

Short abstract
Background
Fraser syndrome is an autosomal recessive disorder characterized primarily by syndactyly, cryptophthalmos, urinary tract anomalies, ambiguous genitalia, and laryngeal anomalies. A 28-year-old man with Fraser syndrome presented with cryptophthalmos, microphthalmia, lacrimal system dysgenesis, and chronic sinusitis.
Objective
The patients’ clinical condition and surgical treatment are described. A literature review was conducted, and articles relevant to the case are presented.
Methods
Case report.
Results
To our knowledge, this is the first published case report of endonasal management of dacryocystoceles in a Fraser syndrome patient. The patient was treated via endoscopic endonasal marsupialization and drainage.
Conclusion
Fraser syndrome patients may initially present to many different specialties as the spectrum of clinical manifestations is broad. Physicians treating these patients should take a collaborative approach to surgical and medical management.
Keywords: Fraser syndrome, dacryocystocele, endoscopic sinus surgery, sinusitis, cryptophthalmos
Introduction
Fraser syndrome is an autosomal recessive disorder characterized primarily by syndactyly, cryptophthalmos, urinary tract anomalies, ambiguous genitalia, and laryngeal anomalies. Here, we present a case report of a 28-year-old man with Fraser syndrome presenting with cryptophthalmos, microphthalmia, lacrimal system dysgenesis, and chronic sinusitis which was treated via endoscopic endonasal marsupialization and drainage of the dacryocystoceles. A literature review was conducted, and articles relevant to the case are presented.
Case Presentation
A 28-year-old man with Fraser syndrome was referred to the Thomas Jefferson University Hospital otolaryngology clinic and the Wills Eye Hospital oculoplastic and orbital surgery clinic for the evaluation of bilateral medial canthal swelling. His medical history on presentation was remarkable for cryptophthalmos, lacrimal system dysgenesis, chronic sinusitis, and asthma. There was no significant family history. He had no lacrimal complaints such as epiphora or punctal discharge. Warm compresses, massage, and a course of oral steroids had been attempted to treat the bilateral medial canthal swelling with no improvement. The patient denied a history of chronic sinusitis but did report frequently noticing a foul smell in his right nostril. He denied nasal obstruction, rhinorrhea, post nasal drip, or anosmia. At the time of presentation, he was taking Flonase and Flovent daily. His Sino-Nasal Outcome Test 22 score at initial visit was 47.
On examination, he was well appearing and had no mental status or cranial nerve dysfunction. Ocular examination revealed bilateral complete cryptophthalmos. The patient had poorly developed ocular adnexa with fusion of skin from eyebrow to his cheek. There was no separation of the eyelids present from birth. He had a history of an attempt at separation of his right eyelids with socket reconstruction with an intent to wear an ocular prosthesis which was unsuccessful. There were no puncta or evidence of proximal lacrimal systems on either side. Large, firm, cystic-feeling masses were palpable in the medial canthus bilaterally. The patient had brisk light perception vision in his right eye through his fused adnexal skin and was no light perception in his left eye. B-scan ultrasounds of his orbits revealed microphthalmia of his right eye and phthisis on the left. The patient had bilateral cryptotia, and his ear canals were narrow but clear. He also had evidence of fusion of the anterior third of his true vocal folds and a breathy voice. His craniofacial abnormalities are shown in Figure 1. Written consent was given by the patient to publish identifiable images.
Figure 1.
