

Abstract
Disulfide heterogeneity and other non-native crosslinks introduced during therapeutic antibody production and storage could have considerable negative effects on clinical efficacy, but tracking these modifications remains challenging. Analysis must also be carried out cautiously to avoid introduction of disulfide scrambling or reduction, necessitating the use of low pH digestion with less specific proteases. Herein we demonstrate that 213 nm ultraviolet photodissociation streamlines disulfide elucidation through bond-selective dissociation of sulfur-sulfur and carbon-sulfur bonds in combination with less specific backbone dissociation. Importantly, both types of fragmentation can be initiated in a single MS/MS activation stage. In addition to disulfide mapping, it is also shown that thioethers and trisulfides can be identified by characteristic fragmentation patterns. The photochemistry resulting from 213 nm excitation facilitates a simplified, two-tiered data processing approach that allows observation of all native disulfide bonds, scrambled disulfide bonds, and non-native sulfur-based linkages in a pepsin digest of Rituximab. Native disulfides represented the majority of bonds according to ion count, but the highly solvent-exposed heavy/light interchain disulfides were found to be most prone to modification. Production and storage methods that facilitate non-native links are discussed. Due to the importance of heavy and light chain connectivity for antibody structure and function, this region likely requires particular attention in terms of its influence on maintaining structural fidelity.
Introduction.
Close to 25% of annual global pharmaceutical sales in 2015 were biological drugs. More than 120 biologics have been approved since 2001, and it was recently estimated that the global market share will rise close to 30% by 2020.1 Given that biologics such as monoclonal antibodies (mAbs) are complex molecules that are most frequently synthesized via recombinant cell technology, there are many questions as to the batch to batch and manufacturer to manufacturer similarity of these molecules.2 This has been highlighted in several case studies that analyzed mAb heterogeneity between Rituximab samples from several manufacturers.3,4 Commonly monitored parameters between batches and vendors include glycosylation profile, charge/size heterogeneity, deamidation, oxidation, as well as cysteinylation, and disulfide scrambling.
Disulfide heterogeneity and degradation in therapeutics is particularly concerning because these crosslinks define three-dimensional structure through covalent bonds. Changes such as disulfide reduction and alternative crosslinks can lead to structural perturbations in antibodies including lower thermostability, spontaneous unfolding, and perturbed antigen affinity.5–6,7,8 There have been a number of bottom-up liquid chromatography mass spectrometry (LCMS) workflows aimed at mapping and characterizing disulfide bond patterns within therapeutic mAbs.9,10 Additionally, several groups have worked towards the detection and characterization of non-native crosslinks such as trisulfides, thioethers and cysteinylation.11,12 Unfortunately, disulfide mapping by mass spectrometry shares many of the same complexities and challenges as encountered with structure-interrogating crosslinking experiments. For example, the number of possible disulfide combinations in a sample scales according to the same formula for peptide crosslinks, (n2+n)/2 where n represents the number of peptide fragments containing a linking residue (for disulfides, cysteine).13 The n2 term in this formula becomes increasingly problematic as the complexity of the digest grows. For example, disuccinimidyl suberate crosslinking followed by tryptic digestion of just 50 randomly selected proteins from the human proteome returns more possible peptide pair combinations than total unique single peptides obtained from digestion of the entire human proteome.14
The quality of information provided by experimental methods can therefore play a crucial role in helping overcome this complexity. Collision-induced dissociation and higher-energy collisional dissociation (CID/HCD) are the most commonly implemented activation methods and can provide useful information, but they also necessitate difficult analyses because cleavage of S-S and C-S bonds are not favored. Fragments from collisional activation are typically b/y ions from one peptide that often remain linked by the disulfide to the other intact peptide.15,16 This complex fragmentation also means that common databases and programs cannot be easily used for analysis.14 To avoid the complication of dealing simultaneously with two different peptide sequences, methods favoring direct dissociation of disulfides have been explored. Several groups have shown that electron-transfer and electron-capture dissociation (ETD/ECD) provide not only sequence information, but also readily cleave S-S bonds, leading to observation of individual peptide pairs and higher confidence in assignments.17–18,19,20,21 Downsides to electron-based dissociation include lengthy activation time, ion charge state dependence, and low dissociation efficiency (i.e. electron transfer/capture with no dissociation is common and sometimes requires another activation stage), although significant progress towards correcting many of these shortcomings has been made.22 Longer activation times reduce the sample complexity that can be interrogated and increase the likelihood that low abundance species will be missed.
