

Abstract
Mitochondrial injury is uniformly observed in several murine models as well as in individuals with diabetic kidney disease (DKD). Although emerging evidence has highlighted the role of key transcriptional regulators in mitochondrial biogenesis, little is known about the regulation of mitochondrial cytochrome c oxidase assembly in the podocyte under diabetic conditions. We recently reported a critical role of the zinc finger Krüppel-like factor 6 (KLF6) in maintaining mitochondrial function and preventing apoptosis in a proteinuric murine model. In this study, we report that podocyte-specific knockdown of Klf6 increased the susceptibility to streptozotocin-induced DKD in the resistant C57BL/6 mouse strain. We observed that the loss of KLF6 in podocytes reduced the expression of synthesis of cytochrome c oxidase 2 with resultant increased mitochondrial injury, leading to activation of the intrinsic apoptotic pathway under diabetic conditions. Conversely, mitochondrial injury and apoptosis were significantly attenuated with overexpression of KLF6 in cultured human podocytes under hyperglycemic conditions. Finally, we observed a significant reduction in glomerular and podocyte-specific expression of KLF6 in human kidney biopsies with progression of DKD. Collectively, these data suggest that podocyte-specific KLF6 is critical to preventing mitochondrial injury and apoptosis under diabetic conditions.
Introduction
Diabetes is the leading cause of chronic kidney disease worldwide (1). A large body of evidence suggests that glomerular injury is the inciting event in the development of diabetic kidney disease (DKD) (2). Although all cellular components of the glomerulus (endothelial cells, mesangial cells, and podocytes) have been implicated in the development and progression of DKD, podocyte loss remains a hallmark feature in DKD (2). Podocytes are terminally differentiated epithelial cells in the glomerulus that are essential for the maintenance of the renal filtration barrier. In DKD, podocyte loss directly hampers the functional capacity to maintain this filtration barrier (3) and is the best predictor of albuminuria and chronic kidney disease in patients with diabetes (4–6). Mitochondrial injury is also uniformly observed in DKD and accompanied by mitochondrial DNA damage and altered expression of genes involved in mitochondrial biogenesis, function, and fragmentation (7–10). Recent studies have demonstrated that respiratory complex (RC) activity is also reduced in diabetic kidneys (8,11). However, the mechanism by which RC activity is reduced in DKD is unclear. Although all RCs are required for oxidative phosphorylation, cytochrome c oxidase (COX; RC IV) is the terminal enzyme of the respiratory chain and catalyzes the transfer of electrons from reduced cytochrome c to molecular oxygen. Furthermore, defective COX assembly has been directly implicated in mitochondrial dysfunction and reduced oxidative phosphorylation in several fatal mitochondrial diseases (12–15). However, to the best of our knowledge, transcriptional regulation of key COX assembly factors has never been investigated in DKD.
Along with others, we have reported an essential role of Krüppel-like factors (KLFs) in glomerular biology (16–18). KLFs are a subtype of zinc-finger transcriptional factors that are involved in several fundamental cellular processes (18,19). We previously reported the essential role of KLF6 in podocyte apoptosis (20). Specifically, we demonstrated that KLF6 is an early-inducible injury response gene that regulates cytochrome c oxidase (COX) expression, thereby abrogating the release of cytochrome c and activation of apoptosis in the setting of cell stress (20). In the setting of cell stress, we also showed that the loss of Klf6 in podocytes reduced the expression of key transcripts involved in mitochondrial replication, transcription, function, and COX assembly (20). Based on these previous studies, we hypothesize that the podocyte-specific loss of KLF6 accelerates DKD by increasing the susceptibility to mitochondrial dysfunction, leading to activation of the intrinsic apoptotic pathway, and eventual podocyte apoptosis. In this study, we report that the podocyte-specific loss of Klf6 in mice renders the kidney susceptible to early DKD. We also observed that the loss of KLF6 increases mitochondrial fragmentation and reduces COX expression. Finally, gene expression arrays from independent cohorts combined with immunostaining of human kidney biopsies demonstrated a significant reduction in glomerular and podocyte-specific KLF6 expression with progression of DKD.
Research Design and Methods
Genotyping of Podocin-Cre Klf6flox/flox Mice
Mice with Klf6 targeting vector (C57BL/6) were previously generated using the targeting strategy as previously described (21). Klf6flox/flox mice were crossed with mice expressing Cre recombinase driven by the podocin promoter (B6.Cg-Tg [NPHS2-cre] 295Lbh/J; The Jackson Laboratory, Bar Harbor, ME). Male offspring expressing Cre with two floxed Klf6 alleles were used as the experimental group (Podocin-Cre Klf6flox/flox). Mice with two wild-type alleles and Cre expression were used as controls (Podocin-Cre Klf6+/+). Genotyping by tail preparation and PCR were performed at 2 weeks of age as described (21). Confirmation of podocyte-specific knockdown and extent of injury was previously described (20).
Streptozotocin-Induced Type 1 Diabetes Murine Model
In the streptozotocin (STZ) model (22), Podocin-Cre Klf6flox/flox and Podocin-Cre Klf6+/+ (12 weeks of age) were administered STZ (50 mg/kg) in 50 mmol/L sodium citrate buffer (pH 5.4) by intraperitoneal injection over the course of 5 days (22,23). On day 14, blood glucose was measured from the tail vein after mice were fasted for 6 h using a OneTouch glucometer (LifeScan, Milpitas, CA) (23). To verify hyperglycemia, repeat fasting blood glucose measurements were taken monthly. Diabetes was defined as sustained fasting blood glucose >250 mg/dL at two distinct time points 2 weeks post–STZ injection (22). Mice were sacrificed 12 weeks posttreatment.
Measurement of Urine Albumin and Creatinine
Urine albumin was measured by ELISA as previously described (Bethyl Laboratories, Inc., Houston, TX). Urine creatinine levels were measured in the same samples using the QuantiChrom Creatinine Assay Kit (DICT-500; BioAssay Systems). The urine albumin excretion rate was expressed as the ratio of albumin to creatinine (23).
Measurement of Glomerular Filtration Rate
Glomerular filtration rate (GFR) in mice was determined using the clearance of fluorescein isothiocyanate–inulin (FITC-inulin) as described in the Animal Models of Diabetic Complications Consortium and Rieg (24). Briefly, anesthetized mice were injected retro-orbitally with dialyzed 5% FITC-inulin (2 μL/g body wt). Blood was collected from the tail at 3, 5, 7, 10, 15, 35, 56, and 75 min after injection. Separated serum was buffered in 0.5 mol/L HEPES, pH 7.4, and fluorescence was measured as described previously (24). GFR was calculated using the two-compartment clearance model with the following equation:
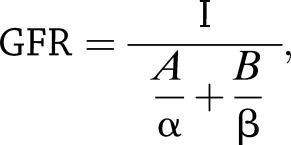 |
where I is the total amount of FITC-inulin delivered in the bolus injection retro-orbitally, A is the y-intercept of the rapid phase of elimination, B is the y-intercept of the slow phase of elimination, α is the decay constant for elimination, and β is the decay constant for distribution.
