoprolol succinate was at steady state following 4 weeks of therapy (data not shown).
Statistical Analysis
Logistic regression was used to predict genotype-phenotype discordance using co-ingestion of CYP2D6 drugs, age, ethnicity, and underlying CYP2D6 genotype as potential covariates. Each potential covariate was assessed univariately as a predictor for genotype-phenotype discordance. Any potential covariate that was found to meet the significance criteria of p < 0.20 was included in the full model to determine potential confounding. Any variable that was found to be insignificant (p > 0.05) using Chi-square likelihood ratio tests was excluded from the final model. This analysis was repeated by combining the genotypes into two groups “Poor/Intermediate” and “Normal/Ultra”. Finally, genotype was used, excluding the lone poor metabolizer genotype subject, to stratify the results based on CYP2D6 co-ingestion.
Logistic regression was used to determine associations between binary variables and potential confounders. Chi-square likelihood ratio tests were used to determine overall significance for predictors. Simple linear regression and multivariate linear regression was used to test associations between continuous outcomes and potential covariates. Negative log transformations of variables were performed for continuous variables whose distributions were non-normally distributed. Variable selection for covariates was carried out as in the primary analysis.
In order to have 80% power to detect a 0.6 difference in the AUC ratio of metabolites with 0.4 standard deviation at the 0.05 alpha level(30), 10 patients in each group were needed.
Supplementary Material
STUDY HIGHLIGHTS
What is the current knowledge on the topic?
CYP2D6 enzyme activity can be decreased by co-ingestion of inhibitors.
What question did this study address?
Does co-ingestion of CYP2D6 enzyme substrates alter enzyme activity or drug effectiveness.
What does this study add to our knowledge?
Co-ingestion of CYP2D6 dependent drugs alters CYP enzyme phenotype from what is predicted by underlying CYP2D6 genotype. This CYP2D6 drug-drug interaction alters metoprolol effectiveness.
How might this change clinical pharmacology or translational science?
Clinicians should consider CYP2D6 drug-drug interaction when prescribing drugs dependent upon this enzyme. Substrate dependent drug-drug interaction at the drug metabolizing enzyme can lead to altered drug effectiveness.
Acknowledgments
Funding Disclosure: Dr. Monte received support from NIH K23 GM110516, NIH R35GM124939 and NIH CTSI UL1 TR001082 for this work. The contents of this work are the sole responsibility of the authors and do not necessarily represents the views of the National Institutes of Health (NIH).
Footnotes
Conflict of Interest: The authors declared no competing interests for this work.
REFERENCES
- 1.Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin Pharmacokinet 2009;48(11):689–723. [DOI] [PubMed] [Google Scholar]
- 2.Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part II. Clin Pharmacokinet 2009;48(12):761–804. [DOI] [PubMed] [Google Scholar]
- 3.Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J 2005;5(1):6–13. [DOI] [PubMed] [Google Scholar]
- 4.Crews KR, Gaedigk A, Dunnenberger HM, Klein TE, Shen DD, Callaghan JT, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype. Clin Pharmacol Ther 2012;91(2):321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hicks JK, Bishop JR, Sangkuhl K, Muller DJ, Ji Y, Leckband SG, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin Pharmacol Ther 2015;98(2):127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hicks JK, Swen JJ, Thorn CF, Sangkuhl K, Kharasch ED, Ellingrod VL, et al. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther 2013;93(5):402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006;368(9536):704. [DOI] [PubMed] [Google Scholar]
- 8.Gressier F, Ellul P, Dutech C, Ait Tayeb Ael K, Monfort J, Corruble E, et al. Serotonin toxicity in a CYP2D6 poor metabolizer, initially diagnosed as a drug-resistant major depression. Am J Psychiatry 2014;171(8):890. [DOI] [PubMed] [Google Scholar]
- 9.Suzumura T, Kimura T, Kudoh S, Umekawa K, Nagata M, Matsuura K, et al. Reduced CYP2D6 function is associated with gefitinib-induced rash in patients with non-small cell lung cancer. BMC Cancer 2012;12:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahern TP, Hertz DL, Damkier P, Ejlertsen B, Hamilton-Dutoit SJ, Rae JM, et al. Cytochrome P-450 2D6 (CYP2D6) Genotype and Breast Cancer Recurrence in Tamoxifen-Treated Patients: Evaluating the Importance of Loss of Heterozygosity. Am J Epidemiol 2017;185(2):75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monte AA, Heard KJ, Campbell J, Hamamura D, Weinshilboum RM, Vasiliou V. The effect of CYP2D6 drug-drug interactions on hydrocodone effectiveness. Acad Emerg Med 2014;21(8):879–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monte AA, Heard KJ, Vasiliou V. Prediction of drug response and safety in clinical practice. J Med Toxicol 2012;8(1):43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker RB, Soberman JE. Effects of paroxetine on the pharmacokinetics and pharmacodynamics of immediate-release and extended-release metoprolol. Pharmacotherapy 2011;31(7):630–41. [DOI] [PubMed] [Google Scholar]
- 14.Gu Q, Dillon CF, Burt VL. Prescription drug use continues to increase: U.S. prescription drug data for 2007–2008. NCHS Data Brief 2010(42):1–8. [PubMed] [Google Scholar]
- 15.Tinetti ME, Bogardus ST Jr., Agostini JV. Potential pitfalls of disease-specific guidelines for patients with multiple conditions. N Engl J Med 2004;351(27):2870–4. [DOI] [PubMed] [Google Scholar]
- 16.Kaufman DW, Kelly JP, Rosenberg L, Anderson TE, Mitchell AA. Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. Jama 2002;287(3):337–44. [DOI] [PubMed] [Google Scholar]
- 17.Labcorp, comprehensive urine drug screen 2017. [cited 2018 May 26, 2018]. Available from: https://www.labcorp.com/test-menu/24306/drug-analysis-profile-comprehensive-urine.