Ultraviolet photodissociation (UVPD) offers different fragmentation capabilities and is increasing in use and popularity.23–24,25 Similar to ETD, UVPD can access higher energy fragmentation channels, but efficiency is largely independent of charge state26 and does not require additional reagents, charge reduction or additional stages of activation to dissociate products. This leads to shorter acquisition times and increased sampling. Additionally, it is possible to modulate fragmentation by manipulating laser wavelength, power, and number of laser pulses.26,27 For example, a single MS2experiment utilizing 193 nm UVPD has the potential to produce more fragmentation than any other dissociation technique due to the non-selective nature of excitation.26,28 The data-rich results from these experiments also necessitate careful analysis.29 Conversely, for unmodified peptides, 266 nm photons strongly favor fragmentation at S-S bonds,30 allowing for direct observation of disulfide bound peptides from a tryptic digest.31 Furthermore, nearby chromophores such as tyrosine and tryptophan have the ability to enhance photodissociation yield of the disulfide bond.32 A characteristic spectrum containing 3 peaks is typically observed: the parent ion and two flanking peaks corresponding to the individual peptides. In related work, it was recently shown that the number of free cysteines in proteins can be evaluated by reaction with benzeneselenol followed by UVPD activation at 266 nm. Homolytic dissociation of the S-Se bond allows for analysis of free cysteine content.33 These methods are convenient and facilitate simple analysis, but generally do not afford sequence information since the peptide backbone does not readily absorb 266 nm photons. 213 nm UVPD offers a unique compromise between the selectivity of 266 nm and the extensive dissociation observed at 193 nm.34 213 nm photons are the proper wavelength to initiate both backbone fragmentation and bond-selective dissociation of S-S and C-S bonds, which produces a characteristic triplet pattern for each peptide from a disulfide-linked pair. Dissociation of the disulfide bond typically is more abundant than other fragmentation pathways when present, but peptide bonds are still dissociated in lower abundance.
Herein we explore the utility of 213 nm UVPD for evaluation of the disulfide heterogeneity in a commercial sample of Rituximab. Low pH digestion utilizing pepsin prior to LCMS/MS ensures that any scrambled or degraded disulfide links are not artefacts of the analysis. While pepsin digests are typically complex,35 characteristic triplet fragmentation patterns allow for selective identification of disulfide pairs within the 213 nm UPVD spectra. Additional fragmentation of the backbone is also observed, allowing for confident assignment of each peptide sequence. The low specificity of pepsin allows for complete digestion of the non-reduced antibody. Trisulfides and thioethers are also detected and provide unique fragmentation patterns that are similar to, but distinct from disulfide bonds. Analysis of the data reveals that a majority of disulfide bonds in Rituximab were intact, but some scrambling and alternative linkages were also present. Deviations from native disulfide links were especially prevalent in the hinge region, suggesting that this area is more prone to modification during antibody production and storage.
Experimental.
Rituximab was obtained from Carbosynth LTD (China) at 25 mg/mL in pH 6.0 phosphate buffer and was either used upon receipt or lyophilized and stored at −20 Co. Non-reducing digestion conditions at pH 2.0 were utilized to suppress disulfide scrambling or other modifications. For digestion, 50 µg of Rituximab was desalted/lyophilized and then 50 µL of 0.01 M HCl and 2 M urea in ultrapure water were added. To this mixture, 5 µL of pepsin resin (Proteochem) was added and the mixture was left at room temperature for 15 hours. Resin was removed by centrifugation, and the supernatant was collected. The digest was then desalted and lyophilized before LCMS.
Mass Spectrometry.
LCMS was performed using an Agilent 1100 HPLC with a 5 µM particle, 300 A porous C4 column from Jupiter (Phenomenex, Torrance CA) with a flow rate of 0.200 mL/min. Injection was followed by washing with 2 % ACN (solvent B) for 5 minutes before data collection. Gradients were ramped as follows: 2 % B at 5 minutes, 25% B at 35 minutes, 60 % at 50 minutes and finally 95 % at 60 minutes. Mass spectra were collected on a modified Velos LTQ Orbitrap which had a quartz window installed in the vacuum housing behind the HCD cell to accept laser pulses in a similar manner to previous modifications of an ion trap.36 Data collection was performed in data dependent acquisition mode with the most intense MS1 precursor being selected for MS2 twice before a 60 s exclusion period. Only 2+ ions or higher charge states were selected for analysis. An automatic gain control setpoint of 1.0 × 106 was used, injection times for MS1 and MS2 were 10 and 40 ms, at resolutions of 15000 and 30000, respectively. UVPD activation was performed on ions in the HCD cell during MS2 for 50 ms using a 1000 Hz, 2.5 µJ/pulse solid state CryLaS laser (Germany) triggered by a delay generator (Berkley Nucleonics, CA) to fire repeatedly during MS2. 50 ms was determined to be the shortest activation time that yielded adequate disulfide cleavage for all examined disulfide-bound peptide pairs.
Data Analysis.
