

Abstract
Background
Liposarcoma is the most common type of soft tissue sarcoma, but its molecular mechanism is poorly defined. This study aimed to identify genes crucial to the pathogenesis of liposarcoma and to explore their functions, related pathways, and prognostic value.
Material/Methods
Differentially expressed genes (DEGs) in the GSE59568 dataset were screened. Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were conducted to investigate the DEGs at the functional level. Protein-protein interaction (PPI) networks and module analysis were applied to identify hub genes from among the DEGs. The GSE30929 dataset was used to validate the relationship between hub genes and the distant recurrence-free survival (DRFS) of liposarcoma patients using Cox model analysis.
Results
A total of 1111 DEGs were identified. GO and KEGG pathway analysis indicated that the DEGs were mainly associated with lipopolysaccharides and pathways in cancer. The PPI network and module analysis identified 10 hub genes from the DEG network. The Cox model identified 3 genes (NIP7, RPL10L, and MCM2) significantly associated with DRFS. The risk score calculated by the Cox model of the NIP7-RPL10L-MCM2 signature could largely predict the 1-, 3-, and 5-year DRFS of liposarcoma patients, and the prognostic value was even higher for subtypes of liposarcoma.
Conclusions
This study identified genes that might play critical roles in liposarcoma pathogenesis as well as a 3-gene-based signature that could be used as a candidate prognostic biomarker for patients with liposarcoma.
MeSH Keywords: Computational Biology; Genes, vif; Liposarcoma; Microarray Analysis
Background
Liposarcoma is the most common soft tissue sarcoma, accounting for approximately 20% of all sarcomas in adults [1]. The most recent World Health Organization classification of soft tissue tumors recognizes 5 categories of liposarcoma: 1) well-differentiated, which includes the adipocytic, sclerosing, and inflammatory subtypes; 2) de-differentiated; 3) myxoid; 4) round cell; and 5) pleomorphic [2]. Surgery remains the primary treatment for localized liposarcoma, while conventional radiotherapy and cytotoxic chemotherapy are often used to treat metastatic liposarcoma. However, the most common types, well-differentiated liposarcoma and de-differentiated liposarcoma, show obvious resistance to conventional radiotherapy and cytotoxic chemotherapy [3].
Liposarcoma subtype is an important determinant of local recurrence and metastatic potential [4], but precise prediction of patient outcomes currently remains difficult for individual patients. Previous studies have conducted microarray analysis to explore genes as potential biomarkers for diagnosis, prognosis, or monitoring of curative effects in liposarcoma, and several genes or gene signatures have been identified [5–7]. However, these studies have several limitations. For example, some of these studies only examined the function of several potential genes, but did not construct gene networks to find hub genes or estimate the prognostic value of those genes. Other studies merely examined the function of individual genes, but did not construct gene signatures to search for more valuable prognostic indicators. Moreover, the results of some studies have not been validated by other studies.
Previously, Iura et al. [8] and Gobble et al. [9] investigated the genes that contribute to liposarcomagenesis using microarray analysis methods; however, neither studies performed a gene-gene interaction analysis to identify key genes, nor did they establish a gene signature to predict the prognosis of liposarcoma. Therefore, the exact genes underlying liposarcoma tumorigenesis remain to be elucidated. In this study, we aimed to identify the genes or gene signature associated with the prognosis of liposarcoma patients by re-analyzing the microarray data from Iura et al. [8] and Gobble et al. [9] studies using bioinformatic analysis methods. We first explored the gene profiles related to liposarcoma pathogenesis and the associated functions and related pathways; then we identified the hub genes of the gene profiles; and finally, we established a gene signature as a new candidate indicator for predicting survival in patients with liposarcoma.
Material and Methods
Microarray data
The gene expression profiles were downloaded as microarray data from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database. The liposarcoma-associated dataset GSE59568 [8] based on GPL13915 3D-Gene Human Oligo chip 25k V2.1 was downloaded from the GEO database, representing a total of 9 human liposarcoma specimens, including 6 myxoid liposarcoma samples and 3 normal adipose tissue samples. To validate gene markers for use as specific signatures for patients with liposarcoma, liposarcoma data (GSE30929, 140 patients) [9] was also downloaded from the database. Approval from an ethics committee was not necessary because the data were freely provided by the GEO database.
Identification of differentially expressed genes (DEGs)
The R statistical software (version 3.4.2) and Bioconductor packages were employed to identify differentially expressed genes (DEGs) among liposarcoma samples and normal samples. The probe data were converted into gene expression data before the pro-process analysis. For the case of a gene corresponding to multiple probe data, an average data was calculated and used as the gene expression value [10]. Also, genes with over 20% [11] missing values were eliminated; otherwise, the data were supplemented with mean values, and the box diagram of the expression value in each sample was drawn before and after pre-processing, then the t-tests were applied to analyze the DEGs between the liposarcoma group and normal using the limma package. A DEG was identified when it met the criteria of false discovery rate (FDR) < 0.05 and |log fold change C| (logFC) ≥2.
