

CONSPECTUS:
Proton-coupled electron transfer (PCET) covers a wide range of reactions involving the transfer(s) of electrons and protons. The best-known PCET reaction, hydrogen atom transfer (HAT), has been studied in detail for more than a century. HAT is generally described as the concerted transfer of a hydrogen atom (H• ≡ H+ + e−) from one group to another, Y + H−X → Y−H + X, but a strict definition of HAT has been difficult to establish. Distinctions are more challenging when the transfer of “H•” involves e− and H+ that transfer to/from spatially distinct sites or even completely separate reagents (multiple-site concerted proton−electron transfer, MS-CPET). MS-CPET reactivity is increasingly proposed in biological and synthetic contexts, and some reactions typically described as HAT more resemble MS-CPET. Despite that HAT and MS-CPET reactions “look different,” we argue here that these reactions lie on a reactivity continuum, and that they are governed by many of the same key parameters. This Account walks the reader across this PCET reactivity continuum, using a series of studies to show the strong similarities of reactions that move protons and electrons in seemingly different ways. To prepare for our stroll, we describe the thermochemical and kinetic frameworks for HAT and MS-CPET. The driving force for a solution HAT reaction is most easily discussed as the difference in the bond dissociation free energies (BDFEs) of the reactants and products. BDFEs can be analyzed as sums of electron and proton transfer steps and can therefore be obtained from pKa and E° values. Even though MS-CPET reactions do not make and break H−X bonds in the same way as HAT, the same thermochemical description can be used with the introduction of an effective BDFE (BDFEeff). The BDFEeff of a reductant/acid pair is the free energy of that pair to form H•, which can be obtained from pKa and E° values in an analogous fashion to a standard BDFE. When the PCET thermochemistry is known, HAT and PCET rate constants can be understood and often predicted using linear free energy relationships (the Brønsted catalysis law) and Marcus theory type approaches. After this background, we walk the reader through a continuum of PCET reactivity. Our journey begins with a study of metalmediated HAT from hydrocarbon substrates to a metal-oxo complex and travels to the MS-CPET end of the reactivity spectrum, involving the transfer of H+ and e− from the hydroxylamine TEMPOH to two completely separate molecules. These examples, and those in between, are all analyzed within the same thermodynamic and kinetic framework. A description of the first examples of MS-CPET with C−H bonds uses the same framework and highlights the importance of hydrogen bonding and preorganization. The examples and analyses show that the reactions along the PCET continuum are more similar than they are different, and that attempts to divide these reactions into subcategories can obscure much of the essential chemistry. We hope that developing the many common features of these reactions will help experts and newcomers alike to explore exciting new territories in PCET reactivity.
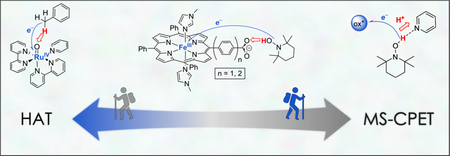
Graphical abstract
INTRODUCTION
Any reaction which involves proton transfer (PT) and electron transfer (ET) can be described as proton-coupled electron transfer (PCET).1 Hydrogen atom transfer (HAT) (eq 1) is perhaps the most well-studied and fundamental PCET mechanism. Over a century of research has highlighted the importance of HAT in chemical synthesis and biologically relevant redox reactions; HAT steps are critical in processes as diverse as hydrocarbon combustion, atmospheric chemistry and enzymatic catalysis.2 HAT is often one of the first reactions taught in organic chemistry classes, as a characteristic reaction of alkanes. The invaluable Landolt-Börnstein compendia of radical reactions lists over 10,000 HAT rate constants.3
| (1) |
Given the long history and pedagogy of HAT, it is initially surprising that its definition is not well established. With the development of broader PCET chemistry over the last decades, different usages and definitions of HAT have appeared in the literature (and will likely appear in this issue).4–6 This Account describes a range of reactions that involve the transfer of 1e− and 1H+ in a single kinetic step (concerted proton–electron transfer, CPET). Some of these reactions “look like” HAT, and some of them do not. Our goal here is not to suggest the adoption of a specific definition for HAT within the larger area of PCET reactivity. Instead, the examples from our work illustrate a continuum of PCET reactivity. The examples are “stops” along a PCET journey to demonstrate the remarkable diversity of reactions that involve transfer of one electron and one proton, and that these reactions are more similar than different.
HAT and Not HAT?
HAT is generally described as the concerted transfer of a proton and an electron from one group to another in a single kinetic step (eq 1). Among the most widely recognized are HAT from hydrocarbons to oxyl radicals or halogen atoms. For example, radical-chain chlorination mechanisms include a step with CI• abstracting H• from a C—H bond, forming a Cl—H bond and a carbon-centered radical (Scheme 1A). These reactions fit the most restrictive definition of HAT: that the proton and electron transfer as a hydrogen atom (H• ≡ H+ + e−) from one H—X bond to form another. Although electrons are delocalized and indistinguishable, our chemical intuition for these reactions indicates that the electron in the C—H bond being cleaved is the electron that is used toform the new H—Cl or O—H bond. The removal of H• leaves a radical in roughly the same stereoelectronic position on the carbon as the C—H bond occupied. For illustrative purposes in this Account, we will call this the “canonical” definition of HAT, but we do not advocate the use of this term to differentiate kinds of HAT.
