

Abstract
Defects in the biosynthesis and/or function of primary cilia cause a spectrum of disorders collectively referred to as ciliopathies. A subset of these disorders is distinguished by profound abnormalities of the skeleton that include a long narrow chest with markedly short ribs, extremely short limbs, and polydactyly. These include the perinatal lethal short-rib polydactyly syndromes (SRPS) and the less severe asphyxiating thoracic dystrophy (ATD), Ellis van Creveld (EVC) syndrome and cranioectodermal dysplasia (CED) phenotypes. To identify new genes and define the spectrum of mutations in the skeletal ciliopathies, we analyzed 152 unrelated families with SRPS, ATD and EVC. Causal variants were discovered in 14 genes in 120 families, including one newly associated gene and two genes previously associated with other ciliopathies. These three genes encode components of three different ciliary complexes; FUZ which encodes a planar cell polarity complex molecule; TRAF3IP1 which encodes an anterograde ciliary transport protein; and LBR which encodes a nuclear membrane protein with sterol reductase activity. The results established the molecular basis of SRPS type IV, in which mutations were identified in four different ciliary genes. The data provide systematic insight regarding the genotypes associated with a large cohort of these genetically heterogeneous phenotypes and identified new ciliary components required for normal skeletal development.
Keywords: Skeletal dysplasia, ciliopathy, cilia, chondrocyte
1. INTRODUCTION
Cilia are microtubule-based organelles that project from the surfaces of almost all cell types and function as receptors for chemosensory, mechanosensory and osmoregulatory signaling (Anderson, et al., 2008). Cilia have conventionally been categorized as motile or primary (non-motile) based on their axonemal structures. Most motile cilia have a 9+2 structure of microtubule doublets while primary cilia generally lack the two central microtubules and therefore have a 9+0 microtubule arrangement. Cilia are generally absent from dividing cells but when they exit the cell cycle a single primary cilium is assembled from the axonemal substructure at the basal body, which anchors the cilium within the cytoplasm (Fliegauf, et al., 2008).
Ciliary proteins are selectively transported into the cilium by intraflagellar transport (IFT), a highly evolutionarily conserved process that serves to transport both structural and signaling components along the ciliary axoneme. Anterograde transport by the IFT-B complex moves cargo from the cytoplasm to the ciliary tip using kinesin motors (Scholey, 2008). The IFT-B anterograde complex consists of 9 core components (IFT22, IFT25, IFT27, IFT46, IFT52, IFT70, IFT74, IFT81, and IFT88) and several peripheral subunits (IFT20, IFT54, IFT57, IFT80, IFT172, IFT38 and IFT56) (Taschner, et al., 2012; Taschner, et al., 2016). In Caenorhabditis elegans, IFT-B subunit mutations primarily block assembly of cilia, leading to cells with truncated or absent cilia (Pan, et al., 2006). Cargo may be unloaded along the cilium and at the tip, and molecules are conveyed back to the cytoplasm by retrograde transport using the IFT-A complex, which is catalyzed by the dynein 2 complex molecular motor (Ishikawa and Marshall, 2011; Taschner, et al., 2012). The IFT-A complex consists of 6 primary components (IFT43, WDR35, IFT122, TTC21B, IFT140, and WDR19) and other ancillary proteins. Knockout or knockdown of IFT-A proteins primarily results in architecturally abnormal cilia with prominent bulges due to accumulation of IFT particles at the tip (Merrill, et al., 2009).
Defects in the biosynthesis and/or function of primary cilia cause a spectrum of disorders collectively referred to as ciliopathies (Badano, et al., 2006; Waters and Beales, 2011). A subset of these disorders have profound abnormalities of the skeleton and present with a characteristic set of phenotypic features that include long narrow chests with markedly short ribs, short limbs and polydactyly. The skeletal ciliopathies are clinically, radiographically and genetically heterogeneous diseases that include the non-lethal phenotypes Ellis-van Creveld syndrome (EVC MIM# 225500) and cranioectodermal dysplasia (CED, also known as Sensenbrenner syndrome MIM# 218330), the more severe asphyxiating thoracic dystrophy (ATD, also known as Jeune syndrome MIM# 208500) phenotype that is lethal in a up to half of the cases, especially those with dynein-2 complex mutations (Baujat, et al., 2013; Tuysuz, et al., 2009), and the perinatal lethal short rib-polydactyly syndromes (SRPS) (Huber and Cormier-Daire, 2012). Based on distinctive radiographic findings, the SRPS have been classified into four subtypes: SRPS type I (Saldino-Noonan Syndrome MIM# 263530), SRPS type II (Majewski Syndrome MIM# 263520), SRPS type III (Verma–Naumoff Syndrome MIM# 263510) and SRPS type IV (Beemer–Langer Syndrome MIM# 269860). Each skeletal ciliopathy can also manifest concomitant abnormalities in the craniofacial, nervous, cardiac, gastrointestinal, and genitourinary systems. All forms of the skeletal ciliopathies are thought to be inherited in an autosomal recessive pattern.
To date mutations in 23 genes have been found in cases of lethal and/or non-lethal skeletal ciliopathies (Mitchison and Valente, 2017). In the dynein 2 motor complex, DYNC2H1, encoding the dynein motor heavy chain (Dagoneau, et al., 2009; Merrill, et al., 2009) is the major locus and mutations in four additional genes, encoding the light intermediate chain (DYNC2LI1 (Taylor, et al., 2015)), intermediate chains (WDR34 (Huber, et al., 2013; Schmidts, et al., 2013c), WDR60 (McInerney-Leo, et al., 2013)) and light chain (TCTEX1D2 (Schmidts, et al., 2015)), have also been reported. Mutations in all six of the genes encoding retrograde transport (IFT-A) components (IFT43 (Arts, et al., 2011), WDR19 (Bredrup, et al., 2011), IFT122 (Walczak-Sztulpa, et al., 2010), TTC21B (Davis, et al., 2011), WDR35 (Mill, et al., 2011) and IFT140 (Perrault, et al., 2012; Schmidts, et al., 2013b)) have also been characterized in these disorders. By contrast, mutations in only a subset of the genes encoding IFT-B complex members, IFT80 (Cavalcanti, et al., 2011), IFT172 (Halbritter, et al., 2013), IFT52 (Zhang, et al., 2016) and IFT81 (Duran, et al., 2016), have been identified among the skeletal ciliopathies. In addition, mutations in two genes (EVC (Ruiz-Perez, et al., 2000), EVC2 (Galdzicka, et al., 2002)) that encode basal body proteins, the NEK1 kinase gene (Thiel, et al., 2011) and the gene encoding its interacting protein C21ORF2 (McInerney-Leo, et al., 2017; Wheway, et al., 2015), the ICK MAP-like kinase gene (Taylor, et al., 2016), the INTU planar cell polarity (PCP) gene (Toriyama, et al., 2016), the KIAA0586 (Alby, et al., 2015) centrosomal protein gene and the CEP120 core centriolar protein gene (Shaheen, et al., 2015) have also been reported. Mutations in several of these genes, including the latter five genes, are rare causes of skeletal ciliopathies, each having been observed in only one or a few families. Allelic heterogeneity in a number of the genes results in phenotypes along a spectrum of skeletal severity ranging from CED to SRPS.
To identify new genes and define the spectrum of mutations that can produce the skeletal ciliopathies, we analyzed 152 unrelated families with SRPS, ATD and EVC from the International Skeletal Dysplasia Registry (ISDR). Causal variants were discovered in 14 genes in 120 families, including one newly associated gene and two genes previously associated with other ciliopathies. These three genes encode components of three different ciliary complexes; FUZ which encodes a planar cell polarity complex molecule; TRAF3IP1 which encodes an anterograde ciliary transport protein; and LBR which encodes a nuclear membrane protein with sterol reductase activity. In addition, a wider phenotypic spectrum was characterized for several of the skeletal ciliopathy genes. Finally, the results established the molecular basis of SRPS type IV, in which we identified mutations in four different ciliary genes. The data have provided systematic insight regarding the genotypes associated with a large cohort of these highly genetically heterogeneous phenotypes and identified new ciliary components required for normal skeletal development.
2. MATERIALS AND METHODS
2.1. Sample selection and clinical categorization
The study was carried out under approved University of California at Los Angeles human subjects protocol 14–000177 and written informed consent was obtained from all subjects and/or their parents as appropriate. All cases were selected from the International Skeletal Dysplasia Registry (ISDR) archive. The diagnosis of each phenotype was primarily radiographic (Supp. Figures S1–4), but clinical data were integrated when available, according to the following criteria:
SRPS type I.
The most severe of the SRPS phenotypes with extreme micromelia and very short, poorly mineralized long bones. The chest was very small, with very short ribs and hypoplastic scapulae. Polydactyly occurred in most cases, usually involving multiple limbs, and there was poor mineralization of the hands. Multiple organ system anomalies and hydrops were common.
SRPS type III.
Similar to but less severe than SRPS type I with small chests and short limbs. Radiographs showed short, horizontal ribs and long bones with lateral metaphyseal spikes. Distal extremities were short and poorly mineralized, especially the carpal and distal phalangeal bones. Polydactyly was a variable feature. Small iliac bones with a trident pelvis and poorly formed scapulae were common as were multiple craniofacial anomalies.
SRPS type II.
Similar to SRPS type III but with smooth ends of the long bones. Common findings included small rounded ilia and rhizomelia with thin or absent fibulae. Femurs were typically rounded at both ends. Rounded tibiae were present in some cases but were not a consistent finding. Polydactyly was universally present.
SRPS type IV.
Findings similar to SRPS type II in that all long bones were short with smooth ends (Yang, et al., 1991). Bending of the long bones, and especially the radii and ulnae, was common and fibulae were thin. Ilia were small. Polydactyly was absent.
ATD.
Affected individuals had long, narrow chests, moderately short ribs, handlebar clavicles, trident acetabulae and short extremities. Brachydactyly was typically present with polydactyly as a variable feature. The ends of the long bones had metaphyseal abnormalities that were either smooth or had lateral spikes. The overall skeletal findings were somewhat similar to all types of SRPS but with less severe skeletal involvement, adjusted for gestational age. For the approximately 50% of individuals who survived, pulmonary compromise, cystic livers and cystic kidneys could occur.