Blood Pressure Monitoring
Blood pressure was measured using the CODA-programmable noninvasive tail-cuff sphygmomanometer (Kent Scientific, Torrington, CT) on conscious mice as previously described (23,25). Mice were initially subjected to an acclimation period of five cycles before blood pressure assessment. Subsequently, blood pressure was measured in each mouse for 60 continuous cycles and an average of systolic blood pressure and diastolic blood pressure quantified as previously described (23,25,26).
Cell Culture
Conditionally immortalized human podocytes were gifts from Dr. Moin Saleem (University of Bristol, Southmead Hospital, Bristol, U.K.). These cells proliferate under permissive conditions (33°C), but differentiate under nonpermissive conditions (37°C). Methods for podocyte cultivation, immortalization, and differentiation were based on previously described protocol (27).
Short Hairpin RNA–Mediated KLF6 Knockdown Using Lentivirus
KLF6 knockdown in human podocytes was performed using the expression arrest GIPZ lentiviral shRNAmir system (Thermo Fisher Scientific, Huntsville, AL) as previously described (20). Cells expressing short hairpin RNA (shRNA) were selected with puromycin for 2 to 3 weeks prior to use in all studies. EV-shRNA serves as empty vector control. Confirmation of KLF6 knockdown was previously shown (20).
LentiORF-KLF6 Overexpression
The LentiORF-KLF6 clone was purchased from Thermo Fisher Scientific, and stable KLF6 overexpression was achieved by lentiviral delivery. Cells expressing LentiORF-KLF6 were selected with blasticidin for 2 to 3 weeks prior to use in all studies. LentiORF-control serves as the GFP control vector. GFP expression and Western blot were performed to confirm KLF6 overexpression as previously shown (20).
Real-time PCR
Total RNA was extracted by using TRIzol (Gibco). First-strand cDNA was prepared from total RNA (1.5 mg) using the SuperScript III First-Strand Synthesis Kit (Life Technologies), and cDNA (1 mL) was amplified using SYBR GreenER qPCR Supermix on ABI QuantStudio 3 (Applied Biosystems). Primers for mouse collagen 1α 1 (Col1α1), α–smooth muscle actin (α-SMA), fibronectin, and CTGF were designed using National Center for Biotechnology Information Primer-BLAST and validated for efficiency before application (Supplementary Table 3). Light-cycler analysis software was used to determine crossing points using the second derivative method. Data were normalized to housekeeping genes (GAPDH or β-actin) and presented as fold increase compared with RNA isolated from the control group using the 2−ΔΔ threshold cycle method.
High-Glucose Treatment of Podocytes in Culture
Human podocytes were differentiated for 14 days at 37°C prior to all experiments. To stimulate hyperglycemic conditions, cells were serum starved for 12 h prior to treatment with 30 mmol/L d-glucose (high glucose [HG]), 25 mmol/L d-mannitol plus 5 mmol/L d-glucose (osmotic control), and 5 mmol/L d-glucose (normal glucose [NG]) for 7–14 days.
Western Blot
Podocytes were lysed with a 2× Laemmli buffer and then quantified using Pierce BCA Protein Assay (23225; Thermo Fisher Scientific). Lysates were subjected to immunoblot analysis using primary antibody as previously described (20) for the target of interest: mouse anti–β-actin (A3854; Sigma-Aldrich, St. Louis, MO), rabbit anti-cleaved caspase-3 (9644; Cell Signaling Technology), and rabbit anti–synthesis of cytochrome c oxidase 2 (SCO2; AB115877; Abcam).
Mitochondrial Fragmentation Studies
To visualize mitochondrial morphology, differentiated human podocytes were incubated with a Rosamine-based MitoTracker probe (Invitrogen) at 100 nmol/L for 30 min. After the incubation period, cells were washed with PBS and fixed with 3.7% formaldehyde in growth medium as previously described (20). Mitochondrial fragmentation was quantified using previously described methods (20,28). Briefly, mitochondrial morphology was assessed in each cell by a blinded investigator into a tubular (>75% of mitochondria with tubular length >5 mm), intermediate (25–75% of mitochondria with tubular length >5 mm), or fragmented pattern (<25% of mitochondria with tubular length >5 mm).
Histopathology and Morphometric Studies by Bright-Field Light Microscopy
Mice were perfused with PBS, and the kidneys were fixed in 10% phosphate-buffered formalin overnight and switched to 70% ethanol prior to processing for histology. Kidney tissue was embedded in paraffin by American Histolabs (Gaithersburg, MD), and 3-μm–thick sections were stained with periodic acid Schiff (PAS; Sigma-Aldrich) and Masson trichrome (23).
Quantification of mesangial area and glomerular volume was performed as described (29,30). In brief, digitized images were scanned, and profile areas were traced using ImageJ 1.26t software (National Institutes of Health; rsb.info.nih.gov/ij). Mean glomerular tuft volume (GV) was determined from mean glomerular cross-sectional area (GA) by light microscopy. GA was calculated based on average area of 30 glomeruli in each group, and GV was calculated based on the following:
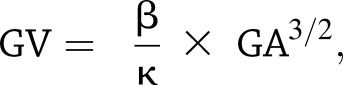 |
where β = 1.38, the shape coefficient of spheres (the idealized shape of glomeruli), and κ = 1.1, the size distribution coefficient.
Mesangial expansion was defined as PAS-positive and nuclei-free area in the mesangium. Quantification of mesangial expansion was based on 30 glomeruli cut at the vascular pole in each group.
Quantification of podocyte number per glomerulus was determined using WT1-stained podocytes. Kidney sections from these mice were initially prepared in identical fashion. Subsequently, 4-μm–thick sections were stained with rabbit anti-WT1 (Novus Biologicals, Littleton, CO) as previously described (31). Counting of podocytes and measurement of glomerular area and volume were performed using ImageJ and by the method described by the Animal Models of Diabetic Complications Consortium (29).
Histopathology and Morphometric Studies by Transmission Electron Microscopy
Mice were perfused with PBS and then immediately fixed in 2.5% glutaraldehyde for electron microscopy (EM). Sections were mounted on a copper grid and photographed under a Hitachi H7650 microscope. Briefly, negatives were digitized, and images with a final magnitude of approximately ×10,000 were obtained (32). The quantification of podocyte effacement was performed as previously described (33). In brief, ImageJ was used to measure the length of the peripheral glomerular basement membrane (GBM), and the number of slit pores overlying this GBM length was counted. The arithmetic mean of the foot process width (WFP) was calculated using the following:
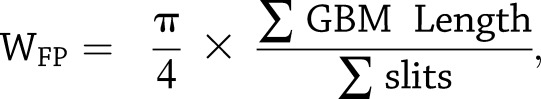 |
where Σ GBM length indicates the total GBM length measured in one glomerulus, Σ slits indicates the total number of slits counted, and 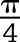 is the correction factor for the random orientation by which the foot processes were sectioned) (33).
is the correction factor for the random orientation by which the foot processes were sectioned) (33).
Quantification of GBM thickness was performed as described (34). The thickness of multiple capillaries was measured in three to four glomeruli per mouse. A mean of 418 measurements was taken per mouse (from podocyte to endothelial cell membrane) at random sites where GBM was displayed in the best cross section.
