

Abstract
Glioblastoma is one of the first tumors where the biological changes accompanying a single epigenetic modification, the methylation of the MGMT gene, were found to be of clinical relevance. The exploration of the epigenomic landscape of glioblastoma has allowed to identify patients carrying a diffuse hypermethylation at gene promoters and with better outcome. Epigenetic and genetic data have led to the definition of major subgroups of glioma and were the basis of the current WHO classification of CNS tumors and of a novel classification based solely on DNA methylation data that shows a remarkable diagnostic precision.The reversibility of epigenetic modifications is considered a therapeutic opportunity in many tumors also because these alterations have been mechanistically linked to the biological characteristics of glioblastoma. Several alterations like IDH1/2 mutations that interfere with “epigenetic modifier” enzymes, the mutations of the histone 3 variants H3.1 and H3.3 that alter the global H3K27me3 levels and the altered expression of histone methyltransferases and demethylases are considered potentially druggable targets in glioma and molecules targeting these alterations are being tested in preclinical and clinical trials. The recent advances on the knowledge of the players of the “epigenetic orchestra” and of their mutual interactions are indicating new paths that may eventually open new therapeutic options for this invariably lethal cancer.
Keywords: glioblastoma, epigenetics, therapy, DNA methylation, histone code
Introduction
Glioblastoma (Glioblastoma Multiforme, GBM) is a rare tumor (Orphanet 360) that, being responsible for 4% of all tumor deaths and with a 5-years survival of 2%, is one of the deadliest human tumors (1) with the median survival ranging from 14 to 24–30 months depending from the molecular subtype of the tumor (2).
GBM, like other tumors, harbors many genetic alterations that interfere with cancer-related pathways (3), however clinical trials targeting molecular alterations in this tumor were largely unsuccessful so far (4–6). In the last 30 years, the only significant improvement in OS occurred with the introduction of Temozolomide (TMZ) in addition to surgery and radiotherapy (7, 8). GBM patients are stratified into two categories according to the methylation status of the O-6-methylguanine-DNA methyltransferase gene (MGMT) that repairs the DNA damages induced by TMZ and the patients whose tumor contains methylated MGMT have an overall survival of 21.7 months compared to the 12.7 months of those carrying unmethylated MGMT (9).
Epigenetic modifications are considered a key mechanism in GBM development (10). Epigenetic inheritance is mediated by the four deeply interconnected layers shown in Figure 1:
1- DNA methylation
2- Histone modifications
3- Chromatin remodeling
4- Non-coding RNA
Figure 1.
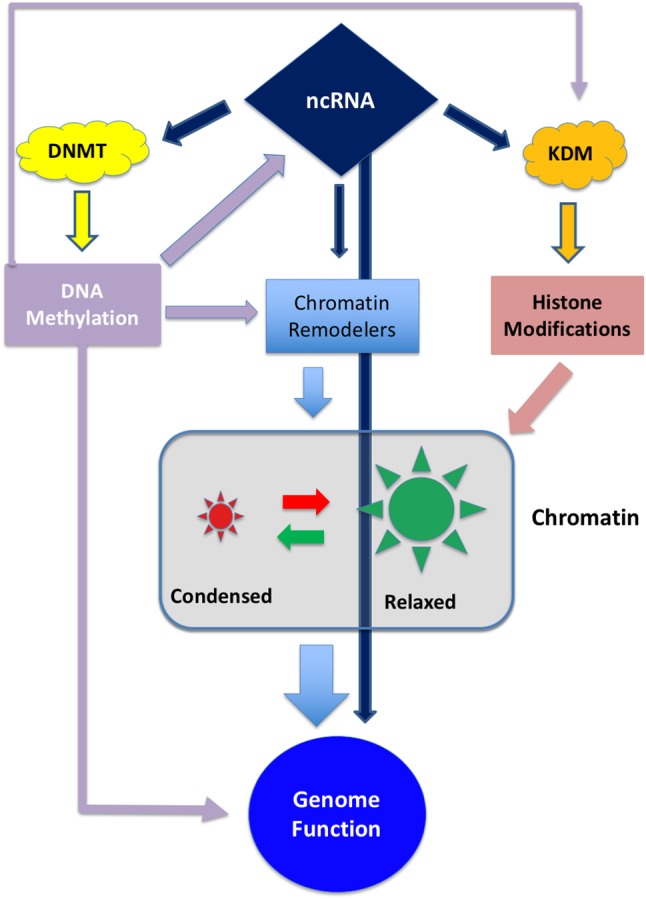
Schematic representation of the interplay between the different epigenetic layers. ncRNA can directly influence genome activity by interfering with transcripts or, indirectly, by degrading transcripts involved in DNA methylation, histone modification or chromatin remodeling. On the other hand ncRNA can be epigenetically inactivated by DNA methylation (11, 12). DNA methylation can directly interfere with gene expression and, indirectly, can regulate the expression of chromatin and histone modifiers.
These layers are controlled by a set of enzymes that act as “writers,” “readers,” and “erasers” that modify their target by adding, removing or regulating the interactions between proteins and DNA. Both DNA methylation and histone modification, along with chaperon molecules, participate to chromatin remodeling thus conferring an exquisite plasticity to the genetic apparatus (13, 14).
The latest WHO classification defines subgroups of glioma integrating genetic and epigenetic criteria (10, 15–18) (Figure 2) and a novel classification of CNS tumors based on DNA methylation data, shows a remarkable diagnostic precision being able to correctly modify the primary diagnosis in 12% of the cases (19). A large number of intrinsically reversible cancer-related modifications that are attractive targets of therapy was unveiled and the present review provides an overview of the most recent preclinical and clinical attempts to defy GBM through epigenetic reprogramming.
