

Abstract
Genetic associations between variants on chromosome 5p13 and 8q24 and gastric cancer (GC) have been previously reported in the Asian population. We aimed to replicate these findings and to characterize the associations at the genome and transcriptome level. We performed a fine‐mapping association study in 1926 GC patients and 2012 controls of European descent using high dense SNP marker sets on both chromosomal regions. Next, we performed expression quantitative trait locus (eQTL) analyses using gastric transcriptome data from 143 individuals focusing on the GC associated variants. On chromosome 5p13 the strongest association was observed at rs6872282 (P = 2.53 × 10−04) and on chromosome 8q24 at rs2585176 (P = 1.09 × 10−09). On chromosome 5p13 we found cis‐eQTL effects with an upregulation of PTGER4 expression in GC risk allele carrier (P = 9.27 × 10−11). On chromosome 8q24 we observed cis‐eQTL effects with an upregulation of PSCA expression in GC risk allele carrier (P = 2.17 × 10−47). In addition, we found trans‐eQTL effects for the same variants on 8q24 with a downregulation of MBOAT7 expression in GC risk allele carrier (P = 3.11 × 10−09). In summary, we confirmed and refined the previously reported GC associations at both chromosomal regions. Our data point to shared etiological factors between Asians and Europeans. Furthermore, our data imply an upregulated expression of PTGER4 and PSCA as well as a downregulated expression of MBOAT7 in gastric tissue as risk‐conferring GC pathomechanisms.
Keywords: eQTL study, gene expression, genetic association study, stomach neoplasms
1. INTRODUCTION
The majority of gastric adenocarcinomas, here called gastric cancer (GC), is sporadic and has a multifactorial and heterogeneous etiology. According to its localization, GC is subdivided into a cardia and noncardia type and according to the histopathological classification of Lauren, GC is subdivided into a diffuse, intestinal or mixed type.1 On the etiological level, inflammation represents an important risk factor, which is mainly caused by Helicobacter pylori (H. pylori) infection.1
In multifactorial diseases, genome‐wide association studies (GWAS) have systematically led to the identification of risk loci involved in disease etiology. Accordingly, GWAS in GC have been carried out in the Asian population, which have led to the identification of risk loci on chromosome 1q22, 3q13, 5p13, 6p21, 7p15, 8q24, and 10q23.2, 3, 4, 5, 6 The risk loci on 5p137 and 8q248, 9 have been confirmed to contribute to GC development also in the European population. However, functional data on the underlying risk genes and on disease conferring pathomechanisms at these loci are scarce. Thus, we aimed to further confirm the role of 5p13 and 8q24 in the etiology of GC in Europeans and to characterize the associations on the genome and transcriptome level. However, it should be noted, that the locus on chromosome 1q22 was also independently replicated in an European GC sample under certain genetic models (dominant and recessive).10 Because no allelic association tests were presented in this study we did not follow up the association at the 1q22 locus.
2. MATERIALS AND METHODS
2.1. Samples
For the fine‐mapping association study at 5p13 and 8q24, we used a sample consisting of 1926 histopathologically confirmed GC patients (680 females, 1246 males) and 2012 ethnically matched controls (985 females, 1027 males) that were not screened for the presence of GC. All study subjects were of European descent and were recruited at four different study sites in Latvia, Lithuania, Spain and Germany. Table S1 provides sample details, including origin of samples and distribution of GC localization (cardia, noncardia) as well as Lauren type (diffuse, intestinal, and mixed). Informed consent was obtained from all participants and approval was obtained from the ethic board of each participating site.
The expression quantitative trait locus (eQTL) analysis was based on 143 individuals of German descent (15 females, 128 males). All subjects were histopathologically diagnosed with intestinal metaplasia of the distal esophagus (Barrett's esophagous) and additionally, endoscopic biopsies of the gastric cardia were obtained during routine surveillance gastroscopies. Absence of pathological changes and H. pylori infection in the gastric mucosa was histopathologically confirmed for all cases. Informed consent was obtained from all participants as well as approval from the responsible ethic board.