Scheme 1.

Illustrations of Concerted Proton-Electron Transfer (CPET) Reactions: (A) “Canonical HAT “; (B) Separated; and (C) Multiple Site
H atom removal from phenols, however, does not clearly fit the canonical definition. Phenols are considered classic hydrogen atom donors, and this is key to the antioxidant action of vitamin E and BHT (butylated-hydroxytoluene).7 Yet the ground state of phenol is planar, so transfer of e−/H+ leaves a lone pair on oxygen where the proton was, while the electron transfers from the aromatic π-system (Scheme 1B1). This was first highlighted in a computational study of phenol/phenoxyl H atom self-exchange, which showed an almost planar transition state with PT between oxygens and ET between the π systems.8 A later study found a lower transition state, with ET between π-stacked aromatic rings coupled to PT between the oxygens.9 This kind of mechanism where e− and H+ appear to travel by different paths to different locations has been found in other computational studies, and is a common enzymatic reaction step. most notably, H atom abstractions by the iron-oxo “compound I” intermediate in cytochromes P450 and related enzymes (Scheme 1B2) are always described as HAT but actually involve substantial e−/H+ separation. Removal of H• from the C—H bond of the substrate does look like canonical HAT, but the proton adds to the oxoforming a hydroxo ligand while the electron fills a “hole” away from the oxo, a mixture of porphyrin radical cation and thiyl radical.10
Thus, reactions that for decades have been described as HAT, such as those of phenols and enzymatic compound I intermediates (Scheme 1B), do not perfectly fall under the canonical definition of HAT in Scheme 1A. We do not bring up these differences to argue that phenol and P450 reactions are not HAT, but rather to point out the challenges in categorizing even very familiar HAT reactions. A theoretical study by Sirjoosingh and Hammes-Schiffer suggested that phenol-phenoxyl is not HAT because the exchange is electronically nonadiabatic,11 but this is difficult to examine experimentally. We favor an expanded definition of HAT that includes essentially all reactions involving transfer of H• from a single donor reagent to a single acceptor reagent, without concern for the electronic molecular orbitals formally involved in the ET component.1,8
A distinction can usually be drawn, however, between such HAT processes and reactions where the particles transfer to (or from) completely separate molecules or sites.12,13 For instance, X—H can be oxidized to X• by proton transfer to a nearby base and electron transfer to a nearby oxidant (Scheme 1C). We term these multiple-site concerted proton—electron transfer (MS-CPET). MS-CPET reactions are greatly facilitated by the formation of a classical hydrogen bond between the proton donor and acceptor. Thus, for instance, MS-CPET reactions involving O—H and N—H bonds are common while we have only recently found the first example involving a C—H bond (see Stop #5 below). This hydrogen bonding effect can be very useful, imparting different chemoselectivity from HAT processes. Still, when comparing reactions of similar bonds, or when taking hydrogen bonding into account, we argue below that even the HAT/MS-CPET distinction does not reflect a core difference in the chemical processes. While MS-CPET reactions “look different” from canonical HAT reactions (Scheme 1A vs C), these are but stops on a reactivity continuum.
Thermochemistry and Kinetics of HAT and MS-CPET Reactions
The disparate reactions in Scheme 1 can be treated with the same thermochemical framework. These solution reactions should be analyzed using free energies (ΔG°) instead of the bond enthalpies more typical of HAT discussions, because ΔG° is directly connected with equilibrium constants, linear free-energy relations (LFERs), and versions of Marcus theory.5 Entropic contributions are often small but can be substantial for transition metal complexes.5 ΔG° for a HAT reaction is the difference between the two bond dissociation free energies, BDFE (X—H) — BDFE (Y—H). Solution BDFEs can be determined from solution pKa and E° values (Scheme 2A).12 A very similar square scheme can be drawn for the combination of an acid and a reductant (Scheme 2B), giving what we have defined an “effective BDFE “,12,13 even though there is no bond that is homolytically cleaved when the proton and electron come from different sites or different reagents. Yet the thermochemical analysis is the same: the sum of a pKa and an E°. This BDFEeff can be used in the same manner to derive the free energy of an MS-CPET reaction that transfers a hydrogen atom equivalent. The use of BDFEeff therefore allows for thermodynamic comparisons of concerted 1e−/1H+ reactions regardless of their form.
Scheme 2. Thermodynamic Cycles (Square Schemes) and Equations for BDFEs of (A) a Single PCET Reagent and (B) a Reductant/Acid Pairaa.

aThe CG,sol constant is in essence ΔG°(H+ + e−→ H) in solvent “sol”. Adapted with permission from ref 13. Copyright 2012 Royal Society of Chemistry.