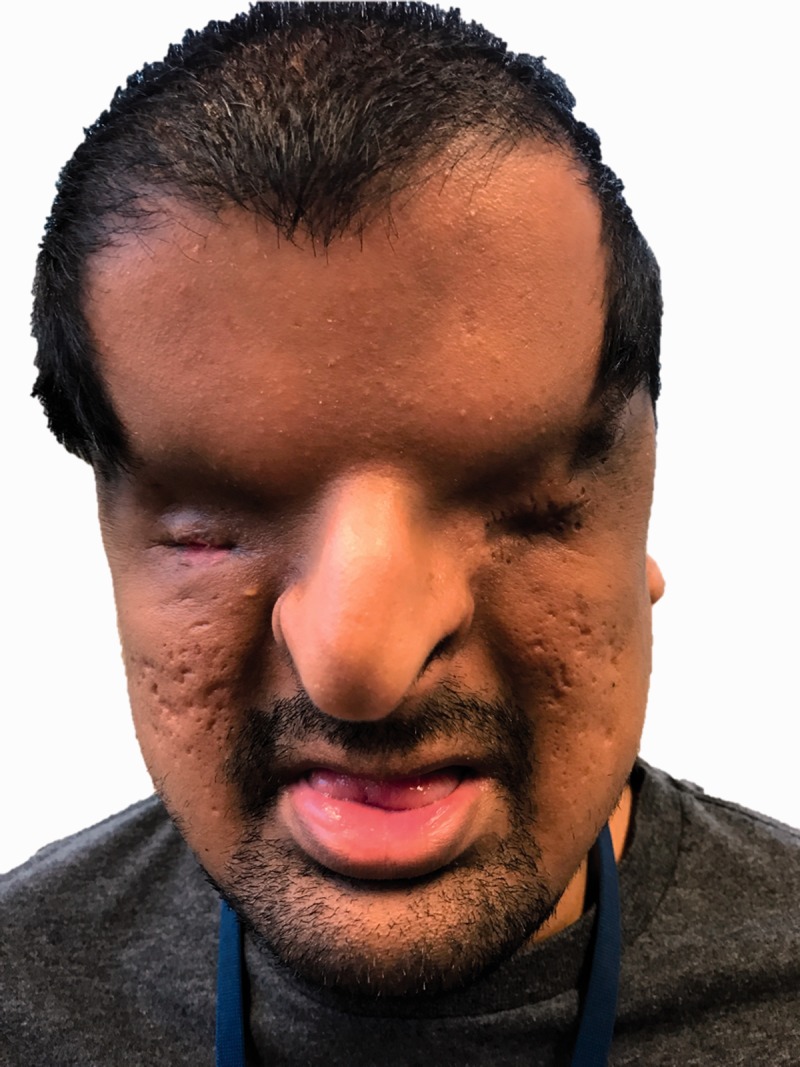
External photo showing cryptophthalmos and a beaked nasal bridge. There was a previous attempt at the creation of eyelids with residual scar.
Flexible nasal endoscopy revealed mucopus streaming from the right osteomeatal complex and a left septal deviation with crowding at the middle meatus. Laryngoscopy revealed fusion of the anterior third of the true vocal folds with a 3-mm posterior glottis gap and a breathy voice.
Computed tomography of the sinuses (Figure 2) and magnetic resonance imaging of the orbits (Figure 3) demonstrated right greater than left bilateral dacryocystoceles containing proteinaceous material extending into the lacrimal ducts. There was also complete opacification of the right maxillary sinus with expansion at the middle meatus.
Figure 2.

Pretreatment-computed tomography of the sinuses demonstrates bilateral dacryocystoceles greater on the right than left.
Figure 3.

Magnetic resonance imaging demonstrates bilateral dacryocystoceles, larger and with a more hyperintense T2 signal on the right. There is a small, shrunken right globe with enophthalmos, and the left globe is deformed with an absent lens.
Based on these findings, he was then taken to the operating room for a bilateral endonasal decompression of dacryocystoceles and a right maxillary antrostomy. An endoscopic approach was favored due to the patient’s monocular status. Due to the agenesis of the patient’s proximal lacrimal drainage system, further interventions prior to lacrimal sac marsupialization were impossible. A conjunctivodacryocystorhinostomy (CDCR) was not indicated, as it would not have added to the described surgery and would have put his barely seeing eye in undue risk as well as implanted a foreign body that carries further risks and complications. These were discussed, and the reported surgery decided upon.