Gene function annotation and pathway enrichment analysis
The gene annotation analysis of DEGs used the DAVID online tool which is freely available (https://david.ncifcrf.gov/). We analyzed 3 Gene Ontology (GO) categories, including Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). A GO category was considered significant enrichment when the P value was less than 0.05. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was also conducted to evaluate the modules at the functional level, and this analysis used the online tool of DAVID. A significant pathway was defined when the P value was less than 0.05.
Protein-protein interaction (PPI) network and module analysis
The protein-protein interaction (PPI) network is often used to identity hub genes which are involved in disease pathogenesis at the protein interaction level. In this study, the Search Tool for the Retrieval of Interacting Genes (STRING) online tool (http://www.string-db.org/) was used to analyze the PPI of DEGs. Cytoscape software (version 3.5) was then used for construction of a PPI network using the data from STRING. Module analysis and GO analysis were then carried out by 2 Cytoscape plug-ins, namely, Molecular Complex Detection (MCODE) and Biological Network Gene Ontology tool (BiNGO), respectively, to illuminate the biological significance of gene modules in liposarcoma.
Construction of distant recurrence-free survival (DRFS) model and receiver operating characteristic curve (ROC) curve
The hub genes from the PPI network were selected as candidate markers and applied to the construction of a distant recurrence-free survival (DRFS) model. We used the linear genes prognostic model to calculate a gene signature prognostic score for 140 patients with liposarcoma based on the survival data from GSE30929. The univariate Cox proportional regression model was applied to identify the hub genes that related to the DRFS, and the significance level was defined if the P value was less than 0.05.
A prognostic risk score was developed to predict the DRFS of liposarcoma. This prognostic risk score was based on a linear combination of the gene expression level weighted by the regression coefficient (β) derived from the univariate Cox regression. The formula of calculation of risk score was as followed: risk score=expression of Gene1×β1Gene1+expression of Gene2×β2Gene2+expression of Genen×βnGenen [12]. We chose the median values of the risk scores to divide the patients into high risk and low risk groups respectively, then the receiver operating characteristic curve (ROC) to predict the 1-, 3-, and 5-year survival of the patients based on the risk score of the gene signature. We also used the Kaplan-Meier curves to estimate the association of high or low risk scores with the survival of patients. A 2-sided P less than 0.05 was regarded as a significantly difference.
Construction of the nomogram
The nomograms for the prediction of the probability of gene signature on liposarcoma were established with the selected independently significant variables, including the significant clinical characteristics, genes and risk scores. The nomograms were visualized by rms package (version 5.1–2) and its auxiliary packages.
Results
Identification of DEGs
Based on the DEG selection criteria (|logFC| ≥2, with FDR <0.05), a total of 1111 DEGs between human liposarcoma tissues and normal adipose tissues were identified after pre-processing the raw data from GSE59568 dataset, and a subset comprising 604 significantly downregulated DEGs and 507 significantly upregulated DEGs was selected for subsequent analysis. The distribution of the upregulated and downregulated DEGs is displayed in Figure 1.
Figure 1.
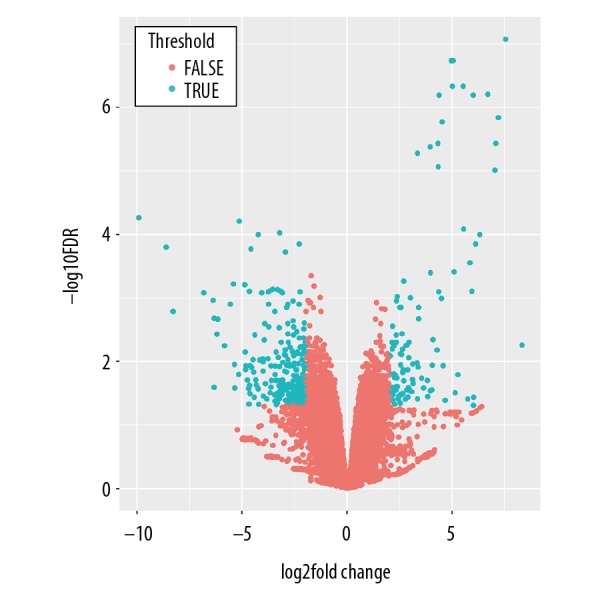
Volcano plot shows the distribution of upregulated and downregulated DEGs. The X-axis indicates the fold change and Y-axis indicating the -log10 FDR value. DEGs upregulated with a fold change >2 and FDR <0.05 are depicted in red, and those downregulated with a fold change >2 and FDR <0.05 are shown in turquoise. DEGs – differentially expressed genes; FDR – false discovery rate.