2.2. Genotyping and expression analyses
For the fine‐mapping association study on 5p13 and 8q24, single‐nucleotide polymorphisms (SNPs) were selected for genotyping with the aim to impute a maximum number of genetic variants within both regions. To define the regions of interest the main recombination hotspots flanking both loci were determined using Haploview Version 4.2 and the HapMap dataset III (Release August 2010).11 Next, all SNPs with a minor allele frequency (MAF) of ≥5% on Illumina Human Core Exome Chip were used to reduce the number of necessary tagging SNPs within both regions. To prove that these SNPs cover the regions sufficiently an imputation on a genome‐wide genotyped test data set12 was performed. Finally, we used the test data set and ensured that all common publicly released SNPs within both regions were imputed with high imputation quality scores (INFO > 0.4). This resulted in 14 SNPs located within 5p13 and 6 SNPs within 8q24 (Table 1) as tagging markers for the respective loci. All 20 SNP markers were genotyped using a Sequenom MassARRAY iPlex Gold® system (Sequenom, San Diego, CA, USA). For quality control (QC), intra‐ and interplate duplicates were included. Furthermore, negative controls (H2O) were added to each 384 well plate in order to exclude contamination. Cluster plots of all SNPs were visually checked and manually corrected if necessary. The postgenotype QC comprised the exclusion of SNPs with Hardy‐Weinberg equilibrium (HWE) P < 1 × 10−04 in controls and P < 1 × 10−06 in patients, a call rate (CR) < 95% as well as exclusion of samples with a CR < 90%. A single marker (rs138377917 on chromosome 8q24) failed QC. Detailed information on primer sequences, genotyping and genotype calling is available upon request.
Table 1.
Case‐control comparison (1926 GC cases, 2012 controls) of genotyped SNPs at chromosomal regions 5p13 and 8q24. One marker (rs138377917 on chromosome 8q24) failed QC and is not shown
| SNP | Chr | Allelea | RR (95% CI) | Combined P‐value |
|---|---|---|---|---|
| rs7716982 | 5 | G/T | 1.06 (0.96‐1.16) | 2.31 × 10−01 |
| rs6893430 | 5 | C/T | 1.15 (1.03‐1.29) | 1.20 × 10−02 |
| rs6861121 | 5 | A/G | 1.07 (0.94‐1.23) | 3.07 × 10−01 |
| rs7726237 | 5 | G/A | 1.12 (1.02‐1.23) | 1.89 × 10−02 |
| rs10737963 | 5 | C/A | 1.08 (0.97‐1.19) | 1.60 × 10−01 |
| rs12523329 | 5 | C/T | 1.06 (0.96‐1.17) | 2.79 × 10−01 |
| rs1002424 | 5 | A/G | 1.16 (1.05‐1.29) | 3.09 × 10−03 |
| rs257009 | 5 | C/T | 1.09 (0.99‐1.20) | 8.39 × 10−02 |
| rs10053664 | 5 | C/T | 1.06 (0.97‐1.16) | 2.23 × 10−01 |
| rs13361707 | 5 | C/T | 1.17 (1.06‐1.29) | 2.29 × 10−03 |
| rs3805486 | 5 | A/G | 1.13 (0.99‐1.29) | 6.20 × 10−02 |
| rs462366 | 5 | T/C | 1.04 (0.94‐1.15) | 4.28 × 10−01 |
| rs6876367 | 5 | C/T | 1.12 (1.01‐1.24) | 2.70 × 10−02 |
| rs2291782 | 5 | A/G | 1.06 (0.96‐1.16) | 2.57 × 10−01 |
| rs2976400 | 8 | A/G | 1.04 (0.95‐1.15) | 3.70 × 10−01 |
| rs2976392 | 8 | A/G | 1.29 (1.18‐1.41) | 2.14 × 10−08 |
| rs2976397 | 8 | T/G | 1.30 (1.19‐1.42) | 7.18 × 10−09 |
| rs12155758 | 8 | A/G | 1.24 (1.13‐1.37) | 1.03 × 10−05 |
| rs1435453 | 8 | C/T | 1.23 (1.12‐1.34) | 6.98 × 10−06 |
Chr, chromosome; CI, confidence interval; RR, relative risk.
The underlined allele represents the GC risk allele.
For the eQTL analysis DNA from 143 donors was extracted from peripheral blood and genotyped genome‐wide using Human OmniExpress‐v1.1 and HumanOmniExpressExome‐v1.2 BeadChips (Illumina, San Diego, CA, USA). The postgenotype QC comprised the exclusion of SNPs with HWE P < 1 × 10−05, minor allele frequency (MAF) <5% or a CR < 98% as well as the exclusion of samples with a CR < 99%. For the expression analysis, total RNA from gastric cardia biopsies was isolated using the AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany). The transcriptome was assessed using the HumanHT‐12v4 Expression BeadChip (Illumina) that targets more than 48 000 transcripts in the refseq database (Build 36.2, release 22). Only probes with a P detection < 0.01 in more than 5% of the samples were included for analysis. Furthermore, all probes were filtered for unique alignment and quality. Probes classified with a perfect or good quality as reported in the R package illuminaHumanv4.db were considered for further analysis.