Figure 2.

Schematic representation of the different subtypes of glioma with the principal molecular and epigenetic characteristics. In this chart are represented the possible carcinogenic evolutions of the precursor cells. The major genetic and epigenetic alterations are reported along with the clinical characteristics of each subtype.
Targeting epigenetic alterations in glioblastoma
Manipulating the epigenome has been lengthy considered a therapeutic opportunity in cancer. The epigenetic landscape of GBM was thoroughly explored, and many epigenetic modifications were mechanistically linked to the biological characteristics of this tumor and some of them were considered as therapeutic targets. At the moment, only the molecules acting on DNA methylation and histone methylation/chromatin remodeling were tested in clinical trials. Manipulation of ncRNA expression is restricted to pre-clinical studies and is not discussed in the present review.
Layer 1: DNA methylation
Methylation of cytosine at C-5 within CpG doublets, is mediated by a set of DNA Methyltransferases that are responsible mainly but not exclusively of maintenance (DNMT1) and de novo DNA methylation (DNMT3a and 3b) to preserve genomic integrity (20). The human genome contains approximately 3 X 107 CpG doublets and although methylation at single doublets may, in principle, have functional consequences (21, 22), the biologically-relevant DNA methylation is that occurring at CpG clusters (CpG islands) in gene promoter regions and inversely correlates with gene transcription (23, 24). Intragenic CpG clusters are generally hypermethylated to prevent spurious initiation particularly at internal promoters.
In GBM DNA methylation is tightly linked to the response to TMZ treatment. The alkylating agent TMZ, the first-line chemotherapy for GB, methylates guanine in position N7 and O6 and Adenine in position N3. O6-methylguanine adducts lead to strand breaks, triggering p53-mediated apoptosis through the Fas/CD95/Apo-1 receptor in p53wt cells or through the mitochondrial pathway in p53mut tumors (25). The action of TMZ is counteracted by the MMR system and by the product of the MGMT gene that repairs the O6 adducts that limit the activity of the drug (26). To mimic the effects of MGMT methylation, synthetic inhibitors of MGMT entered human trials (9, 27). However, several studies revealed that the MGMT inhibitors O6-benzylguanine and PaTrim-2 (Lomeguatrib) did not improve the response rate to TMZ and increased the adverse effects of chemotherapy (27–30).
Inducing TMZ sensitivity in MGMT-unmethylated tumors with other molecules (Resvetrol, oncolytic viruses or by MGMT depletion) was tested only in preclinical models with alternate success (31). The correlation between MGMT methylation and MGMT protein expression is controversial and the lack of correlation seen in recent studies likely depends on the early method utilized for methylation analysis (32–34).
Layer 1: methylator phenotype and IDH1/2 mutations
The discovery of mutations of the Isocitrate Dehydrogenase (IDH) genes and of the DNA hypermethylation signature (Glioma CpG Island Methylator Phenotype: G-CIMP) has led to the definition of a distinct GBM subtype characterized by younger age and improved survival (2, 17, 18, 35) (Figure 2, N. 1). IDH mutations are rare in GBM developing in older patients who usually carry EGFR and PTEN alterations (primary GBM), (Figure 2, N. 2), but are present in a large proportion of low-grade glioma and, along with TP53 mutations, in high-grade glioma that evolved from low–grade tumors (secondary glioblastoma) (35) Figure 2, N. 3 and 4). IDH genes can be mutated at two mutually exclusive sites, R132 (IDH1) or R172 (IDH2) and these mutations have important metabolic consequences and are driving alterations in gliomagenesis. The product of IDH converts Isocitrate into αKetoglutarate (αKG) which is involved in a variety of cellular processes (Supplementary Figure 1). IDH mutants produce 2-Hydroxy Glutarate [2-HG] that is a competitive inhibitor of αKG-dependent dioxygenases including the histone demethylases JHDM1 and KDM4 and the DNA demethylase TET2. Thus, IDH mutations, that are not restricted to brain tumors, result in extensive epigenetic dysregulation including DNA and histone hypermethylation (36, 37) and altered cell differentiation (38). Other IDH mutations were occasionally found but only few of them produce 2-HG (39).
Strategies to target IDH-mutant tumors can be designed to either inactivate the functions of IDH mutants or to block the effects of 2-HG. The treatment with hypomethylating agents of mice xenografted with IDHmut GBM cells resulted in delayed tumor growth and improved survival (40, 41). Along this line phase I and II clinical trials were started to test two formulations of 5-azacytidine (NCT02223052) and the combination decitabine/immunotherapy (NCT02332889) in GBM and other solid tumors.
Normalizing the 2-HG concentration could reverse DNA hypermethylation and release the block of differentiation in IDH-mutated cells. Several inhibitors of mutated IDH1/2 were synthesized and showed to be effective in in vitro models (42–44); this finding was the starting point for a large series of clinical trials to assess the safety and bioavailability of the molecules under investigation in a variety of tumors, mainly AML, MDS and glioma (Supplementary Table I). Preliminary data on the clinical efficacy of IDH inhibitors showed promising results in hematological malignancies opening the way for stringent randomized trials (45–47). As of June 2018, no public data are yet available for glioma patients.
Mutated IDH1/2 can be functionally considered as highly specific tumor-associated neoantigens that could be targeted by immunotherapy; a vaccine targeting mutant IDH1 showed antitumor activity in a glioma animal model opening the possibility of new experimental therapies (48).