GO and KEGG pathway enrichment analysis
The GO functions of the DEGs showed that the most enriched GO term relevant to BP was response to lipopolysaccharide (GO: 0032496, P=3.37E-25), to CC proteinaceous extracellular matrix (GO: 0005578, P=1.53E-05), and to MF protease binding (GO: 0002020, P=0.001). The KEGG pathway analysis revealed that Cancer Pathways (hsa05200, P=0.002) were the most significant pathways of the DEGs. The results are presented in Figure 2.
Figure 2.

(A) GO enrichment analysis of upregulated DEGs in biological processes; (B) KEGG analysis of upregulated DEGs. DEGs – differentially expressed genes; GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes.
Construction of PPI network and subnetwork analysis
Based on the results of STRING online tool analysis of the DEGs, a PPI network was constructed by the Cytoscape software, which comprised 312 nodes and 687 edges. Two plug-ins for the Cytoscape software, MCODE and BiNGO, were used to carry out subnetwork analysis. Ten DEGs with high degrees of connectivity were selected as the hub genes in liposarcoma from the PPI network: PRPL10L, RPS3A, RPS23, RPS3, RPL36, MCM2, WRD12, NIP7, MRPL3, RPL23A, and MK1671P (Figure 3A). The top 3 subnetworks are shown in Figure 3B–3D. The GO enrichment analysis of the top 3 subnetworks is shown in Table 1.
Figure 3.

Module analysis of the PPI network for DEGs using data based on the STRING dataset. (A) The PPI network for the total DEGs, and hub genes located at the edge of the PPI network. (B–D) Functional submodules of the PPI network analyzed by Cytoscape. DEGs – differentially expressed genes; PPI – protein-protein interaction.
Table 1.
Enrichment analysis results of the three modules (GO).
| Term | Description | Count | P value | Genes |
|---|---|---|---|---|
| Module 1 | ||||
| GO: 0003735 | Structural constituent of ribosome | 14 | 2.47E-25 | MRPL3, RPL36, RPL23A, RPL22L1, RPL39, RPL28, RPS3, RPL10L, RPL32, RPS3A, RPL34, RPL26L1, RPS10, RPS23 |
| GO: 0006412 | Translation | 14 | 1.51E-24 | MRPL3, RPL36, RPL23A, RPL22L1, RPL39, RPL28, RPS3, RPL10L, RPL32, RPS3A, RPL34, RPL26L1, RPS10, RPS23 |
| GO: 0006614 | SRP-dependent cotranslational protein targeting to membrane | 11 | 5.20E-21 | RPL32, RPS3A, RPL34, RPL26L1, RPL36, RPS10, RPL23A, RPL39, RPL28, RPS23, RPS3 |
| GO: 0019083 | Viral transcription | 11 | 3.24E-20 | RPL32, RPS3A, RPL34, RPL26L1, RPL36, RPS10, RPL23A, RPL39, RPL28, RPS23, RPS3 |
| GO: 0000184 | Nuclear-transcribed mRNA catabolic process, nonsense-mediated decay | 11 | 6.09E-20 | RPL32, RPS3A, RPL34, RPL26L1, RPL36, RPS10, RPL23A, RPL39, RPL28, RPS23, RPS3 |
| Module 2 | ||||
| GO: 0006364 | rRNA processing | 5 | 8.71E-07 | WDR75, DDX49, BYSL, WDR12, RRP9 |
| GO: 0005730 | Nucleolus | 6 | 4.41E-06 | WDR75, NIP7, BYSL, WDR12, ZNRD1, RRP9 |
| GO: 0044822 | Poly(A) RNA binding | 6 | 2.49E-05 | WDR75, DDX49, NIP7, BYSL, BCCIP, RRP9 |
| GO: 0005654 | Nucleoplasm | 6 | 0.001328 | WDR75, DDX49, BYSL, WDR12, ZNRD1, RRP9 |
| GO: 0042273 | Ribosomal large subunit biogenesis | 2 | 0.010377 | NIP7, WDR12 |
| Module 3 | ||||
| GO: 0045211 | Postsynaptic membrane | 3 | 7.88E-04 | EPHA4, EPHA7, CLSTN2 |
| GO: 0072178 | Nephric duct morphogenesis | 2 | 0.001191 | EPHA4, EPHA7 |
| GO: 0005004 | GPI-linked ephrin receptor activity | 2 | 0.001658 | EPHA4, EPHA7 |
| GO: 0046875 | Ephrin receptor binding | 2 | 0.006147 | EPHA4, EPHA7 |
| GO: 0031594 | Neuromuscular junction | 2 | 0.012018 | EPHA4, EPHA7 |
DRFS model and ROC curve analysis
Because MKI67IP was not significantly expressed in the GSE30929 dataset, we did not select it for subsequent analysis. The Cox regression model revealed that only NIP7, RPL10L, and MCM2 exhibited significant correlation with DRFS in liposarcoma in the GSE30929 dataset, and the regression coefficients were −0.676, −0.703, and 0.868, respectively (Table 2). Thus, we chose NIP7, RPL10L, and MCM2 to construct the DRFS model. We divided the genes into high and low expression groups according to their median expression. As shown in Figure 4, low expression of MCM2 was associated with a better DRFS in liposarcoma compared with higher expression, while higher expression of NIP7 and RPL10L in liposarcoma correlated with poor DRFS results.