Spectra were searched both manually and with the aid of software. Data for analysis by software were centroided, noise-reduced, and converted into mgf or mzML filetypes using software from proteowizard.37 The resulting files were searched with StavroX 38, MeroX39 and Kojak40 for characteristic UVPD fragmentation patterns associated with disulfide containing peptides. Briefly, in silico digest conditions were set such that fragments from 2–25 amino acids in length could be produced with single oxidation of Met as a variable. The ion types a/x b/y c/z were included for scoring. With MeroX, cleavage patterns of S-S, C-S, and S-S-S bonds were utilized as additional parameters in scoring. Returned spectral matches in all programs were then also manually evaluated for correctness.
Results and Discussion.
Given the structural and functional importance of disulfide bonds, a method capable of directly and confidently elucidating disulfide bound pairs from complex digestions is desirable. Figure 1a shows 213 nm UVPD of a disulfide bound peptide pair derived from a pepsin digest of Rituximab. Immediately apparent are peptide fragments SP1 and SP2, resulting from homolytic cleavage of the disulfide bond. The annotation used for individual peptides is shown in Scheme 1. Closer inspection facilitated by the zoomed inset in Figure 1a highlights that UVPD at this wavelength has actually produced two characteristic sets of triplets. Each central peak is flanked by two minor peaks corresponding to either a gain or loss of 32 Da (±S). Importantly, summation of any two complementary dissociation peaks yields the mass of the selected precursor ion, as depicted visually in Scheme 1a. Such distinct MS2 fingerprints allow for rapid data searching and the ability to confidently identify spectra derived from disulfide-bound peptide pairs. The value of bond-selective dissociation was demonstrated previously for a simple protein in disulfide mapping experiments at 266 nm, which produces a set of doublets for each precursor ion.33 Paek and coworkers have also demonstrated that searching MS2 spectra for conserved fragmentation patterns drastically reduces the number of crosslinked candidates.41
Figure 1.

Examples of 213 nm UVPD of disulfide bound peptide pairs extracted from a pepsin digest of Rituximab. a) MS2 of a disulfide link between Cys133 and Cys193 with inset showing the characteristic disulfide triplet pattern. In addition to direct observation of disulfide pairs, sequence information from both chains is also seen. b) UVPD of a disulfide between Cys265 and Cys325 which produces fragments SP1/SP2 and abundant backbone fragmentation. Peptides and fragments are color-coded, i.e. a4 is a normal a4 fragment from P1, whereas a10-S-S-P1 corresponds to the a10 fragment from SP2 with the full sequence of SP1 still attached.
Scheme 1.

Possible complementary fragmentation pairs for a) disulfides b) thioethers and c) trisulfides. Note that depending on the crosslink type, heterolytic and homolytic fragmentation of the crosslink can occur. Simplified notations for each precursor ion and fragment are outlined by red boxes.
In addition to the signature disulfide triplets observed in Figure 1, several sequence ions are also noted for each peptide. A b2 ion from SP1, a y3 ion from SP2 which is still linked to SP1, and a c15-CO2 ion from SP2 are among several low intensity ions. Each of these provides sequence information on SP1 or SP2 and enables a two-tiered data processing approach. In the first pass, all disulfide containing spectra are separated by observation of at least one complementary SP1 and SP2 ion pair. When observed, triplets accompanying these peaks will further confirm the presence of a disulfide. SP1 and SP2 can then be assigned possible identities based on closest matches by mass. In the second pass, data can be further scrutinized for sequence-specific information based only on the preselected candidate identities of SP1 and SP2. Not only does this simplify analysis, it also improves confidence in each assignment. This tiered workflow can be implemented with the recently developed crosslinking analysis program, MeroX.41
To better understand the advantage of this two-tiered approach, consider the MS2 precursor ion in Figure 1a. The deconvoluted mass of this ion is 2428.2251 Da. Using only this mass with an error ≤5 ppm, there would be 22 possible matches based solely on consideration of disulfide linked pairs for a nonspecific digestion of Rituximab. Upon incorporating the masses of the observed ions SP1 and SP2, the number of matches drops down to just two disulfide linked possibilities. Inclusion of backbone fragment ions narrows the search to a single candidate. The power of this workflow is also corroborated by decoy analysis with MeroX. When data is submitted against a reversed Rituximab sequence with the two-tiered approach, 29 identifications above a 5% false discovery rate are obtained. In stark contrast, ignoring the specificity provided by disulfide bond cleavage causes a dramatic shift in scoring and identification. Nearly half of all possible matches are attributable to decoys if bond-selective cleavage is ignored (Supporting Figure S1), and the total number of confident identifications drops to 4. This contrast illustrates the tremendous statistical power that can be leveraged from a small amount of controlled fragmentation.