Immunofluorescence
Kidney sections from each experimental group of mice were prepared in identical fashion. Immunostaining was performed using the primary antibody for target of interest: rabbit anti-KLF6 (AB135783; Abcam), mouse antinestin (MAB353; Millipore), mouse anti–endothelial nitric oxide synthase (eNOS; 610296; BD Biosciences), mouse anti-WT1 (SC7385; Santa Cruz Biotechnology), rabbit anti-cleaved caspase-3 (8172S; Cell Signaling Technology), rabbit anti-cleaved caspase-9 (9505; Cell Signaling Technology), rabbit anti-SCO2 (AB115877; Abcam), rabbit anti-vimentin (D21H3; Cell Signaling Technology), rabbit anti-Col1α1 (AB34710; Abcam), mouse anti-GBM α-SMA rabbit (A5228), anti–translocase of outer mitochondrial membrane 20 (TOMM20; AB78547; Abcam), and mouse anti–cytochrome c (AB13575; Abcam) antibodies. After washing, sections were incubated with the corresponding fluorophore-linked secondary antibody (Alexa Fluor 568 anti-rabbit IgG or Alexa Fluor 488 anti-mouse IgG from Invitrogen). After staining, slides were mounted in Prolong gold antifade mounting media (P36930; Invitrogen) and photographed using a Nikon Eclipse i90 microscope (Nikon, Melville, NY) with a digital camera.
Terminal deoxynucleotidyl TUNEL staining was performed using the In Situ Cell Death Detection Kit as per the manufacturer’s instructions (12156792910; Roche). Briefly, tissue sections were incubated for 15–30 min at room temperature with Proteinase K working solution (10 μg/mL). Subsequently, tissue sections were rinsed twice in PBS, and 50 μL of TUNEL reaction mixture was added to each section. After the incubation period, tissue sections were kept at 37°C in the dark for 60 min prior to repeat PBS wash and mounting.
Quantification of eNOS, KLF6, vimentin, and SCO2 expression in the glomerulus was determined by measuring the percent area stained in the glomerulus in 20 high-power field (×20) digitized images using ImageJ (20). Quantification of TUNEL staining was determined by quantifying the number of TUNEL+ cells per glomerular cross section. Cleaved caspase-3 and cleaved caspase-9 staining in the glomerulus was determined by the percentage of glomeruli with positive staining. Cytosolic cytochrome c expression was determined by quantifying the percentage of glomerular cytochrome c and TOMM20 colocalization by ImageJ.
Human Kidney Biopsies
De-identified human kidney biopsy specimens from Stony Brook Medicine and the University of Utah were obtained for staining of KLF6. Human kidney biopsy specimens were categorized into early-stage (<10%) and late-stage (>50%) chronic tubulointerstitial fibrosis by a renal pathologist (M.P.R.). Control kidney biopsy specimens were acquired from the unaffected pole of kidneys removed due to renal cell carcinoma. Quantification of KLF6 staining in the podocytes was determined by quantifying the ratio of cells that are KLF6+WT1+Hoechst+ to the number of cells that are WT1+Hoechst+ staining using ImageJ 1.26t software.
COX Activity in Kidney Sections
Immunohistochemistry for COX activity was performed as previously described (35,36). Briefly, 4-μm thickness mouse kidney cryosections were allowed to dry at room temperature for 1 h. Bovine catalase (2 μg) was added to prepared incubation solution (1× 3,3′-diaminodbenzidine with 100 μmol/L) as previously described (36). Slides were incubated in 37°C for 40 min in the incubation solution containing bovine catalase, subsequently washed with 0.2 mol/L phosphate buffer (37), and dehydrated in ethanol. Slides were then placed in xylene for 10 min and mounted with Entellan prior to imaging. Quantification of COX activity in the glomerulus was determined by measuring the percent area stained in the glomerulus in 20 high-power field (×20) images using ImageJ (20).
Annexin V and Propidium Iodide With FACS
Differentiated human podocytes were trypsinized and washed with PBS. Cells were initially suspended in 1× Annexin binding buffer (V13246; Thermo Fisher Scientific) and subsequently stained with conjugated Annexin V (559933; BD Pharmingen) was followed by propidium iodide (PI; 556463; BD Pharmingen) as per the manufacturer’s protocol. Cells were incubated in the dark at room temperature for 15 min and then analyzed by an FACSCalibur flow cytometer with data analysis of 10,000 gated events. Quantification was determined as a fold change in percentage of apoptotic cells relative to NG-treated EV-shRNA cells, which was defined as cells with low PI and high Annexin binding.
Extracellular Oxygen Consumption Rate
Differentiated human podocytes were plated at 2,000 cells/well in a black 96-well assay plate (3603; Corning). HG treatment was introduced for 7–14 days according to the specific experimental conditions. Extracellular oxygen consumption rate (OCR) was measured using the OCR Assay Kit (600800; Cayman Chemical) as per the manufacturer’s instructions and as previously described (38). Briefly, a MitoXpress Xtra (600801; Cayman Chemical) phosphorescent oxygen-sensing probe was introduced and sealed with HS Mineral Oil Assay Reagent (660910; Cayman Chemical) to prevent the loss of extracellular oxygen. Fluorescence at 380 nm was read every 3 min with a microplate reader at 37°C for 120 min, and OCR was calculated as the ratio of change in oxygen consumption over time and expressed as a fold change in OCR relative to NG-treated control cells.
COX Activity in Cell Culture
Mitochondria were initially isolated as previously described (39) from differentiated human podocytes after specific experimental conditions. Briefly, 10% digitonin was used for cell permeabilization followed by homogenization with a mannitol buffer supplemented with dithiothreitol and protease inhibitors. To pellet mitochondria, differential centrifugation was performed at 15,000g for 10 min. COX activity was subsequently measured calorimetrically using the COX Human Enzyme Activity Microplate Assay Kit as per the manufacturer’s instructions (ab109909; Abcam). Total mitochondria were normalized per sample by measuring mitochondrial protein concentration using a Bradford assay. COX activity is a product of the initial rate of oxidation of cytochrome c, which is determined by measuring the absorbance at 550 nm every 3 min for 120 min and calculating the change in absorbance over time. Data are expressed as a fold change in COX activity relative to NG-treated control cells.
Statistical Analysis
An unpaired t test was used to compare data between two groups and one-way ANOVA with Tukey posttest to compare data among more than two groups. Because we could not assume normality on some of the other data sets with smaller sample sizes, nonparametric SPSS was performed using the Mann-Whitney test to compare data between two groups and Kruskal-Wallis test with Dunn posttest to compare data among more than two groups. χ2 or Fisher exact test, as appropriate, was used to compare the significance between categorical variables. The exact test used for each experiment is denoted in the figure legends. Data were expressed as mean ± SEM and medians and interquartile ranges (25th and 75th percentiles) or numbers of patients for categorical variables. All experiments were repeated a minimum of three times, and representative experiments are shown. Statistical significance was considered when P < 0.05. All statistical analysis was performed using GraphPad Prism 6.0.
Study Approval
All animal studies conducted were approved by the Stony Brook University Animal Institute Committee. The National Institutes of Health Guide for the Care and Use of Laboratory Animals was followed strictly. The Stony Brook University Institutional Review Board approved the use of archived de-identified human biopsy specimens for immunostaining.