Similar BDFEeff’s can be obtained from different combinations of one-electron oxidant/reductant and acid/base reagents. This provides a thermochemical tunability that can be a powerful tool.12,13 However, there are inherent reagent incompatibilities that limit MS-CPET chemistry. Bases are inherently electron-rich and can readily react with oxidants, which are electron-poor. Similarly, acids can protonate reductants or form H2. These obstacles are not insurmountable, but they often present significant challenges.13 Mitigating these incompatibilities by using photoredox agents has been part of Knowles and others developing MS-CPET reactions for synthetic organic chemistry.14
The thermochemistry of 1e−/1H+ CPET reactions is a foundation for understanding their kinetics. Within a set of related CPET reactions, we have usually but not always observed a correlation of rate constants with equilibrium constants, following the Brønsted catalysis law (eq 2). Using the Eyring equation, this is equivalent to correlating ΔG‡ with ΔG° (eq 3). These two equations give the same unitless slope α. To go beyond this LFER, many CPET studies have used a Marcus-Hush-Levitch-like treatment of rate versus driving force (Figure 1, eq 4). Specifically, we have found that the Marcus cross relation predicts the rate constants for a wide range of inorganic and organic reactions, often within an order of magnitude, as described elsewhere.5 (The cross relation cannot, however, be applied to MS-CPET reactions due to the lack of a measurable CPET self-exchange rate constant for a reductant/acid pair.) most thermal CPET reactions have been studied in the regime of low driving forces, |ΔG°| ≪ 2λ, where a linear Brønsted plot with a ≅ 0.5 is often observed (eq 5). This one-dimensional Marcus treatment is a great simplification of much more complete treatments, notably the multistate continuum theory of Hammes-Schiffer et al. that treats the proton quantum mechanically.6
| (2) |
| (3) |
| (4) |
| (5) |
Figure 1.

Marc theory description of reactions where the intersection of the parabolic reactant and product free energy surfaces gives the free energy of the transition state (ΔG‡) in terms of ΔG° and the intrinsic barrier λ, eq 4.
Walking through a Continuum of Reactivity: From HAT to MS-CPET
We have approached our studies of HAT and now MS-CPET starting from the thermodynamic and kinetic frameworks described above. In the following sections, we will “walk” the continuum between “canonical” HAT and MS-CPET, utilizing examples from our lab to illustrate this reactivity landscape. First, we explore studies where the destinations of the electron and proton are systematically separated from one another. Finally, we examine several systems where the electron and proton are transferred to different reagents. The kinetics and thermodynamics in HAT-like and MS-CPET-like reactions are employed to draw comparisons and illustrate differences.
Stop #1: H yd rogen Atom Abstraction by a Ruthe-nium—Oxo Complex.
The oxidation of C—H bonds by [(bpy)2(py)RuIVO]2+ (RuIVO2+, bpy = bipyridine, py = pyridine) was discovered in the 1980s by Thom as Meyer, one of the founders of the field of PCET.15 Our later studies showed that this reaction proceeds by rate limiting HAT (Figure 2A).16,17 One significant piece of evidence for concerted e− and H+ transfer was a linear correlation of rate constant vs equilibrium constant (rate/driving force relationship, Figure 2B). Such correlations have been frequently used in studies of HAT reactions of C—H bonds, going back at least to the work of Evans and Polanyi in the 1930s.18 Correlations traditionally used log(k) or Ea vs bond dissociation enthalpies (BDEs), as we originally did,16 but they should use BDFEs to determine Keq’s (Figure 2B has been replotted here using BDFEs, causing a shift by a constant amount8). The slope of the plot of ln(kHAT) vs ln(Keq) is 0.49(7), close to the predicted a = 0.5. The rate constant for HAT from toluene to RuIVO2+ was close to the value predicted by the Marcus cross relation, using RuIV(O)/RuIII(OH) and PhCH2·/PhCH3 self-exchange rate constants and the known thermochemistry.
Figure 2.

(A) HAT from toluene to RuIVO2+. (B) Graph showing the dependence of rate constants (ln(kHAT)) on driving force (ln(keq)) for oxidations of hydrocarbons by RuIVO2+. Data from ref 16.
The reaction of RuIVO2+ with the hydrocarbon substrates in Figure 2B is, in our view, an example of canonical HAT. From the perspective of the hydrocarbon, this reaction is classic HAT to form a carbon radical, as in Scheme 1A, above. With toluene, for instance, removal of H• (e−/H+) leaves behind a “hole” (the SOMO) that is mostly located in roughly the same region of space as the original C—H bond, although there is significant SOMO density on the aromatic ring and the formed CH2 group is now planar. From the perspective of RuIVO2+, the proton goes to the oxygen and one could consider that formally the electron goes to the metal center, reducing it to RuIII. However, the electron is transferred into a half-filled Ru—O π* orbital that has significant density on both Ru and O, so the e− and H+ being transferred can be considered to be involved in forming the O—H bond. This is therefore our example of a metal-mediated canonical HAT reaction.