EVC.
Characteristic radiographic findings included shortened ribs (less severe than ATD and SRPS), short, heavy tubular bones and bowed humeri. The pelvis was abnormal with medial and lateral protrusions of the acetabulum. In the postnatal period there was accelerated ossification of the capital femoral epiphyses with hypoplasia of the proximal tibiae. Brachydactyly with cone shaped epiphyses and polydactyly were seen.
2.2. Exome sequencing and variant filtering
Genomic DNA was isolated from peripheral blood and/or cultured fibroblasts or chondrocytes of all probands and available parents and siblings using standard procedures. Library construction and exome sequencing of DNA from each proband was carried out at the University of Washington Center for Mendelian Genomics. The exome sequencing libraries were prepared with the NimbleGen SeqCap EZ Exome Library v2.0 kit and sequenced on the Illumina GAIIx platform with paired-end 50 base pair reads. Reads were mapped to the human reference genome (NCBI build 37) with BWA 0.5.9 (Li and Durbin, 2010) and variants called using the Genome Analysis Toolkit version 3.4 following their best practices recommendations (McKenna, et al., 2010). De-multiplexing, sequence alignment, variant calling and annotation were performed as described previously (Taylor, et al., 2015; Zhang, et al., 2016). Variants were filtered for good quality (covered by ≥10 independent reads) with a minor allele frequency (MAF) <0.005 in the ExAC database (v0.3) (Lek, et al., 2015). Copy number variant analysis was performed by ReCapSeg (McKenna, et al., 2010) and CoNIFER v0.2.2 (Krumm, et al., 2012) with the exome sequencing data. All variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Since these disorders are recessively inherited, samples were initially assessed for compound heterozygosity or homozygosity for mutations in known SRPS genes. Cases heterozygous for single mutations in known SRPS genes were also identified. For these cases we specifically checked coverage for each exon in the identified skeletal ciliopathy genes to exclude the possibility of low coverage leading to missing a second mutation. Sanger sequence analysis was used to validate all variants of interest and to assess appropriate segregation within each family whenever possible. All primer sequences are available upon request. Sequence trace files were aligned and analyzed using Sequencher version 4.6 (Genecodes).
3. RESULTS
3.1. Phenotypic assignment and exome sequence analysis
For each affected individual, clinical and radiographic data were reviewed by three independent investigators. Radiographic criteria (Supp. Figures S1–4) and noted above based on previously published descriptions were employed to differentiate EVC, ATD and SRPS, and the SRPS cases were then further categorized into subtypes (Bonafe, et al., 2015). Visceral abnormalities were observed in a large number of affected individuals, but were not generally used to distinguish among the phenotypes.
Exome sequencing was carried out for 152 affected individuals, consisting of 66 individuals with ATD, 4 with EVC and 82 with lethal phenotypes in the SRPS spectrum. Exome sequencing generated an average of 75X coverage across all samples, with on average 93% of all targeted regions being covered by at least 10X reads and 85% by 20X reads (Supp. Table S1). These data were then filtered to exclude those with a minor allele frequency (MAF) > 0.005 in the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/).
Each family was analyzed under an autosomal recessive model of inheritance. Variants were predicted to be pathogenic based on meeting one or more of the following criteria: 1) was a nonsense, frameshift or splice junction consensus sequence variant in a gene known to underlie a skeletal ciliopathy; 2) was observed among multiple families in our cohort or among published cases; 3) segregated according the recessive inheritance model; 4) altered either highly evolutionarily conserved amino acid residue (s) or conserved functional protein domains; 5) met computational predictions of functional impact using GERP++, PolyPhen2 and CADD (Adzhubei, et al., 2010; Davydov, et al., 2010; Kircher, et al., 2014). All predicted pathogenic variants were confirmed by Sanger sequence analysis of amplified genomic DNA. We identified homozygosity or compound heterozygosity for predicted pathogenic mutations in 14 genes in 120 (79%) of the 152 families studied (Supp. Table S2).
3.2. New loci for the skeletal ciliopathies
We identified a case of SRPS (ISDR reference number R11–569) due to homozygosity for a frameshift deletion mutation in FUZ (Supp. Figure S5). FUZ is a component of a newly identified planar cell polarity regulatory module, formed by Inturned (encoded by INTU), Fuzzy (FUZ) and WDPCP, which governs ciliogenesis in vertebrates (Gray, et al., 2009; Park, et al., 2006). The FUZ mutation (c.98_111+9del) is predicted to lead to the deletion of five amino acids as well as the splice donor for exon 1, likely to represent a loss-of-function mutation. The phenotype, which was initially identified by prenatal ultrasound, was most similar to SRPS type II but with extreme polydactyly of all four limbs (Fig. 1). A hypoplastic left ventricle, ventricular septal defect, hypoplastic kidneys, midline facial cleft, thickened nuchal fold and dilated third ventricle were also noted. The phenotype was similar to Fuz knockout mice, which had severe developmental abnormalities that included a malformed sternum, ribs and long bones, polydactyly, and incompletely penetrant rostral neural tube closure defects (Gray, et al., 2009). Although dominant mutations in FUZ have been reported to cause isolated neural tube defects (Seo, et al., 2015), the SRPS carrier parents had no reported abnormalities.
FIGURE 1.
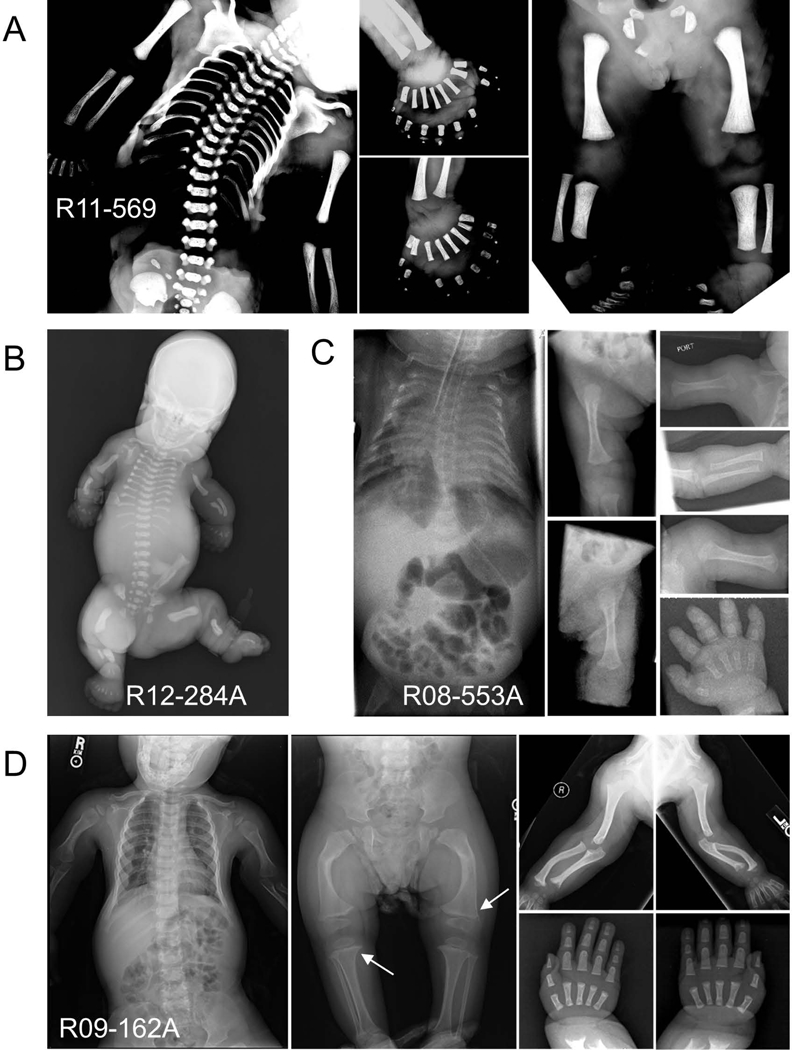
Radiographic phenotypes of the SRPS probands with mutations in FUZ, TRAF3IP1 and LBR. (a) FUZ mutations in SRPS type II (R11–569, 24 weeks) showing a long, narrow chest, moderately short ribs, short long bones, hypoplastic tibiae and extreme polydactyly of all four limbs. (b) TRAF3IP1 mutations in SRPS type II (R12–284A, neonatal) showing short long bones, very short horizontal ribs and long narrow chest, severe micromelia and polydactyly of all limbs of the affected individuals. (c) TRAF3IP1 mutations in ATD (R08–553A, neonatal) showing a bell-shaped thorax, moderately short ribs, handlebar clavicles, micromelia of all long bones and brachydactyly. (d) LBR mutations associated with ATD (R09–162A, 2y 9m) showing moderately short horizontal ribs, short limbs, metaphyseal abnormalities (white arrows) and no polydactyly.
Compound heterozygosity for mutations in TRAF3IP1 was discovered in two cases, one with SRPS type II and one with a lethal form of ATD (Supp. Figure S5). TRAF3IP1 is an anterograde ciliary transport protein. Both affected individuals each carried a TRAF3IP1 splice acceptor mutation, one predicting use of an alternative splice acceptor and the other predicting exon skipping and a frameshift, together with a damaging missense change. The SRPS type II case (R12–284A; c.988–1G>C; c.169G>A, p.Glu57Lys) was identified by prenatal ultrasound at 23 weeks gestation. In addition to severe micromelia and polydactyly of all limbs, ultrasound examination revealed enlarged, echogenic kidneys, mild oligohydramnios, generalized skin edema and a thickened nuchal fold. The brain showed an enlarged cisterna magna and ventriculomegaly. The heart was reported as structurally normal but with a pericardial effusion. The infant was delivered at term and died in the immediate perinatal period. The radiographic phenotype was typical for SRPS type II with smooth ends of the long bones and polydactyly of all limbs (Fig. 1).