While fragmentation in Figure 1a is less abundant than that seen using 193 nm,42 the resulting spectrum is simple to analyze and easily provides sufficient sequence information for unambiguous assignment. The majority of spectra obtained from disulfide bound peptides pairs are similar to that shown in Figure 1a (see Supporting Information). However, certain backbone fragmentation channels can be competitive, as illustrated in Figure 1b. The peptide pair linked by Cys265 and Cys325 of the heavy chain yields backbone dissociation comparable to the yield of the SP1/SP2 fragments. Both sequences contain proline (ISRTPEVTCVVVD, YKCKVSNKALPAPIE), which is known to facilitate backbone dissociation at 213 nm, producing intense b+2, a+2, and complementary y-2 ions.43 Figures S2 and S6 have examples of this unusual fragmentation which results in observation of yn-2 and yn doublets. Although the spectrum in Figure 1b does not stand out as a disulfide bound pair, the software easily makes a correct assignment because the SP1 and SP2 masses are still observed. Furthermore, if desired, the proclivity for proline fragmentation at 213 nm could be incorporated into scoring algorithms for analysis of extremely complex samples. In addition, the spectrum in Figure 1b still contains relatively few peaks, facilitating data analysis. Overall, the results in Figure 1 demonstrate that UVPD at 213 nm yields data conducive to the rapid and confident identification of disulfide bound peptide pairs in mAbs.
Interestingly, 213 nm UVPD also revealed spectra with similar features to those found in disulfides that did not match any disulfide bound peptides pairs. For example, in Figure 2a selective dissociation yields a SP1/P2 fragment pair lacking the expected accompanying triplets or even complementary SP1/SP2 ions. Consideration of the precursor mass suggests a thioether between Cys371 and Cys429 of the heavy chain. The generic fragments that would be expected from UVPD of a thioether are shown in Scheme 1b. Cleavage on either side of the C-S bond can theoretically yield doublets for each P1-S-P2 pair. This is not observed in Figure 2a, most likely because C-S bond cleavage is less favorable than S-S bond dissociation.36 In any case, the selective dissociation is again supplemented with backbone sequence fragmentation for more confident assignment. Figure 2b represents another example of a disulfide-like spectrum that doesn’t match any disulfide pairs. In this case, a triplet is observed, suggesting a disulfide is present. Indeed, the data match a trisulfide interchain crosslink between Cys213 of the light chain and Cys224 of the heavy chain. Trisulfide bonds can yield triplets (possibly even quadruplets), as illustrated in Scheme 1c. The SP2 ion presents as a singlet with an abundant loss of CO2 (likely due to location of the cysteine at the C-terminus). Importantly, bond-selective dissociation still creates signature spectra for these unusual modifications, facilitating identification and assignment. Thioethers are thought to form under basic conditions where the additional sulfur is lost during formation of the crosslink. Thioethers are non-reducible and decrease the length between cysteine residues by ~1.5 Å, which may be sufficient to influence functionality in certain cases. Trisulfides result from insertion of an additional sulfur atom into the disulfide bond, increasing the length between cysteine residues by ~1.5 Å relative to a disulfide. Trisulfide formation has been shown to track closely with free H2S levels, which can be elevated by certain hybridoma fermentation conditions.44 The increased bond length between cysteine residues results in higher reactivity, as is well documented in a previous study examining trisulfides in mAbs.45
Figure 2.

Examples of alternative crosslink fragmentation following UVPD activation. MS/MS of a) thioether and b) trisulfide
The dissociation chemistry illustrated in Figures 1 and 2 is powerful for identification of sulfur-containing crosslinks, both native and non-native. However, care must be taken during the experiment to avoid introduction of non-native crosslinks or loss of native crosslinks. Although digestion below pH 7 will reduce disulfide scrambling, it has been demonstrated that significantly lower pH is required to eliminate all scrambling in antibodies.46,21Also, while IgGs should not contain free cysteines, non-zero levels have been detected in all four sub-classes, which can have deleterious effects on mAb function.47 The choice of pH and protease is crucial during digestion as free cysteine is reactive down to pH 4.48 High reactivity has also been observed in our own work where disulfides or cysteine containing peptides in pH 5 buffered solution were found to be partially scrambled/crosslinked after incubation at 37oC for 12 hours (data not shown). Furthermore, while specific proteases produce predictable and reproducible digestion patterns, fewer and larger fragments are generated than with less specific proteases. This results in digested fragments having a higher chance of falling outside the normal m/z range, being less activatable by some MS2 methods, and lower probability for observing sequences of potential interest since cleavage sites are restricted to one or two specific residues. The peptide pairs shown in Figures 1 and 2 were obtained at pH 2.0 following digestion of Rituximab by pepsin. This pH insures very minimal scrambling, but also necessitates the use of pepsin, which offers less selective digestion relative to many proteases. The lack of selectivity is both a blessing and a curse. Proteins with disulfide bonds intact are less amenable to digestion than proteins that have been reduced and capped. Therefore, as protease specificity increases, the likelihood for successful digestion decreases because proteases with high specificity are of necessity more selective and less likely to find suitable binding sites. This outcome is illustrated in Supplemental Figure S8, which shows LCMS chromatograms for digestion at pH 6.5 with trypsin and pH 2.0 with pepsin. Visual inspection reveals that digestion is more complete with pepsin, which is confirmed by quantitative analysis. Tryptic peptide mapping produces only 32% sequence coverage and does not reveal all 8 native disulfides even with the power of bond-selective dissociation afforded by 213 nm UVPD.