2.3. Fine‐mapping and eQTL analyses
For the fine‐mapping association analysis, all genotyped SNPs were used for the imputation of additional markers at both regions. For this purpose, IMPUTE213 was used utilizing the 1000 Genomes Phase 3 data as reference.14 After imputation, all SNPs with an info score >0.7 were further processed, which resulted in 478 SNPs on 5p13 and 315 SNPs on 8q24. For each of the four study sites, association analysis of genotyped and imputed SNPs was performed by SNPTEST v2.5.15 A fixed‐effects meta‐analysis was performed to combine the results across study sites. We additionally performed genotype‐phenotype (GxP) analyses and stratified our cases according to tumor localization (cardia, noncardia) as well as histopathological Lauren type (diffuse, intestinal, or mixed). Moreover, pairwise linkage disequilibrium (LD) between markers was determined using SNAP16 and the 1000 Genomes Pilot 1 data from the European population.14
For the eQTL analysis genotypes of 568 265 autosomal SNPs and expression intensity data from 11 900 probes were used from all 143 probands. The expression data were quantile normalized and eQTLs were mapped using a linear regression model implemented in the MatrixEQTL R package.17 The associations were corrected for the top five principal components and eQTL effects with a false discovery rate (FDR) of <0.05 were considered as significant. In addition, eQTLs with a distance of <1 Mb to the corresponding probes were considered as cis‐regulatory variants. Vice versa, SNPs with a distance of >1 Mb to the corresponding probes were classified as trans‐eQTLs.
3. RESULTS
3.1. Association and eQTL findings on chromosome 5p13
Of all genotyped SNPs at 5p13 5 variants showed significant GC association (Table 1). Table S2 shows the association findings within each study site. The strongest association in the entire sample was observed for rs13361707 (P = 2.29 × 10−03, RR = 1.17 (95% CI = 1.06‐1.29)). The fine‐mapping at this region included 478 imputed SNPs, of which 28 showed GC association. The strongest association was observed for rs6872282 (P = 2.53 × 10−04, RR = 1.22 (95% CI = 1.09‐1.35), risk allele C, opposite allele T, see Table S3). Of note, rs13361707 and rs6872282 are in high LD with r 2 = 0.92. Figure 1A provides the regional association plot at 5p13 and shows that all associated variants encompass the coding regions of the genes PTGER4, TTC33, and PRKAA1. The GxP analysis at 5p13 using GC tumor localization and histopathological Lauren type as strata did not lead to an association improvement. Table S4 shows all GxP results for rs6872282, the strongest associated marker in the entire analysis. In addition, a conditional analysis using rs6872282 did not reveal any independent disease association (P > 0.5, data not shown).
Figure 1.
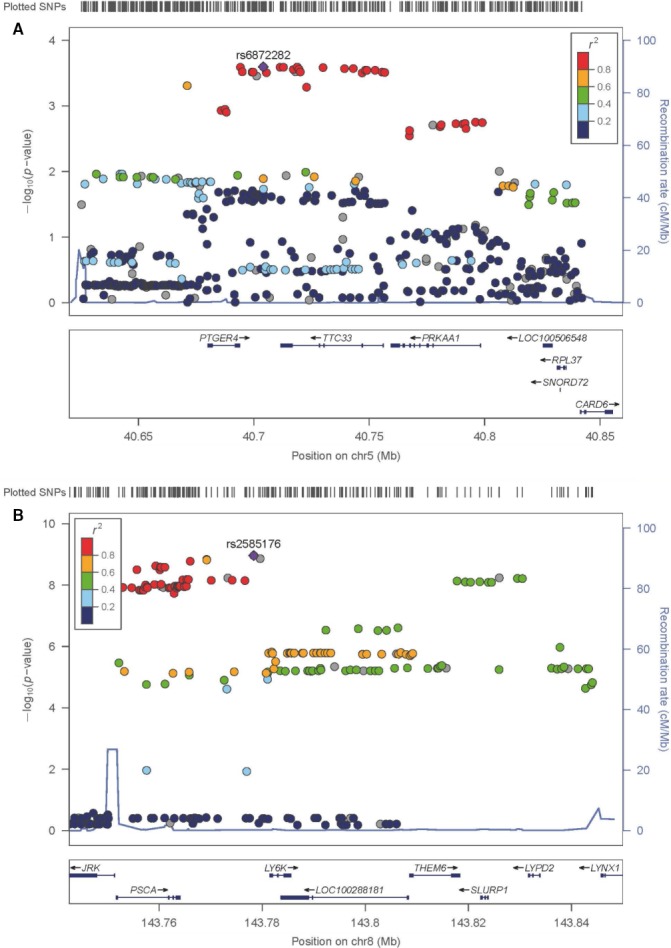