The individual with ATD (R08–553A; c.1368–1delG; c.1358C>G, p.Ser453Cys) was delivered by emergency caesarian section at 26 weeks gestation due to preterm labor and breech footling presentation. At birth she exhibited respiratory distress and was ventilated. Hydrocephalus and a grade IV intraventricular hemorrhage were identified, requiring placement of a shunt. A bell-shaped thorax, micromelia of all long bones and brachydactyly were noted. Radiographs were diagnostic for ATD and showed a narrow chest and handlebar clavicles, a mild lateral slope of the proximal humeri, flat acetabulae, spurring of the sciatic notch, short metacarpals and phalanges, and a normal spine (Fig. 1). The infant died 5 months after birth. TRAF3IP1 missense mutations have been reported in nephronophthisis with extrarenal abnormalities [MIM# 616629] (Bizet, et al., 2015), but have not previously been associated with ATD or SRPS.
Compound heterozygosity for missense mutations in the gene encoding the lamin B receptor (LBR) were associated with a non-lethal form of ATD (Fig. 1, Supp. Fig S5). LBR is a multifunctional inner nuclear membrane protein that both anchors the lamina and heterochromatin to the nuclear membrane and has sterol reductase activity. The missense changes (c.1174G>A, p.Gly392Arg and c.1535G>A, p.Arg512Gln) altered highly conserved amino acids in the C-terminal hydrophobic transmembrane domain responsible for sterol reductase activity (Schuler, et al., 1994). In silico predictions were deleterious for both changes and only the p.Arg512Gln variant was found in online variant databases, and at an extremely low frequency (rs754049402:G>A, MAF=4.172e-05). Thus this ATD phenotype appears to be allelic with Greenberg dysplasia, a lethal skeletal disorder resulting from loss of sterol reductase activity (Best, et al., 2003; Borovik, et al., 2013; Clayton, et al., 2010; Hoffmann, et al., 2002; Waterham, et al., 2003).
3.3. The molecular basis of SRPS type IV
SRPS type IV was first described by Beemer and Langer (Beemer, et al., 1983) and among forms of SRPS is most radiographically similar to SRPS type II (Hennekam, 1991; Yang, et al., 1991), suggesting the hypothesis that these two forms of SRPS might have a common molecular basis (Hennekam, 1991). We identified recessively inherited mutations in four independent SRPS type IV cases in the following genes: NEK1, TTC21B, WDR19 and IFT80 (Supp. Table S2). With the exception of WDR19, for which the SRPS case in our cohort is the only described SRPS case with mutations in the gene, we also identified mutations in the other three genes in both SRPS type II and SRPS type IV families. Our findings thus establish the molecular basis of SRPS type IV and demonstrate that it is allelic to SRPS type II.
3.4. Dynein 2 complex mutations are the major cause of SRPS phenotypes
Pathogenic variants in genes encoding dynein 2 complex components were discovered in 81 families, including 73 with mutations in DYNC2H1. Mutations in DYNC2H1 were found in 43 SRPS families (41% of all SRPS families), including 40 with SRPS type III. DYNC2H1 mutations explained one SRPS type II family, confirming (El Hokayem, et al., 2012) that mutations in the gene are a rare cause of this SRPS subtype. The remaining two SRPS families had SRPS type I, concordant with our previous findings of 3 SRPS type I families with DYNC2H1 mutations (Badiner et al., 2016). Thirty ATD families (45% of all ATD families) had mutations in DYNC2H1, similar to previously published frequency estimates in ATD families of Northern European ancestry (Baujat, et al., 2013; Schmidts, et al., 2013a).
Among the families, 110 different pathogenic mutations in DYNC2H1 were identified (Supp. Table S2), 91 (83%) of which were novel and 67 of which were missense changes (Fig. 2). The majority of the missense mutations clustered in the N-terminal tail (DHC_N1), AAA2–4 ATPase domains and a conserved C-terminal domain, indicating the importance of maintaining the functional integrity of these domains. No case was found with two loss-of-function mutations, a genotype which has been speculated to lead to early embryonic lethality (Ocbina, et al., 2011).
FIGURE 2.

Mutations in DYNC2H1 in cases of ATD and SRPS. The structure of the human DYNC2H1 protein showing the locations of the 110 mutations described in this study. Font color indicates mutations associated with ATD (black), SRPS type I (fuchsia), SRPS type II (blue) and SRPS type III (red). Mutations frequencies are denoted with the number of circles and the types of mutations are distinguished with colors. The figure also includes 11 mutations from 6 previously published cases with mutations in the gene (Badiner, et al., 2016; Merrill, et al., 2009). Conserved protein domain localizations were taken from Schmidts et al., 2013 (Schmidts, et al., 2013a) with six AAA+ domains, an MT-binding stalk, an N-terminal tail (DHC_N1), a linker domain (DHC_N2) and a conserved C-terminal domain (Dynein_heavy). The plot was generated with Protein Paint.
Mutations in the genes encoding other dynein 2 motor complex components were also found to be causative for SRPS and ATD (Supp. Table S2). Pathogenic mutations were identified in 8 families, including mutations in WDR34 (one SRPS type III and three ATD families) and WDR60 (two ATD and two SRPS type III families). WDR34 and WDR60 are both members of the WD repeat protein family and ten of the eleven missense changes altered the WD40 motifs (Fig. 3), likely altering the protein-protein interactions mediated by these repeats (Li and Roberts, 2001).
FIGURE 3.

Mutations identified in dynein motor and IFT-A proteins. The structures of the human dynein motor (WDR34 and WDR60) and IFT-A (IFT140, WDR19, WDR35 and TTC21B) proteins showing the locations of the mutations identified in this study. Mutation frequencies are denoted with the number of circles, and the mutation types are distinguished with colors as described in Figure 1. The figure includes 10 mutations from 7 previously published cases with mutations in these genes (Duran, et al., 2017; Huber, et al., 2013). Conserved protein domain regions were identified using the UniProt and InterPro websites.
3.5. Mutations in IFT-A complex genes
The IFT-A complex cooperates with the dynein motor to mediate retrograde transport (Taschner, et al., 2012). Mutations in the genes encoding four of the six IFT-A complex components (see Fig. 5), including the genes encoding two core proteins (WDR19 and IFT140) and two peripheral (satellite) proteins (WDR35 and TTC21B), were identified in 14 (9%) of the families (Supp. Table S2). All three families with IFT140 mutations had ATD. One individual was homozygous for a frameshift mutation, indicating that loss of this IFT-A core protein results in a non-lethal ATD phenotype. Similarly, five of the six families with WDR19 mutations had ATD, including three non-lethal cases. The sixth case fit the phenotype of SRPS type IV, one of four cases in our cohort with this rare form of SRPS.
FIGURE 5.

Ciliary proteins mutated among the skeletal ciliopathies. The complexes in which each protein participates are shown with the mutated components identified by red asterisks.
Mutations affecting the three peripheral IFT-A components resulted in a broader spectrum of disease including non-lethal ATD and lethal SRPS phenotypes. TTC21B mutations explained one family each with SRPS types II and IV. WDR35 mutations in SRPS type IV further illustrate the locus heterogeneity underlying this SRPS subtype (see above). Similar to WDR34 and WDR60, missense mutations primarily corresponded to WD40 repeats within IFT140, WDR19 and WDR35 (Fig. 3), underscoring the functional importance of these domains.
3.6. Mutations in IFT-B complex genes
The IFT-B anterograde complex is powered by kinesin motors and consists of 9 salt stable core components (IFT22, IFT25, IFT27, IFT46, IFT52, IFT70, IFT74, IFT81, and IFT88) and several peripheral subunits (IFT20, TRAF3IP1, IFT57, IFT80, IFT172) (Taschner, et al., 2012; Taschner, et al., 2016). IFT80 was the first IFT gene found to be associated with a skeletal ciliopathy (ATD), and mutations in IFT80 have been identified in both ATD and SRPS type III patients (Beales, et al., 2007; Cavalcanti, et al., 2011). In our cohort, IFT80 mutations were found in families with ATD, SRPS type II and SRPS type IV, expanding the phenotypic spectrum due to mutations in the gene. Most were missense changes residing in the WD40 repeat containing domains (Supp. Table S2). In one case of SRPS type II (R10–594), the affected individual was homozygous for a p.Gly241Arg missense variant. This change was previously reported by Cavalcanti et al. in a mixed Latin-European and African SRPS family from Brazil (Cavalcanti, et al., 2011) and the family we studied was African American, suggesting the allele may be prevalent among SRPS cases with an African genetic background. As described above, mutations were found in TRAF3IP1 in both ATD and SRPS.
3.7. EVC and EVC2 mutations can result in severe skeletal abnormalities
Mutations in EVC and EVC2 are known to result in recessively inherited EVC syndrome (Galdzicka, et al., 2002; Ruiz-Perez, et al., 2000) and autosomal dominant Weyers Acrofacial Dysostosis (Ye, et al., 2006). In our cohort, causative EVC mutations were found in four families and EVC2 mutations in six families. Significant skeletal abnormalities were identified by prenatal ultrasound in six of the families and the pregnancies were terminated in the late second trimester. Of the five newborn cases (including one recurrence), three died of respiratory insufficiency in the immediate neonatal period. Among the affected individuals, fourteen different mutations were identified, most of which are predicted to lead to loss of function, including nine that were previously reported in EVC cases (Ruiz-Perez and Goodship, 2009). Clinical information was limited for some of the cases, but four individuals had structural heart defects, 2 cases had hypoplastic nails, and frenulum was seen in seen one case. For both the EVC and EVC2 cases there was a consistent radiographic phenotype consisting of a narrow chest with relatively well developed ribs, reverse campomelia of the humeri along with a distinctive configuration of the radius and ulna, brachydactyly with well-ossified digits, heavy-appearing bones in the lower extremities albeit with thin and short fibulae, and in some cases accelerated ossification of the calcanei, knee epiphyses and/or capital femoral epiphyses (Fig. 4). Thus while the skeletal phenotype among the cases with EVC/EVC2 mutations appeared to be more severe than might be expected for EVC, elements of the EVC phenotype were evident. Our findings also illustrate the difficulty of phenotype assessment in the fetal period, particularly in the second trimester. Radiographic findings in the EVC/EVC2 cases delivered in the late second trimester were more similar to ATD (two individuals) or SRPS type II (four individuals) than EVC and mutations in EVC/EVC2 would not have been predicted by postmortem assessment alone. Thus, phenotypic overlap among the skeletal ciliopathies, particularly in the second trimester, highlights the importance of mutation identification in cases ascertained during the prenatal period.