Layer 2: histone modifications
Histones are subject to modifications that could either repress or activate transcription (Supplementary Figure 2). More than 100 enzymes act in concert to assemble a “code” of histone modifications that define the transcriptional properties of a given gene (49) determining drug response (50) and the development of cancer and other diseases (10, 51–53).
The rapid acquisition of drug resistance is a major cause of treatment failure in GBM (54) and could be explained by the development of epigenetically poised cells that undergo chromatin remodeling and display transient drug resistance (55–57).
Histone acetylation
The addition of acetyl groups to certain lysines of H3 and H4 weakens the interaction between the core histones and DNA favoring the accessibility of the transcription apparatus. Deacetylation removes the acetyl groups provoking chromatin condensation and gene inactivation (49, 58). Acetylation and deacetylation are dynamic processes mediated by histone acetyltransferases (HAT) and histone deacetylases (HDAC) that maintain the balanced state of acetylation. Gain of HDAC expression has been found in many tumors, including GBM, and inhibitors of HDAC (HDACi) have been extensively explored for GBM therapy. HDACi have a large spectrum of antitumor activity and six HDACi have been approved by FDA: Vorinostat (11 studies concluded and 3 ongoing), Romidepsin (one study concluded), Belinostat (one study ongoing), Panobinostat (2 studies terminated before completion), Valproic acid (two studies terminated before completion and two recruiting) and Entimostat (no studies yet) (59) (https://clinicaltrials.gov, June 2018). Preclinical studies have shown that HDACi are very effective against GB cells, but the results of the clinical trials were largely disappointing. In adult patients Vorinostat was utilized as single agent and in combination with standard or biological therapies and in one study (NCT00238303) prolonged disease stabilization in a small subset of patients when used as single agent (60) but its addition to the standard radio/chemotherapy did not improve survival (61). Phase I/II trials with Romidepsin, with Panobinostat and anti-VEGF, or with Vorinostat and the proteasome inhibitor Bortezomib were either ineffective or toxic and were discontinued (62–64). Panobinostat however, is now being tested as a radiosensitizing molecule with promising results (65). Along this line phase II studies demonstrated that the addition of valproic acid to the standard radio/chemotherapy or to radiotherapy alone improved survival (66, 67). Randomized trials are necessary to confirm this finding.
Histone methylation
Histone methylation was discovered along with histone acetylation (58) but its function remained obscure for many years because methylation does not change the DNA/protein interactions and it seemed an irreversible modification. With the discovery of LSD1 (KDM1), the first histone demethylase, it became clear that histone methylation is a reversible process (68) mediated by approximately 30 enzymes subdivided into distinct classes, and linked to a variety of physiological and pathological conditions including cancer, cardiovascular diseases, abnormal immune response and neurological disorders (51, 69). Histone methylation involves certain lysine and arginine of H3 and H4 and can either activate or repress transcription (Supplementary Figure 2).
In glioblastoma, histone methylation has distinct implications in pediatric and adult patients. Histone variant H3.3 (H3F3A) marks active chromatin domains and in pediatric tumors can be mutated at two sites: lysine 27 (K27M) and glycine 34 (G34R/V) (70, 71) (Figure 2, N. 5 and 6). In pediatric glioma the K27M mutation is restricted mostly to midline tumors whereas G34R/V is prevalent in hemispheric gliomas. K27M decreases methylation at K27 leading to transcriptional activation. G34R/V is associated with the redistribution of the activation mark H3K36 methylation and results in the upregulation of the oncogene MYCN (72) whose exogenous overexpression initiates glioma formation during development (73). H3F3A-K27M also inhibits the PRC2-EZH2 axis (2, 74), that acts as a histone methyltransferase, leading to the generalized loss of H3K27 methylation and to the CpG hypomethylator phenotype (CHOP) whose consequence is the aberrant activation of gene expression (75).
The methylation of H3K27 is regulated by PRC2-EZH2 methylases and by the UTX (KDM6A) and KDM6B demethylases; the effect of the K27M mutation could be reversed by inhibiting H3K27 demethylation. In an experimental model of diffuse intrinsic pontine glioma (DPIG), the small molecule GSK J4 was utilized to inhibit the activity of KDM6B (76). It was found that GSK J4 passes the Blood Brain Barrier and prolongs survival of mice xenografted with H3K27 tumors but not that of mice carrying WT H3.3 or G34R/V tumors. Although GSK J4 has proven to be effective in in vivo tumor models as single agent or synergically with HDACi (76–78), as of June 2018, clinical trials employing this or similar molecules have not been launched yet.
Targeting EZH2 is another mechanism to modulate histone methylation and to reverse tumor growth (79). Several FDA-approved EZH2 inhibitors are available (Tazemetostat, CPI-1205, GSK2816126) and others are in advanced pre-clinical testing. More than 20 trials that include EZH2 inhibition are reported in the Clinical Trial database mostly aimed at hematological disorders. As of June 2018, most studies are still ongoing and recruiting. However, studies with Tazemetostat (NCT03213665; NCT03217253) were suspended because of adverse events and one study with GSK2816126 (NCT02082977), was interrupted because of insufficient evidences of clinical response.
Mutations of H3.3-H3FA are uncommon in adult GBM where H3.3 can be functionally inactivated by the MLL5 gene that is overexpressed in GBM stem cultures (80). Finally, it was found that GSK J4, like in pediatric GBM, has strong suppressive effects on cell viability and self-renewal properties (80, 81).