The lacrimal bone and mucosa was taken down endonasally using a Stryker Sonopet™. The congenitally enlarged lacrimal sacs were then punctured using an 11-blade knife and marsupialized to ensure appropriate drainage. On the right, mucoid material was expressed, and on the left, serous fluid was encountered. At the time of surgery, polypoid inflammation was noted in the right maxillary sinus. Postoperatively, he had resolution of the foul odor and otherwise felt normal. At 2- and 6-month follow-up, he was noted to be doing well with no evidence of recurrence. His lacrimal sacs remained well marsupialized into the nasal cavities. Endonasal imaging from 6-month follow-up is shown in Figure 4.
Figure 4.

Endoscopic view of the right lacrimal fossa 6 months post operatively. The left side of the picture demonstrates a well marsupialized lacrimal fossa into the nasal cavity.
Discussion
Fraser syndrome was first described in 2 pairs of siblings by George Fraser in 1962.1 It is a rare disorder: a large epidemiological study in a European population showed a frequency of 0.2 cases of Fraser syndrome per 100 000 births.2 A 15% to 25% incidence of consanguinity in families with Fraser syndrome has been reported.3,4
Fraser syndrome has been localized to chromosome 4q21 and linked to mutations in the FRAS1, FREM2, and GRIP1 genes that disrupt epithelial–mesenchymal interactions.5,6 These mutations are related to failure of programmed cell necrosis or to defects in epidermal adhesion, which results in the formation of large blisters during embryonic development.7,8
These abnormalities in development are reflected in a wide spectrum of clinical manifestations. The most recent diagnostic criteria for Fraser syndrome were outlined in 2007 by van Haelst et al., as an update to the original criteria published in 1986.4,9 A diagnosis of Fraser syndrome should fulfill 3 major criteria, 2 major criteria and 2 minor, or 1 major criteria and 3 minor (Table 1).9
Table 1.
Diagnostic Criteria for Fraser Syndrome.
| Major criteria | Minor criteria |
|---|---|
| Syndactyly | Anorectal defects |
| Cryptophthalmos spectrum | Dysplastic ears |
| Ambiguous genitalia | Skull ossification defects |
| Urinary tract abnormalities | Umbilical abnormalities |
| Laryngeal and tracheal anomalies | Nasal anomalies |
| Positive family history |
Fraser syndrome encompasses a wide variety of craniofacial abnormalities, which fall into both major and minor diagnostic criteria. In a review of 68 cases, Gattuso et al. found that craniofacial abnormalities were present in every case.10 Cryptophthalmos spectrum is present in 83% to 93% of cases and is considered to be one of the most important diagnostic criteria for Fraser syndrome.3,4,10,11 Other ocular findings include eyelid coloboma, microphthalmos, absence of eyelashes/eyebrows, anterior segment anomalies, and small orbits. Lacrimal duct defects, as seen in this case, are a less common manifestation but have been estimated to be present in 9% of patients.10 Ear anomalies seen in Fraser syndrome include atresia of external auditory canal, low set ears, microtia, or absent pinnae.4,12 Nose anomalies include a beak-like nose, as seen in this case, as well as choanal atresia, small nares, and coloboma of the alae nasa.4,12 Skeletal defects are common and frequently include the orbit and skull. Another common craniofacial abnormality, in about one-third of patients, is extended hair growth on the forehead from the lateral forehead to the lateral eyebrow.10
Although uncommon, lacrimal system abnormalities similar to those seen in our case have been described in the literature. Ali et al.13 report finding 5 cases of Fraser syndrome associated with lacrimal system anomalies. They present the case of a Fraser syndrome patient with bilateral complex congenital nasolacrimal duct obstruction, for which endoscopic CDCR was recommended.13 Although not in the context of Fraser syndrome, successful endoscopic marsupialization of a dilated lacrimal sac in the setting of proximal lacrimal agenesis has been described in 1 patient.14 The success of these cases lends support to the surgical management used in this case and for future patients.