FIGURE 4.
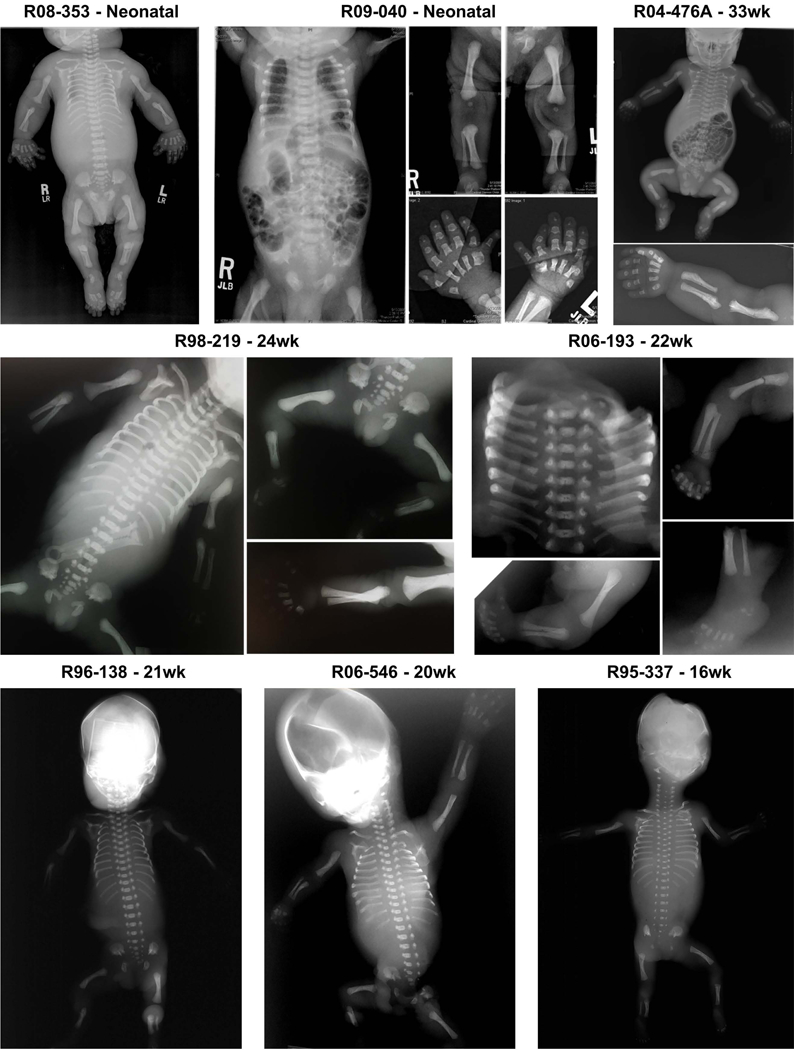
Radiographic phenotype of the probands with EVC and EVC2 mutations. At later gestational ages, an EVC radiographic phenotype consisting of short long bones, moderately short horizontal ribs and a long narrow chest, a trident acetabulum, reverse campomelia of the humeri along with a distinctive configuration of the radius and ulna, polydactyly and brachydactyly with well ossified digits was evident. At earlier ages, the phenotypes more closely resembled SRPS and ATD. ISDR reference numbers and gestational ages in weeks are indicated for each individual.
3.8. Unsolved cases
We did not identify biallelic causative mutation(s) in 32 (21%) families. A single known or likely pathogenic mutation was identified in 22 cases and there were 6 cases in which there were two or more pathogenic variants (one per gene) in genes known to underlie a skeletal ciliopathy (Supp. Table S3). While we did not detect copy number variations indicative of small deletions within the dataset, it remains possible that small deletions comprising a single exon were missed. Thus, in only 4/152 families was there no candidate pathogenic variant found.
4. DISCUSSION
4.1. The genetic architecture of the skeletal ciliopathies
This study identified homozygosity or compound heterozygosity for mutations in 14 different ciliary genes in 120/152 (79%) genetically independent families with ATD, EVC and all subtypes of SRPS. Mutations in one new gene and two genes previously associated with other ciliopathies, FUZ, TRAF3IP and LBR, were found to be mutated in these disorders, further illustrating the diversity of genes and ciliary complexes whose functions are required for normal skeletal development. The data also establish the molecular basis of SRPS type IV and demonstrate that it is allelic with SRPS type II. Previous publications of SRPS and ATD cases from the ISDR have included 22 additional families with mutations in 9 ciliary genes (DYNC2H1, WDR34, IFT52, ICK, INTU, IFT81, DYNC2LI1, IFT43, WDR35), including 6 genes not represented among the current set of families (Duran, et al., 2016; Huber, et al., 2013; Taylor, et al., 2016; Taylor, et al., 2015; Toriyama, et al., 2016; Zhang, et al., 2016). Mutations have also been reported in families in this spectrum of disease in six additional genes, C21ORF2 (McInerney-Leo, et al., 2017; Wheway, et al., 2015), TCTEX1D2 (Gholkar, et al., 2015), IFT172 (Halbritter, et al., 2013), KIAA0586 (Alby, et al., 2015), CEP120 (Shaheen, et al., 2015) or IFT122 (Walczak-Sztulpa, et al., 2010), bringing the number of known skeletal ciliopathy genes to twenty six. A model of the ciliary complexes showing the components mutated in the disorders is shown in Fig. 5.
4.2. Newly identified skeletal ciliopathy loci
FUZ is one of the core components of the CPLANE regulatory module that controls cilia assembly and PCP signaling (Zilber, et al., 2013). A role for FUZ in skeletal development was recognized when Fuz knockout mice had abnormalities resembling a severe skeletal ciliopathy with extreme polydactly (Gray, et al., 2009). In a family with an SRPS type II-like phenotype including extreme polydactyly there was homozygosity for a loss-of-function FUZ mutation, suggesting that the function of FUZ is conserved in human skeletal development. Furthermore, mutations in INTU, another CPLANE network core component (Toriyama, et al., 2016), have been identified in SRPS with the signature skeletal abnormality of extreme polydactyly, highlighting the critical role of the CPLANE complex in skeletal development, with a particularly important function in distal limb patterning.
TRAF3IP1 is a peripheral anterograde IFT component essential for cilia formation and skeletal defects were observed in Traf3ip1 gene-trapped mice (Berbari, et al., 2011). We identified one ATD and one SRPS type II case resulting from compound heterozygosity for TRAF3IP1 mutations, with polydactyly in the SRPS case. TRAF3IP1 mutations have been previously reported in Senior-Loken syndrome [MIM# 616629], a disorder primarily characterized by kidney abnormalities, but skeletal findings including polydactyly, brachydactyly and epiphyseal dysplasia were present in the reported cases (Bizet, et al., 2015). Heterogeneity in clinical presentation among cases with TRAF3IP1 mutations might be explained by the effects of the distinct mutations on anterograde transport, as most NPH patients were homozygous for missense mutations, in contrast to our cases that resulted from compound heterozygosity for one loss-of-function and one missense mutation.
We identified an individual with ATD associated with compound heterozygosity for two missense mutations in LBR, encoding the lamin B receptor. The phenotype thus appears to be allelic with Greenberg dysplasia, a lethal skeletal disorder resulting from disruption of the sterol reductase activity of the protein. Our results suggest a new link between LBR and cilia, and we speculate that disruption of sterol modification of key signaling proteins, including sonic hedgehog, that are essential for cilia function and for normal skeletal development is the underlying basis for the phenotype.
4.3. Locus heterogeneity in SRPS type IV
Causal variants in four genes, NEK1, TTC21B, WDR19 and IFT80, were discovered in 4 cases of SRPS type IV (Beemer, et al., 1983). Mutations in IFT122 have also been identified in a case of SRPS type IV (Silveira, et al., 2017). Mutations in NEK1, TTC21B, and IFT80 were also discovered in cases of SRPS type II both in our cohort and as reported by others (Cavalcanti, et al., 2011; El Hokayem, et al., 2012; Mill, et al., 2011), demonstrating that these two forms of SRPS can be allelic, as hypothesized in prior publications (Hennekam, 1991).
4.4. DYNC2H1 is the major locus for SRPS and ATD
Mutations affecting the dynein 2 transport motor accounted for 53% of all families in our cohort. We identified 110 different DYNC2H1 variants, 91 of which have not been reported previously. DYNC2H1 mutations were by far the most frequent overall cause of the skeletal ciliopathies studied, accounting for 48% of the families screened and with wide clinical variability ranging from ATD to SRPS types I, II and III (but not SRPS type IV). DYNC2H1 mutations have been previously reported in 5 SRPS type II, 8 SRPS type III and 48 ATD cases, and the gene has been recognized as the major locus for ATD (Baujat, et al., 2013; Schmidts, et al., 2013a). We identified mutations in DYNC2H1 in 30/66 ATD cases. The results also demonstrate that DYNC2H1 is the main locus for SRPS type III (Dagoneau, et al., 2009), with 40/49 cases resulting from mutations in the gene. Furthermore, all SRPS type I cases in the cohort had DYNC2H1 mutations (Badiner, et al., 2016), supporting the current classification of SRPS types I and III together and suggest DYNC2H1 as the main gene to be tested in new cases with these diagnoses.
Defects in the dynein 2 motor result in ciliary architecture abnormalities that lead to abnormal synthesis and/or maintenance of the axoneme by IFT to form the mature primary cilium (Merrill, et al., 2009). As demonstrated by knockdown experiments in C. elegans and mice (Ocbina and Anderson, 2008; Signor, et al., 1999), and in cells cultured from DYNC2H1 mutation cases (Merrill, et al., 2009), dynein defects can lead to structurally abnormal cilia. DYNC2H1 encodes the dynein heavy chain and is the ATPase motor within the dynein complex (Pazour, et al., 1999). The motor functions by binding and hydrolysis of ATP (Kon, et al., 2004) and 32 of the 67 identified missense mutations localized to the nucleotide binding domains. Modeling the elimination of nucleotide binding at AAA2–4 domains in D. discoideum and S. cerevisiae cytoplasmic dynein (Cho, et al., 2008; Kon, et al., 2004) showed a severe slowdown in the microtubule sliding activity of the protein, implying dysfunctional motor activity. Enrichment of DYNC2H1 missense mutations in the motor domain supports the hypothesis that disruption of motor integrity interferes with proper anterograde and retrograde IFT activity and mechanistically contributes to the pathology seen in the skeletal ciliopathies.