Several histone demethylases are constitutively or transiently overexpressed in adult GBM. LSD1 (KDM1) is FAD-monoamine oxidase that demethylates several lysine of H3 (K4, K9, K27, and K36). KDM1 interacts with non-histone substrates and inhibits p53 activity by demethylating K370me1 and by inhibiting the interaction with the coactivator 53BP1 (82). Inhibitors of KDM1 derive mainly from MAO inhibitors utilized in the clinical practice and are strong suppressors of tumor cell proliferation in vitro and in animal models (83). Most KDMi are non-selective for KDM1 and have additional irreversible activity on MAO. As of June 2018, three molecules were approved by FDA for clinical utilization (GSK2879552, IMG-7289 and INCB059872) in addition to the antidepressants Tranylcypromine and Phenelzine whose antitumor activity is being explored in phase I trials. Some of these trials were prematurely concluded because of toxicity and low efficacy while others are still ongoing.
In GBM, recurrence occurs from residual cells at the margin of resection that rapidly acquire radio- and chemo-resistance during treatment and cannot be efficiently counteracted by other drugs.
The induction of drug resistance is accompanied by the overexpression of several KDM genes. Indeed it was shown that upon treatment, a restricted population of slow-cycling cells undergo epigenetic, thus reversible, changes that result in drug resistance and sustained tumor growth (55, 56). A key effector of this mechanism is the H3K4 demethylase KDM5A gene whose exogenous expression or inactivation mimics drug resistance and sensitivity in different tumors including GBM (55, 56, 84, 85). Overall many pre-clinical and clinical evidences indicate that the entire KDM5 family, as well as other KDMs are emerging targets in cancer therapy (69, 86–89).
The pan-KDM inhibitor JIB 04 is maximally active against KDM5A but, at lower efficacy, inactivates also other KDMs found overexpressed in TMZ-resistant GBM cells (85) and has a strong antitumor effect (90). JIB 04 was utilized in a model of acquired TMZ resistance and shown to ablate TMZ-resistant cells, to synergize with TMZ at clinically-relevant concentrations and finally, in a pilot experiment, to have promising activity in vivo (91). Similar effects were obtained with CPI-455, a selective inhibitor of KDM5 (92) but at a concentration difficult to reach in vivo (91, 92). Similarly, NSCLC cells that acquired resistance to taxane/platinum combinations became sensitive to JIB 04 and GSK J4 that reverted, at least in part, the transcriptional program of resistant cells to that of drug-naïve cells and synergize with standard chemotherapy as JIB 04 and Temozolomide (93). As of June 2018, none of these promising molecules is being tested in clinical trials.
Layer 3: chromatin remodeling
Changing chromatin conformation regulates accessibility to transcription factors, to the DNA replication and repair machineries. The proper chromatin conformation is determined by histones and their modifications and by the chromatin remodeling complexes that include the histone modifiers described in section Layer 2: Histone Modifications and the ATP-dependent chromatin remodeling complexes (SWI/SNF; ISWI; CHD and INO80) (94–96). These complexes include many components that play essential and redundant roles in normal cells and that are variably altered in most human cancers (97). Because of their complexity, chromatin remodelers are very difficult targets for drug discovery and the identification of their synthetic inhibitors is still in its infancy (94). The tumor suppressor SWI/SNF complex was the first chromatin remodeler discovered, is mutated in more than 20% of the tumors (97, 98) and is involved in the maintenance of stemness in glioma cells (99). The effects of SWI/SNF inactivation can be counteracted by inhibitors of the TK pathway and of NF-kB (100, 101); however, as outlined previously, these targeted therapies were unsuccessful in GBM patients. PARP-1 polymerase is involved in chromatin remodeling mechanisms through histone modification and inhibition of the ISWI complex (102). Two PARP inhibitors (Oliparib and Veliparib) were recently licensed by the FDA for ovarian cancer treatment and several other experimental molecules are undergoing extensive testing in humans and in animal models (103). For GBM, the NIH Clinical Trials Database reports seven ongoing or completed trials with Olaparib (104, 105) (NCT01390571, NCT03212274, NCT02974621) Veliparib (NCT02152982, NCT03581292, NCT01514201) and with BSI-201 (NCT00687765). The results of these studies were not yet disclosed.
Targeting of histone chaperon molecules in glioblastoma is just beginning to be explored, however promising results in animal models were obtained by targeting FACT, a nucleosome reorganization protein (106) with CBL0137 in combination with TMZ (107).
Conclusions
Despite all the progresses in medicine, the median survival of GBM patients has not substantially improved, likely because this tumor rapidly becomes radio- and chemo-resistant and infiltrates the surrounding brain tissue making impossible the complete surgical eradication. To overcome this deadlock many experimental therapies were devised but none of them met the expected results. Epigenetic modifications are gaining strong relevance in glioblastoma because they can be either clinical biomarkers for the optimal stratification and classification of the patients and because they can be also potential drug targets as suggested by many preclinical trials. Molecules with epigenetic effects can potentially modulate the plasticity of the tumor environment in glioma and may drive the changes of the epigenomic environment restoring or rendering more susceptible the tumor cells to standard chemotherapy rather than be used as a monotherapy. In this respect the timing and the scheduling of the epigenetics and cytotoxic drugs could be crucial for the best clinical result and should be carefully defined on the basis of the chemical, biological and cellular effect of these treatments (91). Certainly, the addition of proteomic and metabolomic approaches to the extensive epigenomic and transcriptomic studies already conducted will have the capacity to unveil the inner mechanisms of glioma biology allowing the design of more effective drugs.
Author contributions
MR conceived the idea of this mini-review article and wrote the first draft. BB, MP, and MR equally participated to the final writing of the article.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer XC and the handling editor declared their shared affiliation at the time of the review.