Table 2.
Correlation between DRFS and hub gene expression in liposarcoma of GSE30929 dataset.
| Gene | Node degree | Crude HR (95% CI)* | Crude P | Coefficient β** |
|---|---|---|---|---|
| NIP7 | 24 | 0.509 (0.281–0.920) | 0.025 | −0.676 |
| RPS3 | 24 | 1.296 (0.734–2.290) | 0.371 | 0.260 |
| MRPL3 | 22 | 1.230 (0.696–2.173) | 0.477 | 0.207 |
| RPL10L | 22 | 0.495 (0.272–0.900) | 0.021 | −0.703 |
| RPL23A | 22 | 0.708 (0.401–1.251) | 0.235 | −0.345 |
| RPS23 | 19 | 0.566 (0.314–1.019) | 0.058 | −0.569 |
| RPS3A | 19 | 0.823 (0.465–1.455) | 0.502 | −0.195 |
| MCM2 | 18 | 2.383 (1.292–4.393) | 0.005 | 0.868 |
| RPL36 | 18 | 0.752 (0.423–1.336) | 0.331 | −0.285 |
| WDR12 | 18 | 1.658 (0.929–2.960) | 0.087 | 0.506 |
The GSE30929 do not have the MKI67IP expression data;
low gene expression was the reference group;
derived from the univariate Cox proportional hazards regression analysis in PDAC patients.
DRFS – distant recurrence-free survival.
Figure 4.

Kaplan-Meier survival curves for liposarcoma patients with high and low expression of mRNA with regard to distant recurrence-free survival. (A) MCM2; (B) NIP7; (C) RPL10L.
The risk score for each patient was calculated based on the regression coefficient of the 3 genes. By applying the median as the cutoff point, 140 patients with liposarcoma were classified into the high-risk group (n=70) and the low-risk group (n=70). The heatmap shows that the protective genes had high expression in the low-risk group, while the risky genes exhibit high expression in high-risk group (Figure 5A). The patients in the high-risk group exhibited significantly worse DRFS than those in the low-risk group (Figure 5B). The risk score could largely predict the 1-, 3-, and 5-year DRFS of patients with liposarcoma, as the value of the area under the ROC curve (AUC) was 0.745, 729, and 0.677, respectively (Figure 5C).
Figure 5.

The prognostic performance of the 3-mRNA signature of liposarcoma. (A) Patient survival status and time distributed by risk score (upper); risk score curve of the 3-mRNA signature (middle); heatmap of 3-mRNA signature from liposarcoma patients (low). (B) The prognostic performance of the risk score shown by the time-dependent receiver operating characteristic (ROC) curve for predicting the 1-, 3-, and 5-year DRFS. (C) The Kaplan-Meier test of the risk score for the overall survival. DRFS – distant recurrence-free survival.
A nomogram was visualized by rms and its auxiliary packages based on the subtypes of liposarcoma, MCM2, NIP7, RPL10L, and risk scores, and demonstrated that the risk scores contributed the most risk points, whereas the subtypes of liposarcoma and 3 genes contributed much less (Figure 6).
Figure 6.

A nomogram predicting 1-, 3- and 5-year DRFS, and comparing 3 gene-signature with risk score. DRFS, distant recurrence-free survival.
Prognostic value analysis for subtypes of liposarcoma
The GSE30929 dataset included 5 subtypes of liposarcoma. Hence, we performed subtype analysis on patients with liposarcoma. We found that significantly different survival rates only occurred in the round cell subtype, with a Log-Rank P-value of 0.031; subtype AUC analysis revealed the 1- and 3-year AUCs of the myxoid subtype and the 3-year AUC of myxoid/round cell to be over 0.8, indicating a high prognostic value (see Table 3).