4.5. IFT-A complex components are mutated more frequently than IFT-B components
We identified mutations in four of the six genes known to underlie ATD and/or SRPS subtypes. Overall, 14 of the cases (9%) were found with IFT-A gene mutations, with the majority of the missense mutations located in the highly conserved WD40 domains involved in intracellular trafficking, cargo recognition and protein binding (Fig. 2). No mutations were identified in IFT122, encoding the WDR10 protein that plays an important role in assembly and maintenance of cilia, although mutations in the gene are associated with CED and SRPS type IV. Previous work identified mutations in IFT-A components in cases of CED or ATD, but the results presented here expand the phenotypic series to include ATD and lethal SRPS. Overall, variants in genes encoding all IFT-A members can affect the skeleton, further illuminating the dependence of skeletogenesis on IFT-A function.
We identified causal variants in only 6/152 families and only 2 of the 14 genes (TRAF3IP1 and IFT80) that encode IFT-B components. In the total ISDR skeletal ciliopathy cohort there was also one family with unclassified SRPS due to IFT52 mutations (Zhang, et al., 2016) and one family each with ATD and SRPS type III due to IFT81 mutations (Duran, et al., 2016), so overall 9/175 (5%) families studied had IFT-B defects. It is unclear why the genes encoding anterograde IFT components are less frequently mutated than retrograde molecules in the SRPS-ATD spectrum of disease. Although the IFT-A complex components are quite large proteins whose genes present a large mutational target size, identification of mutations in only a small fraction genes encoding IFT-B components in ATD/SRPS suggest that anterograde function is less closely tied to skeletogenesis than IFT-A.
4.6. For what reason do mutations in these genes primarily affect the skeleton?
Among the ciliopathy disorders, only a subset have a primary effect on the skeleton, suggesting that the proteins encoded by the skeletal ciliopathy loci have particularly important roles in the function of primary cilia in chondrocytes (Scherft and Daems, 1967), but exactly why the skeleton is so severely affected remains unknown. Defective sonic hedgehog signaling during skeletal pattern formation appears to be the mechanism underlying polydactyly, and disruption of proper Hh signaling has been observed in fibroblasts from SRPS fibroblasts (Duran, et al., 2016; Taylor, et al., 2015). Altered Indian hedgehog signaling is likely to be responsible in part for the defects in endochondral ossification that result in structural alterations of the growth plate and defective bone growth. Indian hedgehog is a key signaling ligand for chondrocyte differentiation, particularly the transition from proliferative to hypertrophic chondrocytes (St-Jacques, et al., 1999). Chondrocyte differentiation was disrupted in IFT80 silenced bone marrow-derived stromal cells (Wang, et al., 2013) but primary cilia also regulate other signaling pathways in the skeleton, including the fibroblast growth factor, platelet-derived growth factor, Wnt, TGF-β, and Notch pathways (Clement, et al., 2013; Ezratty, et al., 2011; Neugebauer, et al., 2010; Schneider, et al., 2005; Simons, et al., 2005). In addition, primary cilia have mechanosensory functions in bone cells that are important in bone maintenance (He, et al., 2016) and there are specific orientations of cilia in osteocytes and chondrocytes (Uzbekov, et al., 2012), but how these processes are influenced in the skeletal cilia disorders is unknown. Despite the observation that IFT80 was found to be highly expressed in the mouse tibia growth plate during chondrogenic differentiation (Wang, et al., 2013), human fetal chondrocyte RNAseq data (unpublished) have not shown the skeletal ciliopathy genes to be more highly expressed in chondrocytes than in other tissues.
It is also possible that the preferential effect on the skeleton might be related to specific cargo molecules carried by the proteins encoded by the mutated genes and the complexes in which they participate. IFT proteins are largely conserved and contain protein-protein interaction domains such as tetratrico peptide repeats (TPRs), WD40 β-propellers and coiled-coils. A subset of these domains/proteins are required for IFT complex formation while others may be involved in IFT motor and/or cargo binding (Taschner, et al., 2011). However, it remains unresolved how specific cargo molecules are acquired and released. IFT motor proteins and many axonemal proteins move as cargo during cilia assembly via anterograde IFT to the ciliary tip (Craft, et al., 2015; Wren, et al., 2013), and cilia length can be regulated by adjusting the amount of precursors entering the cilia, but how cells integrate cilia length information is also unknown (Craft, et al., 2015; Lechtreck, 2015). It has been demonstrated that different WDR35 mutations can result in distinct ciliary protein fates (Mill, et al., 2011), as IFT88 protein accumulation was observed around the basal body in WDR35 SRPS fibroblasts while IFT88 clustered in the distal part of the ciliary axoneme and cilia tip in WDR35 CED fibroblasts. These correlations suggest that the different phenotypes might reflect differences in cilia assembly and function. In addition, a recent study showed selective cargo transport activity of WDR35 in regulating cilium assembly in human RPE1 cells (Fu, et al., 2016). Further investigation of IFT particles and specific cargos would provide deeper insight into the mechanisms that dictate the effects on skeletogenesis among the skeletal ciliopathies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the families for the generosity of their participation. We are also grateful to the following clinicians, genetic counselors and other health professionals who referred the cases described in this manuscript to the International Skeletal Dysplasia Registry between 1986–2015 and apologize to all individuals of whom we are not aware but who also participated in the care of the families: Mary-Alice Abbot, Tullie Ackerly, Carol Anderson, Brad Angle, Niki Armstrong, Art Aylsworth, Peter Baker, Matthew Basham, Erawati Bawle, Dorothy Beazley, Karen Beck, Robert Bendon, Rebecca Bent, Maryann Berg, Joann Bergoffen, Lynne Bird, Karin Blakemore, Steven Bleyl, Maureen Bocian, Joann Bodurtha, Robert Bonebrake, Christina Botti, Stephen Braddock, Steve Brown, Carol Brown-Elliot, Jeanette Camacho, Jack Campbell, Robert Campbell, Dru Carlson, Manuel Castillo, Francis Chang, William Chang, Albert Chen, Robert Cotterill, Carol Crowe, Cynthia Curry, George Davis, Debra Day-Salvatore, Catherine Dean, Robert Debbs, Larry Dennis, Maria Descartes, Paul Dicker, David Ding, Patricia Dix, Emily Doherty, Michael Donlan, Alan Donnenfield, Christopher Dotson, Karen Drake, Morris Edelman, Charles Epstein, Linda Ernst, Prab Gill, George Gilson, Mahin Golabi, Marcel Goldberger, Orlando Gonzalez, Laura Gorski, Ian Grable, Dorothy Grange, Kathy Grange, Robert Greenstein, Catherine Griswold, Art Grix, Nikki Hajdeek, Tony Hampton, Emily Hanson, Marc Heller, Joseph Hersch, Daniel Hersh, Douglas Hershey, Alice Hirata, George Hoganson, Jacob Hogue, Jeffrey Horbar, Y. Edward Hsia, Rachel Humphrey, LaDonna Immken, Heather Irwin, Erin Jarvis, Tamison Jewett, Marilyn Jones, Jack Jung, Madelyn Kahn, Ghizala Kaleem, Lilian Kaminsky, Ariana Kariminejad, Marion Kaufman, Chantal Kelly, Laura Keppen, Salman Kirmani, Joyce Kobori, David Lee, Anna Lehman, Michael Levine, Lawrence Lilien, Jeffrey Lipshitz, Mark Maberry, Geoffrey Machin, Neil Mandsager, Bruno Maranda, Dora Mayen Molina, Gilda Mayen, James McGrath, Tracy McGregor, Lynn McLean, Wendy Meschino, Natalie Mierowitz, Thomas Miles, David Miller, John Moeschler, Dora Molina, Stephen Myers, Thomas Myles, Jose Negron-Soto, Roger Newman, Clay Nichols, Allisan Nixdorf-Miller, Malforzata Nowaczyk, John O’Brien, Richard Oloya, Berrin Ozturk, Rich Pauli, Joseph Payman, Margaret Pearson, Sam Pepkowitz, Mark Peters, Kathleen Pfleghaar, Solveig Pfluger, Jean Pfotenhaues, Julie Platt, Marvin Platt, Rhonda Pollack Spiro, Phillip Potter, Chitra Prasad, Susan Presnell, Peter Pride, Tracy Prosen, Faisal Qureshi, Omar Rahman, Linda Randolph, Judith Ranells, Cheryl Reid, Douglas Richards, David Rimoin, Patricia Robertson, Haynes Robinson, Becky Rogers, Curtis Rogers, Gary Roloson, Anthony Royek, Orion Rust, Vasiliki Saitas, Irwin Schafer, Lisa Schimmenti, Michael Schneider, Dena Selby, Claire Singletary, Victoria Siu, Susan Sklower Brooks, Melvin Smith, Ana Spence, Jeffrey Spencer, Rhonda Spiro, Gregory Stamps, Harvey Stern, Devi Sureddi, Norman Tacktill, Kamer Tezcan, George E. Tiller, George R. Tiller, Audrey Toda, Elizabeth Tompson, Lisa Troyer, Alicia Vaglio, Peter Van Dorsten, Nithiwat Vatanavicharn, Keith Vaux, Angel Velez Rodriguez, Joseph Wagstaff, Robert Wallerstein, Ronald Wapner, William Wilcox, Kara Winthrow, Jacob Wolstein, Paul Wong, Sarah Wong and Catherine Yeagley.