Footnotes
Funding. This study was supported by the Italian Ministry of Health, Core Grant Ricerca Corrente to the IRCCS Ospedale Policlinico San Martino, by the grant from the Fondazione Compagnia di San Paolo–Torino: Terapie innovative per il glioblastoma and by the grant Associazione Italiana Ricerca sul Cancro (AIRC) 5 × 1000 N. 21073: Epigenetic modeling/remodeling of cancer metastases and tumor immune contexture to improve efficacy of immunotherapy.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2018.00448/full#supplementary-material
IDH1/2 pathways. Metabolic pathways involving IDH1 (cytoplasmic) and IDH2 (mitochondrial). IDH1/2 (wt) converts Isocitrate into αKetoglutarate (αKG) while the mutated forms convert Isocitrate into 2-hydroxyglutarate that competitively inhibits αKG-dependent dioxygenases including the histone demethylases JHDM1 and KDM4 and the DNA demethylase TET2
Schematic representation of the major H3 and H4 modifications and their functional role. In green and red the activating and the repressive modifications, respectively.
Clinical trials employing FDA-approved IDH1/2 inhibitors.
References
- 1.Hanif F, Muzaffar K, Perveen k, Malhi S, Simjee S. Glioblastoma multiforme: a review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pacific J Cancer Prev. (2017) 18:3–9. 10.22034/APJCP.2017.18.1.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell (2012) 22:425–37. 10.1016/j.ccr.2012.08.024 [DOI] [PubMed] [Google Scholar]
- 3.McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis GM, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455:1061–8. 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bastien JIL, McNeill KA, Fine HA. Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date: molecular characterizations of Glioblastoma. Cancer (2015) 121:502–16. 10.1002/cncr.28968 [DOI] [PubMed] [Google Scholar]
- 5.Chen R, Cohen AL, Colman H. Targeted therapeutics in patients with high-grade gliomas: past, present, and future. Curr Treatment Opt Oncol. (2016) 17:42. 10.1007/s11864-016-0418-0 [DOI] [PubMed] [Google Scholar]
- 6.Cai X, Sughrue ME. Glioblastoma: new therapeutic strategies to address cellular and genomic complexity. Oncotarget (2018) 9:9540–54. 10.18632/oncotarget.23476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 8.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. 10.1016/S1470-2045(09)70025-7 [DOI] [PubMed] [Google Scholar]
- 9.Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. (2005) 352:997–1003. 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- 10.Gusyatiner O, Hegi ME. Glioma epigenetics: from subclassification to novel treatment options. Semin Cancer Biol. (2017) 51:50–8. 10.1016/j.semcancer.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 11.Parodi F, Carosio R, Ragusa M, Di Pietro C, Maugeri M, Barbagallo D, et al. Epigenetic dysregulation in neuroblastoma: a tale of miRNAs and DNA methylation. Biochim Biophys Acta (2016) 1859:1502–14. 10.1016/j.bbagrm.2016.10.006 [DOI] [PubMed] [Google Scholar]
- 12.Maugeri M, Barbagallo D, Barbagallo C, Banelli B, Di Mauro S, Purrello F, et al. Altered expression of miRNAs and methylation of their promoters are correlated in neuroblastoma. Oncotarget (2016) 7:83330–41. 10.18632/oncotarget.13090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carén H, Pollard SM, Beck S. The good, the bad and the ugly: epigenetic mechanisms in glioblastoma. Mol Aspects Med. (2013) 34:849–62. 10.1016/j.mam.2012.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maleszewska M, Kaminska B. Deregulation of histone-modifying enzymes and chromatin structure modifiers contributes to glioma development. Future Oncol. (2015) 11:2587–601. 10.2217/fon.15.171 [DOI] [PubMed] [Google Scholar]
- 15.Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321:1807–12. 10.1126/science.1164382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. 10.1016/j.ccr.2009.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG Island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell (2010) 17:510–22. 10.1016/j.ccr.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wesseling P, Capper D. WHO 2016 classification of gliomas. Neuropathol Appl Neurobiol. (2018) 44:139–50. 10.1111/nan.12432 [DOI] [PubMed] [Google Scholar]
- 19.Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature (2018) 555:469–74. 10.1038/nature26000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu F, Mao C, Ding Y, Rui C, Wu L, Shi A, et al. Molecular and enzymatic profiles of mammalian DNA methyltransferases: structures and targets for drugs. Curr Med Chem. (2010) 17:4052–71. 10.2174/092986710793205372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthri Rheum. (2008) 58:2686–93. 10.1002/art.23758 [DOI] [PubMed] [Google Scholar]
- 22.Pogribny IP, Pogribna M, Christman JK, James SJ. Single-site methylation within the p53 promoter region reduces gene expression in a reporter gene construct: possible in vivo relevance during tumorigenesis. Cancer Res. (2000) 60:588–94. [PubMed] [Google Scholar]
- 23.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. (2003) 349:2042–54. 10.1056/NEJMra023075 [DOI] [PubMed] [Google Scholar]