Regional association plots of GC associations at chromosome 5p13 (A) and chromosome 8q24 (B). SNP association results are shown as ‐log P. The most significant associated SNP—rs6872282 at 5p13 (A), rs2585176 at 8q24 (B)—is shown as solid diamond. Pair‐wise correlation (r 2) between the most significant associated SNP and the other SNPs in a 500 kb flanking region is illustrated by the color scheme. The blue spikes show the estimated recombination rates. All annotated genes in both regions are shown at the bottom and their reading direction is given by arrows
The eQTL analysis at 5p13 was initially restricted to SNPs with r 2 > 0.8 to the most associated GC variant and revealed cis‐eQTL effects for the expression of PTGER4 in gastric tissue (P = 9.27 × 10−11 for rs10074991). Here, an upregulated expression of the transcript was observed in GC risk allele carrier (Figure 2A). We then tested all 5p13 SNPs for PTGER4 eQTL effects, which revealed that rs10074991 represents the strongest eQTL for the expression of PTGER4 at this locus. Finally, we did not observe any trans‐eQTL effects using GC associated risk variants at 5p13 and an FDR < 0.05.
Figure 2.
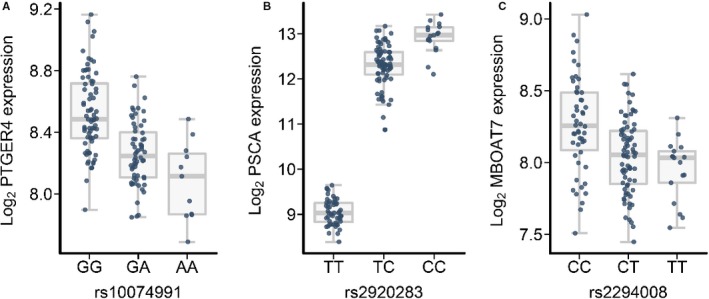
eQTL effects for GC associated variants at chromosomal regions 5p13 and 8q24. Log2 gene expression, error bars for median log2 expression and standard deviation are shown as box plot (y axis) sorted by SNP genotype (x‐axis) with the common allele on the left. The individual log2 gene expression is indicated by small dots in blue (y‐axis). A, cis‐eQTL (rs10074991, risk allele G) for the expression of PTGER4 at 5p13 (P = 9.27 × 10−11, regression slope (β) = −56.18). B, cis‐eQTL (rs2920283, risk allele C) for the expression of PSCA (P = 2.17 × 10−47, β = 3958.10). C, trans‐eQTL (rs2294008, risk allele T) for the expression of MBOAT7 (P = 1.99 × 10−09, β = −40.57)
3.2. Association and eQTL findings on chromosome 8q24
Of all genotyped SNPs at 8q24 4 variants showed significant GC association (Table 1). Table S2 shows the association findings within each study site. The strongest association in the entire sample was observed for rs2976397 (P = 7.18 × 10−09, RR = 1.30 (95% CI = 1.19‐1.42)). The fine‐mapping at 8q24 included 315 imputed SNPs. Of them 42 SNPs showed GC association with rs2585176 being the most associated variant (P = 1.09 × 10−09, RR = 1.34 (95% CI = 1.22‐1.47), risk allele T, opposite allele A, see Table S3). Notably, rs2976397 and rs2585176 are in high LD with r 2 = 0.90. Figure 1B shows the regional association plot at 8q24 and shows that all associated variants are located close to the genes JRK, PSCA, and LY6K. The GxP analysis at 8q24 using GC tumor localization and histopathological Lauren type as strata did not lead to the identification of a particular GC subtype with predominant association. Table S5 shows all GxP results for rs2585176, the strongest associated marker in the entire analysis. In addition, a conditional analysis using rs2585176 did not reveal any independent disease association (P > 0.5, data not shown).