This study was supported in part by the National Institute of Dental and Craniofacial Research (NIDCR) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health under Award Numbers RO1DE01957, R01AR062651 and RO1AR066124. S.P.T. was supported in part by NIH Training Grant in Genomic Analysis and Interpretation Award T32 HG002536. Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW CMG) which is funded by the National Human Genome Research Institute (NHGRI) and the National Heart, Lung and Blood Institute (NHLBI) Award 1U54HG006493. We also thank the March of Dimes, the Joseph Drown Foundation and the Orthopaedic Institute for Children for their support of the International Skeletal Dysplasia Registry.
Footnotes
DISCLOSURE STATEMENT
The authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nature Methods 7(4):248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alby C, Piquand K, Huber C, Megarbane A, Ichkou A, Legendre M, Pelluard F, Encha-Ravazi F, Abi-Tayeh G, Bessieres B, El Chehadeh-Djebbar S, Laurent N, Faivre L, Sztriha L, Zombor M, Szabo H, Failler M, Garfa-Traore M, Bole C, Nitschke P, Nizon M, Elkhartoufi N, Clerget-Darpoux F, Munnich A, Lyonnet S, Vekemans M, Saunier S, Cormier-Daire V, Attie-Bitach T, Thomas S. 2015. Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome. American Journal of Human Genetics 97(2):311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CT, Castillo AB, Brugmann SA, Helms JA, Jacobs CR, Stearns T. 2008. Primary cilia: Cellular sensors for the skeleton. Anatomical Record-Advances in Integrative Anatomy and Evolutionary Biology 291(9):1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts HH, Bongers EMHF, Mans DA, van Beersum SEC, Oud MM, Bolat E, Spruijt L, Cornelissen EAM, Schuurs-Hoeijmakers JHM, de Leeuw N, Cormier-Daire V, Brunner HG, Knoers NVAM, Roepman R. 2011. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. Journal of Medical Genetics 48(6):390–395. [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. 2006. The ciliopathies: An emerging class of human genetic disorders. Annual Review of Genomics and Human Genetics 7:125–148. [DOI] [PubMed] [Google Scholar]
- Badiner N, Taylor SP, Forlenza K, Lachman RS, University of Washington Center for Mendelian G, Bamshad M, Nickerson D, Cohn DH, Krakow D. 2016. Mutations in DYNC2H1, the cytoplasmic dynein 2, heavy chain 1 motor protein gene, cause short-rib polydactyly type I, Saldino-Noonan type. Clin Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baujat G, Huber C, El Hokayem J, Caumes R, Claire DNT, David A, Delezoide AL, Dieux-Coeslier A, Estournet B, Francannet C, Kayirangwa H, Lacaille F, Le Bourgeois M, Martinovic J, Salomon R, Sigaudy S, Malan V, Munnich A, Le Merrer M, Sang KHLQ, Cormier-Daire V. 2013. Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families. Journal of Medical Genetics 50(2):91–98. [DOI] [PubMed] [Google Scholar]
- Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, Johnson C, Irving M, Elcioglu N, Winey M, Tada M, Scambler PJ. 2007. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nature Genetics 39(6):727–729. [DOI] [PubMed] [Google Scholar]
- Beemer FA, Langer LO Jr., Klep-de Pater JM, Hemmes AM, Bylsma JB, Pauli RM, Myers TL, Haws CC 3rd. 1983. A new short rib syndrome: report of two cases. Am J Med Genet 14(1):115–23. [DOI] [PubMed] [Google Scholar]
- Berbari NF, Kin NW, Sharma N, Michaud EJ, Kesterson RA, Yoder BK. 2011. Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Developmental Biology 360(1):66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best S, Salvati F, Kallo J, Garner C, Height S, Thein SL, Rees DC. 2003. Lamin B-receptor mutations in Pelger-Huet anomaly. British Journal of Haematology 123(3):542–544. [DOI] [PubMed] [Google Scholar]
- Bizet AA, Becker-Heck A, Ryan R, Weber K, Filhol E, Krug P, Halbritter J, Delous M, Lasbennes MC, Linghu B, Oakeley EJ, Zarhrate M, Nitschke P, Garfa-Traore M, Serluca F, Yang F, Bouwmeester T, Pinson L, Cassuto E, Dubot P, Elshakhs NAS, Sahel JA, Salomon R, Drummond IA, Gubler MC, Antignac C, Chibout S, Szustakowski JD, Hildebrandt F, Lorentzen E, Sailer AW, Benmerah A, Saint-Mezard P, Saunier S. 2015. Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. 2015. Nosology and Classification of Genetic Skeletal Disorders: 2015 Revision. American Journal of Medical Genetics Part A 167(12):2869–2892. [DOI] [PubMed] [Google Scholar]
- Borovik L, Modaff P, Waterham HR, Krentz AD, Pauli RM. 2013. Pelger-Huet Anomaly and a Mild Skeletal Phenotype Secondary to Mutations in LBR. American Journal of Medical Genetics Part A 161(8):2066–2073. [DOI] [PubMed] [Google Scholar]
- Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbo M, Filhol E, Bole-Feysot C, Nitschke P, Gilissen C, Haugen OH, Sanders JSF, Stolte-Dijkstra I, Mans DA, Steenbergen EJ, Hamel BCJ, Matignon M, Pfundt R, Jeanpierre C, Boman H, Rodahl E, Veltman JA, Knappskog PM, Knoers NVAM, Roepman R, Arts HH. 2011. Ciliopathies with Skeletal Anomalies and Renal Insufficiency due to Mutations in the IFT-A Gene WDR19. American Journal of Human Genetics 89(5):634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalcanti DP, Huber C, Sang KHL, Baujat G, Collins F, Delezoide AL, Dagoneau N, Le Merrer M, Martinovic J, Mello MFS, Vekemans M, Munnich A, Cormier-Daire V. 2011. Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. Journal of Medical Genetics 48(2):88–92. [DOI] [PubMed] [Google Scholar]
- Cho C, Reck-Peterson SL, Vale RD. 2008. Regulatory ATPase sites of cytoplasmic dynein affect processivity and force generation. Journal of Biological Chemistry 283(38):25839–25845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton P, Fischer B, Mann A, Mansour S, Rossier E, Veen M, Lang C, Baasanjav S, Kieslich M, Brossuleit K, Gravemann S, Schnipper N, Karbasyian M, Demuth I, Zwerger M, Vaya A, Utermann G, Mundlos S, Stricker S, Sperling K, Hoffmann K. 2010. Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein. Nucleus-Austin 1(4):354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement CA, Ajbro KD, Koefoed K, Vestergaard ML, Veland IR, de Jesus MPRH, Pedersen LB, Benmerah A, Andersen CY, Larsen LA, Christensen ST. 2013. TGF-beta Signaling Is Associated with Endocytosis at the Pocket Region of the Primary Cilium. Cell Reports 3(6):1806–1814. [DOI] [PubMed] [Google Scholar]
- Craft JM, Harris JA, Hyman S, Kner P, Lechtreck KF. 2015. Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. Journal of Cell Biology 208(2):223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagoneau N, Goulet M, Genevieve D, Sznajer Y, Martinovic J, Smithson S, Huber C, Baujat G, Flori E, Tecco L, Cavalcanti D, Delezoide AL, Serre V, Le Merrer M, Munnich A, Cormier-Daire V. 2009. DYNC2H1 Mutations Cause Asphyxiating Thoracic Dystrophy and Short Rib-Polydactyly Syndrome, Type III. American Journal of Human Genetics 84(5):706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, Muzny DM, Young AC, Wheeler DA, Cruz P, Morgan M, Lewis LR, Cherukuri P, Maskeri B, Hansen NF, Mullikin JC, Blakesley RW, Bouffard GG, Gyapay G, Rieger S, Tonshoff B, Kern I, Soliman NA, Neuhaus TJ, Swoboda KJ, Kayserili H, Gallagher TE, Lewis RA, Bergmann C, Otto EA, Saunier S, Scambler PJ, Beales PL, Gleeson JG, Maher ER, Attie-Bitach T, Dollfus H, Johnson CA, Green ED, Gibbs RA, Hildebrandt F, Pierce EA, Katsanis N, Progra NCS. 2011. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nature Genetics 43(3):189–U28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. 2010. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP plus. Plos Computational Biology 6(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran I, Taylor SP, Zhang W, Martin J, Qureshi F, Jacques SM, Wallerstein R, Lachman RS, Nickerson DA, Bamshad M, Cohn DH, Krakow D. 2017. Mutations in IFT-A satellite core component genes IFT43 and IFT121 produce short rib polydactyly syndrome with distinctive campomelia. Cilia 6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran I, Taylor SP, Zhang WJ, Martin J, Forlenza KN, Spiro RP, Nickerson DA, Bamshad M, Cohn DH, Krakow D. 2016. Destabilization of the IFT-B cilia core complex due to mutations in IFT81 causes a Spectrum of Short-Rib Polydactyly Syndrome. Scientific Reports 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hokayem J, Huber C, Couve A, Aziza J, Baujat G, Bouvier R, Cavalcanti DP, Collins FA, Cordier MP, Delezoide AL, Gonzales M, Johnson D, Le Merrer M, Levy-Mozziconacci A, Loget P, Martin-Coignard D, Martinovic J, Mortier GR, Perez MJ, Roume J, Scarano G, Munnich A, Cormier-Daire V. 2012. NEK1 and DYNC2H1 are both involved in short rib polydactyly Majewski type but not in Beemer Langer cases. Journal of Medical Genetics 49(4):227–233. [DOI] [PubMed] [Google Scholar]
- Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E. 2011. A Role for the Primary Cilium in Notch Signaling and Epidermal Differentiation during Skin Development. Cell 145(7):1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. 2008. When cilia go bad: cilia defects and ciliopathies (vol 8, pg 880, 2007). Nature Reviews Molecular Cell Biology 9(1):88–88. [DOI] [PubMed] [Google Scholar]
- Fu WX, Wang L, Kim S, Li J, Dynlacht BD. 2016. Role for the IFT-A Complex in Selective Transport to the Primary Cilium. Cell Reports 17(6):1505–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdzicka M, Patnala S, Hirshman MG, Cai JF, Nitowsky H, Egeland JA, Ginns EI. 2002. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Molecular Genetics and Metabolism 77(4):291–295. [DOI] [PubMed] [Google Scholar]