- 18.Von Moltke LL, Greenblatt DJ, Granda BW, Duan SX, Grassi JM, Venkatakrishnan K, et al. Zolpidem metabolism in vitro: responsible cytochromes, chemical inhibitors, and in vivo correlations. Br J Clin Pharmacol 1999;48(1):89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bijl MJ, Visser LE, van Schaik RH, Kors JA, Witteman JC, Hofman A, et al. Genetic variation in the CYP2D6 gene is associated with a lower heart rate and blood pressure in beta-blocker users. Clin Pharmacol Ther 2009;85(1):45–50. [DOI] [PubMed] [Google Scholar]
- 20.Zineh I, Beitelshees AL, Gaedigk A, Walker JR, Pauly DF, Eberst K, et al. Pharmacokinetics and CYP2D6 genotypes do not predict metoprolol adverse events or efficacy in hypertension. Clin Pharmacol Ther 2004;76(6):536–44. [DOI] [PubMed] [Google Scholar]
- 21.Fux R, Morike K, Prohmer AM, Delabar U, Schwab M, Schaeffeler E, et al. Impact of CYP2D6 genotype on adverse effects during treatment with metoprolol: a prospective clinical study. Clin Pharmacol Ther 2005;78(4):378–87. [DOI] [PubMed] [Google Scholar]
- 22.Ben S, Cooper-DeHoff RM, Flaten HK, Evero O, Ferrara TM, Spritz RA, et al. Multiplex SNaPshot-a new simple and efficient CYP2D6 and ADRB1 genotyping method. Hum Genomics 2016;10:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther 2008;83(2):234–42. [DOI] [PubMed] [Google Scholar]
- 24.Jurica J, Bartecek R, Zourkova A, Pindurova E, Sulcova A, Kasparek T, et al. Serum dextromethorphan/dextrorphan metabolic ratio for CYP2D6 phenotyping in clinical practice. J Clin Pharm Ther 2012;37(4):486–90. [DOI] [PubMed] [Google Scholar]
- 25.Oh KS, Park SJ, Shinde DD, Shin JG, Kim DH. High-sensitivity liquid chromatography-tandem mass spectrometry for the simultaneous determination of five drugs and their cytochrome P450-specific probe metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2012;895–896:56–64. [DOI] [PubMed] [Google Scholar]
- 26.Frank D, Jaehde U, Fuhr U. Evaluation of probe drugs and pharmacokinetic metrics for CYP2D6 phenotyping. Eur J Clin Pharmacol 2007;63(4):321–33. [DOI] [PubMed] [Google Scholar]
- 27.Shen H, He MM, Liu H, Wrighton SA, Wang L, Guo B, et al. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos 2007;35(8):1292–300. [DOI] [PubMed] [Google Scholar]
- 28.VandenBrink BM, Foti RS, Rock DA, Wienkers LC, Wahlstrom JL. Prediction of CYP2D6 drug interactions from in vitro data: evidence for substrate-dependent inhibition. Drug Metab Dispos 2012;40(1):47–53. [DOI] [PubMed] [Google Scholar]
- 29.Kerry NL, Somogyi AA, Bochner F, Mikus G. The role of CYP2D6 in primary and secondary oxidative metabolism of dextromethorphan: in vitro studies using human liver microsomes. Br J Clin Pharmacol 1994;38(3):243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niwa T, Murayama N, Yamazaki H. Comparison of cytochrome P450 2D6 and variants in terms of drug oxidation rates and substrate inhibition. Curr Drug Metab 2011;12(5):412–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.