The eQTL analysis at 8q24 was initially restricted to SNPs with r 2 > 0.8 to the most associated GC variant and revealed cis‐eQTL effects for the expression of PSCA (P = 2.17 × 10−47). The lead eQTL is rs2920283 and an upregulated PSCA expression in gastric tissue was observed in GC risk allele carrier (Figure 2B). As for 5p13, we then tested all 8q24 SNPs for PSCA eQTL effects, which revealed that rs2920283 represents the strongest eQTL for the expression of PSCA at this locus. Finally, we tested for trans‐eQTL effects using GC associated risk variants at 8q24 with r 2 > 0.8 to the lead GC SNP. Here, we observed a regulatory effect for the expression of MBOAT7 in gastric tissue, which is located on chromosome 19q13. The most significant trans‐eQTL was rs2294008 (P = 1.99 × 10−09) and led to a downregulated expression of MBOAT7 in GC risk allele carrier [Figure 2C]. Notably, the lead cis‐ and trans‐eQTLs are in perfect LD (r 2 = 1.00) pointing to a common regulatory effect. However, it should be noted that the statistical significance of the cis‐eQTL is much higher than for the trans‐eQTL.
4. DISCUSSION
The GC associations at 5p13 and 8q24 have been initially reported in the Asian population.2, 4 It has subsequently been shown that both loci also play a role in GC etiology in European populations.7, 8, 9 In the present study, we aimed to further replicate the GC association in the largest so far analyzed European cohort as well as to refine and characterize the association signals on the genome and transcriptome level.
On chromosome 5p13 all GC associated variants were in high LD (r 2 > 0.8) to the most associated marker (rs6872282) and in high LD (r 2 > 0.8) to all risk SNPs previously reported at this locus in Asians2, 18, 19, 20, 21, 22, 23 and Europeans.7 Thus, together with previous findings, our data provide evidence that one or more of these SNPs represent(s) the true GC conferring variant(s) in both the Asian and European population. Of the implicated SNPs rs3805495 is in perfect LD to the leading GC risk variant rs6872282 (r 2 = 1) and represents an interesting functional SNP. According to RegulomeDB24 this variant is predicted to alter motifs for several transcription factors and to affect histone modification in digestive tissues including gastric mucosa. In addition, we found that the GC risk alleles of the associated SNPs lead to an upregulated PTGER4 expression in gastric tissue, which represents a plausible GC pathomechanism at this locus. To our knowledge, this eQTL effect has not been reported so far and thus needs further replication in independent gastric tissue samples.
Also on the functional level, PTGER4 represents an interesting candidate for GC. The gene encodes the prostaglandin E2 (PGE2) receptor 4 (EP4), which mediates cellular responses to PGE2. PGE2 is derived from arachidonic acids through the enzymatic activity of cyclooxygenase‐2 (COX‐2). It has already been shown that the COX‐2/PGE2 pathway plays a key role in generation of the inflammatory microenvironment in GC and represents a downstream effector of Toll‐like receptor (TLR) activation that is—among others—triggered through H. pylori infection,25 the most prominent environmental GC risk factor. Interestingly, one study with H. pylori positive and negative GC cases and controls found a gene‐environment interaction between 5p13 risk SNPs and H. pylori infection that showed association with GC.18
Genetic variability at 5p13 also contributes to other diseases with inflammatory components, including Crohn's disease,26, 27, 28, 29, 30, 31 ulcerative colitis,32, 33, 34 ankylosing spondylitis35 and multiple sclerosis.36, 37, 38 Furthermore, cis‐regulatory effects involving the expression of PTGER4 in some of these traits have been described previously in lymphoblastoid cell lines.39 However, all SNPs and eQTLs implicated in these studies are only in weak LD (r 2 < 0.3) to the GC variants and PTGER4‐eQTLs identified in the present study (see Table S6). The findings point to PTGER4 as risk gene for different diseases with inflammatory components. However, more complex pathomechanisms with disease‐ and/or tissue‐specific effects on the PTGER4 expression regulation seem to be present at this locus.
On chromosome 8q24 all GC associated variants were in high LD (r 2 > 0.8) to the most associated marker (rs2585176) and in high LD (r 2 > 0.8) to all risk SNPs previously reported at this locus in Asians and Europeans (summarized in Ref. 40). As for 5p13, this provides evidence that shared etiological factors between Asians and Europeans contribute to GC risk at this locus and that one or more of the identified SNPs represent(s) the true GC conferring variant(s). Of the implicated SNPs rs2294008 (C>T, Met1Thr) represents an interesting functional variant, which is located in the translation starting site of PSCA, is in high LD (r 2 = 0.86) with the lead GC SNP and also the strongest trans‐eQTL for the expression of MBOAT7 (see below). This SNP leads to an alternative PSCA splice form on the protein level41 and thus has been favored as true GC risk variant at this locus. In addition, rs2585183 represents an interesting functional variant according to RegulomeDB.24 The variant is in high LD to the leading GC risk SNP (r 2 = 0.90) and is predicted to affect histone modification in digestive tissues including gastric mucosa.