Results
Diabetic Podocin-Cre Klf6flox/flox Mice Exhibit an Increase in Glomerular and Interstitial Injury
To ascertain whether the podocyte-specific loss of Klf6 increases glomerular and interstitial injury under diabetic conditions, we used the previously generated and validated Podocin-Cre Klf6flox/flox and Podocin-Cre Klf6+/+ mice on the C57BL/6 background (20). Although treatment with STZ induces diabetes, C57BL/6 mice are typically resistant to STZ-induced DKD (23,40). Initially, we confirmed that STZ induced a sustained increase in serum glucose measurements in both the Podocin-Cre Klf6flox/flox and Podocin-Cre Klf6+/+ mice at 4, 8, and 12 weeks after STZ treatment (Supplementary Table 1). Diabetic Podocin-Cre Klf6flox/flox mice exhibited a significant increase in albuminuria and kidney weight as compared with all other groups (Fig. 1A and B and Supplementary Table 1). In addition, the diabetic Podocin-Cre Klf6flox/flox mice demonstrated a significant increase in GFR as compared with all other groups, consistent with hyperfiltration observed in early-stage DKD (Fig. 1C). No significant differences in albuminuria, kidney weight, or GFR were noted between the diabetic and wild-type Podocin-Cre Klf6+/+ mice. Tail-cuff manometry of blood pressure did not reveal a significant difference in the systolic blood pressure, diastolic blood pressure, or mean arterial pressure between groups, suggesting that the observed changes were not due to hypertension (Supplementary Fig. 1). PAS staining revealed an increase in glomerular hypertrophy, mesangial expansion, and glomerular eNOS expression with a reduction in podocyte number in the diabetic Podocin-Cre Klf6flox/flox mice as compared with all other groups (Fig. 2A–D). Furthermore, the diabetic Podocin-Cre Klf6flox/flox mice exhibited an increase in GBM thickness and foot process effacement as compared with all other groups by transmission EM (Fig. 2A, E, and F). Interestingly, Podocin-Cre Klf6flox/flox mice also exhibited a decrease in podocyte number and increase in GBM thickness as compared with Podocin-Cre Klf6+/+ mice, independent of diabetic conditions (Fig. 2D and E). In addition, Masson trichrome staining showed an increase in glomerular collagen expression in the diabetic Podocin-Cre Klf6flox/flox mice as compared with all other groups (Fig. 3A and B). Similarly, we also observed an increase in glomerular vimentin in the diabetic Podocin-Cre Klf6flox/flox mice as compared with all other groups (Fig. 3A and C). To assess for interstitial fibrosis, we initially stained for interstitial Col1α1 and α-SMA and measured Col1α1, α-SMA, fibronectin, and CTGF mRNA expression from all four groups. Interestingly, only Col1α1 mRNA and protein expression was increased in diabetic Podocin-Cre Klf6flox/flox mice as compared with all other groups, with no significant changes in expression of other markers between the groups (Supplementary Fig. 2). Combined, these findings suggest that the podocyte-specific loss of Klf6 contributes to the development of early DKD under diabetic conditions.
Figure 1.

Diabetic Podocin-Cre Klf6flox/flox mice exhibit hyperfiltration and an increase in albuminuria. Podocin-Cre Klf6flox/flox (Pod-Cre Klf6fl/fl) and Podocin-Cre Klf6+/+ mice were treated with STZ and vehicle at 12 weeks of age. Decapsulated kidney weight and total body weight were measured, and urine was collected from each experimental group 12 weeks posttreatment. A: Urine albumin-to-creatinine (cr) ratio is shown at 12 weeks post–STZ treatment (n = 10; **P < 0.01 vs. all other groups, Kruskal-Wallis test with Dunn posttest). B: Kidney weight/body weight (g/g) ratio is shown (n = 10/group; *P < 0.05; **P < 0.01, Kruskal-Wallis test with Dunn posttest). Prior to being euthanized, mice were injected retro-orbitally with dialyzed FITC-inulin to measure GFR using the two-compartment clearance model. C: GFR (μL/min) at 12 weeks post–STZ treatment is shown (n = 6/group; *P < 0.05 vs. all other groups, Kruskal-Wallis test with Dunn posttest). Inset: clearance of FITC-inulin over time as measured by fluorescence.
Figure 2.

Diabetic Podocin-Cre Klf6flox/flox (Pod-Cre Klf6fl/fl) mice demonstrate increased glomerular injury. A: Representative images of paraffin-embedded sections stained with PAS are shown at low power (×10) and high power (×40) to demonstrate changes in mesangial expansion, glomerular volume, and tubulointerstitial changes. Ultrastructural changes are also shown at original magnification ×10,000 by transmission EM to show changes in podocyte structure and GBM. Red arrowheads indicate upright foot processes, and red arrow shows foot process effacement. Scale bars = 100 μm. Quantification of glomerular volume (B), mesangial expansion (C), glomerular eNOS staining (D), and podocyte number (E; number of WT1+ cells per glomerulus) is shown (n = 20 glomeruli/mouse, n = 6 mice/group; *P < 0.05; **P < 0.01, Kruskal-Wallis test with Dunn posttest). Quantification of ultrastructural changes: GBM thickness (F) and podocyte effacement (G; measurement by foot process width between slit diaphragm) is also shown (n = 3 mice/group; *P < 0.05; **P < 0.01, Kruskal-Wallis test with Dunn posttest). AU, arbitrary units.
Figure 3.

Diabetic Podocin-Cre Klf6flox/flox (Pod-Cre Klf6fl/fl) mice exhibit increased glomerular extracellular matrix deposition. A: Representative images of paraffin-embedded sections stained with Masson trichrome are shown at low power (×10) and high power (×40) to demonstrate changes in glomerular collagen deposition. Immunofluorescence staining for vimentin was also performed, and representative images are shown. Scale bars = 100 μm. Quantification of changes by ImageJ is shown for glomerular collagen (B) and glomerular vimentin (C). n = 20 glomeruli/mouse, n = 6 mice/group; **P < 0.01, Kruskal-Wallis test with Dunn posttest. AU, arbitrary units.
Diabetic Podocin-Cre Klf6flox/flox Mice Exhibit Increased Apoptosis in the Glomeruli
To ascertain the mechanism by which podocyte-specific loss of Klf6 induces glomerular injury under diabetic conditions, we initially sought to identify changes in podocyte number under diabetic conditions. The diabetic Podocin-Cre Klf6flox/flox mice demonstrated a significant reduction in podocyte number as compared with diabetic Podocin-Cre Klf6+/+ mice (Fig. 2E), suggesting that a loss of podocytes might be contributing to the development of early DKD in these mice. We previously reported that the loss of Klf6 in podocytes increases the susceptibility to mitochondrial injury with activation of the intrinsic apoptotic pathway under cell stress (20). Because several cell types exist in the kidney, we used immunostaining techniques to demonstrate the spatial changes in mitochondrial injury and apoptosis specifically in the glomerulus. Interestingly, the Podocin-Cre Klf6flox/flox mice exhibited a significant increase in TUNEL+ cells per glomerular cross section as well as an increase in the percentage of positive glomeruli with cleaved caspase-3 and cleaved caspase-9 staining as compared with the Podocin-Cre Klf6+/+ mice under diabetic conditions (Fig. 4A–C), suggesting activation of the intrinsic apoptotic pathway. To assess whether these changes are specific to the podocyte, we costained for cleaved caspase-3 with nestin, a podocyte-specific marker previously reported to maintain its expression in the setting of glomerular injury (41). Representative images shown in Supplementary Fig. 3 demonstrate the colocalization of nestin with cleaved caspase-3, suggesting activation of apoptotic pathway in the podocyte.