- 24.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. (2011) 25:1010–22. 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roos WP, Batista LFZ, Naumann SC, Wick W, Weller M, Menck CFM, et al. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene (2007) 26:186–97. 10.1038/sj.onc.1209785 [DOI] [PubMed] [Google Scholar]
- 26.Cai S, Xu Y, Cooper RJ, Ferkowicz MJ, Hartwell JR, Pollok KE, et al. Mitochondrial targeting of human O6-methylguanine DNA methyltransferase protects against cell killing by chemotherapeutic alkylating agents. Cancer Res. (2005) 65:3319–27. 10.1158/0008-5472.CAN-04-3335 [DOI] [PubMed] [Google Scholar]
- 27.Quinn JA, Desjardins A, Weingart J, Brem H, Dolan ME, Delaney SM, et al. Phase I trial of temozolomide plus O 6 -benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. (2005) 23:7178–87. 10.1200/JCO.2005.06.502 [DOI] [PubMed] [Google Scholar]
- 28.Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, et al. Phase II trial of temozolomide plus O6 -benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol. (2009) 27:1262–7. 10.1200/JCO.2008.18.8417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, et al. Phase I trial of temozolomide plus O6-benzylguanine 5-day regimen with recurrent malignant glioma. Neuro Oncol. (2009) 11:556–61. 10.1215/15228517-2009-007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tawbi HA, Villaruz L, Tarhini A, Moschos S, Sulecki M, Viverette F, et al. Inhibition of DNA repair with MGMT pseudosubstrates: phase I study of lomeguatrib in combination with dacarbazine in patients with advanced melanoma and other solid tumours. Br J Cancer (2011) 105:773–7. 10.1038/bjc.2011.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan C-H, Liu W-L, Cao H, Wen C, Chen L, Jiang G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. (2013) 4:e876. 10.1038/cddis.2013.388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oberstadt MC, Bien-Möller S, Weitmann K, Herzog S, Hentschel K, Rimmbach C, et al. Epigenetic modulation of the drug resistance genes MGMT, ABCB1 and ABCG2 in glioblastoma multiforme. BMC Cancer (2013) 13:617. 10.1186/1471-2407-13-617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah N, Lin B, Sibenaller Z, Ryken T, Lee H, Yoon J-G, et al. Comprehensive analysis of MGMT promoter methylation: correlation with MGMT expression and clinical response in GBM. PLoS ONE (2011) 6:e16146. 10.1371/journal.pone.0016146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uno M, Oba-Shinjo SM, Camargo AA, Moura RP, Aguiar PH, de Cabrera HN, et al. Correlation of MGMT promoter methylation status with gene and protein expression levels in glioblastoma. Clinics (2011) 66:1747–55. 10.1590/S1807-59322011001000013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. (2009) 360:765–73. 10.1056/NEJMoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19:17–30. 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature (2012) 483:479–83. 10.1038/nature10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483:474–8. 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, et al. Identification of additional IDH mutations associated with oncometabolite R(–)-2-hydroxyglutarate production. Oncogene (2012) 31:2491–8. 10.1038/onc.2011.416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borodovsky A, Salmasi V, Turcan S, Fabius AWM, Baia G, Eberhart CG, et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget (2013) 4:1737–47. 10.18632/oncotarget.1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget (2013) 4:1729–36. 10.18632/oncotarget.1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science (2013) 340:626–30. 10.1126/science.1236062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science (2013) 340:622–6. 10.1126/science.1234769 [DOI] [PubMed] [Google Scholar]
- 44.Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C, et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood (2015) 125:296–303. 10.1182/blood-2013-10-533604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stein EM, Altman JK, Collins R, DeAngelo DJ, Fathi AT, Flinn I, et al. AG-221, an oral, selective, first-in-class, potent inhibitor of the idh2 mutant metabolic enzyme, induces durable remissions in a phase i study in patients with idh2 mutation positive advanced hematologic malignancies. Blood (2014) 124:115 10.1158/2159-8290.CD-16-1034 [DOI] [Google Scholar]
- 46.Fan B, Le K, Manyak E, Liu H, Prahl M, Bowden CJ, et al. Longitudinal Pharmacokinetic/Pharmacodynamic Profile of AG-120, a Potent Inhibitor of the IDH1 Mutant Protein, in a Phase 1 Study of IDH1-Mutant Advanced Hematologic Malignancies. Available online at: https://ash.confex.com/ash/2015/webprogramscheduler/Paper82908.html (Accessed June 24, 2018).
- 47.DiNardo C, de Botton S, Pollyea DA, Stein EM, Fathi AT, Roboz GJ, et al. Molecular Profiling and Relationship with Clinical Response in Patients with IDH1 Mutation-Positive Hematologic Malignancies Receiving AG-120, a First-in-Class Potent Inhibitor of Mutant IDH1, in Addition to Data from the Completed Dose Escalation Portion of the Phase 1 Study. Available online at: https://ash.confex.com/ash/2015/webprogramscheduler/Paper82957.html (Accessed June 24, 2018).