In contrast, digestion with pepsin at pH 2.0 easily yields identification of all native disulfide bonds (disulfide coverage is shown in Figure 3). 65% sequence coverage is obtained, which is reasonable given that singly charged ions were omitted with the expectation that all crosslinked peptides would be present in the 2+ charge state or higher. A full list of native peptide pairs identified by UVPD is given in Table 1. Due to the non-specific nature of pepsin digestion, the same disulfide links were often identified in various peptide pairs. In other words, SP1 and SP2 often varied in length for the same pairs of cysteine residues. This inherent multiplicity highlights the utility of bond-selective dissociation for identifying peaks of interest from a complex mixture.
Figure 3.

Sequence and disulfide coverage for the peptic digestion of Rituximab. LCMS-MS identified regions of sequence and crosslinks are labeled by red text or lines, respectively. The identified hinge region at HC230-HC230 and HC233–233 disulfides are marked by red asterisks
Table 1.
Correctly linked disulfide pairs observed by LCMS. Lowercase m denotes oxidized methionine
| Mass | Peptide 1 | Peptide 2 | Link |
|---|---|---|---|
| 1474.65 | TSEDSAVYYC | SCK | HC22-HC96 |
| 2670.26 | EDSAVYYCAR | GAELVKPGASVKmSC | |
| 2296.14 | AVYYCAR | VKPGASVKMSCKASG | |
| 3095.46 | AVYYCARST | VKPGASVKmSCKASGYTFTS | |
| 1854.91 | YYCARS | PGASVKmSCKA | |
| 2691.33 | YCARST | LVKPGASVKmSCKASGYTF | |
| 2666.28 | PSSSLGTQTYICNV | AALGCLVKDYF | HC148-HC204 |
| 1441.7 | YICN | GTAALGCLVK | |
| 1799.87 | YICNV | SGGTAALGCLVKD | |
| 2688.15 | EPKSCDKTHTCP | EPKSCDKTHTCP | HC230-HC230 |
| 1645.65 | HTCPPCPA | HTCPPCPA | +HC233-HC233 |
| 1255.58 | CPAPEL | CPAPEL | |
| 5408.70 | STYRVVSVLTVLHQDWLNGKEYKCK | DTLMISRTPEVTCVVVDVSHED | HC265-HC325 |
| 3954.03 | NGKEYKCKVSNKALPAP | RTPEVTCVVVDVSHEDPEV | |
| 1501.76 | GKEYKCK | VTCVVV | |
| 3075.66 | YKCKVSNKALPAPIE | ISRTPEVTCVVVD | |
| 2594.36 | KCKVS | KPKDTLmISRTPEVTCVV | |
| 1119.49 | FSCSVM | LTCL | HC371-429 |
| 2366.05 | FSCSVmHEAL | TCLVKGFYPSD | |
| 3026.44 | FSCSVmHEALHNHYTQKSLSLSPG | TCL | |
| 1752.79 | SCSVM | TCLVKGFYPSD | |
| 2594.35 | SRVEAEDAATYYCQQWTSNP | CR | LC23-LC87 |
| 1155.69 | YYC | VTMTCR | |
| 1774.76 | CQQWTSN | KVTMTCRA | |
| 2315.17 | VVCL | TLSKADYEKHKVYACE | LC133-LC193 |
| 2101.04 | VVCL | SKADYEKHKVYACE | |
| 2707.36 | VVCLLNN | TLSKADYEKHKVYACE | |
| 2216.1 | VCL | KHKVYACEVTHEGLSSP | |
| 2704.31 | VCLLNNF | TLSKADYEKHKVYACE | |
| 2687.32 | KVYAC | EPPSDEELKSGTASVVCLLD | |
| 2370.9 | KVDKKAEPKSC | VTKSFNRGEC | LC213-HC224 |
| 3953.97 | VNHKPSNTKVDKKAEPKSC | HQGLSSPVTKSFNRGEC |
Misconnected pairs, including thioethers and trisulfides, are listed in Table 2. The relative abundances of each type of crosslink, as determined by extracted ion chromatogram (EIC) peak areas, are shown in Figure 4. The results suggest that roughly 87% of the sample was found in correctly linked form with 8% corresponding to disulfide scrambling. As expected, thioether and trisulfide links only account for ~5% of the total. The 25 mg/mL Rituximab sample was received from the manufacturer in a pH 6.0 solution of phosphate buffer. This relatively high pH and unknown temperature variations during transit may account for some of the disulfide scrambling.