- Gholkar AA, Senese S, Lo YC, Capri J, Deardorff WJ, Dharmarajan H, Contreras E, Hodara E, Whitelegge JP, Jackson PK, Torres JZ. 2015. Tctex1d2 associates with short-rib polydactyly syndrome proteins and is required for ciliogenesis. Cell Cycle 14(7):1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray RS, Abitua PB, Wlodarczyk BJ, Szabo-Rogers HL, Blanchard O, Lee I, Weiss GS, Liu KJ, Marcotte EM, Wallingford JB, Finnell RH. 2009. The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nature Cell Biology 11(10):1225–U153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, McInerney-Leo AM, Krug P, Filhol E, Davis EE, Airik R, Czarnecki PG, Lehman AM, Trnka P, Nitschke P, Bole-Feysot C, Schueler M, Knebelmann B, Burtey S, Szabo AJ, Tory K, Leo PJ, Gardiner B, McKenzie FA, Zankl A, Brown MA, Hartley JL, Maher ER, Li CM, Leroux MR, Scambler PJ, Zhan SH, Jones SJ, Kayserili H, Tuysuz B, Moorani KN, Constantinescu A, Krantz ID, Kaplan BS, Shah JV, Hurd TW, Doherty D, Katsanis N, Duncan EL, Otto EA, Beales PL, Mitchison HM, Saunier S, Hildebrandt F, Consortium UK. 2013. Defects in the IFT-B Component IFT172 Cause Jeune and Mainzer-Saldino Syndromes in Humans. American Journal of Human Genetics 93(5):915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Leong DJ, Zhuo Z, Majeska RJ, Cardoso L, Spray DC, Goldring MB, Cobelli NJ, Sun HB. 2016. Strain-induced mechanotransduction through primary cilia, extracellular ATP, purinergic calcium signaling, and ERK1/2 transactivates CITED2 and downregulates MMP-1 and MMP-13 gene expression in chondrocytes. Osteoarthritis and Cartilage 24(5):892–901. [DOI] [PubMed] [Google Scholar]
- Hennekam RCM. 1991. Short Rib Syndrome - Beemer Type in Sibs. American Journal of Medical Genetics 40(2):230–233. [DOI] [PubMed] [Google Scholar]
- Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Muller D, Vaya A, Aznar J, Ware RE, Cruz NS, Lindner TH, Herrmann H, Reis A, Sperling K. 2002. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly). Nature Genetics 31(4):410–414. [DOI] [PubMed] [Google Scholar]
- Huber C, Cormier-Daire V. 2012. Ciliary disorder of the skeleton. American Journal of Medical Genetics Part C-Seminars in Medical Genetics 160c(3):165–174. [DOI] [PubMed] [Google Scholar]
- Huber C, Wu SL, Kim AS, Sigaudy S, Sarukhanov A, Serre V, Baujat G, Sang KHLQ, Rimoin DL, Cohn DH, Munnich A, Krakow D, Cormier-Daire V. 2013. WDR34 Mutations that Cause Short-Rib Polydactyly Syndrome Type III/Severe Asphyxiating Thoracic Dysplasia Reveal a Role for the NF-kappa B Pathway in Cilia. American Journal of Human Genetics 93(5):926–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF. 2011. Ciliogenesis: building the cell’s antenna. Nature Reviews Molecular Cell Biology 12(4):222–234. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics 46(3):310–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon T, Nishiura M, Ohkura R, Toyoshima YY, Sutoh K. 2004. Distinct functions of nucleotide-binding/hydrolysis sites in the four AAA modules of cytoplasmic dynein. Biochemistry 43(35):11266–11274. [DOI] [PubMed] [Google Scholar]
- Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, Coe BP, Quinlan AR, Nickerson DA, Eichler EE, Project NES. 2012. Copy number variation detection and genotyping from exome sequence data. Genome Research 22(8):1525–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF. 2015. IFT-Cargo Interactions and Protein Transport in Cilia. Trends in Biochemical Sciences 40(12):765–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O’Donnell-Luria A, Ware J, Hill A, Cummings B, Tukiainen T, Birnbaum D, Kosmicki J, Duncan L, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Cooper D, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki M, Levy Moonshine A, Natarajan P, Orozco L, Peloso G, Poplin R, Rivas M, Ruano-Rubio V, Ruderfer D, Shakir K, Stenson P, Stevens C, Thomas B, Tiao G, Tusie-Luna M, Weisburd B, Won H-H, Yu D, Altshuler D, Ardissino D, Boehnke M, Danesh J, Roberto E, Florez J, Gabriel S, Getz G, Hultman C, Kathiresan S, Laakso M, McCarroll S, McCarthy M, McGovern D, McPherson R, Neale B, Palotie A, Purcell S, Saleheen D, Scharf J, Sklar P, Patrick S, Tuomilehto J, Watkins H, Wilson J, Daly M, MacArthur D. 2015. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Roberts R. 2001. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cellular and Molecular Life Sciences 58(14):2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney-Leo AM, Schmidts M, Cortes CR, Leo PJ, Gener B, Courtney AD, Gardiner B, Harris JA, Lu YP, Marshall M, Scambler PJ, Beales PL, Brown MA, Zankl A, Mitchison HM, Duncan EL, Wicking C, Consortium UK. 2013. Short-Rib Polydactyly and Jeune Syndromes Are Caused by Mutations in WDR60. American Journal of Human Genetics 93(3):515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney-Leo AM, Wheeler L, Marshall MS, Anderson LK, Zankl A, Brown MA, Leo PJ, Wicking C, Duncan EL. 2017. Homozygous variant in C21orf2 in a case of Jeune syndrome with severe thoracic involvement: Extending the phenotypic spectrum. American Journal of Medical Genetics Part A 173(6):1698–1704. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill AE, Merriman B, Farrington-Rock C, Camacho N, Sebald ET, Funari VA, Schibler MJ, Firestein MH, Cohn ZA, Priore MA, Thompson AK, Rimoin DL, Nelson SF, Cohn DH, Krakow D. 2009. Ciliary Abnormalities Due to Defects in the Retrograde Transport Protein DYNC2H1 in Short-Rib Polydactyly Syndrome. American Journal of Human Genetics 84(4):542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MAM, Keighren M, Bahlo M, Bromhead CJ, Budd P, Aftimos S, Delatycki MB, Savarirayan R, Jackson IJ, Amor DJ. 2011. Human and Mouse Mutations in WDR35 Cause Short-Rib Polydactyly Syndromes Due to Abnormal Ciliogenesis. American Journal of Human Genetics 88(4):508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison HM, Valente EM. 2017. Motile and non-motile cilia in human pathology: from function to phenotypes (vol 241, pg 294, 2016). Journal of Pathology 241(4):564–564. [DOI] [PubMed] [Google Scholar]
- Neugebauer JM, Amack JD, Peterson AG, Bisgrove BW, Yost HJ. 2010. FGF signalling during embryo development regulates cilia length in diverse epithelia (vol 458, pg 651, 2009). Nature 463(7279). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina PJR, Anderson KV. 2008. Intraflagellar Transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Developmental Dynamics 237(8):2030–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina PJR, Eggenschwiler JT, Moskowitz I, Anderson KV. 2011. Complex interactions between genes controlling trafficking in primary cilia. Nature Genetics 43(6):547–U75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XY, Ou GS, Civelekoglu-Scholey G, Blacque OE, Endres NF, Tao L, Mogilner A, Leroux MR, Vale RD, Scholey JM. 2006. Mechanism of transport of IFT particles in C-elegans cilia by the concerted action of kinesin-II and OSM-3 motors. Journal of Cell Biology 174(7):1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Haigo SL, Wallingford JB. 2006. Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nature Genetics 38(3):303–311. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Witman GB. 1999. The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J Cell Biol 144(3):473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F, Salih MAM, Gerber S, Delphin N, Bigot K, Orssaud C, Silva E, Baudouin V, Oud MM, Shannon N, Le Merrer M, Roche O, Pietrement C, Goumid J, Baumann C, Bole-Feysot C, Nitschke P, Zahrate M, Beales P, Arts HH, Munnich A, Kaplan J, Antignac C, Cormier-Daire V, Rozet JM. 2012. Mainzer-Saldino Syndrome Is a Ciliopathy Caused by IFT140 Mutations. American Journal of Human Genetics 90(5):864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Goodship JA. 2009. Ellis-van Creveld Syndrome and Weyers Acrodental Dysostosis Are Caused by Cilia-Mediated Diminished Response to Hedgehog Ligands. American Journal of Medical Genetics Part C-Seminars in Medical Genetics 151c(4):341–351. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, King L, Francomano C, Freisinger P, Spranger S, Marino B, Dallapiccola B, Wright M, Meitinger T, Polymeropoulos MH, Goodship J. 2000. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nature Genetics 24(3):283–286. [DOI] [PubMed] [Google Scholar]
- Scherft JP, Daems WT. 1967. Single Cilia in Chondrocytes. Journal of Ultrastructure Research 19(5–6):546–&. [DOI] [PubMed] [Google Scholar]