- 48.Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature (2014) 512:324–7. 10.1038/nature13387 [DOI] [PubMed] [Google Scholar]
- 49.Jenuwein T, Allis CD. Translating the histone code. Science (2001) 293:1074–80. 10.1126/science.1063127 [DOI] [PubMed] [Google Scholar]
- 50.Ivanov M, Kacevska M, Ingelman-Sundberg M. Epigenomics and interindividual differences in drug response. Clin Pharmacol Therapeut. (2012) 92:727–36. 10.1038/clpt.2012.152 [DOI] [PubMed] [Google Scholar]
- 51.Rotili D, Mai A. Targeting histone demethylases: a new avenue for the fight against cancer. Genes Cancer (2011) 2:663–79. 10.1177/1947601911417976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. (2007) 1:19–25. 10.1016/j.molonc.2007.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu K, Liu Y, Lau JL, Min J. Epigenetic targets and drug discovery Part 2: histone demethylation and DNA methylation. Pharmacol Therapeut. (2015) 151:121–40. 10.1016/j.pharmthera.2015.04.001 [DOI] [PubMed] [Google Scholar]
- 54.Kitange GJ, Mladek AC, Carlson BL, Schroeder MA, Pokorny JL, Cen L, et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res. (2012) 18:4070–9. 10.1158/1078-0432.CCR-12-0560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell (2010) 141:69–80. 10.1016/j.cell.2010.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan H, Chen X, Zhang Q, Qin J, Li H, Liu C, et al. Drug-tolerant cancer cells show reduced tumor-initiating capacity: depletion of CD44+ cells and evidence for epigenetic mechanisms. PLoS ONE (2011) 6:e24397. 10.1371/journal.pone.0024397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell (2010) 141:583–94. 10.1016/j.cell.2010.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. (1964) 51:786–94. 10.1073/pnas.51.5.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoon S, Eom GH. HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J. (2016) 52:1–11. 10.4068/cmj.2016.52.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. (2009) 27:2052–8. 10.1200/JCO.2008.19.0694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Galanis E, Anderson SK, Miller CR, Sarkaria JN, Jaeckle K, Buckner JC, et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: results of Alliance N0874/ABTC 02. Neuro Oncol. (2018) 20:546–56. 10.1093/neuonc/nox161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iwamoto FM, Lamborn KR, Kuhn JG, Wen PY, Alfred Yung WK, Gilbert MR, et al. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: north american brain tumor consortium study 03-03. Neuro Oncol. (2011) 13:509–16. 10.1093/neuonc/nor017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. (2012) 14:215–21. 10.1093/neuonc/nor198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee EQ, Reardon DA, Schiff D, Drappatz J, Muzikansky A, Grimm SA, et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro Oncol. (2015) 17:862–7. 10.1093/neuonc/nou350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi W, Palmer JD, Werner-Wasik M, Andrews DW, Evans JJ, Glass J, et al. Phase I trial of panobinostat and fractionated stereotactic re-irradiation therapy for recurrent high grade gliomas. J Neurooncol. (2016) 127:535–9. 10.1007/s11060-016-2059-3 [DOI] [PubMed] [Google Scholar]
- 66.Krauze AV, Myrehaug SD, Chang MG, Holdford DJ, Smith S, Shih J, et al. A Phase 2 study of concurrent radiation therapy, temozolomide, and the histone deacetylase inhibitor valproic acid for patients with glioblastoma. Int J Radiat Oncol Biol Phys. (2015) 92:986–92. 10.1016/j.ijrobp.2015.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenfeld MR, Chamberlain MC, Grossman SA, Peereboom DM, Lesser GJ, Batchelor TT, et al. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol. (2010) 12:1071–7. 10.1093/neuonc/noq071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell (2004) 119:941–53. 10.1016/j.cell.2004.12.012 [DOI] [PubMed] [Google Scholar]
- 69.Cloos PAC, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. (2008) 22:1115–40. 10.1101/gad.1652908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature (2012) 482:226–31. 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- 71.Talbert PB, Henikoff S. Histone variants–ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. (2010) 11:264–75. 10.1038/nrm2861 [DOI] [PubMed] [Google Scholar]
- 72.Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. (2013) 3:512–9. 10.1158/2159-8290.CD-12-0426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell (2012) 21:601–13. 10.1016/j.ccr.2012.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lewis PW, Müller MM, Matthew S, Koletsky, Cordero F, Lin S, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science (2013) 340:857–61. 10.1126/science.1232245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, et al. Reduced H3K27me3 and DNA Hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell (2013) 24:660–72. 10.1016/j.ccr.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 76.Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med. (2014) 20:1394–6. 10.1038/nm.3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature (2014) 514:513–7. 10.1038/nature13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mathur R, Sehgal L, Havranek O, Köhrer S, Khashab T, Jain N, et al. Inhibition of demethylase KDM6B sensitizes diffuse large B-cell lymphoma to chemotherapeutic drugs. Haematologica (2017) 102:373–80. 10.3324/haematol.2016.144964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer (2006) 6:846–56. 10.1038/nrc1991 [DOI] [PubMed] [Google Scholar]
- 80.Gallo M, Coutinho FJ, Vanner RJ, Gayden T, Mack SC, Murison A, et al. MLL5 orchestrates a cancer self-renewal state by repressing the histone variant H3.3 and globally reorganizing chromatin. Cancer Cell (2015) 28:715–29. 10.1016/j.ccell.2015.10.005 [DOI] [PubMed] [Google Scholar]
- 81.Sui A, Xu Y, Li Y, Hu Q, Wang Z, Zhang H, et al. The pharmacological role of histone demethylase JMJD3 inhibitor GSK-J4 on glioma cells. Oncotarget (2017) 8:68591–8. 10.18632/oncotarget.19793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, et al. p53 is regulated by the lysine demethylase LSD1. Nature (2007) 449:105–8. 10.1038/nature06092 [DOI] [PubMed] [Google Scholar]