Figure 4.

Diabetic Podocin-Cre Klf6flox/flox (Pod-Cre Klf6fl/fl) mice exhibit increased mitochondrial injury and activation of intrinsic apoptotic pathway. TUNEL staining was performed on paraffin-embedded sections from diabetic Podocin-Cre Klf6flox/flox and Podocin-Cre Klf6+/+ mice. A: Quantification of TUNEL+ cells per GA is shown. Immunostaining for cleaved caspase-3 (B) and cleaved caspase-9 (C) was also performed and quantified by measuring the percent of glomeruli with positive staining. D: Immunohistochemistry for COX activity was performed, and ImageJ was used to quantify glomerular COX activity in each group. E: Immunostaining for SCO2 was also performed. Representative images of glomerular SCO2 staining are shown in the top panel. Quantification of glomerular SCO2 expression was determined by ImageJ and is shown in the bottom panel. F: Immunostaining for TOMM20 and cytochrome c was performed to determine the release of cytochrome c from the mitochondria, and representative images are shown. White arrowheads indicate loss of cytochrome c and TOMM20 colocalization. Right panel shows quantification of colocalization. n = 20 glomeruli/mouse, n = 6 mice/group. *P < 0.05; **P < 0.01, Kruskal-Wallis test with Dunn posttest. Scale bars = 100 μm.
We previously demonstrated that SCO2, a mitochondrial membrane-bound metallochaperone critical for COX assembly, is transcriptionally regulated by KLF6 (20). We observed a reduction in glomerular COX activity and SCO2 expression in the Podocin-Cre Klf6flox/flox mice as compared with the Podocin-Cre Klf6+/+ mice under diabetic conditions (Fig. 4D and E). Interestingly, there was a trend toward a reduction in glomerular COX activity and SCO2 expression in the Podocin-Cre Klf6flox/flox mice as compared with the Podocin-Cre Klf6+/+ mice with buffer treatment, but it did not reach statistical significance. To further demonstrate the activation of the intrinsic apoptotic pathway in the glomeruli in diabetic Podocin-Cre Klf6flox/flox mice, we assessed for mitochondrial stability by measuring the cytosolic release of cytochrome c from the mitochondria by costaining cytochrome c with the mitochondria-specific marker TOMM20 protein. We observed a significant increase in cytosolic cytochrome c expression in the diabetic Podocin-Cre Klf6flox/flox mice as compared with all other groups (Fig. 4F). These data suggest that the knockdown of Klf6 in podocytes increases the susceptibility to podocyte loss, which might be due to activation of the intrinsic apoptotic pathway under diabetic conditions.
Loss of KLF6 in Podocytes Increases Mitochondrial Injury and Apoptosis Under HG Conditions
Because cells other than podocytes can contribute to glomerular injury under diabetic conditions, we used differentiated human podocytes in culture with KLF6 knockdown (KLF6-shRNA) (20) to further explore the mechanism by which podocyte-specific loss of KLF6 activates the intrinsic apoptotic pathway under HG conditions. Efficiency and specificity of KLF6 knockdown in cultured human podocytes has been previously reported (20). Differentiated EV-shRNA (control) and KLF6-shRNA human podocytes were treated with HG (30 mmol/L), NG (5 mmol/L), and mannitol (5 mmol/L glucose plus 25 mmol/L mannitol) for 7 days. HG-treated KLF6-shRNA human podocytes exhibited a significant increase in cleaved caspase-3 expression as compared with HG-treated EV-shRNA human podocytes (Fig. 5A and B). No significant changes were observed under mannitol conditions (Supplementary Fig. 4A). We also validated these findings by staining for Annexin V and PI and performing FACS to quantify the percentage of apoptotic cells (Fig. 5C). We previously reported that KLF6 regulates SCO2 under cell stress to prevent mitochondrial injury, with a reduction in SCO2 expression in human podocytes with KLF6 knockdown (20). Furthermore, HG-treated KLF6-shRNA podocytes showed an increase in cells with mitochondrial fragmentation as compared with HG-treated EV-shRNA human podocytes (Fig. 5D). In addition, HG-treated KLF6-shRNA podocytes exhibited a reduction in extracellular OCR and COX activity as compared with HG-treated EV-shRNA human podocytes (Fig. 5E and F). Collectively, these findings suggest that the knockdown of KLF6 in cultured human podocytes increases the susceptibility to mitochondrial injury, leading to apoptosis under HG conditions.
Figure 5.

Knockdown of KLF6 in podocytes exacerbates mitochondrial injury and apoptosis under HG conditions. Differentiated EV-shRNA and KLF6-shRNA human podocytes were initially treated under HG and NG conditions for 7 days. A: Western blot for cleaved caspase-3 was performed, and representative images of three independent experiments are shown. B: Quantification of cleaved caspase-3 expression by densitometry (n = 3, Kruskal-Wallis test with Dunn post hoc test; *P < 0.05; **P < 0.01). C: To quantify apoptosis, Annexin V/PI staining in combination with FACS was performed (n = 3, Kruskal-Wallis test with Dunn post hoc test; **P < 0.01). D: Rosamine-based MitoTracker probe was used to assess mitochondrial structure and fragmentation. The representative images of six independent experiments are shown in the top panel (×20). Mitochondrial staining is indicated by tubular (red arrows) and fragmented (red arrowheads) pattern. The bottom panel shows the scoring of mitochondrial morphology from 100 podocytes in each group (n = 3; *P < 0.05 compared with EV-shRNA cells, two-way ANOVA test with Tukey posttest). Scale bars = 100 μm. Extracellular OCR (E) and COX activity (F) were measured and expressed as fold change relative to untreated EV-shRNA podocytes. n = 3; *P < 0.05; **P < 0.01, Kruskal-Wallis test with Dunn post hoc test. AU, arbitrary units.
Overexpression of KLF6 in Podocytes Attenuates Mitochondrial Injury and Apoptosis Under HG Conditions
Because the loss of KLF6 in podocytes increases the susceptibility to mitochondrial injury and apoptosis under HG conditions, we hypothesized that the overexpression of KLF6 will ameliorate these changes. We used the differentiated human podocytes with (LentiORF-KLF6) and without (LentiORF-control) stable overexpression of KLF6 (20). We previously confirmed the specificity of KLF6 overexpression in cultured human podocytes (20). Differentiated LentiORF-control and LentiORF-KLF6 human podocytes were treated with NG, HG, and mannitol for 14 days. HG-treated LentiORF-KLF6 human podocytes exhibited a decrease in cleaved caspase-3 expression with some restoration in mitochondrial fragmentation as compared with HG-treated LentiORF-control human podocytes (Fig. 6A and B and Supplementary Fig. 4B). Contrary to the loss of SCO2 expression with KLF6 knockdown (20), HG-treated LentiORF-KLF6 podocytes showed an increase in SCO2 expression, extracellular OCR, and COX activity as compared with HG-treated LentiORF-control podocytes (Fig. 6C–E). Combined, these data suggest that KLF6 is critical to preventing mitochondrial injury and apoptosis under HG conditions.