- Schmidts M, Arts HH, Bongers EMHF, Yap Z, Oud MM, Antony D, Duijkers L, Emes RD, Stalker J, Yntema JBL, Plagnol V, Hoischen A, Gilissen C, Forsythe E, Lausch E, Veltman JA, Roeleveld N, Superti-Furga A, Kutkowska-Kazmierczak A, Kamsteeg EJ, Elcioglu N, van Maarle MC, Graul-Neumann LM, Devriendt K, Smithson SF, Wellesley D, Verbeek NE, Hennekam RCM, Kayserili H, Scambler PJ, Beales PL, Knoers NVAM, Roepman R, Mitchison HM, Uk10k. 2013a. Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. Journal of Medical Genetics 50(5):309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts M, Frank V, Eisenberger T, al Turki S, Bizet AA, Antony D, Rix S, Decker C, Bachmann N, Bald M, Vinke T, Toenshoff B, Di Donato N, Neuhann T, Hartley JL, Maher ER, Bogdanovic R, Peco-Antic A, Mache C, Hurles ME, Joksic I, Guc-Scekic M, Dobricic J, Brankovic-Magic M, Bolz HJ, Pazour GJ, Beales PL, Scambler PJ, Saunier S, Mitchison HM, Bergmann C, 10k U. 2013b. Combined NGS Approaches Identify Mutations in the Intraflagellar Transport Gene IFT140 in Skeletal Ciliopathies with Early Progressive Kidney Disease. Human Mutation 34(5):714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts M, Hou Y, Cortes CR, Mans DA, Huber C, Boldt K, Patel M, van Reeuwijk J, Plaza JM, van Beersum SE, Yap ZM, Letteboer SJ, Taylor SP, Herridge W, Johnson CA, Scambler PJ, Ueffing M, Kayserili H, Krakow D, King SM, Uk10K, Beales PL, Al-Gazali L, Wicking C, Cormier-Daire V, Roepman R, Mitchison HM, Witman GB. 2015. TCTEX1D2 mutations underlie Jeune asphyxiating thoracic dystrophy with impaired retrograde intraflagellar transport. Nat Commun 6:7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts M, Vodopiutz J, Christou-Savina S, Cortes CR, McInerney-Leo AM, Emes RD, Arts HH, Tuysuz B, D’Silva J, Leo PJ, Giles TC, Oud MM, Harris JA, Koopmans M, Marshall M, Elcioglu N, Kuechler A, Bockenhauer D, Moore AT, Wilson LC, Janecke AR, Hurles ME, Emmet W, Gardiner B, Streubel B, Dopita B, Zankl A, Kayserili H, Scambler PJ, Brown MA, Beales PL, Wicking C, Duncan EL, Mitchison HM, Uk10k. 2013c. Mutations in the Gene Encoding IFT Dynein Complex Component WDR34 Cause Jeune Asphyxiating Thoracic Dystrophy. American Journal of Human Genetics 93(5):932–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. 2005. PDGFR alpha alpha signaling is regulated through the primary cilium in fibroblasts. Current Biology 15(20):1861–1866. [DOI] [PubMed] [Google Scholar]
- Scholey JM. 2008. Intraflagellar transport motors in cilia: moving along the cell’s antenna. Journal of Cell Biology 180(1):23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler E, Lin F, Worman HJ. 1994. Characterization of the Human Gene Encoding Lbr, an Integral Protein of the Nuclear-Envelope Inner Membrane. Journal of Biological Chemistry 269(15):11312–11317. [PubMed] [Google Scholar]
- Seo JH, Zilber Y, Babayeva S, Liu JJ, Kyriakopoulos P, De Marco P, Merello E, Capra V, Gros P, Torban E. 2015. Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans (vol 20, pg 4324, 2011). Human Molecular Genetics 24(13):3893–3893. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Schmidts M, Faqeih E, Hashem A, Lausch E, Holder I, Superti-Furga A, Mitchison HM, Almoisheer A, Alamro R, Alshiddi T, Alzahrani F, Beales PL, Alkuraya FS, Consortium UK. 2015. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Human Molecular Genetics 24(5):1410–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signor D, Wedaman KP, Orozco JT, Dwyer ND, Bargmann CI, Rose LS, Scholey JM. 1999. Role of a class DHC1b dynein in retrograde transport of IFT motors and IFT raft particles along cilia, but not dendrites, in chemosensory neurons of living Caenorhabditis elegans. Journal of Cell Biology 147(3):519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira KC, Moreno CA, Cavalcanti DP. 2017. Beemer-Langer syndrome is a ciliopathy due to biallelic mutations in IFT122. American Journal of Medical Genetics Part A 173(5):1186–1189. [DOI] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G. 2005. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nature Genetics 37(5):537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. 1999. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation (vol 13, pg 2072, 1999). Genes & Development 13(19):2617–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Bhogaraju S, Lorentzen E. 2012. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation 83(2):S12–S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Bhogaraju S, Vetter M, Morawetz M, Lorentzen E. 2011. Biochemical Mapping of Interactions within the Intraflagellar Transport (IFT) B Core Complex IFT52 BINDS DIRECTLY TO FOUR OTHER IFT-B SUBUNITS. Journal of Biological Chemistry 286(30):26344–26352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Weber K, Mourao A, Vetter M, Awasthi M, Stiegler M, Bhogaraju S, Lorentzen E. 2016. Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin-binding IFT-B2 complex. Embo Journal 35(7):773–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SP, Bosakova MK, Varecha M, Balek L, Barta T, Trantirek L, Jelinkova I, Duran I, Vesela I, Forlenza KN, Martin JH, Hampl A, University of Washington Center for Mendelian G, Bamshad M, Nickerson D, Jaworski ML, Song J, Ko HW, Cohn DH, Krakow D, Krejci P. 2016. An inactivating mutation in intestinal cell kinase, ICK, impairs hedgehog signaling and causes short rib-polydactyly syndrome. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SP, Dantas TJ, Duran I, Wu SL, Lachman RS, Nelson SF, Cohn DH, Vallee RB, Krakow D, Geno UWCM. 2015. Mutations in DYNC2LI1 disrupt cilia function and cause short rib polydactyly syndrome. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, Zenker M, Zahnleiter D, Stoss H, Beinder E, Abou Jamra R, Ekici AB, Schroder-Kress N, Aigner T, Kirchner T, Reis A, Brandstatter JH, Rauch A. 2011. NEK1 Mutations Cause Short-Rib Polydactyly Syndrome Type Majewski. American Journal of Human Genetics 88(1):106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, Tabler JM, Drew K, Kelly MR, Kim S, Park TJ, Braun DA, Pierquin G, Biver A, Wagner K, Malfroot A, Panigrahi I, Franco B, Al-Lami HA, Yeung Y, Choi YJ, University of Washington Center for Mendelian G, Duffourd Y, Faivre L, Riviere JB, Chen J, Liu KJ, Marcotte EM, Hildebrandt F, Thauvin-Robinet C, Krakow D, Jackson PK, Wallingford JB. 2016. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet 48(6):648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuysuz B, Baris S, Aksoy F, Madazli R, Ungur S, Sever L. 2009. Clinical Variability of Asphyxiating Thoracic Dystrophy (Jeune) Syndrome: Evaluation and Classification of 13 Patients. American Journal of Medical Genetics Part A 149a(8):1727–1733. [DOI] [PubMed] [Google Scholar]
- Uzbekov RE, Maurel DB, Aveline PC, Pallu S, Benhamou CL, Rochefort GY. 2012. Centrosome Fine Ultrastructure of the Osteocyte Mechanosensitive Primary Cilium. Microscopy and Microanalysis 18(6):1430–1441. [DOI] [PubMed] [Google Scholar]
- Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A, Szczepanska M, Krawczynski M, Zachwieja J, Zwolinska D, Beales PL, Ropers HH, Latos-Bielenska A, Kuss AW. 2010. Cranioectodermal Dysplasia, Sensenbrenner Syndrome, Is a Ciliopathy Caused by Mutations in the IFT122 Gene. American Journal of Human Genetics 86(6):949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CD, Yuan X, Yang SY. 2013. IFT80 is essential for chondrocyte differentiation by regulating Hedgehog and Wnt signaling pathways. Experimental Cell Research 319(5):623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, Mooyer P, van Noort G, Kelley RI, Wilcox WR, Wanders RJA, Hennekam RCM, Oosterwijk JC. 2003. Autosomal recessive HEM/greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol Delta(14)-reductase deficiency due to mutations in the lamin B receptor gene. American Journal of Human Genetics 72(4):1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, Beales PL. 2011. Ciliopathies: an expanding disease spectrum. Pediatric Nephrology 26(7):1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway G, Schmidts M, Mans DA, Szymanska K, Nguyen TMT, Racher H, Phelps IG, Toedts G, Kennedy J, Wunderlich KA, Sorusch N, Abdelhamed ZA, Natarajan S, Herridge W, van Reeuwijk J, Horn N, Boldt K, Parry DA, Letteboer SJF, Roosing S, Adams M, Bell SM, Bond J, Higgins J, Morrison EE, Tomlinson DC, Slaats GG, van Dam TJP, Huang LJ, Kessler K, Giessl A, Logan CV, Boyle EA, Shendure J, Anazi S, Aldahmesh M, Al Hazzaa S, Hegele RA, Ober C, Frosk P, Mhanni AA, Chodirker BN, Chudley AE, Lamont R, Bernier FP, Beaulieu CL, Gordon P, Pon RT, Donahue C, Barkovich AJ, Wolf L, Toomes C, Thiel CT, Boycott KM, McKibbin M, Inglehearn CF, Stewart F, Omran H, Huynen MA, Sergouniotis PI, Alkuraya FS, Parboosingh JS, Innes AM, Willoughby CE, Giles RH, Webster AR, Ueffing M, Blacque O, Gleeson JG, Wolfrum U, Beales PL, Gibson T, Doherty D, Mitchison HM, Roepman R, Johnson CA, Consortium UK, Geno UWCM. 2015. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nature Cell Biology 17(8):1074–U483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren KN, Craft JM, Tritschler D, Schauer A, Patel DK, Smith EF, Porter ME, Kner P, Lechtreck KF. 2013. A Differential Cargo-Loading Model of Ciliary Length Regulation by IFT. Current Biology 23(24):2463–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SS, Roth JA, Langer LO. 1991. Short Rib Syndrome Beemer-Langer Type with Polydactyly - a Multiple Congenital-Anomalies Syndrome. American Journal of Medical Genetics 39(3):243–246. [DOI] [PubMed] [Google Scholar]
- Ye XQ, Song GT, Fan MW, Shi LS, Jabs EW, Huang SZ, Guo RQ, Bian Z. 2006. A novel heterozygous deletion in the EVC2 gene causes Weyers acrofacial dysostosis. Human Genetics 119(1–2):199–205. [DOI] [PubMed] [Google Scholar]
- Zhang W, Taylor SP, Nevarez L, Lachman RS, Nickerson DA, Bamshad M, University of Washington Center for Mendelian Genomics C, Krakow D, Cohn DH. 2016. IFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilber Y, Babayeva S, Seo JH, Liu JJ, Mootin S, Torban E. 2013. The PCP effector Fuzzy controls cilial assembly and signaling by recruiting Rab8 and Dishevelled to the primary cilium. Mol Biol Cell 24(5):555–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.