Table 2.
Observed unique peptide pairs composed of alternate crosslinks. Thioether and trisulfide linkages marked with asterisks are also paired at incorrect linkage sites.
| Mass | Peptide 1 | Peptide 2 | Link Sites |
|---|---|---|---|
| Incorrectly Linked | |||
| 2763.48 | NEVSLTCLVK | YKCKVSNKALPAPIE | HC148-HC325 |
| 2329.19 | CLV | DWLDGKEYKCKVSDKAL | HC148-HC325 |
| 3915.87 | PPSDEQLKSGTASVVCLLN | ASPGEKVTMTCRASSSVSY | LC133-LC23 |
| Thioether | |||
| 2315.17 | TKVDKKAEPKSCDKTHT | PKSC | HC224-HC224 * |
| 3397.68 | PSRDELTKNQVSLTCLVK | WQQGNFCSVM | HC371-HC429 |
| Trisulfide | |||
| 3953.99 | SRDELTKNQVSLTCLVKGFYP | STYRVVSVLTVHQDWLNGKEYKCK | HC265-HC325 |
| 1472.65 | KSC | KAEPKSCDKT | HC224-HC224 * |
| 2670.28 | LSSPVTKSFNRGEC | AEPKSCDKTH | LC213-HC224 |
Figure 4.
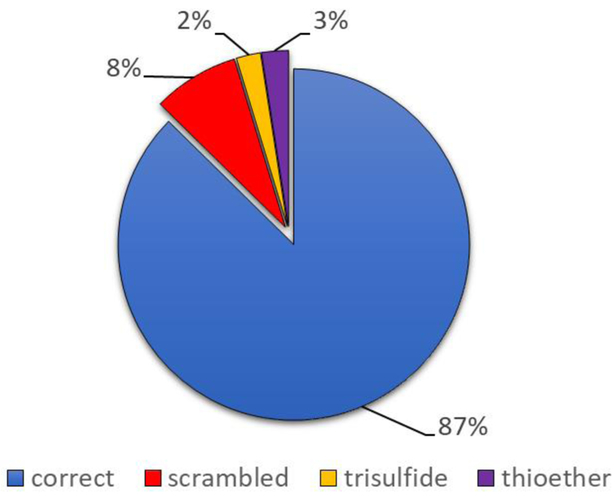
Extent of disulfide heterogeneity as approximated by EIC intensities.
It is interesting to note that more than half of the tabulated thioether and trisulfide modifications are related to the interchain disulfide between Cys224 of the heavy chain and Cys213 of the light chain. In addition, the native HC224-LC213 disulfide is one of the least observed pairs in our data (seen only twice). The implications of each mislink are not immediately clear in terms of mAb function except for the thioether between HC224-HC224 which may result from a complete loss of heavy-light chain connectivity. It is not a stretch to imagine this loss would dramatically affect mAb structure and function. To explore potential reasons for observation of the apparent increased reactivity of interchain cysteine pairs, the IgG1 structure was examined in more detail. The relative locations of native interchain disulfide bonds in an analogous murine IgG1 mAb are presented in Figure 5 (PDB: 1IGY). Upon inspection of the crystal structure, it is apparent that the unstructured hinge region is likely flexible due to lack of any secondary structure and absence of flanking structures. The reported challenges associated with crystallizing this IgG subclass are also consistent with flexibility.49 Increased motion at the hinge region should also result in higher solvent accessibility and therefore greater susceptibility to chemical modification. Indeed, reports of selective partial reduction of IgG1 in the hinge region and its use for attaching antibody-drug conjugate payloads both support the notion of increased chemical reactivity.50,51 In addition, solvent accessibility is also consistent with the common practice of utilizing proteolytic enzymes like pepsin or papain for separation of Fab and Fc regions, which requires cleavage just below and just above the hinge region, respectively. These results from the literature and our own observations suggest that the hinge regions are more reactive and should be given careful consideration during the production, formulation, and storage of therapeutic antibodies.
Figure 5.

Crystal structure of a full IgG1 mAb. Heavy chain portions are illustrated in green while the light chains are blue. Zoomed inset of the hinge region shows both the heavy-light interchain disulfide links HC224-HC213 and heavy-heavy interchain links HC230-HC230 and HC233-HC233. These links are numbered as they would appear in Rituximab for illustrative purposes.