Figure 6.

Overexpression of KLF6 in podocytes attenuates mitochondrial injury and apoptosis under HG conditions. Differentiated LentiORF-KLF6 and LentiORF-control human podocytes were initially treated under HG and NG conditions for 14 days. A: Western blot for cleaved caspase-3 was performed, and representative images of three independent experiments are shown in the top panel. The bottom panel shows the quantification of cleaved caspase-3 by densitometry. B: Rosamine-based MitoTracker probe was used to assess mitochondrial structure and fragmentation. The representative images of six independent experiments are shown in the left panel (×20). Mitochondrial staining is indicated by tubular (arrows) and fragmented (arrowheads) patterns. The right panel shows the scoring of mitochondrial morphology from 100 podocytes in each group (n = 3; *P < 0.05, two-way ANOVA test with Tukey posttest). Scale bars = 100 μm. C: Western blot for SCO2 was also performed, and representative images of three independent experiments are shown in the top panel. The bottom panel shows the quantification of SCO2 by densitometry. Extracellular OCR (D) and COX activity (E) were measured and expressed as fold change relative to untreated LentiORF-control podocytes. n = 3; *P < 0.05; **P < 0.01, Kruskal-Wallis test with Dunn posttest. AU, arbitrary units.
Glomerular KLF6 Expression Is Reduced in Late-Stage DKD
To assess the role of KLF6 in human DKD, we initially interrogated the glomerular expression of KLF6 in previously reported expression arrays from isolated glomeruli in human kidney biopsies with DKD as compared with healthy donor nephrectomies (42,43). KLF6 expression was significantly reduced in microdissected glomeruli from kidney biopsies with DKD as compared with donor nephrectomies in two independent cohorts (Fig. 7A and B). To ascertain whether glomerular KLF6 protein expression is also reduced in DKD, we obtained kidney tissue from 12 control subjects (healthy donor nephrectomies) and kidney biopsies from 7 early-stage DKD and 17 late-stage DKD specimens. Clinical characteristics of the participants at the time of the kidney biopsy are shown in Supplementary Table 2. Immunostaining and quantification for glomerular KLF6 protein expression in kidney biopsies from controls, early-stage DKD, and late-stage DKD validated the findings from these independent expression arrays (Fig. 7C and D). Because previous studies have reported significant podocyte injury and loss with DKD progression (2,4,5), the reduction in KLF6 expression might, in part, be secondary to podocyte loss. To control for podocyte loss, we immunostained for WT1 with KLF6 and quantified the ratio of KLF6+WT1+ cells to WT1+ cells in the glomeruli. We observed a decrease in podocyte-specific KLF6 expression in kidney biopsies with early-stage DKD as compared with control specimens, which was significantly exacerbated in late-stage DKD specimens (Fig. 7E). Collectively, these findings suggest that podocyte and glomerular KLF6 expression is reduced with the progression of DKD.
Figure 7.

Reduced KLF6 expression in human kidney biopsies with fibrosis. A and B: Previously reported gene expression arrays from Woroniecka et al. (42) and Ju et al. (43) were used to examine KLF6 expression in RNA from microdissected glomerular compartments of kidney biopsies with DKD as compared with healthy living donor nephrectomies (**P < 0.01, unpaired t test). C: Immunostaining for KLF6 and Hoechst was performed in kidney biopsies with early-stage DKD (n = 7) and late-stage DKD (n = 17) and in control specimens (n = 12), and representative images are shown. Dashed white line was traced to show the glomerular region. Scale bars = 100 μm. D: ImageJ was used to quantify the intensity of KLF6 expression in all three groups. E: To determine podocyte-specific expression of KLF6, immunostaining for KLF6 and WT1 was performed, and each glomerulus was selected and quantified for KLF6 staining in the podocytes as a ratio of KLF6+WT1+ cells to WT1+ cells. **P < 0.01, Kruskal-Wallis test with Dunn posttest. AU, arbitrary units.
Discussion
Mitochondrial injury in the podocyte is a hallmark feature in the progression of DKD. Several molecules have been described to play an important role in mitochondrial DNA damage, biogenesis, dynamics, and function under diabetic conditions (2,7,9). Although KLFs have not previously been implicated in DKD, several laboratories have demonstrated a critical role of KLFs in various aspects of mitochondrial structure and function (16,20,44,45). In this study, we identify that KLF6 is a critical mediator of mitochondrial injury in the podocytes under diabetic conditions. This was demonstrated by the following: 1) podocyte-specific knockdown of Klf6 increased the susceptibility to DKD with mitochondrial injury and eventual podocyte loss in a DKD-resistant mouse strain (C57BL/6), and 2) shRNA-mediated KLF6 knockdown in human podocytes resulted in increased mitochondrial fragmentation and enhanced apoptosis under HG conditions. Interestingly, induction of KLF6 under HG conditions increased the expression of SCO2, a key factor in COX assembly, while reducing apoptosis under HG conditions. Finally, we observed that glomerular and podocyte-specific KLF6 expression was significantly reduced with progression of DKD in human kidney biopsy specimens.
As previously described, podocyte-specific loss of Klf6 results in modest glomerular injury on the C57BL/6 background (20). Although the podocyte-specific knockdown of Klf6 was sufficient to induce some histological changes (podocyte loss and GBM thickness), it was not sufficient to induce other histological changes (glomerular volume, mesangial expansion, and foot process effacement) as well as functional changes in the glomerulus (albuminuria and hyperfiltration) and mitochondrial injury until exposure to diabetic conditions. These findings suggest a threshold of podocyte injury in the Podocin-Cre Klf6flox/flox mice, which is exacerbated under diabetic conditions. In addition, we postulate that podocyte injury that results from knockdown of Klf6 might contribute to excess eNOS production, leading to glomerular hyperfiltration and increased glomerular volume due to podocyte–endothelial cross talk in the setting of diabetes. Previous studies have demonstrated that eNOS and endothelial injury play a critical role in glomerular hyperfiltration and glomerular volume by modulating hemodynamic changes (46–48). Furthermore, we postulate that Podocin-Cre Klf6flox/flox mice exposed to diabetic conditions for longer duration might further exacerbate GBM thickness and podocyte loss, resulting in further worsening of kidney function. Several factors might also contribute to the lack of severe glomerular injury in these mice at baseline, including the mosaicism of the Cre recombinase as well as the resistant nature of C57BL/6 background strain to extensive DKD (40). Additional studies using alternate podocyte-specific promoters driving Cre recombinase or backcrossing the mice to a susceptible strain are necessary to confirm these hypotheses. Alternatively, use of Crispr/Cas9 to knock out Klf6 in the podocytes will ultimately be required to determine the extent by which Klf6 is required to prevent podocyte injury. In addition, inducible knockout of Klf6 in adult mice needs to be performed to assess whether the changes observed in this study are not primarily due to Klf6 knockdown during development. Nonetheless, as reported in other proteinuric murine models (20), podocyte-specific loss of Klf6 increases the susceptibility to DKD in the STZ murine model. These data further support that podocyte-specific Klf6 is required to prevent mitochondrial injury in the setting of cell stress. Interestingly, other members of the KLF family have been recently reported to play a critical role in mitochondrial biogenesis in other tissue types (45). Furthermore, specificity proteins (paralogs of KLFs) have been described to regulate cytochrome c subunit genes in primary neurons (49). The focus of future studies will involve demonstrating the overlapping and antagonistic roles of these zinc fingers regulating mitochondrial biogenesis and dynamics in the podocyte.