Moreover, we found that the risk alleles of the GC associated markers lead to an upregulated expression of PSCA. The presence of this eQTL in gastric tissue is supported by GTEx (version 7, dbGaP accession phs000424.v7.p2), where the strongest eQTL in our study (rs2920283) shows cis‐regulatory effects for PSCA with P = 8.1 × 10−33. Furthermore, cis‐eQTL effects involving the presented variants and PSCA have been shown previously in healthy gastric and gastric tumor tissue.42 Thus, the PSCA eQTL has been independently validated and represents a plausible GC pathomechanism at 8q24.
Also on the functional level PSCA represents an interesting candidate for GC development. The gene encodes a glycosylphosphatidylinositol‐anchored cell‐surface protein with highest expression in tumor cells and seems to play a role in multiple cellular processes, including immune‐modulation, cell adhesion, proliferation and survival.43 The PSCA risk alleles of the GC associated SNPs also mediate risk for chronic atrophic gastritis,9, 44 an H. pylori ‐induced precursor lesion of GC. In contrast, the opposite alleles at the same variants lead to duodenal ulcer,41, 44, 45, 46 which is also induced by H. pylori infection, but characterized by severe antral mucosa inflammation and protective GC effects.47, 48, 49 Thus, it has been hypothesized that gastric H. pylori infection leads to chronic atrophic gastritis and GC in patients with GC risk alleles at PSCA variants, whereas carrier of the opposite alleles tend to develop severe antral mucosa inflammation and duodenal ulcer.41, 44, 45, 46
The same regulatory variants on 8q24 that represent cis‐eQTLs for the expression of PSCA lead to a downstream downregulated MBOAT7 expression in gastric tissue, which suggests that MBOAT7 might be also of relevance in GC etiology. Interestingly, MBOAT7 encodes an enzyme with lysophosphatidylinositol acyltransferase activity and has been implicated in anti‐inflammatory processes through the regulation of arachidonic acid‐derived prostaglandin (PG) levels.50 Recently, it has been shown that MBOAT7 plays a pivotal role in hepatic inflammation and fibrosis in patients with hepatitis C infection51 and alcohol‐related liver cirrhosis.52 Thus, MBOAT7 might also contribute to GC susceptibility via biological pathways that are involved in inflammation. However, although the expression of PSCA and MBOAT7 is regulated by the same SNPs, a cellular connection between them has not been described so far.
In conclusion, our study has confirmed the role of 5p13 and 8q24 in the etiology of GC in Europeans and has characterized the associations on the genome and transcriptome level. Whereas the eQTL effect of PSCA has been described before, the eQTL effects involving PTGER4 and MBOAT7 have not been described before in the context of GC. Future work is now required as to whether the same eQTL effects are present in the remaining gastric regions or other tissues with relevance in GC development. Additionally, functional work is required to prove that the observed eQTL effects play a role in GC development rather than represent epiphenomena with no relevance for GC development. In this context, our fine‐mapping association and eQTL study with altered gastric expression of PTGER4, PSCA, and MBOAT7 in GC risk allele carriers may serve as impetus.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
The authors thank all patients and controls for participating in this study. In addition, the authors thank Prof. Bernd Pötzsch (Institute of Experimental Hematology and Transfusion Medicine, University of Bonn) for help with collecting DNA samples from anonymous blood donors. This work contains substantial parts of the doctoral thesis of S.K.M.H.
Heinrichs SKM, Hess T, Becker J, et al. Evidence for PTGER4, PSCA, and MBOAT7 as risk genes for gastric cancer on the genome and transcriptome level. Cancer Med. 2018;7:5057–5065. 10.1002/cam4.1719
Funding Information
M.V. and J.S. received support for this work from the Deutsche Forschungsgemeinschaft (DFG), individual grant SCHU 2596/6‐1. S.K.M.H. received support for this work from the BONFOR program of the University of Bonn. M.M.N. is a member of the DFG funded Excellence Cluster ImmunoSensation. UG and JK were funded by the European Social Fund according to the activity “Improvement of researchers” qualification by implementing world‐class R&D projects of Measure No. 09.3.3‐LMT‐K‐712‐01‐0130. This study has been funded by Instituto de Salud Carlos III through the project FIS PI15/00331, co‐funded by European Union (ERDF/ESF, “Investing in your future”), and CIBER de enfermedades hepáticas y digestivas (CIBERehd).