Because of their rarity and lack of uniformity in signs and symptoms, patients with Fraser syndrome should be managed on a case-by-case basis. Life expectancy of these patients is variable. Major causes of early mortality include laryngeal stenosis or atresia and bilateral renal agenesis or obstructive uropathy.3,11 Patients may require a tracheostomy at birth, and the main cause of perinatal mortality is airway involvement.15 Patients who survive past age 10, as in this case report, are less likely to have major phenotypic abnormalities.3 Patients who survive past infancy should have surgical corrections of abnormalities, such as syndactyly, when possible.12
Conclusion
To our knowledge, this is the first published case report of endonasal management of dacryocystoceles in a Fraser syndrome patient. Fraser syndrome patients may initially present to many different specialties as the spectrum of clinical manifestations is broad. Physicians treating these patients should take a collaborative approach to surgical and medical management.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Not required, as this was a case report and thus was not put through an Institutional Review Board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
Informed written consent was obtained.
References
- 1.Fraser GR. Our genetical ‘load’. A review of some aspects of genetical variation. Ann Hum Genet. 1962; 25(4):387–415. [Google Scholar]
- 2.Barisic I, Odak L, Loane M, et al. Fraser syndrome: epidemiological study in a European population. Am J Med Genet A. 2013; 161A(5):1012–1018. [DOI] [PubMed] [Google Scholar]
- 3.Slavotinek AM, Tifft CJ. Fraser syndrome and cryptophthalmos: review of the diagnostic criteria and evidence for phenotypic modules in complex malformation syndromes. J Med Genet. 2002; 39(9):623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986; 25(1):85–98. [DOI] [PubMed] [Google Scholar]
- 5.McGregor L, Makela V, Darling SM, et al. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet. 2003; 34(2):203–208. [DOI] [PubMed] [Google Scholar]
- 6.Vogel MJ, van Zon P, Brueton L, et al. Mutations in GRIP1 cause Fraser syndrome. J Med Genet. 2012; 49(5):303–306. [DOI] [PubMed] [Google Scholar]
- 7.Short K, Wiradjaja F, Smyth I. Let’s stick together: the role of the Fras1 and Frem proteins in epidermal adhesion. IUBMB Life. 2007; 59(7):427–435. [DOI] [PubMed] [Google Scholar]
- 8.Pavlakis E, Chiotaki R, Chalepakis G. The role of Fras1/Frem proteins in the structure and function of basement membrane. Int J Biochem Cell Biol. 2011; 43(4):487–495. [DOI] [PubMed] [Google Scholar]
- 9.van Haelst MM, Scambler PJ, Fraser Syndrome Collaboration Group, Hennekam RC. Fraser syndrome: a clinical study of 59 cases and evaluation of diagnostic criteria. Am J Med Genet A. 2007; 143A(24):3194–3203. [DOI] [PubMed] [Google Scholar]
- 10.Gattuso J, Patton MA, Baraitser M. The clinical spectrum of the Fraser syndrome: report of three new cases and review. J Med Genet. 1987; 24(9):549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyd PA, Keeling JW, Lindenbaum RH. Fraser syndrome (cryptophthalmos-syndactyly syndrome): a review of eleven cases with postmortem findings. Am J Med Genet. 1988; 31(1):159–168. [DOI] [PubMed] [Google Scholar]
- 12.Mina MM, Greenberg C, Levin B. ENT abnormalities associated with Fraser syndrome: case report and literature review. J Otolaryngol. 1988; 17(5):233–236. [PubMed] [Google Scholar]
- 13.Ali MJ, Gupta S, Patel A, Naik M. Lacrimal drainage anomalies in Fraser syndrome. Ophthal Plast Reconstr Surg. 2018; 34(1):92–93. [DOI] [PubMed] [Google Scholar]
- 14.Ali MJ, Singh S, Naik M. Endoscopic features of a lacrimal sac in a case of punctal and canalicular agenesis. Ophthal Plast Reconstr Surg. 2017; 33(2):153–154. [DOI] [PubMed] [Google Scholar]
- 15.Alvaréz-Neri H, Morán VF, De La Torre C, Villamor P, Penchyna Grub J. Airway features in Fraser syndrome: case report and literature review. Int J Pediatr Otorhinolaryngol Extra. 2017; 18(Supplement C):16–18. [Google Scholar]