Previous studies have demonstrated mitochondrial fragmentation with reduced COX activity in DKD (8). We previously reported that KLF6 transcriptionally regulates the expression of nuclear-encoded COX assembly gene SCO2 in the podocyte (20). In this study, we observe that podocyte-specific loss of KLF6 results in reduced SCO2 expression and COX activity under diabetic conditions. Conversely, overexpression of KLF6 in cultured human podocytes increased SCO2 expression regardless of hyperglycemic conditions. Interestingly, studies demonstrate a reduction in COX expression as well other RC activity in kidneys from diabetic mice (8). However, the mechanism by which RC activity is reduced in DKD remains unresolved. Although DNA damage may reduce the generation of intermediate subunits and lead to a decrease in RC activity, it remains unclear if the kinetics of RC assembly are perturbed under diabetic conditions. Although all RCs are required for oxidative phosphorylation, COX is the terminal enzyme of the respiratory chain and catalyzes the transfer of electrons from reduced cytochrome c to molecular oxygen. Furthermore, defective COX assembly has been directly implicated in mitochondrial dysfunction with altered oxidative phosphorylation in several fatal mitochondrial diseases (12–15). In addition, defective assembly will permit accumulation of precursors, which might increase oxidative stress, as previously reported in SCO2 deficiency (50). Therefore, additional studies are required to investigate the mechanism(s) by which knockdown of SCO2 results in defective COX assembly under diabetic conditions.
We recognize the pitfalls associated with the use of an STZ-induced diabetic murine model. For instance, administration of STZ has previously been reported to cause nephrotoxicity secondary to lipophilic derivatives of STZ (51,52), hence podocyte injury independent of STZ-induced diabetes cannot be excluded. Therefore, future studies will focus on validating our findings in other murine models of DKD. In addition, our efforts will also focus on whether restoring Klf6 expression specifically in the podocyte might improve mitochondrial injury and reverse podocyte loss in murine models of DKD.
In a small cohort of archived human kidney biopsies, we observe that podocyte-specific KLF6 expression is significantly reduced with progression of DKD. Interestingly, independent expression arrays from isolated glomerular fractions also confirm these findings (42,43). These findings require validation in a larger cohort with additional clinical parameters to stratify for potential confounders that might also contribute to podocyte injury and changes in KLF6 expression. Furthermore, the mechanism by which KLF6 is regulated under diabetic conditions remains unclear, especially because the significant reduction in KLF6 is more pronounced in later stages of DKD. Consequently, studies on posttranslational mechanisms of KLF6 regulation under diabetic conditions will be a focus of future investigations. In addition, immunostaining for KLF6 in human kidney biopsies demonstrates its expression in the glomerular as well as the tubulointerstitial compartments. Other laboratories have demonstrated that tubular KLF6 might be involved in tubulointerstitial disease in models of renal fibrosis (53,54), suggesting a cell context–dependent role of KLF6. Similarly, cell context–dependent roles of KLF6 have been demonstrated in the liver and the heart (55,56). Because the tubulointerstitial compartment also plays an important role in the progression of DKD, future studies will also need to explore the tubule-specific role of KLF6 under diabetic conditions.
In conclusion, these data suggest that podocyte-specific KLF6 is important to prevent mitochondrial injury, podocyte loss, and eventual glomerular injury in DKD. These data also validate our previous studies that the podocyte-specific loss of KLF6 increases the susceptibility to podocyte apoptosis under cell stress. To the best of our knowledge, this is the first study to demonstrate that changes in regulation of cytochrome assembly gene expression play an important role in the development of podocyte injury under diabetic conditions.
Supplementary Material
Article Information
Funding. This work was supported by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grants DK102519 and DK112984 (to S.K.M.) and U.S. Department of Veterans Affairs merit grant 1I01BX003698 (to S.K.M.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. S.J.H., J.M.V., Y.G., V.L., S.E.P., A.R.L., J.L., M.P.R., and S.K.M. participated in the conduct of the study, researched and analyzed data, and assisted in preparation of the manuscript. D.B., V.W.Y., J.C.H., and S.K.M. contributed to the design, researched data, and revised the manuscript critically for important intellectual content. S.J.H., J.M.V., and S.K.M. are the guarantors of this work and, as such, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db17-0958/-/DC1.
References
- 1.Levin A, Stevens PE, Bilous RW, et al. Kidney Disease: Improving Global Outcomes (KDIGO) CKD work group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl 2013;3:1–150 [Google Scholar]
- 2.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest 2014;124:2333–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006;55:225–233 [PubMed] [Google Scholar]
- 4.Pagtalunan ME, Miller PL, Jumping-Eagle S, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 1997;99:342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia 1999;42:1341–1344 [DOI] [PubMed] [Google Scholar]
- 6.Steffes MW, Schmidt D, McCrery R, Basgen JM; International Diabetic Nephropathy Study Group . Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int 2001;59:2104–2113 [DOI] [PubMed] [Google Scholar]
- 7.Wang W, Wang Y, Long J, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab 2012;15:186–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dugan LL, You YH, Ali SS, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 2013;123:4888–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long J, Badal SS, Ye Z, et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest 2016;126:4205–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma K, Karl B, Mathew AV, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol 2013;24:1901–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stieger N, Worthmann K, Teng B, et al. Impact of high glucose and transforming growth factor-β on bioenergetic profiles in podocytes. Metabolism 2012;61:1073–1086 [DOI] [PubMed] [Google Scholar]
- 12.Nijtmans LG, Taanman JW, Muijsers AO, Speijer D, Van den Bogert C. Assembly of cytochrome-c oxidase in cultured human cells. Eur J Biochem 1998;254:389–394 [DOI] [PubMed] [Google Scholar]
- 13.Shoubridge EA. Cytochrome c oxidase deficiency. Am J Med Genet 2001;106:46–52 [DOI] [PubMed] [Google Scholar]
- 14.Pecina P, Houstková H, Hansíková H, Zeman J, Houstek J. Genetic defects of cytochrome c oxidase assembly. Physiol Res 2004;53(Suppl. 1):S213–S223 [PubMed] [Google Scholar]
- 15.Leary SC, Cobine PA, Kaufman BA, et al. The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab 2007;5:9–20 [DOI] [PubMed] [Google Scholar]