Heinrichs, Hess, Leja, Kupcinskas, García‐González, Venerito and Schumacher authors contributed equally to this work.
REFERENCES
- 1. Hartgrink HH, Jansen EPM, van Grieken NCT, van de Velde CJH. Gastric cancer. Lancet. 2009;374:477‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shi Y, Hu Z, Wu C, et al. A genome‐wide association study identifies new susceptibility loci for non‐cardia gastric cancer at 3q13.31 and 5p13.1. Nat Genet. 2011;43:1215‐1218. [DOI] [PubMed] [Google Scholar]
- 3. Jin G, Ma H, Wu C, et al. Genetic variants at 6p21.1 and 7p15.3 are associated with risk of multiple cancers in han chinese. Am J Hum Genet. 2012;91:928‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sakamoto H, Yoshimura K, Saeki N, et al. Genetic variation in PSCA is associated with susceptibility to diffuse‐type gastric cancer. Nat Genet. 2008;40:730‐740. [DOI] [PubMed] [Google Scholar]
- 5. Abnet CC, Freedman ND, Hu N, et al. A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nat Genet. 2010;42:764‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Z, Dai J, Hu N, et al. Identification of new susceptibility loci for gastric non‐cardia adenocarcinoma: pooled results from two Chinese genome‐wide association studies. Gut. 2017;66:581‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Helgason H, Rafnar T, Olafsdottir HS, et al. Loss‐of‐function variants in ATM confer risk of gastric cancer. Nat Genet. 2015;47:906‐910. [DOI] [PubMed] [Google Scholar]
- 8. Sala N, Muñoz X, Travier N, et al. Prostate stem‐cell antigen gene is associated with diffuse and intestinal gastric cancer in Caucasians: results from the EPIC‐EURGAST study. Int J Cancer. 2012;130:2417‐2427. [DOI] [PubMed] [Google Scholar]
- 9. Lochhead P, Frank B, Hold GL, et al. Genetic variation in the prostate stem cell antigen gene and upper gastrointestinal cancer in white individuals. Gastroenterology. 2011;140:435‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palmer AJ, Lochhead P, Hold GL, et al. Genetic variation in C20orf54, PLCE1 and MUC1 and the risk of upper gastrointestinal cancers in Caucasian populations. Eur J Cancer Prev. 2012;21(6):541‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263‐265. [DOI] [PubMed] [Google Scholar]
- 12. Gharahkhani P, Fitzgerald RC, Vaughan TL, et al. Genome‐wide association studies in oesophageal adenocarcinoma and Barrett's oesophagus: a large‐scale meta‐analysis. Lancet Oncol. 2016;17:1363‐1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet. 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. 1000 Genomes Project Consortium , Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526:68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome‐wide association studies by imputation of genotypes. Nat Genet. 2007;39:906‐913. [DOI] [PubMed] [Google Scholar]
- 16. Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, De Bakker PIW. SNAP: a web‐based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938‐2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353‐1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eom S‐Y, Hong S‐M, Yim D‐H, et al. Additive interactions between PRKAA1 polymorphisms and Helicobacter pylori CagA infection associated with gastric cancer risk in Koreans. Cancer Med. 2016;5:3236‐3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu N, Wang Z, Song X, et al. Genome‐wide association study of gastric adenocarcinoma in Asia: a comparison of associations between cardia and non‐cardia tumours. Gut. 2016;65:1611‐1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang JY, Kim DH, Ji YI, et al. Recapitulation of previous genome‐wide association studies with two distinct pathophysiological entities of gastric cancer in the Korean population. J Hum Genet. 2013;58:233‐235. [DOI] [PubMed] [Google Scholar]
- 21. Li M, Huang L, Qiu H, et al. Helicobacter pylori infection synergizes with three inflammation‐related genetic variants in the GWASs to increase risk of gastric cancer in a Chinese population. PLoS ONE. 2013;8:e74976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiu L‐X, He J, Cheng L, et al. Genetic variant of PRKAA1 and gastric cancer risk in an Eastern Chinese population. Oncotarget. 2015;6:42661‐42666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song HR, Kim HN, Kweon SS, et al. Genetic variations in the PRKAA1 and ZBTB20 genes and gastric cancer susceptibility in a Korean population. Mol Carcinog. 2013;52:155‐160. [DOI] [PubMed] [Google Scholar]