Conclusions.
Given the nature and purpose of biological therapeutics, it is self-evident that reliable characterization is imperative. The current work demonstrates that 213 nm UVPD can facilitate identification and characterization of disulfide bonds in complex molecules such as antibodies. 213 nm UVPD not only provides useful sequence information, but also preferentially and distinctly fragments S-S and C-S bonds for easy and confident crosslink identification. Disulfides, thioethers and trisulfides are all discernible due to unique fragmentation patterns offered by each link type using 213 nm UVPD. While scrambled disulfides and alternative crosslinks are also detected, they appear to be in relatively low abundance. Because of this, it is also clear that careful control of pH at all stages of sample storage and handling is necessary to minimize scrambling. In particular, our results suggest the interchain disulfide linkage between HC224-LC213 is susceptible to modification. Using data gathered from these types of UVPD experiments has potential to further our understanding of IgG1 native biological structure and reactivity, eventually resulting in improvements to mAb-based therapeutics.
Supplementary Material
Acknowledgements
The authors are grateful for funding from the NIH (NIGMS grant R01GM107099).
Footnotes
Supporting Information
Additional mass spectra and LC chromatograms are provided.
References.
- 1.Rogstad S, Faustino A, Ruth A, Keire D, Boyne M and Park J, J. Am. Soc. Mass Spectrom, 2017, 28, 786–794. [DOI] [PubMed] [Google Scholar]
- 2.Kunert R and Reinhart D, Appl. Microbiol. Biotechnol, 2016, 100, 3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck A, Diemer H, Ayoub D, Debaene F, Wagner-Rousset E, Carapito C, Van Dorsselaer A and Sanglier-Cianférani S, TrAC Trends Anal. Chem, 2013, 48, 81–95. [Google Scholar]
- 4.Nupur N, Chhabra N, Dash R and Rathore AS, MAbs, 2018, 10, 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu H and May K, MAbs, 2012, 4, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kikuchi H; Goto Y; Hamaguchi K Biochemistry 1986, 25 (8), 2009. [DOI] [PubMed] [Google Scholar]
- 7.Lacy ER; Baker M; Brigham-Burke M Anal. Biochem 2008, 382 (1), 66. [DOI] [PubMed] [Google Scholar]
- 8.Ouellette D; Alessandri L; Chin A; Grinnell C; Tarcsa E; Radziejewski C; Correia I Anal. Biochem 2010, 397 (1), 37. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Lu Q, Wu SL, Karger BL and Hancock WS, Anal. Chem, 2011, 83, 3133–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lakbub JC, Clark DF, Shah IS, Zhu Z, Su X, Go EP, Tolbert TJ and Desaire H, Anal. Methods, 2016, 8, 6046–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu S, Wen D, Weinreb PH, Sun Y, Zhang L, Foley SF, Kshirsagar R, Evans D, Mi S, Meier W and Pepinsky RB, Anal. Biochem, 2010, 400, 89–98. [DOI] [PubMed] [Google Scholar]
- 12.McSherry T, McSherry J, Ozaeta P, Longenecker K, Ramsay C, Fishpaugh J and Allen S, MAbs, 2016, 8, 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rappsilber J, J. Struct. Biol, 2011, 173, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yılmaz Ş, Shiferaw GA, Rayo J, Economou A, Martens L and Vandermarliere E, Mass Spectrom. Rev, DOI: 10.1002/mas.21559. [DOI] [PubMed] [Google Scholar]
- 15.Liang Z, McGuinness KN, Crespo A and Zhong W, J. Am. Soc. Mass Spectrom, 2018, 29, 1–10. [DOI] [PubMed] [Google Scholar]
- 16.Jones LM; Zhang H; Cui W; Kumar S; Sperry JB; Carroll JA; Gross ML J. Am. Soc. Mass Spectrom 2013, 24 (6), 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK and McLafferty FW, J. Am. Chem. Soc, 1999, 121, 2857–2862. [Google Scholar]
- 18.Cole SR, Ma X, Zhang X and Xia Y, J. Am. Soc. Mass Spectrom, 2012, 23, 310–320. [DOI] [PubMed] [Google Scholar]
- 19.Liu F, van Breukelen B and Heck AJR, Mol. Cell. Proteomics, 2014, 13, 2776–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones LM, Zhang H, Cui W, Kumar S, Sperry JB, Carroll JA and Gross ML, J. Am. Soc. Mass Spectrom, 2013, 24, 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rush MJP, Riley NM, Westphall MS and Coon JJ, Anal. Chem, 2018, 90, 8946–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fort KL; Cramer CN; Voinov VG; Vasil’Ev YV; Lopez NI; Beckman JS; Heck AJR J. Proteome Res 2018, 17 (2), 926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fung YME, Kjeldsen F, Silivra OA, Chan TWD and Zubarev RA, Angew. Chem. Int. Ed, 2005, 44, 6399–6403. [DOI] [PubMed] [Google Scholar]
- 24.Brodbelt JS, Chem. Soc. Rev, 2014, 43, 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reilly JP, Mass Spectrom. Rev, 2009, 28, 425–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodbelt JS, Anal. Chem, 2016, 88, 30–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becher S; Spengler B; Heiles S Eur. J. Mass Spectrom 2018, 24 (1), 54. [DOI] [PubMed] [Google Scholar]
- 28.Julian RR, J. Am. Soc. Mass Spectrom 2017, 28, 1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL and Brodbelt JS, J. Am. Chem. Soc, 2013, 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ly T and Julian RR, J. Am. Chem. Soc, 2008, 130, 351–358. [DOI] [PubMed] [Google Scholar]
- 31.Agarwal A, Diedrich JK and Julian RR, Anal. Chem, 2011, 83, 6455–6458. [DOI] [PubMed] [Google Scholar]
- 32.Hendricks NG, Lareau NM, Stow SM, Mclean JA and Julian RR, J. Am. Chem. Soc, 2015, 136, 13363–13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker WR; Brodbelt JS J. Am. Soc. Mass Spectrom 2016, 27 (8), 1344. [DOI] [PubMed] [Google Scholar]
- 34.Talbert LE, and Julian RR, J. Am. Soc. Mass Spectrom, 2018, 29, 1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahn J, Cao MJ, Yu YQ and Engen JR, Biochim. Biophys. Acta - Proteins Proteomics, 2013, 1834, 1222–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ly T and Julian RR, J. Am. Chem. Soc, 2010, 132, 8602–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kessner D, Chambers M, Burke R, Agus D and Mallick P, Bioinformatics, 2008, 24, 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Götze M; Pettelkau J; Schaks S; Bosse K; Ihling CH; Krauth F; Fritzsche R; Kühn U; Sinz AJ Am. Soc. Mass Spectrom 2012, 23 (1), 76. [DOI] [PubMed] [Google Scholar]
- 39.Götze M, Pettelkau J, Fritzsche R, Ihling CH, Schäfer M and Sinz A, J. Am. Soc. Mass Spectrom, 2014, 26, 83–97. [DOI] [PubMed] [Google Scholar]
- 40.Hoopmann MR, Zelter A, Johnson RS, Riffle M, Maccoss MJ, Davis TN and Moritz RL, J. Proteome Res, 2015, 14, 2190–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi S, Jeong J, Na S, Lee HS, Kim HY, Lee KJ and Paek E, J. Proteome Res, 2010, 9, 626–635. [DOI] [PubMed] [Google Scholar]
- 42.Cotham VC and Brodbelt JS, Anal. Chem, 2016, 88, 4004–4013. [DOI] [PubMed] [Google Scholar]
- 43.Girod M, Sanader Z, Vojkovic M, Antoine R, MacAleese L, Lemoine J, Bonacic-Koutecky V and Dugourd P, J. Am. Soc. Mass Spectrom, 2015, 26, 432–443. [DOI] [PubMed] [Google Scholar]
- 44.Kshirsagar R, McElearney K, Gilbert A, Sinacore M and Ryll T, Biotechnol. Bioeng, 2012, 109, 2523–2532. [DOI] [PubMed] [Google Scholar]
- 45.Liu R; Chen X; Dushime J; Bogalhas M; Lazar AC; Ryll T; Wang L MAbs 2017, 9 (3), 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sung W-C, Chang C-W, Huang S-Y, Wei T-Y, Huang Y-L, Lin Y-H, Chen H-M and Chen S-F, Biochim. Biophys. Acta - Proteins Proteomics, 2016, 1864, 1188–1194. [DOI] [PubMed] [Google Scholar]
- 47.McSherry T, McSherry J, Ozaeta P, Longenecker K, Ramsay C, Fishpaugh J and Allen S, MAbs, 2016, 8, 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trivedi M, Laurence J and Siahaan T, Curr. Protein Pept. Sci, 2009, 10, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rayner LE, Hui GK, Gor J, Heenan RK, Dalby PA and Perkins SJ, J. Biol. Chem, 2015, 290, 8420–8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu H, Chumsae C, Gaza-Bulseco G, Hurkmans K and Radziejewski CH, Anal. Chem, 2010, 82, 5219–5226. [DOI] [PubMed] [Google Scholar]
- 51.Sochaj AM, Świderska KW and Otlewski J, Biotechnol. Adv, 2015, 33, 775–784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.