Stop #2: A Stroll with Increasing the Distance between Electron and Proton.
This section will show how increasing distances between the proton and electron accepting/donating sites challenges the HAT vs PCET distinction, using reactions of ruthenium and iron complexes to illustrate the progression.
The ruthenium (II)-imidazole complex RuII(acac)2(py-imH) in Figure 3A acts as a net hydrogen-atom donor to the TEMPO nitroxyl radical, forming the TEMPO—H bond.19 The reaction reaches completion in minutes under typical room-temperature conditions. The electron can be considered to transfer from a dπ (t2g) orbital of the d6 RuII reactant (to form the d5 t2g6 RuIII product) and the proton comes from the imidazole N—H bond, leaving behind a nitrogen lone pair. Thus, formally, the electron and proton “come from” different places within the ruthenium complex. While the Ru dπ electron back-bonds and delocalizes into the π orbitals of the imidazole ligand, this π orbital is orthogonal to the reactant N—H bond. Thus, as a first approximation, we can say that the Ru center provides an electron that is ~4.2 Å away from the imidazole N—H bond that provides the proton. Despite this separation, there is a significant “thermodynamic coupling”12 between the Ru center E 1/2 and the imidazole N—H pKa: the E1/2 shifts significantly with deprotonation, and the pKa shifts significantly with oxidation. A related iron-porphyrin-imidazolate reacts similarly, in the opposite direction: oxidizing TEMPOH to TEMPO with proton addition to the N-lone pair of the imidazolate coupled to reduction of the FeIII center.20
Figure 3.

CPET reactions with acid/base sites distant from the metal center. Estimated metal-to-basic atom distances are given in the boxes at right. (A) Transfer of 1e−/1H+ from RuII(acac)2(py-imH) to TEMPO. (B) Transfer of 1e−/1H+ from TEMPOH to Ru-pyCO2− (n = 0) and RupyPhCO2− (n = 1) yields the reduced RuII/carboxylic acid complex. (C) Transfer of 1e−/1H+ from TEMPOH to iron porphyrin complexes with carboxylate oxygen atoms distant from the iron center.
In these and the other reactions in this section, we chose as one reaction partner the 1e−/1H+ redox couple of neutral radical TEMPO and the reduced hydroxylamine TEMPOH. TEMPO/TEMPOH is a very convenient reagent pair because under many conditions it only undergoes concerted 1e−/1H+ transfer (CPET/HAT). TEMPOH has a weak O—H bond (66.5 kcal/mol in MeCN), and the possible stepwise intermediates TEMPOH•+ and TEMPO− are very high in energy.12 Analyses using this thermochemical bias show that all of the reactions in this section (and throughout this Account) follow the CPET pathway.
The ruthenium terpyridine-carboxylate complexes in Figure 3B readily abstract H• from TEMPOH, to give TEMPO and the reduced and protonated RuII product. The distance between the carboxylate oxygens and the Ru center are 6.9 Å for the n = 0 compound and 11.2 Å for the n = 1 compound, with an additional phenyl spacer. Unlike the ruthenium imidazole complexes described above, there is very little thermodynamic coupling12 between the carboxylic acid and the Ru center. Still, PT to the carboxylate is concerted with ET to the ruthenium (CPET), because of the preferences of the TEMPOH reagent.21,22
The iron porphyrin-benzoate and -biphenyl carboxylate systems in Figure 3C have ~10 and 14 Å separations between the iron and the carboxylate.20,23 Yet reactions with TEMPOH or ascorbate occur within minutes or seconds, respectively. While computational studies of these two reactions show that a full description is complex,23 the qualitative conclusion is clear: concerted 1e−/1H+ transfers occur readily even at large separations. This conclusion was previously suggested by studies of enzymes such as ribonucleotide reductase (though it is more challenging to demonstrate concerted mechanisms in enzymatic systems).2,24
The series of reactions in Figures 2 and 3 thus vary the distance between the atom that holds the proton and the redox-active metal center from 1.8 to 14 Å. At the short end, it is easy to describe the reactions as HAT. At the larger distances between the H+ and e− accepting sites, however, these reactions do not “look like” HAT. They are short-range proton transfers concerted with long-range electron transfers. Of this set, rate/driving force correlations were examined only for Ru-pyCO2−. These reactions show little dependence of kCPET on the BDFE of the H atom donor,25 and it is not clear why they differ from the other studies presented here.
Stop #3: MS-CPET with Intramolecular Proton Transfer Across a Hydrogen Bond: Phenol Oxidation.
Our journey now takes a turn to reactions in which an intramolecular proton transfer is coupled to electron transfer to the reaction partner. Because the proton and electron transfer to separate reagents, these reactions are MS-CPET and cannot be described as HAT.
Oxidations of phenols with an intramolecular, hydrogen-bonded base (Figure 4A) have been studied by many groups,26,27 in part to understand the behavior of the redox active tyrosine-histidine pair in photosystem II. We have used phenol-pyridines,28 -imidazoles,29 and -amines30 to investigate the effects of oxidant strength, base strength, proton transfer distance and structure on reaction rates and intrinsic barriers. In this Account, we discuss only the effects of modulating the driving force for oxidation of phenol-pyridines with a methylene spacer, HOArCH2pyX (Figure 4B). Changing the substituent on the base (X = 4-NMe2, 4-CH3, H, and 5-CF3) varied the PT portion of the MS-CPET energetics, and using differently substituted triarylaminium oxidants [N(C6H4Y)3]+• varied the ET portion. The E1/2 for MS-CPET oxidation of HOArCH2pyX varied with the pyridine substituent roughly as much as expected from just the change in the pyX basicity.28 Thus, the phenol portion of HOArCH2pyX is not significantly affected by the pyridine substituent, due to the methylene spacer keeping the two fragments electronically separated.
Figure 4.

(A) MS-CPET oxidation of phenol-base compounds. (B) Phenol-pyridines HOArCH2pyX. (C) Plot of log(k) vs log(Keq) for the reaction of different HOArCH2pyX with various [N(C6H4Y)3]+• Parts (B) and (C) reproduced with permission from ref 28. Copyright 2012 American Chemical Society.
The MS-CPET rate constants for oxidation of HOArCH2pyX varied simply with the overall driving force, ΔG°MS-CPET, determined from the E1/2 values of the oxidant and the phenol (Figure 4C). The Brønsted slope for this homologous series was found to be a = 0.54 (5). This is typical of our studies of phenol-base oxidations, which usually have linear Brønsted plots with α values that are close to the 0.5 predicted by Marcus theory for reactions with |ΔG°| ≪ λ (eq 4 above).28–30 The one exception was a study with photoexcited oxidants, in which ΔG°MS-CPET approached —λ (eq 4).31
In the HOArCH2pyX system, it is notable that log(k) varies by roughly the same magnitude with ΔG°MS-CPET whether the driving force is changed through the proton transfer component (pyX, a = 0.57(6)) or the electron transfer [N(C6H4Y)3+•, a ≅ 0.48(5)] portions of the process (Figure 4C). The implications of the similarity of these slopes are discussed in the next stop of our journey.
Stop #4: Three-Component MS-CPET Reactions.
Our PCET journey in this direction ends with a termolecular reaction, the oxidation of TEMPOH to TEMPO by synchronous transfer of an electron to an external oxidant and a proton to an external base. The transfer of e− and H+ intermolecularly to different reagents is the logical extension of the bimolecular reactions in Stops 2—4, with separated e− and H+.
Oxidation of the hydroxylamine TEMPOH to its neutral radical TEMPO by pyridine bases and ferrocenium oxidants (Figure 5A) typically occurs within seconds at room temperature.32 The formally termolecular reaction occurs via a rapidly formed TEMPO-H⋯py hydrogen-bonded adduct, which was quantified by IR spectroscopy. The rate-determining step is then PT across the hydrogen bond to the pyridine concerted with ET to the ferrocenium (Fc+). Rate constants were determined by optical monitoring of the decay of the Fc+. The H+ and e− that originate from the same O—H bond in TEMPOH are transferred to two separate molecules. As discussed in Stop 2, the properties of TEMPOH direct the reaction to an MS-CPET mechanism.12
Figure 5.

(A) MS-CPET from TEMPOH to pyridine bases and ferrocenium (Fc+) oxidants. (B) Plot of ln(kMS CPET) vs ln(Keq). Part (B) reproduced with permission from ref 32. Copyright 2017 American Chemical Society.
The free energies of the H+ and e− transfer components of ΔG°MS-CPET were independently tuned using substituted pyridines and ferroceniums.32 Each pyridine/ferrocenium pair has an effective BDFE, following the thermochemical framework described above (Scheme 2). This BDFEeff then gives the driving force for the overall reaction for each oxidant/base combination (ΔG°MS-CPET, Keq). The ln(kMS-CPET) values varied linearly with ln(Keq), with an a of 0.46 (2) (Figure 5B). This analysis showed that the rate constant varied with ΔG°MS-CPET in essentially the same way regardless of whether the driving force was changed through the H+ or e− transfer energetics. A similar result was recently described for a metal hydride system.33
The equal effects of the e− and H+ energetics on the rate constant, with a slope of roughly 0.5, implies that the rate- determining step involves the concerted transfer of the two particles, and that this transfer is synchronous. The position of the transition state along the reaction coordinate must be such that the reorganizations to facilitate the proton and electron transfers are comparable. Following the more general discussion of Grunwald,34 we feel that this similar effect of free energy changes on the MS-CPET rate constant is a good experimental definition of a synchronous process.
Thus, the TEMPOH/TEMPO termolecular system, which has the most separated character of the systems described in this Account, shows the same kinetic and thermodynamic behavior as observed along the continuum from HAT to separated CPET reactions. The predictive framework developed for HAT-like reactivity can be applied to these very different-looking MS-CPET reactions.
Stop #5: MS-CPET Involving C—H bonds: Circum-venting the Requirement for a Hydrogen Bond.
We have recently reported the first examples of MS-CPET for the activation and formation of C—H bonds, a previously undescribed reaction class.35 At the outset of this Account, we showed examples of PCET at C—H bonds that generally fell into definitions for HAT. However, all of the examples of MS-CPET given above, and all prior reports of MS-CPET reactions, require the initial formation of a hydrogen bond. Our recent report demonstrates that the appropriate positioning of a proton donor/acceptor moiety can serve to prealign the proton transfer coordinate in the absence of a strong hydrogen bond, thereby facilitating both oxidative cleavage and reductive formation of C—H bonds via MS-CPET.35 We believe that our development of the first MS-CPET reactions of C—H bonds will prove to be a path into an unexplored region of the PCET landscape.
The C—H bond in the fluorenyl-benzoate (1−) is rapidly cleaved upon addition of outersphere oxidants, with formation of the related lactone, as shown in Figure 6A.35 Mechanistic studies indicated that the rate-determining step is MS-CPET to form a reactive carbon-centered radical, by PT to the carboxylate base concerted with ET to the oxidant. The fluorenyl radical then rapidly undergoes oxidative deprotonation and cyclization to form the lactone. Kinetic studies showed clean second-order behavior for a range of oxidants, from the very strong [N(C6H4Br)3]•+ to the very weak Fe[C5(CH3)5]2+. The rate constants show the linear dependence on driving force that is now familiar to the reader (Figure 6B). However, the Brønsted slope of this dependence is very small, a = 0.21, much lower than the a ~ 0.5 in other systems discussed here. Thus, while the reactions studied had equilibrium constants ranging over ~ 1020, the rate constants vary by little more than 104. The origin of this unusual shallow dependence is currently under investigation.
Figure 6.

(A) MS-CPET oxidation of the fluorenyl-benzoate 1−. (B) Plot of log(kMS-CPET) vs log(Keq). Part (A) reproduced and part (B) adapted with permission from ref 35. Copyright The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC) http://creativecommons.org/licenses/bync/4.0/.
To our knowledge, all prior examples of MS-CPET (from our group and others) involve the initial formation of a classical hydrogen bond.14 The oxidation of 1− by MS-CPET is an exception to this rule. We believe that 1− can be oxidized by MS-CPET due to the carboxylate base being sterically positioned to be close to the transferring proton. Similarly, our related reductive MS-CPET reactions that form C—H bonds35 involve acids that are positioned close to the carbon that accepts the proton. In all of these cases, we propose that the role of the structural positioning is to align the proton transfer coordinate. In MS-CPET reactions of O—H or N—H bonds, this alignment is accomplished automatically by the hydrogen bond. We also previously suggested that a hydrogen bond in general accelerates CPET processes, by facilitating the proton transfer portion. One example is the faster HAT reactions of oxyl radicals with O—H vs C—H bonds at similar driving force (very roughly 104 faster).5 This double effect of the hydrogen bond, alignment and facilitating PT, appears to be common across the HAT to MS-CPET landscape. While the proposed positioning requirement for C—H bond MS-CPET could limit its scope in synthetic systems, biological active sites often have precisely positioned acid/base cofactors. It is therefore likely that enzymatic systems utilize MS-CPET mechanisms to make and break C—H bonds, and we have suggested a specific example, the hydrogenation of a porphyrin C = C bond in the biosynthesis of chlorophyll.35
CONCLUSIONS
In this Account, we have taken the reader on a journey from “canonical” hydrogen atom transfer (HAT) reactions to processes in which the proton and electron transfer to or from different parts of a molecule, or even to/from different molecules (multiple site-concerted proton—electron transfer, MS-CPET). Our stops on this journey are exemplars along a continuum of reactivity for PCET, and they indicate that these reactions have a great deal in common. Attempts to designate subclasses of concerted 1e−/1H+ transfer reactions are often problematic, and they can obscure the essential chemistry rather than illuminate it. MS-CPET reactions may “look” very different from HAT but many of the same thermodynamic and kinetic parameters hold across the whole range of these 1e−/1H+ reactions.
Emphasizing the similarities of HAT, MS-CPET and other concerted 1e−/1H+ transfers will facilitate the development of a unifying, predictive framework. For instance, the work developed here shows that H—X bond dissociation free energies (BDFEs) and effective BDFEs for reductant/acid pairs can be utilized in a similar fashion. most of these reactions follow the rate/driving force relationships derived from a simple Marcus theory treatment, allowing quantitative predictions to be made, especially within a group of similar reactions.
PCET reactions span disciplines from synthetic chemistry to biology to materials chemistry. We hope that identifying the HAT to MS-CPET reactivity spectrum will open new vistas of fundamental, impactful PCET research on important processes. One example is our very recent discovery of MS-CPET reactivity of C—H bonds (Stop 5), which challenges the generalizations that MS-CPET occurs only at a strong hydrogen-bonded interface, and that inert C—H bonds can only undergo HAT. Fundamental advances in PCET have applications from organic synthesis14 to enzymology10 to electrocatalysis.36 Our wanderings in the diverse PCET landscape have even led us and others to apply PCET concepts to interfacial reactions at nanoscale materials.37 We hope that readers who have now made this journey with us will be similarly inspired to explore new PCET territory.
ACKNOWLEDGMENTS
The authors thank all of our co-workers, colleagues, and collaborators whose contributions are represented here, especially Dr. Maraia E. Ener. JMM thanks the late Dr. Carol Creutz for her advice to focus on ΔG° not ΔH°. We thank the NIH (GM50422) and NSF GRFP (to B.K.) for financial support.
Biographies
Julia W. Darcy holds a B.S. in Chemistry and a B.A. in Environmental Studies from Worcester Polytechnic Institute. She is currently a Ph.D. candidate in the Department of Chemistry at Yale University. Her current research involves designing novel systems for the fundamental study of PCET processes.
Brian Koronkiewicz received his B.S. in Biochemistry at University of the Sciences and his M.S. in Chemistry at the University of Chicago. He is currently a Ph.D. candidate in the Department of Chemistry at Yale University. His research involves biomimetic MS-CPET.
Giovanny A. Parada received his B.S. and M.S. in Chemistry at the National University of Colombia and Ph.D. at Uppsala University. He is currently a postdoctoral associate at Yale University. His current research involves photoinduced MS-CPET.
James M. Mayer did undergraduate research at Hunter College with Edwin Abbott, and with William Klemperer while earning his A.B. at Harvard. He completed a Ph.D. at Caltech under the direction of John Bercaw in 1982, and after two years as a Visiting Scientist at DuPont moved to the University of Washington. In 2014, he moved to Yale University, where he is currently Charlotte Fitch Roberts Professor of Chemistry. His interests in redox chemical reactions span inorganic, organometallic, bioinorganic, and physical organic chemistry as well as chemistry at the nanoscale.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Mayer JM; Rhile IJ Thermodynamics and kinetics of proton-coupled electron transfer: stepwise vs. concerted pathways. Biochim. Biophys. Acta, Bioenerg 2004, 1655, 51–58. [DOI] [PubMed] [Google Scholar]
- (2).Dempsey JL; Winkler JR; Gray HB Proton-Coupled Electron Flow in Protein Redox Machines. Chem. Rev 2010, 110, 7024–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Radical Reaction Rates in Liquids; Fischer H, Ed.; Landolt-Börnstein New Series; Springer-Verlag: New York; Vol. 13 (1994) and Vol. 18 (1997). [Google Scholar]
- (4).Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- (5).Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res 2011, 44, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hammes-Schiffer S; Stuchebrukhov AA Theory of Coupled Electron and Proton Transfer Reactions. Chem. Rev 2010, 110, 6939–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Burton G; Ingold KU Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function. Acc. Chem. Res 1986, 19, 194–201. [Google Scholar]
- (8).Mayer JM; Hrovat DA; Thomas JL; Borden WT Proton-Coupled Electron Transfer versus Hydrogen Atom Transfer in Benzyl/Toluene, Methoxyl/Methanol, and Phenoxyl/Phenol Self-Exchange Reactions. J. Am. Chem. Soc 2002, 124, 11142–11147. [DOI] [PubMed] [Google Scholar]
- (9).DiLabio GA; Johnson E R Lone pair—π and π– π interactions play an important role in proton-coupled electron transfer reactions. J. Am. Chem. Soc 2007, 129, 6199–6203. [DOI] [PubMed] [Google Scholar]
- (10).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(lV)hydroxide pKa and the Role of Thiolate Ligation in C—H Bond Activation by Cytochrome P450. Science 2013, 342, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sirjoosingh A; Hammes-Schiffer S Proton-Coupled Electron Transfer versus Hydrogen Atom Transfer: Generation of Charge-Localized Diabatic States. J. Phys. Chem. A 2011, 115, 2367–2377. [DOI] [PubMed] [Google Scholar]
- (12).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Waidmann CR; Miller AJM; Ng C-WA; Scheuermann ML; Porter TR; Tronic TA; Mayer JM Using combinations of oxidants and bases as PCET reactants: thermochemical and practical considerations. Energy Environ. Sci 2012, 5, 7771–7780. [Google Scholar]
- (14).Gentry EC; Knowles R R Synthetic applications of proton-coupled electron transfer. Acc. Chem. Res 2016, 49, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Thompson MS; Meyer TJ Mechanisms of oxidation of 2-propanol by polypyridyl complexes of ruthenium (III) and ruthenium (IV). J. Am. Chem. Soc 1982, 104, 4106–4115. [Google Scholar]
- (16).Bryant JR; Mayer JM Oxidation of C—H Bonds by [(bpy)2(py)RuIVO]2+ Occurs by Hydrogen Atom Abstraction. J. Am. Chem. Soc 2003, 125, 10351–10361. [DOI] [PubMed] [Google Scholar]