Table 3.
Prognostic value analysis of gene-signature for the subtype of liposarcoma.
| Log-rank P value | 1-year AUC* | 3-year AUC | 5-year AUC | |
|---|---|---|---|---|
| Well-differentiated | 0.227 | 0.692 | 0.399 | 0.567 |
| Dedifferentiated | 0.075 | 0.655 | 0.622 | 0.645 |
| Round cell | 0.031 | 0.665 | 0.813 | 0.813 |
| Myxoid | 0.487 | 0.920 | 0.920 | 0.461 |
| Pleomorphic | 0.449 | 0.647 | 0.745 | 0.433 |
AUC – area under the curve.
Comparison of the results between microarray data studies and our study
Compared with the previous studies that analyzed the gene profile in the liposarcoma using microarray data, our study lacked cell validated experiment and tissue validated experiment, however, we conducted a PPI network analysis gene signature analysis for the DEG, which were not performed in the previous 2 studies (see Table 4). In addition, we did a validated analysis for a microarray analysis results using another microarray data, which also increased the reliable of our results.
Table 4.
Comparison the results between previous study and our study.
| Iura et al. [8] | Gobble et al. [9] | Our study | |
|---|---|---|---|
| DEGs identification | Yes | Yes | Yes |
| GO analysis | No | No | Yes |
| Pathway analysis | No | Yes | Yes |
| PPI network analysis | No | No | Yes |
| Gene signature analysis | No | No | Yes |
| COX regression analysis | No | No | Yes |
| Cell validated experiment | Yes | Yes | No |
| Tissue validated experiment | Yes | No | No |
| Survival analysis | Yes | Yes | Yes |
| Subtype analysis | Yes | No | Yes |
DEGs – differentially expressed genes; GO – Gene Ontology; PPI – Protein–protein interaction.
Discussion
Many genes are involved in the tumorigenesis of cancers, and some could serve as critical biomarkers for diagnosis, monitoring therapy, and determining the prognosis of cancers. To date, the molecular mechanism of liposarcoma pathogenesis remains unclear. In addition, there is an imperative need for prognostic factors that can reliable pinpoint the outcomes in patients with liposarcoma [13]. Recently, several genomic analyses studies reported a molecular catalogue that significant related to the liposarcoma tumorigenesis and outcome [14–17]. Tap et al. [18] identified chromosomal and genetic abnormalities in well-differentiated and de-differentiated liposarcoma using an oligonucleotide array-based comparative genomic hybridization approach. Crago et al. [19] evaluated the copy number alterations (CNAs) of 55 patients with well-differentiated and 52 patients with de-differentiated liposarcoma using an arrays method. Hoffman et al. [20] analyzed patients who presented with localized or metastatic myxoid liposarcoma and found that the receptor tyrosine kinase encoded by the AXL gene was a prognosticator of disease-specific survival in univariate analysis. De Cecco et al. [21] conducted gene expression profiling and immunohistochemical analyses of specimens of pure myxoid (ML) and pure round cell (RC) liposarcomas and revealed that the YY1/c-MYC/HDAC2 axis, cell cycle-related MKNK2, and stemness-related MSX1 were involved in maintaining RC variant cells in a fast-cycling and undifferentiated state. These studies indicated that there are several valuable genes or copy numbers that are associated with the genomic complexity of liposarcoma and could be key to the tumorigenesis and prognosis of liposarcoma.
Notably, there is little knowledge about the gene signature and the DRFS in patients with liposarcoma. Gobble et al. [9] calculated a risk score for each patient using 588 genes and found that patients with low risk scores had a 3-year DRFS of 83% versus 45% for high risk score patients; they also showed that TOP2A, PTK7, and CHEK1 were overexpressed in liposarcoma samples of all 5 subtypes and in liposarcoma cell lines. Saâda-Bouzid et al. [22] showed that the amplification of HMGA2 was associated with the atypical lipomatous tumor/well-differentiated liposarcoma histological type and a good prognosis, whereas CDK4 and JUN amplifications were associated with de-differentiated liposarcoma histology and a bad prognosis. In the present study, to identify the genes crucial to liposarcoma tumorigenesis and define genes significantly related to the prognosis of patients, we conducted comprehensive analysis of 2 microarray datasets. We screened DEGs and identified the associated functions and pathways. We then identified hub genes of the DEGs, which were key to liposarcoma tumorigenesis. Finally, we identified a 3-gene signature, including 1 protective gene (MCM2) and 2 risky genes (NIP7 and RPL10L) that could independently predict DRFS in patients with liposarcoma. These results provided deep insights into the mechanism of liposarcoma tumorigenesis.
However, no consistent genes have been verified by previous studies, which could potentially be due, at least in part, to differing detection methods and sample sizes. Compared to the previous studies, our study showed that the expression of 3 genes (MCM2, NIP7, and RPL10L) could act as an independent risk factor for liposarcoma patients. Moreover, the risk score of this 3-gene signature could be an indicator for patients in the clinical setting. In addition, our study examined the prognostic value of the MCM2-NIP7-RPL10L signature in subtypes of liposarcoma, and found that the prognostic value of this signature was even better in some subtypes of liposarcoma than in our overall analysis of liposarcoma. Therefore, our results provided a new indicator for the prediction of DRFS in patients with liposarcoma.
The roles of Nip7, RPL10L, and MCM2 have been investigated in several studies. Medvedev et al. analyzed the amino acid sequences of the Nip7 proteins from 35 archaeal species to identify positions containing mutations specific to the hydrostatic pressure and temperature of archaeal habitats. They found that adaptation to temperature changes by the Nip7 protein caused more pronounced modifications in sequence and structure than did pressure changes [23].
Another study found complexes of molecular masses in the range of 40S–80S. Downregulation of Nip7 affects cell proliferation, which is consistent with an important role for Nip7 in rRNA biosynthesis in human cells [24]. However, there is little evidence of any role in the pathogenesis of cancer.
A previous study observed that RPL10L deficiency could disturb ribosome biogenesis in late-prophase spermatocytes and prohibit the transition from prophase into metaphase of the first meiotic division, resulting in male infertility [25]. RPL10 is also a tumor suppressor gene, and the protein it encodes is known as the tumor suppression protein QM [26]. RPL10L was expressed in 76% of a large ovarian tumor panel, 84% in papillary serous cancers, 76% in tumors with mixed histology, and 44% in endometrioid tumors [27]. RPL10L can also be downregulated by TMEM9 and is involved in the cell invasion, migration, and adhesion of hepatoma cells [28].
MCM2 has been shown to be overexpressed in many human malignancies, and is an important target for cancer chemotherapy [29]. In human malignant fibrous histiocytomas (MFHs), a study observed that MCM2 expression correlated with cell proliferation rather than apoptosis of MFHs, and that the expression was ubiquitous in proliferating cells, regardless of P53 expression of [30]. In a study using radio-hyperthermo-chemotherapy (RHC) to treat sarcomas, researchers found high pre-RHC MCM2 and high post-RHC growth indices to be significant unfavorable prognostic factors [31]. High expression of MCM2 has also been associated with poor prognosis in primary localized myxofibrosarcomas [32].
Although the results of our study could have an important impact on the clinical setting, several limitations to our study should be noted. First, due to the limited number of suitable microarray datasets, we only selected GSE59568 to conduct gene function analysis and identify hub genes and we selected GSE30929 for the prognostic value analysis of gene signatures, which might undermine the robustness of the results. Second, the sample size of GSE59568 was small, with only 6 myxoid liposarcoma samples and 3 normal adipose tissue samples; a larger sample size of liposarcoma tissues with other subtypes of liposarcoma is needed to validate our results. Third, the type of liposarcoma in GSE59568 was myxoid liposarcoma; the dataset lacked other subtypes of liposarcoma. Hence, our results should be confirmed in a study using other subtypes of liposarcoma. Fourth, overall survival time is an important endpoint for patients; however, the GSE30929 dataset only provided DRFS data, which lead to this study merely calculating the risk factors of gene signatures associated with DRFS. Thus, other survival indices, such as overall survival time, should be analyzed in a future study. Fifth, the GSE59568 and GSE30929 data were based on the microarray technique. Thus, the results of this study lack any functional validation; other experimental techniques are needed to verify our findings. Despite these limitations, our current study has identified 1111 DEGs via a whole genome expression level screening and 9 hub genes by using a bioinformatics method. We then constructed a 3-gene DRFS prognostic signature of liposarcoma patients. These findings provide insight into tumorigenesis of liposarcoma and might have a clinical utility for liposarcoma diagnosis and decision-making in liposarcoma management.
Conclusions
This study analyzed the genome-wide gene expression profiles of liposarcoma, identified a gene profile that was crucial to the tumorigenesis of liposarcoma, and identified the hub genes of the gene profile. We also identified a 3-gene-signature, which could serve as a crucial indicator for DRFS of patients with liposarcomas. However, due to the limitations in our study, our findings still need to be verified in large cohort studies, and in studies using cells and animal experiments to validate these findings.
Abbreviations
- DEGs
differentially expressed genes
- GO
Gene Ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- DRFS
distant recurrence-free survival
- ROC
receiver operating characteristic curve
- FDR
false discovery rate
- NIP7
nucleolar pre-rRNA processing protein 7
- RPL10L
ribosomal protein L10 like
- MCM2
minichromosome maintenance complex component 2 (MIM number: 116945)
- RPS3A
ribosomal protein S3A (MIM number: 180478)
- RPL36
ribosomal protein L36
- MRPL3
mitochondrial ribosomal protein L3 (MIM number: 607118)
- TMEM9
transmembrane protein 9 (MIM number: 616877
Footnotes
Source of support: This work has been financially supported by The Innovation Project of Guangxi Graduate Education (Grant No. YCBZ2018034)
Conflict of interest
None.
References
- 1.Dei Tos AP. Liposarcoma: New entities and evolving concepts. Ann Diagn Pathol. 2000;4:252–66. doi: 10.1053/adpa.2000.8133. [DOI] [PubMed] [Google Scholar]
- 2.Jo VY, Fletcher CD. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology. 2014;46:95–104. doi: 10.1097/PAT.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 3.Patel RB, Li T, Liao Z, et al. Recent translational research into targeted therapy for liposarcoma. Stem Cell Investig. 2017;4:21. doi: 10.21037/sci.2017.02.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalal KM, Kattan MW, Antonescu CR, et al. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244:381–91. doi: 10.1097/01.sla.0000234795.98607.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daigeler A, Klein-Hitpass L, Chromik MA, et al. Heterogeneous in vitro effects of doxorubicin on gene expression in primary human liposarcoma cultures. BMC Cancer. 2008;8:313. doi: 10.1186/1471-2407-8-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishii T, Kohashi K, Ootsuka H, et al. Comparison between retroperitoneal leiomyosarcoma and dedifferentiated liposarcoma. Pathol Res Pract. 2017;213:634–38. doi: 10.1016/j.prp.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 7.Kashima T, Halai D, Ye H, et al. Sensitivity of MDM2 amplification and unexpected multiple faint alphoid 12 (alpha 12 satellite sequences) signals in atypical lipomatous tumor. Mod Pathol. 2012;25:1384–96. doi: 10.1038/modpathol.2012.90. [DOI] [PubMed] [Google Scholar]
- 8.Iura K, Kohashi K, Hotokebuchi Y, et al. Cancer-testis antigens PRAME and NY-ESO-1 correlate with tumour grade and poor prognosis in myxoid liposarcoma. J Pathol Clin Res. 2015;1:144–59. doi: 10.1002/cjp2.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gobble RM, Qin LX, Brill ER, et al. Expression profiling of liposarcoma yields a multigene predictor of patient outcome and identifies genes that contribute to liposarcomagenesis. Cancer Res. 2011;71:2697–705. doi: 10.1158/0008-5472.CAN-10-3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin S, Kim J, Arafat D, Gibson G. Effect of normalization on statistical and biological interpretation of gene expression profiles. Front Genet. 2012;3:160. doi: 10.3389/fgene.2012.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liew AW, Law NF, Yan H. Missing value imputation for gene expression data: Computational techniques to recover missing data from available information. Brief Bioinform. 2011;12:498–513. doi: 10.1093/bib/bbq080. [DOI] [PubMed] [Google Scholar]
- 12.Bao ZS, Li MY, Wang JY, et al. Prognostic value of a nine-gene signature in glioma patients based on mRNA expression profiling. CNS Neurosci Ther. 2014;20:112–18. doi: 10.1111/cns.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nassif NA, Tseng W, Borges C, et al. Recent advances in the management of liposarcoma. F1000Res. 2016;5:2907. doi: 10.12688/f1000research.10050.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koczkowska M, Lipska-Zietkiewicz BS, Iliszko M, et al. Application of high-resolution genomic profiling in the differential diagnosis of liposarcoma. Mol Cytogenet. 2017;10:7. doi: 10.1186/s13039-017-0309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanojia D, Nagata Y, Garg M, et al. Genomic landscape of liposarcoma. Oncotarget. 2015;6:42429–44. doi: 10.18632/oncotarget.6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egan JB, Barrett MT, Champion MD, et al. Whole genome analyses of a well-differentiated liposarcoma reveals novel SYT1 and DDR2 rearrangements. PLoS One. 2014;9:e87113. doi: 10.1371/journal.pone.0087113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louis-Brennetot C, Coindre JM, Ferreira C, et al. The CDKN2A/CDKN2B/CDK4/CCND1 pathway is pivotal in well-differentiated and dedifferentiated liposarcoma oncogenesis: an analysis of 104 tumors. Genes Chromosomes Cancer. 2011;50:896–907. doi: 10.1002/gcc.20909. [DOI] [PubMed] [Google Scholar]
- 18.Tap WD, Eilber FC, Ginther C, et al. Evaluation of well-differentiated/de-differentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridization. Genes Chromosomes Cancer. 2011;50:95–112. doi: 10.1002/gcc.20835. [DOI] [PubMed] [Google Scholar]
- 19.Crago AM, Socci ND, DeCarolis P, et al. Copy number losses define subgroups of dedifferentiated liposarcoma with poor prognosis and genomic instability. Clin Cancer Res. 2012;18:1334–40. doi: 10.1158/1078-0432.CCR-11-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman A, Ghadimi MP, Demicco EG, et al. Localized and metastatic myxoid/round cell liposarcoma: Clinical and molecular observations. Cancer. 2013;119:1868–77. doi: 10.1002/cncr.27847. [DOI] [PubMed] [Google Scholar]
- 21.De Cecco L, Negri T, Brich S, et al. Identification of a gene expression driven progression pathway in myxoid liposarcoma. Oncotarget. 2014;5:5965–77. doi: 10.18632/oncotarget.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saada-Bouzid E, Burel-Vandenbos F, Ranchere-Vince D, et al. Prognostic value of HMGA2, CDK4, and JUN amplification in well-differentiated and dedifferentiated liposarcomas. Mod Pathol. 2015;28:1404–14. doi: 10.1038/modpathol.2015.96. [DOI] [PubMed] [Google Scholar]
- 23.Medvedev KE, Kolchanov NA, Afonnikov DA. Identification of residues of the archaeal RNA-binding Nip7 proteins specific to environmental conditions. J Bioinform Comput Biol. 2017;15:1650036. doi: 10.1142/S0219720016500360. [DOI] [PubMed] [Google Scholar]
- 24.Morello LG, Hesling C, Coltri PP, et al. The NIP7 protein is required for accurate pre-rRNA processing in human cells. Nucleic Acids Res. 2011;39:648–65. doi: 10.1093/nar/gkq758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang L, Li T, Zhang X, et al. RPL10L Is required for male meiotic division by compensating for RPL10 during meiotic sex chromosome inactivation in mice. Curr Biol. 2017;27:1498–505. doi: 10.1016/j.cub.2017.04.017. [DOI] [PubMed] [Google Scholar]
- 26.Stalberg P, Grimfjard P, Santesson M, et al. Transfection of the multiple endocrine neoplasia type 1 gene to a human endocrine pancreatic tumor cell line inhibits cell growth and affects expression of JunD, delta-like protein 1/preadipocyte factor-1, proliferating cell nuclear antigen, and QM/Jif-1. J Clin Endocrinol Metab. 2004;89:2326–37. doi: 10.1210/jc.2003-031228. [DOI] [PubMed] [Google Scholar]
- 27.Rohozinski J, Anderson ML, Broaddus RE, et al. Spermatogenesis associated retrogenes are expressed in the human ovary and ovarian cancers. PLoS One. 2009;4:e5064. doi: 10.1371/journal.pone.0005064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Ran Y, Xiong Y, et al. Effects of TMEM9 gene on cell progression in hepatocellular carcinoma by RNA interference. Oncol Rep. 2016;36:299–305. doi: 10.3892/or.2016.4821. [DOI] [PubMed] [Google Scholar]
- 29.May M, Burger M, Otto W, et al. Ki-67, mini-chromosome maintenance 2 protein (MCM2) and geminin have no independent prognostic relevance for cancer-specific survival in surgically treated squamous cell carcinoma of the penis. BJU Int. 2013;112:E383–90. doi: 10.1111/j.1464-410X.2012.11735.x. [DOI] [PubMed] [Google Scholar]
- 30.Osaki M, Yamashita H, Shomori K, et al. Expression of minichromosome maintenance-2 in human malignant fibrous histiocytomas: Correlations with Ki-67 and P53 expression, and apoptosis. Int J Mol Med. 2002;10:161–68. [PubMed] [Google Scholar]
- 31.Matsubara T, Eimoto T, Okabe M, et al. Proliferation and apoptosis of tumour cells before and after neoadjuvant therapy for high-grade extremity sarcomas: Divergent associations with tumour response and prognosis. Histopathology. 2008;52:706–16. doi: 10.1111/j.1365-2559.2008.03015.x. [DOI] [PubMed] [Google Scholar]
- 32.Huang HY, Kang HY, Li CF, et al. Skp2 overexpression is highly representative of intrinsic biological aggressiveness and independently associated with poor prognosis in primary localized myxofibrosarcomas. Clin Cancer Res. 2006;12:487–98. doi: 10.1158/1078-0432.CCR-05-1497. [DOI] [PubMed] [Google Scholar]