- 16.Kopp JB. Loss of Krüppel-like factor 6 cripples podocyte mitochondrial function. J Clin Invest 2015;125:968–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mallipattu SK, Estrada CC, He JC. The critical role of Krüppel-like factors in kidney disease. Am J Physiol Renal Physiol 2017;312:F259–F265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bialkowska AB, Yang VW, Mallipattu SK. Krüppel-like factors in mammalian stem cells and development. Development 2017;144:737–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McConnell BB, Yang VW. Mammalian Krüppel-like factors in health and diseases. Physiol Rev 2010;90:1337–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mallipattu SK, Horne SJ, D’Agati V, et al. Krüppel-like factor 6 regulates mitochondrial function in the kidney. J Clin Invest 2015;125:1347–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leow CC, Wang BE, Ross J, et al. Prostate-specific Klf6 inactivation impairs anterior prostate branching morphogenesis through increased activation of the Shh pathway. J Biol Chem 2009;284:21057–21065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu KK, Huan Y. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc Pharmacol 2008;Chapter 5:Unit 5.47. [DOI] [PubMed] [Google Scholar]
- 23.Mallipattu SK, Liu R, Zhong Y, et al. Expression of HIV transgene aggravates kidney injury in diabetic mice. Kidney Int 2013;83:626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rieg T. A high-throughput method for measurement of glomerular filtration rate in conscious mice. J Vis Exp 2013;75:e50330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kume S, Uzu T, Araki S, et al. Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. J Am Soc Nephrol 2007;18:2715–2723 [DOI] [PubMed] [Google Scholar]
- 26.Mallipattu SK, Gallagher EJ, LeRoith D, et al. Diabetic nephropathy in a nonobese mouse model of type 2 diabetes mellitus. Am J Physiol Renal Physiol 2014;306:F1008–F1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He JC, Lu TC, Fleet M, et al. Retinoic acid inhibits HIV-1-induced podocyte proliferation through the cAMP pathway. J Am Soc Nephrol 2007;18:93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang H, Gao Q, Peng X, et al. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell 2011;20:376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanden SK, Wiggins JE, Goyal M, Riggs LK, Wiggins RC. Evaluation of a thick and thin section method for estimation of podocyte number, glomerular volume, and glomerular volume per podocyte in rat kidney with Wilms’ tumor-1 protein used as a podocyte nuclear marker. J Am Soc Nephrol 2003;14:2484–2493 [DOI] [PubMed] [Google Scholar]
- 30.Awazu M, Omori S, Ishikura K, Hida M, Fujita H. The lack of cyclin kinase inhibitor p27(Kip1) ameliorates progression of diabetic nephropathy. J Am Soc Nephrol 2003;14:699–708 [DOI] [PubMed] [Google Scholar]
- 31.Schiffer M, Mundel P, Shaw AS, Böttinger EP. A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem 2004;279:37004–37012 [DOI] [PubMed] [Google Scholar]
- 32.Mallipattu SK, Liu R, Zheng F, et al. Kruppel-like factor 15 (KLF15) is a key regulator of podocyte differentiation. J Biol Chem 2012;287:19122–19135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koop K, Eikmans M, Baelde HJ, et al. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol 2003;14:2063–2071 [DOI] [PubMed] [Google Scholar]
- 34.Reiniger N, Lau K, McCalla D, et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 2010;59:2043–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H, Brosel S, Acin-Perez R, et al. Analysis of mouse models of cytochrome c oxidase deficiency owing to mutations in Sco2. Hum Mol Genet 2010;19:170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross JM. Visualization of mitochondrial respiratory function using cytochrome c oxidase/succinate dehydrogenase (COX/SDH) double-labeling histochemistry. J Vis Exp 2011;57:e3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearse AD, Marks R. Measurement of section thickness in quantitative microscopy with special reference to enzyme histochemistry. J Clin Pathol 1974;27:615–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhdanov AV, Ward MW, Prehn JH, Papkovsky DB. Dynamics of intracellular oxygen in PC12 cells upon stimulation of neurotransmission. J Biol Chem 2008;283:5650–5661 [DOI] [PubMed] [Google Scholar]
- 39.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol 1996;264:484–509 [DOI] [PubMed] [Google Scholar]
- 40.Brosius FC III, Alpers CE, Bottinger EP, et al.; Animal Models of Diabetic Complications Consortium . Mouse models of diabetic nephropathy. J Am Soc Nephrol 2009;20:2503–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perry J, Ho M, Viero S, Zheng K, Jacobs R, Thorner PS. The intermediate filament nestin is highly expressed in normal human podocytes and podocytes in glomerular disease. Pediatr Dev Pathol 2007;10:369–382 [DOI] [PubMed] [Google Scholar]
- 42.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes 2011;60:2354–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ju W, Greene CS, Eichinger F, et al. Defining cell-type specificity at the transcriptional level in human disease. Genome Res 2013;23:1862–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamoto J, Ikeda Y, Iguchi H, et al. A Kruppel-like factor KLF15 contributes fasting-induced transcriptional activation of mitochondrial acetyl-CoA synthetase gene AceCS2. J Biol Chem 2004;279:16954–16962 [DOI] [PubMed] [Google Scholar]
- 45.Liao X, Zhang R, Lu Y, et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest 2015;125:3461–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Veelken R, Hilgers KF, Hartner A, Haas A, Böhmer KP, Sterzel RB. Nitric oxide synthase isoforms and glomerular hyperfiltration in early diabetic nephropathy. J Am Soc Nephrol 2000;11:71–79 [DOI] [PubMed] [Google Scholar]
- 47.Sigmon DH, Gonzalez-Feldman E, Cavasin MA, Potter DL, Beierwaltes WH. Role of nitric oxide in the renal hemodynamic response to unilateral nephrectomy. J Am Soc Nephrol 2004;15:1413–1420 [DOI] [PubMed] [Google Scholar]
- 48.Helal I, Fick-Brosnahan GM, Reed-Gitomer B, Schrier RW. Glomerular hyperfiltration: definitions, mechanisms and clinical implications. Nat Rev Nephrol 2012;8:293–300 [DOI] [PubMed] [Google Scholar]
- 49.Johar K, Priya A, Dhar S, Liu Q, Wong-Riley MT. Neuron-specific specificity protein 4 bigenomically regulates the transcription of all mitochondria- and nucleus-encoded cytochrome c oxidase subunit genes in neurons. J Neurochem 2013;127:496–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sung HJ, Ma W, Wang PY, et al. Mitochondrial respiration protects against oxygen-associated DNA damage. Nat Commun 2010;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kraynak AR, Storer RD, Jensen RD, et al. Extent and persistence of streptozotocin-induced DNA damage and cell proliferation in rat kidney as determined by in vivo alkaline elution and BrdUrd labeling assays. Toxicol Appl Pharmacol 1995;135:279–286 [DOI] [PubMed] [Google Scholar]
- 52.Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2008;51:216–226 [DOI] [PubMed] [Google Scholar]
- 53.Holian J, Qi W, Kelly DJ, et al. Role of Kruppel-like factor 6 in transforming growth factor-beta1-induced epithelial-mesenchymal transition of proximal tubule cells. Am J Physiol Renal Physiol 2008;295:F1388–F1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi W, Chen X, Holian J, Tan CY, Kelly DJ, Pollock CA. Transcription factors Krüppel-like factor 6 and peroxisome proliferator-activated receptor-gamma mediate high glucose-induced thioredoxin-interacting protein. Am J Pathol 2009;175:1858–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banck MS, Beaven SW, Narla G, Walsh MJ, Friedman SL, Beutler AS. KLF6 degradation after apoptotic DNA damage. FEBS Lett 2006;580:6981–6986 [DOI] [PubMed] [Google Scholar]
- 56.Bechmann LP, Vetter D, Ishida J, et al. Post-transcriptional activation of PPAR alpha by KLF6 in hepatic steatosis. J Hepatol 2013;58:1000–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.