- (17).Bryant JR; Matsuo T; Mayer JM Cumene Oxidation by cis-[RuIV(bpy)2(py)(O)]2+, Revisited. Inorg. Chem 2004, 43, 1587–1592. [DOI] [PubMed] [Google Scholar]
- (18).Evans MG; Polanyi M Inertia and driving force of chemical reactions. Trans. Faraday Soc 1938, 34, 11–24. [Google Scholar]
- (19).Wu A; Masland J; Swartz RD; Kaminsky W; Mayer JM Synthesis and Characterization of Ruthenium Bis(β-diketonato) Pyridine-Imidazole Complexes for Hydrogen Atom Transfer. Inorg. Chem 2007, 46, 11190–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Warren JJ; Mayer JM Hydrogen Atom Transfer Reactions of Iron-Porphyrin-Imidazole Complexes as Models for Histidine-Ligated Heme Reactivity. J. Am. Chem. Soc 2008, 130, 2774–2776. [DOI] [PubMed] [Google Scholar]
- (21).Manner VW; DiPasquale AG; Mayer JM Facile Concerted Proton-Electron Transfers in a Ruthenium Terpyridine-4′-Carboxylate Complex with a Long Distance Between the Redox and Basic Sites. J. Am. Chem. Soc 2008, 130, 7210–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Manner VW; Mayer JM Concerted Proton-Electron Transfer in a Ruthenium Terpyridyl-Benzoate System with a Large Separation between the Redox and Basic Sites. J. Am. Chem. Soc 2009, 131, 9874–9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Warren JJ; Menzeleev AR; Kretchmer JS; Miller TF; Gray HB; Mayer JM Long-Range Proton-Coupled Electron-Transfer Reactions of Bis(imidazole) Iron Tetraphenylporphyrins Linked to Benzoates. J. Phys. Chem. Lett 2013, 4, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Minnihan EC; Nocera DG; Stubbe J Reversible, Long-Range Radical Transfer in E. coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res 2013, 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Manner VW Concerted proton-electron transfer reactions of ruthenium and cobalt complexes. Ph.D. Thesis, University of Washington, 2009. [Google Scholar]
- (26).Costentin C; Robert M; Saveant J-M Electrochemical and homogeneous proton-coupled electron transfers: Concerted pathways in the one-electron oxidation of a phenol coupled with an intramolecular amine-driven proton transfer. J. Am. Chem. Soc 2006, 128, 4552–4553. [DOI] [PubMed] [Google Scholar]
- (27).Zhang M-T; Irebo T; Johansson O; Hammarström L Proton-coupled electron transfer from tyrosine: a strong rate dependence on intramolecular proton transfer distance. J. Am. Chem. Soc 2011, 133, 13224–13227. [DOI] [PubMed] [Google Scholar]
- (28).Markle TF; Tronic TA; DiPasquale AG; Kaminsky W; Mayer JM Effect of basic site substituents on concerted proton- electron transfer in hydrogen-bonded pyridyl-phenols. J. Phys. Chem. A 2012, 116, 12249–12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Markle TF; Rhile IJ; DiPasquale AG; Mayer JM Probing concerted proton-electron transfer in phenol-imidazoles. Proc. Natl. Acad. Sci U. S. A 2008, 105, 8185–8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Markle TF; Rhile IJ; Mayer JM Kinetic effects of increased proton transfer distance on proton-coupled oxidations of phenol-amines. J. Am. Chem. Soc 2011, 133, 17341–17352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Schrauben JN; Cattaneo M; Day TC; Tenderholt AL; Mayer JM Multiple-Site Concerted Proton-Electron Transfer Reactions of Hydrogen-Bonded Phenols Are Nonadiabatic and Well Described by Semiclassical Marcus Theory. J. Am. Chem. Soc 2012, 134, 16635–16645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Morris WD; Mayer JM Separating proton and electron transfer effects in three-component concerted proton-coupled electron transfer reactions. J. Am. Chem. Soc 2017, 139, 10312–10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bourrez M; Steinmetz R; Ott S; Gloaguen F; Hammarström L Concerted proton-coupled electron transfer from a metal-hydride complex. Nat. Chem 2015, 7, 140–145. [DOI] [PubMed] [Google Scholar]
- (34).Grunwald E Structure-energy relations, reaction mechanism, and disparity of progress of concerted reaction events. J. Am. Chem. Soc 1985, 107, 125–133. [Google Scholar]
- (35).Markle TF; Darcy JW; Mayer JM A new strategy to efficiently cleave and form C—H bonds using proton-coupled electron transfer. Sci. Adv 2018, 4, eaat5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Costentin C; Robert M; Savéant J-M Electrochemical Approach to the Mechanistic Study of Proton-Coupled Electron Transfer. Chem. Rev 2010, 110, PR1–PR40. [DOI] [PubMed] [Google Scholar]
- (37).Schrauben JN; Hayoun R; Valdez CN; Braten M; Fridley L; Mayer JM Titanium and Zinc Oxide Nanoparticles Are Proton-Coupled Electron Transfer Agents. Science 2012, 336, 1298. [DOI] [PubMed] [Google Scholar]