- 83.Sareddy GR, Nair BC, Krishnan SK, Gonugunta VK, Zhang Q, Miyata N, et al. KDM1 is a novel therapeutic target for the treatment of gliomas. Oncotarget (2013) 4:18–28. 10.18632/oncotarget.725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hou J, Wu J, Dombkowski A, Zhang K, Holowatyj A, Boerner JL, et al. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am J Transl Res. (2012) 4:247–56. [PMC free article] [PubMed] [Google Scholar]
- 85.Banelli B, Carra E, Barbieri F, Würth R, Parodi F, Pattarozzi A, et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle (2015) 14:3418–29. 10.1080/15384101.2015.1090063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Nat Acad Sci USA. (2012) 109:8971–6. 10.1073/pnas.1119836109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gursoy-Yuzugullu O, Carman C, Serafim RB, Myronakis M, Valente V, Price BD. Epigenetic therapy with inhibitors of histone methylation suppresses DNA damage signaling and increases glioma cell radiosensitivity. Oncotarget (2017) 8:24518–32. 10.18632/oncotarget.15543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harmeyer KM, Facompre ND, Herlyn M, Basu D. JARID1 histone demethylases: emerging targets in cancer. Trends Cancer (2017) 3:713–25. 10.1016/j.trecan.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shokri G, Doudi S, Fathi-Roudsari M, Kouhkan F, Sanati M-H. Targeting histone demethylases KDM5A and KDM5B in AML cancer cells: a comparative view. Leuk Res. (2018) 68:105–11. 10.1016/j.leukres.2018.02.003 [DOI] [PubMed] [Google Scholar]
- 90.Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. (2013) 4:2035. 10.1038/ncomms3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banelli B, Daga A, Forlani A, Allemanni G, Marubbi D, Pistillo MP, et al. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of temozolomide-resistant glioblastoma cells. Oncotarget (2017) 8:34896–910. 10.18632/oncotarget.16820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vinogradova M, Gehling VS, Gustafson A, Arora S, Tindell CA, Wilson C, et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat Chem Biol. (2016) 12:531–8. 10.1038/nchembio.2085 [DOI] [PubMed] [Google Scholar]
- 93.Dalvi MP, Wang L, Zhong R, Kollipara RK, Park H, Bayo J, et al. Taxane-Platin-Resistant Lung Cancers Co-develop Hypersensitivity to JumonjiC Demethylase Inhibitors. Cell Rep. (2017) 19:1669–84. 10.1016/j.celrep.2017.04.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mayes K, Qiu Z, Alhazmi A, Landry JW. ATP-dependent chromatin remodeling complexes as novel targets for cancer therapy. Adv Cancer Res. (2014) 183–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tyagi M, Imam N, Verma K, Patel AK. Chromatin remodelers: we are the drivers!! Nucleus (2016) 7:388–404. 10.1080/19491034.2016.1211217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Becker PB, Workman JL. Nucleosome remodeling and epigenetics. Cold Spring Harb Perspect Biol. (2013) 5:a017905. 10.1101/cshperspect.a017905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen JH, Herlong FR, Stroehlein J, Mishra L. Mutations of chromatin structure regulating genes in human malignancies. Curr Protein Peptide Sci. (2016) 17:411–37. 10.2174/1389203717666160122120008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Helming KC, Wang X, Roberts CWM. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell (2014) 26:309–17. 10.1016/j.ccr.2014.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hiramatsu H, Kobayashi K, Kobayashi K, Haraguchi T, Ino Y, Todo T, et al. The role of the SWI/SNF chromatin remodeling complex in maintaining the stemness of glioma initiating cells. Sci Rep. (2017) 7:889. 10.1038/s41598-017-00982-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang PH. Targeting SWI/SNF mutant cancers with tyrosine kinase inhibitor therapy. Exp Rev Anticancer Ther. (2017) 17:1–3. 10.1080/14737140.2017.1257941 [DOI] [PubMed] [Google Scholar]
- 101.Kobayashi K, Hiramatsu H, Nakamura S, Kobayashi K, Haraguchi T, Iba H. Tumor suppression via inhibition of SWI/SNF complex-dependent NF-κB activation. Sci Rep. (2017) 7:11772. 10.1038/s41598-017-11806-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krishnakumar R, Kraus WL. The PARP Side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell (2010) 39:8–24. 10.1016/j.molcel.2010.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kamel D, Gray C, Walia JS, Kumar V. PARP inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: an update of clinical trials. Curr Drug Targets (2018) 19:21–37. 10.2174/1389450118666170711151518 [DOI] [PubMed] [Google Scholar]
- 104.Fulton B, Short SC, James A, Nowicki S, McBain C, Jefferies S, et al. PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin Trans Radiat Oncol. (2018) 8:12–6. 10.1016/j.ctro.2017.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Halford SER, Cruickshank G, Dunn L, Erridge S, Godfrey L, Herbert C, et al. Results of the OPARATIC trial: a phase I dose escalation study of olaparib in combination with temozolomide (TMZ) in patients with relapsed glioblastoma (GBM). J Clin Oncol. (2017) 35:2022 10.1200/JCO.2017.35.15_suppl.2022 [DOI] [Google Scholar]
- 106.Winkler DD, Luger K. The histone chaperone FACT: Structural insights and mechanisms for nucleosome reorganization. J Biol Chem. (2011) 286:18369–74. 10.1074/jbc.R110.180778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barone TA, Burkhart CA, Safina A, Haderski G, Gurova KV, Purmal AA, et al. Anticancer drug candidate CBL0137, which inhibits histone chaperone FACT, is efficacious in preclinical orthotopic models of temozolomide-responsive and -resistant glioblastoma. Neuro Oncol. (2016) 141:186–96. 10.1093/neuonc/now141 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
IDH1/2 pathways. Metabolic pathways involving IDH1 (cytoplasmic) and IDH2 (mitochondrial). IDH1/2 (wt) converts Isocitrate into αKetoglutarate (αKG) while the mutated forms convert Isocitrate into 2-hydroxyglutarate that competitively inhibits αKG-dependent dioxygenases including the histone demethylases JHDM1 and KDM4 and the DNA demethylase TET2
Schematic representation of the major H3 and H4 modifications and their functional role. In green and red the activating and the repressive modifications, respectively.
Clinical trials employing FDA-approved IDH1/2 inhibitors.