- 24. Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Echizen K, Hirose O, Maeda Y, Oshima M. Inflammation in gastric cancer: interplay of the COX‐2/prostaglandin E2 and Toll‐like receptor/MyD88 pathways. Cancer Sci. 2016;107:391‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Consortium TWTCC . Genome‐wide association study of 14 000 cases of seven common diseases and 3 000 shared controls. Nature. 2007;447:661‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barrett JC, Hansoul S, Nicolae DL, et al. Genome‐wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franke A, McGovern DPB, Barrett JC, et al. Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jostins L, Ripke S, Weersma RK, et al. Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Julià A, Domènech E, Ricart E, et al. A genome‐wide association study on a southern European population identifies a new Crohn's disease susceptibility locus at RBX1‐EP300. Gut. 2013;62:1440‐1445. [DOI] [PubMed] [Google Scholar]
- 31. Weersma RK, Stokkers PCF, Cleynen I, et al. Confirmation of multiple Crohn's disease susceptibility loci in a large Dutch‐Belgian cohort. Am J Gastroenterol. 2009;104:630‐638. [DOI] [PubMed] [Google Scholar]
- 32. Latiano A, Palmieri O, Latiano T, et al. Investigation of multiple susceptibility loci for inflammatory bowel disease in an Italian cohort of patients. PLoS ONE. 2011;6:e22688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McGovern DPB, Gardet A, Törkvist L, et al. Genome‐wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anderson CA, Boucher G, Lees CW, et al. Meta‐analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Evans DM, Spencer CCA, Pointon JJ, et al. Interaction between ERAP1 and HLA‐B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA‐B27 in disease susceptibility. Nat Genet. 2011;43:761‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matesanz F, González‐Pérez A, Lucas M, et al. Genome‐wide association study of multiple sclerosis confirms a novel locus at 5p13.1. PLoS ONE. 2012;7:e36140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell‐mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Jager PL, Jia X, Wang J, et al. Meta‐analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome‐wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:0538‐0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qin Z, Tang J, Li X, et al. Association between PSCA gene polymorphisms and the risk of cancer: an updated meta‐analysis and trial sequential analysis. Oncotarget. 2017;8:51766‐51778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tanikawa C, Urabe Y, Matsuo K, et al. A genome‐wide association study identifies two susceptibility loci for duodenal ulcer in the Japanese population. Nat Genet. 2012;44:430‐434. [DOI] [PubMed] [Google Scholar]
- 42. Sung H, Hu N, Yang HH, et al. Association of high‐evidence gastric cancer susceptibility loci and somatic gene expression levels with survival. Carcinogenesis. 2017;38:1119‐1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raff AB, Gray A, Kast WM. Prostate stem cell antigen: a prospective therapeutic and diagnostic target. Cancer Lett. 2009;277:126‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ichikawa H, Sugimoto M, Uotani T, et al. Influence of prostate stem cell antigen gene polymorphisms on susceptibility to Helicobacter pylori‐associated diseases: a case‐control study. Helicobacter. 2015;20:106‐113. [DOI] [PubMed] [Google Scholar]
- 45. García‐González MA, Bujanda L, Quintero E, et al. Association of PSCA rs2294008 gene variants with poor prognosis and increased susceptibility to gastric cancer and decreased risk of duodenal ulcer disease. Int J Cancer. 2015;137:1362‐1373. [DOI] [PubMed] [Google Scholar]
- 46. Tanikawa C, Matsuo K, Kubo M, et al. Impact of PSCA variation on gastric ulcer susceptibility. PLoS ONE. 2013;8:e63698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Graham DY. Helicobacter pylori infection in the pathogenesis of duodenal ulcer and gastric cancer: a model. Gastroenterology. 1997;113:1983‐1991. [DOI] [PubMed] [Google Scholar]
- 48. Correa P, Piazuelo MB. Natural history of Helicobacter pylori infection. Dig Liver Dis. 2008;40:490‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis. 2012;13:2‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gijón MA, Riekhof WR, Zarini S, Murphy RC, Voelker DR. Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J Biol Chem. 2008;283:30235‐30245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thabet K, Asimakopoulos A, Shojaei M, et al. MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C. Nat Commun. 2016;7:12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buch S, Stickel F, Trépo E, et al. A genome‐wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol‐related cirrhosis. Nat Genet. 2015;47:1443‐1448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials