

Abstract
Cardiovascular diseases (CVD) account for nearly half of all deaths in Europe and almost 30% of global deaths. Despite the improved clinical management, cardiovascular mortality is predicted to rise in the next decades due to the increasing impact of aging, obesity, and diabetes. The goal of emerging cardiovascular nanomedicine is to reduce the burden of CVD using nanoscale medical products and devices. However, the development of novel multicomponent nano-sized products poses multiple technical, ethical, and regulatory challenges, which often obstruct their road to successful approval and use in clinical practice. This review discusses the rational design of nanoparticles, including safety considerations and regulatory issues, and highlights the steps needed to achieve efficient clinical translation of promising nanomedicinal products for cardiovascular applications.
Keywords: Cardiovascular nanomedicine , Clinical translation , Nanoparticle design , Nanosafety , Regulatory issues
1. Introduction
Cardiovascular nanomedicine aims to improve diagnosis and treatment of cardiovascular diseases (CVD), which are responsible for the majority of deaths worldwide.1 Regarding diagnostics, the goal is to move the current imaging agents to a new level allowing the detection and characterization of CVD at an early stage. The therapeutic aim is to move forward from conventional drug therapies that lead to full systemic exposure to targeted drug delivery using nanosystems that minimize the systemic side-effects and enhance drug localization and efficacy in atherosclerotic and thrombotic lesions. With hybrid nanoparticles (so called ‘theranostics’) one could combine imaging and treatment, to enable monitoring patients’ responses to therapy.
The possible applications of nanoparticles in the management of CVD range from ultra-sensitive monitoring of cardiovascular markers, through detection and characterization of plaques and aneurysms, in situ detection of thrombosis, imaging of inflammation in myocardial infarction (MI), to the targeted delivery of atheroprotective, or thrombolytic drugs (Figure 1).2–10 Cell labelling with nanoparticles for cell-based therapies can also be envisioned to enhance stent endothelialisation and improve myocardial regeneration.14–16
Figure 1.

Possible applications of nanoparticles (NP) for diagnosis and therapy in CVD patients. The used photoacoustic and MR images are reproduced with permission from Blázquez R et al.11; from Wu C et al.12; and from Yilmaz et al.13
However, bringing a medicinal product into the clinical arena is a challenging and time/cost-consuming process. Extensive in vitro and in vivo preclinical studies are required before first-in-man clinical safety trials can be initiated. In the USA, the Food and Drug Administration (FDA) is the responsible supervising agency to decide if a drug or a medical device is allowed to enter clinical trials and whether, upon completion of the clinical development programme, it will be approved for marketing. In Europe, the European Medicines Agency (EMA) is the regulatory body responsible for drug market approval. The classical process of drug development, testing, and approval is estimated to take around 10–15 years, with costs of roughly around 1 billion USD per product according to some estimates.17 Both the EMA and the FDA have stated that no new regulations are needed for approval or commercialization of nanomedicines, considering that the existing regulatory framework is valid and accurate. There is, however, a need for elaboration of the regulatory framework to accommodate for special safety and quality aspects that complex nanotechnology products can entail, and a need to improve technical guidance documents used for the application and implementation of existing regulatory frameworks.18
A standardized definition of ‘nanoparticle’ varies among organizations and countries. Although a specified size limit is not always relevant for scientific or medical applications, it is needed for regulatory purposes and is defined (albeit differently) both in the EU and in the USA. The definition implemented in the EU states that nanomaterial is a ‘natural, incidental, or manufactured material containing particles, in an unbound state, or as an aggregate, or as an agglomerate and where, for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1–100 nm’, but exceptions are possible to the percentage and the upper limit of 100 nm, especially in the pharmaceutical sector.19 The FDA defines nanomaterial as any material with at least one dimension smaller than 1000 nm and a nanoparticle as an object with all three external dimensions in the 1–100 nm size range.
Despite the costs and regulatory obstacles, about 250 nanomedicinal products, mostly for cancer treatment, were listed by FDA as approved, or were in different phases of clinical trials in 2013,20 whereby the market is dominated by liposomal and polymeric nanomedicines.17,20,21 Medicinal nanosystems in the form of e.g. liposomes (i.e. Doxil®, AmBisome®), or PEG-conjugated proteins (Adagen®, Neulasta®) have already been granted marketing authorization within the EU and the USA under the existing pharmaceutical legislation. As for any medicinal product, the authorities evaluate any marketing application by the established principles of benefit/risk analysis, rather than solely on the basis of the technology per se. The majority of nanosystems approved thus far were relatively simple and aimed at improving stability, half-life, bioavailability, and safety of existing drugs. It is likely that some new nanotechnology products that reach clinical trials will gain in complexity, as the technology gradually advances and treatment goals become ever more ambitious. According to the current EU directives, the decision whether a nanomedicinal product is a medicine or a medical device, which determines the applicable regulatory regime, is based on the principal mode of action. With medical devices, the mode of action is physical (mechanical or chemical) while a medicinal product acts by pharmacological, immunological, or metabolic means.22 However, future nanomedical products may span the regulatory boundaries between medicinal products and medical devices, and for those nanomedicines which have a complex mode of action, this decision may prove difficult as their activity might depend on both physicochemical/mechanical and pharmacological properties.23,24
As compared with the vast number of experimental research reports focusing on cardiovascular applications of nanoparticles that have been published in the recent years (reviewed in Refs25–28), and the reported clinical trials remain very scarce in this field. This is likely due to the rational design of non-cytotoxic nanosystems and hurdles related to scale up, good manufacturing practice (GMP)-grade production, quality control, and full pre-clinical assessments which are required before clinical studies can be started (Figure 2). Based in particular on the experience from the European Commission funded NanoAthero project (‘Nanomedicine for target-specific imaging and treatment of atherothrombosis—Development and initial clinical feasibility’ http://www.nanoathero.eu/), this review addresses the main translational steps and challenges that cardiovascular nanomedicines encounter on their development from bench to bedside.
Figure 2.
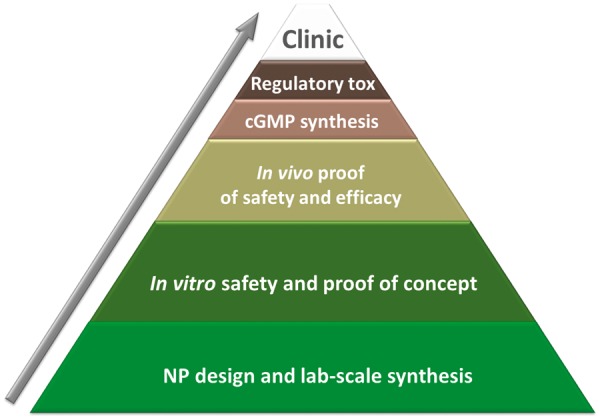
Clinical translation scheme. In vitro studies on imaging and drug-delivery nanosystems produced in the laboratory scale represent the largest shelves in the pyramid. The number of nanomedicinal products reaching and passing the regulatory and toxicological hurdle to enter clinical trials remains very low.
2. Translation hurdle: clinical trials in cancer vs. CVD
The search for clinical trials on the homepage clinicaltrials.gov of the US government dealing with ‘nanoparticles’ and ‘cardiovascular diseases’ delivered 13 results, whereas the search for ‘nanoparticles’ and ‘cancer’ lists 176 performed or ongoing clinical trials. Although not all clinical trials can be found on clinicaltrials.gov, this indicates the considerable challenge in putting cardiovascular nanomedicines on the road to clinical trials when compared with anti-cancer nanodrugs. The 13 listed CVD trials include contrast agents for improved imaging of cardiovascular inflammation and enhanced diagnosis of acute coronary syndrome, nanodrugs for prevention of restenosis after revascularization, and plasmonic photo-thermal therapy (PPTT) of atherosclerotic plaques. Among the completed and published trials, several were related to the clinical use of iron oxide nanoparticles for improved detection and characterization of atherosclerotic plaques [ferumoxtran (Sinerem®)29–31] or aortic aneurysms [ferumoxtran (Sinerem®)5] and detection of inflammation in MI [ferumoxytol (Feraheme®)13,32,33] Recently, in association with the NanoAthero project, a clinical study was done by the group of Stroes et al.,34 investigating the utility of ferumoxytol for carotid plaque imaging. Concerning therapeutic application of nanoparticles in CVD, only a few studies have been reported so far. Some examples are thus briefly highlighted. In the BLAST trial, the safety and anti-restenotic efficacy of a single intravenous bolus of liposomal alendronate, which transiently modulates monocyte function, was examined in patients undergoing bare metal stent placement.35 An angiographic assessment of late lumen loss at 6 months post-implantation demonstrated that the treatment effectively reduced the late loss in the inflammatory patient subgroup, but not in the entire liposomal treatment cohort. The NANOM first-in-man trial, published in 2015, investigated the feasibility of atherosclerosis treatment by reducing the total atheroma volume with PPTT.36 Silica-gold nanoparticles with photothermal properties were delivered on bioengineered artery patch containing stem cells, or via an intravenous catheter under magnetic guidance, followed by irradiation using near-infrared laser.36 This clinical trial showed that both forms of administration were superior to stenting and that photothermal destruction of atheroma tissue resulted in a reduction of the plaque volume down to 37.8% of initial plaque burden, whereas stenting resulted in 52.9% reduction of plaque burden.36 It is however questionable, to what extent this technique may prove applicable in the clinical routine everywhere. Within the NanoAthero project, a clinical trial was also performed to test the treatment of atherosclerotic plaques with a targeted nanomedicine containing prednisolone (Nanocort®). In that study, prednisolone was encapsulated into liposomal nanoparticles, which increased the plasma half-life of the drug.37 After systemic intravenous infusion in an antecubital vein, it was demonstrated that the nanoparticles were localized in the macrophages isolated from atherosclerotic plaques harvested from the iliac arteries, thereby lending proof-of-concept that intravenous liposomes can successfully target inflammatory cells within the atheroma. The clinical studies did not demonstrate that the delivered prednisolone have an anti-inflammatory effect measured by positron emission tomography (PET) imaging in the atherosclerotic lesions,38 which might be related to in vitro observations that macrophages become lipotoxic, exemplified by enhanced lipid loading, ER stress, and apoptosis.39 Summarizing the previous clinical trials, it is clear that the application of nanomedicine in CVD patients is still in its infancy and great effort will likely be needed to enforce a clinical breakthrough.
3. Rational design of nanosystems: safety by design
Imaging is a crucial aspect for risk stratification as well as for the selection of subsequent therapy and follow-up monitoring. Nanoparticulate contrast agents have been shown to improve the detection and characterization of CVD, but their application in potentially healthy subjects raises a particularly high-safety hurdle. Nanoparticle-based drug delivery systems are an attractive platform to improve the efficacy and reduce the systemic toxicity of cardiovascular drugs. For therapeutic applications, novel nanoparticle formulations including drug-carrying liposomes, lipid nanoparticles, superparamagnetic iron oxide nanoparticles (SPIONs), and polyacrylates are currently being developed by our groups and others.37,40–45 Additional modifications, including functionalization with targeting ligands and/or imaging agents are often necessary to locally deliver the therapeutic nanoparticles and to monitor the effect of the treatment. This can result in a complex multicomponent nanosystem (Figure 3), which will require many synthesis and manufacturing steps as well as a range of quality controls, which leads to a tremendous cost increase.46
Figure 3.
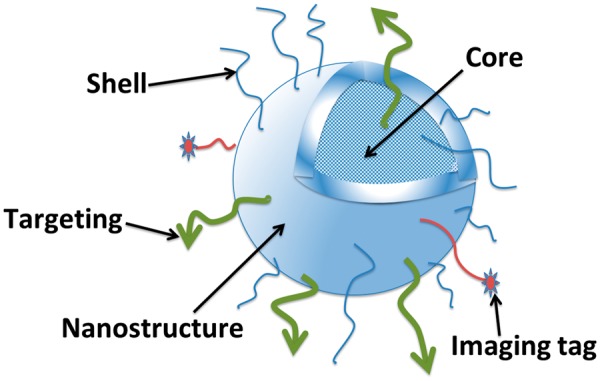
Example design of nanoparticles to achieve a targeted drug delivery and/or imaging agent. Due to the small size of nanoparticles, their surface area is large. This provides various possibilities of surface modifications e.g. by coating in order to stabilize and prevent aggregation of nanoparticles, and to allow conjugation of ligands or drugs.
A more rational design of the particles may partly help to reduce costs and increase chances of achieving successful clinical translation. Setting aims, i.e. selecting the disease process to be addressed and the intended application of nanosystem (diagnostics vs. therapeutic) is the first step in the development process. The design and production at the lab scale must be followed by a complete physicochemical characterization of nanosystems, using the available specifically adapted evaluation methods.47,48 As different techniques for nanoparticle characterization are available (e.g. transmission electron microscopy, Fourier transform infrared spectroscopy, and dynamic light scattering), each of them featuring its own advantages and limitations, the characterization data obtained with several different measurement methods should be compared with ensure reliable results.
In-house physicochemical characterization and storage stability evaluation of produced batches limits the costs in the early development stage. Within the NanoAthero project, nanoparticle characterization and stability evaluation were performed in parallel in-house by nanoparticle providers and also by a selected independent partner, in order to demonstrate comparability and allow for reproducibility validation. In this respect, the Nanotechnology Characterization Laboratory (NCL) in the US (https://ncl.cancer.gov/), and more recently its European counterpart, European Nanomedicine Characterization Laboratory (EU-NCL, http://www.euncl.eu/), provide independent trans-disciplinary testing infrastructures covering a large set of preclinical validated characterization assays (physical, chemical, in vitro, and in vivo biological testing).
Among the parameters to consider when designing a nanosystem is the chemical composition, which is often the most critical feature that affects nanoparticle toxicity.49,50 Further, particle surface charge, indicated by zeta potential, has a strong influence on nanoparticle stability in suspension and in vivo toxicity.51,52 Size is another critical factor that affects the behaviour and biological safety of nanoparticles.53 For example, nanoparticles with hydrodynamic diameter smaller than 10 nm have been reported to cause undesirable effects by passing through the blood–brain barrier, and nanoparticles with diameter less than 5 nm are rapidly cleared by the kidneys, which dramatically reduces their circulation time.52 Particle size and shape are also likely to affect their margination, extravasation, and penetration through vascular walls, particularly in larger vessels relevant to CVD (reviewed in Refs54,55). Previous ex vivo studies in whole blood model showed enhanced margination of micro- compared with nano-sized particles and the dependence of this effect on a high-aspect ratio of particles.56,57 In these investigations, nanorods did not display enhanced margination compared with nanospheres,57 indicating that binding of nanoparticles to arterial endothelium may require a margination-enhancing design and/or active targeting. Nanoparticle agglomeration is another factor with strong adverse consequences in vivo.58 Agglomeration is influenced by the particle composition, size, and zeta potential, but also extrinsic factors, e.g. temperature, as well as pH, osmotic strength, and the presence of serum. As aggregated nanoparticles are no longer nano-sized, they undergo a rapid recognition by the reticuloendothelial system (RES) and are cleared by the liver or spleen. Moreover, their presence in the circulation may cause serious undesirable side-effects, such as clogging blood or lymphatic vessels.59 Prevention of agglomeration is therefore required for designing a stable, clinically safe nanosystem. In this respect, PEGylation of nanoparticles appears effective in reducing their agglomeration. By creating a hydrophilic layer around the nanoparticles, PEGylation also provides a strong steric barrier to opsonin adsorption,60 opposing nanoparticle recognition by the RES, and increasing their circulation half-life. Other methods to reduce particle agglomeration explored within the NanoAthero project included coating of SPIONs with cross-linked dextran or fatty acids44,61 and brush-like coating of polymer nanoparticles with polysaccharides (dextran and fucoidan8). Careful attention should also be given to the protein corona which forms on the surface of the nanoparticles when they interact with plasma, since this can affect their toxicity and efficacy.62,63 Taken together, detailed and standardized characterization can facilitate the prediction of nanoparticle performance in physiological conditions and is mandatory to consider before any given nanosystem can enter the preclinical in vitro and in vivo testing stages.
4. Candidate selection: a multi-criteria decision process
In the selection of the best candidate nanosystems for imaging and therapy of atherothrombosis many factors should be carefully considered. Within the NanoAthero project, a decision tree was established based on the physical and biochemical characteristics of the nanosystems developed in this project (Figure 4). The most important selection criteria are briefly outlined below with short commentaries.
Figure 4.

Candidate selection criteria. ADME, absorption, distribution, metabolism, and elimination; GRAS, generally recognized as safe; PDI, polydispersity index.
4.1 Product physicochemistry
Physicochemical properties of the nanoparticles are the critical determinants of their safety and in vivo performance. Intensive efforts are being developed by the NCL and EU-NCL initiatives, as well as by different research groups, including the NanoAthero consortium, to propose standardized methods for measurements of key parameters such as particle diameter and zeta potential.64–66 Apart from the parameters listed in the Section 3 (diameter, charge, and polydispersibility), pH and osmolarity of the final dispersion for long-term storage and for injection should be considered when selecting suitable candidates. In case of nanosystems containing drugs or contrast agents, encapsulation/binding efficiency and the amount of drug or contrast agent per particle are the important selection parameters.
4.2 Ingredient quality (safety, sterilisability, and pyrogen content)
The quality of the starting materials is an important point to be considered. Preferably, raw materials with existing pharmacopoeia reference (i.e. Ph.Eur., USP) or medical-grade substances should be used for nanoparticle synthesis. The safety of ingredients can be confirmed using the GRAS (Generally Recognized as Safe) Substances Database (https://www.accessdata.fda.gov/scripts/fdcc/? set=SCOGS).
The final drug product should obviously be sterile. Sterilisability of the produced nanoparticles must therefore be ensured, which is in most cases achieved with (redundant) sterile filtration through <0.2 μm filters right before filling into the sterile dosage units. Depending on the chemical/biological components and the production process, the final nanosystem may contain bacterial endotoxins,67 which can cause adverse effects upon in vivo administration, potentially leading to the organ damage. The FDA-recommended high-sensitivity bacterial endotoxin LAL test (limulus amoebocyte lysate assay) is commonly used in preclinical pharmaceutical development, but many nanoparticles interfere with the assay.67,68 In our project, there was a case of endotoxin contamination of an additive, the commercially available bovine serum albumin used as a coating to improve the biocompatibility of one type of SPIONs. This resulted in unexpected inflammatory effects of these particles and additionally necessitated a complete and very costly purification of the synthesis unit to avoid cross-contamination. To overcome this problem, clinical grade serum albumin of human origin was used for further development of these particles.
4.3 Manufacturability (process, cost of goods)
The manufacturing process of a nanomedicinal product may involve a multi-step procedure requiring a number of excipients, which can drastically increase the cost and represents an additional production hurdle. The research and development methods often involve a low-volume production and scaling up the process may entail serious difficulties for some nanoparticles, and be easier for others.69 For instance, scaled-up production of lipid nanoparticles is relatively easy to implement, and has been documented for more than 25 years in the medical field.70 Apart from this, the costs and availability of raw materials must be considered, as well as the batch-to-batch reproducibility of physicochemical characteristics.
4.4 Stability
Although it is not strictly required for human application, long-term stability on storage of nanosytems is a prerequisite for a nanoproduct to be marketable. Ideally, the shelf life should be equal to or longer than 6 months. The parameters to consider include colloidal stability and chemical stability of drugs and excipients on storage, but also potential leakage of drug or contrast agent from the nanoparticles. Within NanoAthero, the standardized physicochemical characterization of nanosystems was performed at 1 month post-preparation date and—to determine the long-term particle stability—the subsequent measurements were performed after 3, 6, and 12 months of storage at 4°C in the respective nanoparticle dilution media. The acceptable variation was set to 10% diameter variation, and 20% polydispersity index variation at PDI of maximally 0.25.
4.5. Toxicity/biocompatibility
One of the factors that may influence the particle behaviour and toxicity is stability in biological fluids (serum-containing cell culture media, plasma, whole blood).71 Analysis of nanoparticle agglomeration in plasma and blood is therefore mandatory. The screening of nanosystems should first be done in vitro, to assess the potential toxicity of nanoparticles towards blood cells, other first-contact cells (e.g. endothelial cells in the case of intravenous application) and the actual target cells. Other undesired effects, including haemolytic reactions, platelet, and complement activation, reactive oxygen species production can relatively quickly be evaluated by in vitro tests.44,72 After in vitro screening to select the constructs with adequate haemo- and biocompatibility, proof of principle studies in in vivo models and GMP-compliant manufacturing process are required, that are followed by regulatory toxicity studies in animals, usually rats and mice (see Section 7.2).73
4.6. Efficacy
To some extent, the in vivo performance and potential efficacy of nanosystems can be predicted with in vitro or ex vivo models or phantoms,44,74,75 but the ultimate preclinical proof of efficacy requires an animal model of disease. In the NanoAthero project, the characterized nanosystems containing imaging agents (radionuclides, iron oxides, micellar formulations containing gadolinium) were tested in appropriate animal models: mouse or rabbit models of atherosclerosis and a rat model of thrombosis.8 Dedicated small animal magnetic resonance imaging (MRI) coils and a 3T MRI system were used for imaging. To verify the accumulation of the nanosystems in the diseased region, histological analysis of the imaged sections was performed post-mortem. Single photon emission computed tomography/computed tomography (SPECT/CT) and PET analyses, including the grafting of tracers was also performed, as well as in vivo and ex vivo fluorescence imaging after nanoparticle labelling.
For therapeutic purposes, the nanosystems containing compounds with anti-inflammatory activity were pre-screened in vitro74 and then tested in the apolipoprotein-E (apoE)-knockout mouse model,41 followed by selection of promising candidates.
4.7. Pharmacokinetics and biodistribution
The determination of pharmacokinetics (PK) and biodistribution is usually done by the detection of particle- and/or drug-bound radiolabels in animal tissues harvested at different time points. Radiolabelling (3H or other radionuclides) allows imaging of the biodistribution of nanoparticles on tissue sections in rodents and the quantification of the percent of injected dose in different organs and body fluids. Full-body autoradiography or selected tissues sampling for well counting should identify the main-targeted organs (usually liver, kidney, and spleen). Biodistribution estimation derived from in vivo nuclear imaging using nanoparticles labelled with gamma- or positron-emitters radionuclides is an alternative. While the accuracy of the measurement is lower when compared with direct tissue sampling, this approach allows for iterative assessments in a single animal, enabling either a marked reduction of the number of animals sacrificed for a given experimental protocol, and/or an increase in the number of measurements.76 Fluorescence imaging, having the advantages of lower cost, detection below cellular level by microscopy techniques, is another alternative, but its main limitation is that it is not truly quantitative (semi-quantitative analysis).
In case of nanoparticles containing a drug payload, the drug can be labelled with 14C. By using dual 14C and 3H-detection, parallel quantification and comparison of the biodistribution between the free drug and nanoparticle-conjugated drug is possible.45,77Ex vivo validation by autoradiography allows localizing a radioactive material within particular tissues or cells with high sensitivity and quantitative estimation of the delivered amount of (nano)drug.78,79 These data are of critical importance to determine the ability of nanoparticles to target and deliver their drug payload to particular tissues, but the expense and the efforts required for these investigations are considerable.
4.8. Clinical « acceptability »
Novel nanodrugs are commonly greeted with a degree of concern and reserve in fear of their potential nanotoxicity, unless the nanosystem carrier is well established and/or carries an approved drug. Acceptance is usually less of an issue in high-medical-need indications.80 Quite obviously also the administration route is a factor of importance, whereby oral administration is preferred by patients.81
However, parenteral administration and in particular intravenous injection is often the only feasible way cardiovascular contrast agents and nanodrugs should be given, which requires admission of a patient to a hospital or outpatient clinic, and significantly increases the costs.
5. In vitro proof of safety and efficacy
Nanomedicine offers unique possibilities in terms of CVD management, but despite these exciting possibilities, it is clear that nanomedicines can also entail new and sometimes unforeseen risks. The impact of nanoparticles on biological pathways and their toxic effects on the human body can be difficult to predict. Due to the interference of the particles with the traditional photometric cytotoxicity assays, routinely used tests such as lactate dehydrogenase assay or 3-(4, 5dimethylthiazol-2-yl)2, 5diphenyltetrazolium bromide assay can produce false-positive or false-negative results.82,83 Toxicity of engineered nanoparticles can be over- or underestimated due to their influence on absorbance of light in the visible spectrum, quenching of fluorescence, or even adsorption of the dye to their surface.83 Suitable in vitro assays must thus be chosen and validated to enable a meaningful in vitro toxicity evaluation. Several organizations, including the NCL or the International Organization for Standardization underscore the importance of a general standardization of in vitro toxicity assessment within nanotechnology and nanomedicine.47 Here, an essential point is the batch-to-batch reproducibility, because safety or efficacy evaluations are not reliable if batch-to-batch reproducibility is insufficient.
Full biocompatibility (including haemo-, cyto-, and immune compatibility) of the nanosystems is absolutely essential, as the target population of CVD patients may be prone to critical responses to any incompatibility. Therefore, developing a systematic workflow to analyse the biological effects of nanoparticles under standardized conditions is particularly relevant. All nanosystems intended for intravascular administration should be tested for their potential toxicity towards primary human endothelial cells. Using two complementary methods for long-term in vitro monitoring in parallel is recommended, as one single method may increase the risk of bias. In our opinion, real-time cell analysis and live-cell microscopy represent the suitable methods for parallel testing of the toxicity of nanoparticles in static in vitro conditions.66 Importantly, no interference resulting from the presence of nanoparticles should be detectable by real-time cell analysis in the absence of cells, which was indeed the case in our studies. This clearly underscores the suitability of the techniques, we used for the future standardized nanotoxicology studies. Beyond analysis of nanoparticle effects on cell viability in static culture conditions, investigating the effects of circulating nanoparticles on endothelial monolayer under physiological-like shear stress conditions allows performing the in vitro assays in dynamic conditions corresponding to the physiological environment of endothelial cells.66 Among the nanosystems evaluated positively in cell-compatibility studies, selected candidates should undergo detailed analyses to exclude haemolysis, coagulation, platelet activation and aggregation, leucocyte activation, and complement activation.44
Concerning possible compounds in evaluation for therapeutic applications, an in vitro screening setup for selected promising compounds/formulations and their potential athero-protective effects should be established. For instance, within NanoAthero we selected a range of in vitro assessments that address several pivotal pathological pathways in atherosclerotic plaques. These assays revealed that pterostilbene, simvastatin, and the liver X receptor agonist T0901317 were the most promising atheroprotective compounds to be integrated into nanosystems for plaque therapy74 and three simvastatin-loaded nanocarriers, including high-density lipoprotein nanoparticles, PEGylated liposomes, and polymeric micelles, were subsequently evaluated in vivo, in apoE-deficient mice.41
6. Preclinical animal models
Multiple animal models are available that address CVD in different species, including rodents, and larger animals (rabbits, pigs, and non-human primates).84,85 To date, genetically engineered hyperlipidaemic mice are among the most widely used models of atherosclerosis, but several transgenic,87 models in alternative species (rat and pig) have also been created.86,87 While mouse models of atherosclerosis are inexpensive and highly valued as a tool to identify the molecular mechanisms of the disease that can be targeted by novel (nano)medicines, they lack multifactorial background of atherosclerosis and have limited predictive value as the lipid profile and metabolism of mice, as well as the plaque composition are different from humans.88 Additional drawback is the small size of these animals, which limits the availability of biological samples, as well as the possibility of morphological and functional imaging of atherosclerosis. Despite of these drawbacks, mice still represent a model of choice for initial drug testing or biodistribution studies, and the continuing efforts to develop transgenic models, e.g. apoE3Leiden/cholesteryl ester transfer protein (CETP) mice,89 aim at better reproduction of human disease characteristics.
To promote clinical translation of emerging nanomedicinal products, larger animal models suitable for interventional procedures and imaging are advantageous. Rabbits represent a cost-efficient model of atherosclerosis with similarities to human lipoprotein profile, CETP expression, and size large enough to allow tissue sampling and imaging in clinically used scanners. The limitation of the rabbit model is that lesion complications observed in humans (haemorrhage, ulcerations, and thrombosis) are usually absent and their foam cell and macrophage load is increased compared with human plaques. Pigs and non-human primates represent two atherosclerosis models considered optimal to reflect the disease in patients, because of their similarities to humans in terms of metabolism, cardiovascular anatomy, and physiology. Human-like complex plaque morphology90 and instability traits have been reported in these animals.91,92 Despite the disadvantages of the large animal models, including the great expense and ethical considerations, these models allow the best extrapolation of findings to humans, thus contributing to the development of emerging therapies.
Detailed recommendations on design and performing animal studies in common models of atherosclerosis have been recently published in a statement of American Heart Association.85
7. In vivo safety: a prerequisite for approval
Toxicology assessment of nanomedicines in vivo is in principle not very different from conventional drug products, albeit that specific potential nanomedicine-related safety issues in humans need to be looked for in special animal models. These are listed below.
7.1. Complement activation-related pseudoallergy assessment
To characterize, predict, and prevent pseudoallergic reactions to nanomedicines, which often arise following their first intravenous administration, EMA recommends the detection of the Complement activation-related pseudoallergy (CARPA). The unique in vivo porcine model of CARPA allows evaluation of the risk of—otherwise unpredictable—acute cardiopulmonary distress, which can be severe or occasionally lethal, and therefore, unacceptable for CVD patients.93 The CARPA tests in pigs should include both single dose and repeat-dose administration, corresponding to the predicted use of the final nanosystem.94 The candidates that passed the CARPA evaluation successfully without inducing hypersensitivity reaction (i.e. were CARPA-negative) can subsequently enter the regulatory toxicity studies.44,72
Additionally, many nanomedicines undergoing development or approved as products include a coating to improve stability, minimize aggregation, and prolong circulation time. The presence of coating has the potential to impact on bio-molecular and cellular interactions of nanoparticles upon in vivo administration. For example, naturally occurring anti-PEG antibodies (IgM) have been detected in nearly 25% of healthy donors with no known exposure to PEG, indicating a growing prevalence of PEG exposure (e.g. in cosmetics or processed foods) and an increased risk of immunogenicity/antigenicity.95 Anti-PEG antibodies may also lead to increased clearance of PEGylated nanomedicines upon administration, thus reducing their biological activity. Although no specific animal models have been recommended for testing PEG antigenicity, it is important to monitor the patients for the presence of anti-PEG antibodies prior to and during the administration of PEGylated nanomedicines.96
7.2. Regulatory toxicity studies
Prior to clinical trials, the authorities require preclinical safety and PK evaluation in animals under good laboratory practice (GLP) regulations. As such, the regulatory toxicity studies are commonly outsourced to an approved and fully equipped Contract Research Organization (CRO). The non-clinical safety assessment for marketing approval of a pharmaceutical usually includes pharmacology studies, general toxicity studies, toxicokinetic (TK), and non-clinical pharmacokinetic studies, reproduction toxicity studies and genotoxicity studies. The non-clinical safety studies should be adequate to characterize potential adverse effects that might occur under the conditions of the clinical trial to be supported. The choice of the more adequate panel of studies to address safety of novel nanosystems should be guided mainly by the dosage expected to be used in humans and also by the specific characteristics of the nanoparticles.
The following toxicology tests are the main ones required before nanomedicinal product trials in humans: (i) safety pharmacology, a core battery according to ICHS7A, ICHS7B including the assessment of effects on cardiovascular, respiratory and central nervous systems (QT prolongation, respiratory function, and Irwin Test); (ii) TK and PK studies to determine plasma PK and elimination, as well as the validation of analysis methods in relevant species for repeated dose studies; (iii) acute toxicity studies, mainly based on single dose or expanded acute toxicity studies in two mammalian non-primate species. Animals are monitored over 14 days for body weight, organ weight indices, as well as behavioural, biochemical, and histopathological changes. The maximum tolerated dose and the no observed adverse effect level should be obtained; (iv) repeated dose toxicity studies over 2 weeks (minimum duration), in two mammalian species (rodent and non-rodent); (v) local tolerance studies in rabbit, using routes relevant to the proposed clinical administration route; (vi) genotoxicity studies including gene mutation (Ames Test) and chromosomal damage test (human lymphocytes). The requirement to execute this complete panel of tests is related to the effective dose that it is supposed to be used. For instance, not all these tests are required for a PET/SPECT nanosystem for imaging using microdoses and a single injection.
More specific studies should be taken into account with respect to different peculiarity of the investigated nanosystems (i.e. iron determination for iron-based nanosystems, rate and location of a drug released by a liposome system, etc.). All the above mentioned studies are necessary for the evaluation of benefits and risks for patients, but due to the large costs of procedures, often constitute a first major financial hurdle for a given nanoproduct.
8. Production scale-up and GMP-compliant synthesis
Another major hurdle to overcome in the process of approval of a nanomedicine for clinical use relates to its scale-up and production under GMP regulation. At many academic institutes, adequate facilities and expertise for scaled-up production and manufacturing under GMP are lacking, and therefore, these activities need to be outsourced to a fully licensed manufacturer capable of handling nanomedicinal products. Very often significant pharmaceutical development has to be done before a process can be scaled up and brought under GMP. A major issue with nanomedicinal products is their sterilization, where one is mostly condemned to sterile filtration through 0.2 μm filters or—if particles are around or larger than 200 nm—needs to implement an aseptic manufacturing method, which comes with its own challenges.
Besides a robust manufacturing process, also the quality control, which includes the release specifications of the product and the implementation of the full set of characterization assays has to be prepared. All assays have to be verified or qualified before GMP manufacturing. Finally one must ensure that containers, closures and packaging material are of the right quality and fully compatible with the product.
9. Preparation of regulatory dossiers for local/national authorizations of clinical trials involving nanomedicines
For all pharmaceutical/medicinal products, non-clinical and clinical information has to be compiled in the format of an Investigational Medicinal Product Dossier (IMPD) and an Investigator’s Brochure (see below). This documentation is required for clinical trial approval, as well as for the final product dossier and usually requires the specific expertise of a dedicated academic or industrial CRO.
In order to inject any nanomedicinal product (ranging from a macromolecular assembly to a complex nanoparticulate structure loaded with drug and/or contrast agent) in humans, several steps have to be followed, in accordance with the specific guidelines (manufacture of sterile medicines, manufacture of experimental drugs and manufacture of radiopharmaceuticals). The first step involves the IMPD preparation. This document compiles all information related to the drug substance (Part S) and the investigational medical product under test (Part P). The drug substance can be a natural or a synthetic compound, and the product is the nanoformulation of the drug.
Part S describes (i) the origin and the structure of the drug substance, (ii) its manufacturing process and process controls, (iii) the control of materials and critical steps, (iv) its composition and the impurities, (v) the full control of the drug substance (specifications, analytical procedures, validation of analytical procedures, batch analyses, and justification of specifications), (vi) the container closure system, and (vii) the stability under long term and accelerated storage conditions. Part P describes the nanoformulation of the drug and the pharmaceutical development, as well as the same information as in Part S except an additional specific control of excipients. For the development of a radiopharmaceutical for PET or SPECT imaging-based diagnostics, an additional IMPD Part P has to be completed. The reason for this is that the cold nanoformulation of the drug described in the IMPD Parts S and P is not the final product to be injected into humans, so that the final formulation with the added radionuclide is considered as a new medicinal product under test.
The second document to be completed is the Investigator’s Brochure composed of five chapters that assemble all the non-clinical and clinical information available about the investigational product that is relevant in the outlook of administration to human subjects. The first chapter is a short review that deals with the biological properties of the medical product and their effects in humans in accordance with the medical indications. The second chapter is a summary of the main results of the IMPD Part S and P. The third chapter contains a scientific description of all the preclinical results (in vitro and in vivo pharmacology; biodistribution, PK, and dosimetry, if necessary). In addition, a special focus is placed on a battery of toxicology studies: acute oral toxicology, extended single dose toxicity, CARPA, and genotoxicity. The fourth chapter compiles all the data obtained previously in humans with the use of the investigated medical product. The last chapter provides the investigators with a guidance summarizing the information essential for a clinical study (therapeutic indications, posology and administration route, contra-indications, special warnings, and reference safety information).
Finally, the interventional research protocol (study protocol) and a dedicated document dealing with information to be specified for clinical trials on first administration in humans (if necessary) have to be prepared. The study protocol should indicate the scientific justification for the trial, the objectives, a description of the trial, the procedure of the trial and eligibility criteria, the treatment administered to study participants, and the efficacy assessment. Furthermore, this document must contain information regarding regulatory issues such as specific committees for the trial, safety assessment (risks and restrictions added by the study), data management, statistical aspects, quality control and insurance, ethical and legal considerations, as well as funding and insurance issues.
The national competent authority issues a clinical trial authorization upon reviewing study protocol and the Investigator’s Brochure. The content and format of the protocol must comply with Community guideline on Good Clinical Practice (CPMP/ICH/135/95). In parallel, the subject information leaflet and informed consent form are prepared, informing the patients on the nature, scope and possible consequences of the study. These must be in language and terms understandable to the participants. Local authorization of clinical trials is obtained through institutional ethical committees based on the submitted study protocols, the case report forms, subject information leaflet, and informed consent forms, according to the National and European legislation (Helsinki, National codes of Public Health, the principles GCP, Bioethics law, European Dir. 95/46/CE) (Figure 5).
Figure 5.

List of regulatory documents to prepare and submit for national authorization of Phase 1 clinical trials.
10. Clinical adoption challenges
The implementation of new technologies in healthcare faces multiple challenges, including institutional interests, availability of appropriate infrastructure and clinical skills for preparing patients and their treatment, the administration and the monitoring of treatment outcomes, the enrolment of patients in the clinical trials and the evaluation and acceptance endpoints,97 and last but certainly not least, country-specific reimbursement structures and affordability. Thus, recent experience with the biologicals (PCSK9-antibodies) has taught that even highly effective and safe interventions98 will be implemented at a low pace, if the price is considered to be out of balance with the offered advantages. Studies in several EU member states concerning the attitudes of the public to nanomedicine revealed a global support, because nanotechnology in medicine is expected to bring medical progress, but the potential for safety risks is also often cited.99,100 However, the perception of risks differs very significantly between the use of industrial nanomaterials (generally of inorganic nature) and the use of medical nanomaterials. Despite the fact that the regulation, control and approval between the two categories of nanomaterials are very different, with an incomparably more stringent regulation in nanomedicine, the implementation of technologies that involve significant use of manufactured nanoparticles may face resistance from those patients who perceive nanotechnologies to be associated with unseen future risks.101 This implies a need to engage with the public and especially with patients’ organizations as the introduction of nanomedicinal products proceeds.102 Interaction should be sought already at an early stage with relevant patients’ organizations and also with medical staff who would eventually become the end users of the nanomedicines. The contact to patients' organizations can be established before or during designing clinical trials, but care is needed to ensure that the safety aspects are sensitively handled, and that sufficient support and information is provided to patients and carers.
To create a platform for dissemination among the patients’ communities and the general public, within the scope of the NanoAthero project Edinetics Ltd. developed the Democs card game entitled ‘Nanomedicine for Atherosclerosis’, which provides an inexpensive and entertaining way to engage, inform and discuss the benefits and risks of novel nanomedicines with a broader public.
11. Summary and conclusions
The potential clinical impact of nanotechnology in terms of CVD diagnosis, management and risk assessment to ultimately reduce the global disease burden cannot be overestimated. The translation of basic studies into clinical trials clearly represents the biggest challenge in this field, because developing and bringing a novel nanomedical product to the clinic is a process that involves multidisciplinary efforts of biologists, chemists, pharmacists, bio-engineers and clinicians (Figure 6), and requires strong expertise in safety issues, healthcare structures, GMP-compliant production and marketing.
Figure 6.
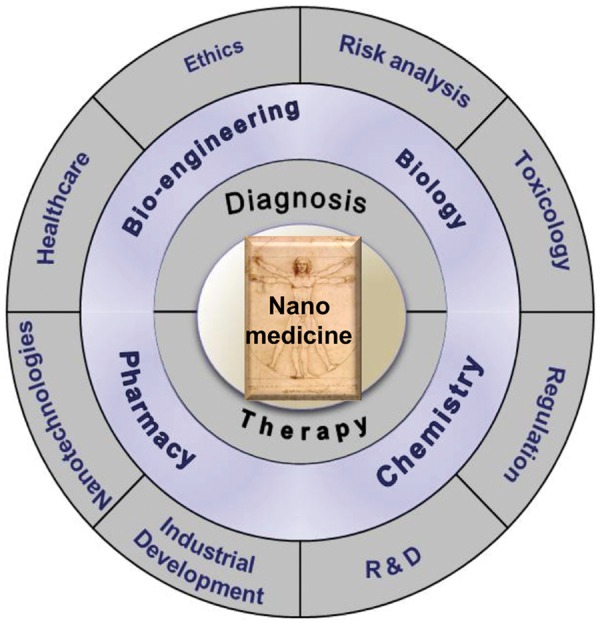
Multidisciplinarity in nanomedicine. Bringing the nanomedicinal product into the clinic requires co-operative efforts of experts from different areas of science, technology, healthcare, and industry.
Whereas about 20% of the approved nanodrugs are indicated for the treatment of cancer, according to current estimates,17,20 cardiovascular nanomedicines represent about 1% of the market. While approved anti-cancer nanomedicines generally alter the toxicological profile of the encapsulated drugs in patients, they do not really enhance local antitumor efficacy, which allegedly results from poor, erratic and heterogenous drug delivery in tumour tissues.103 Indeed, given the high medical need, the translational hurdle is generally lower for anti-cancer nanomedicines, but the absence of a real breakthrough in terms of improved drug delivery and anti-tumour effect may slow down the momentum in the development of nanomedicines for other indications. It is clear that the clinical relevance of nanomedicine, both in oncology and cardiology, will depend on rational design of particles for which sufficient delivery to target tissues can be ensured.103 For this purpose, extensive fundamental studies on nanoparticle interactions with vascular endothelium in the presence of blood cells will be essential to determine the relationship between physicochemical properties of nanoparticles and their delivery efficiency. In terms of safety, cardiovascular nanomedicine is further expected to benefit from standardized definitions and clear guidelines, but also from reliable, interference-free assays serving as nanotoxicity screening tools.
Addressing the key steps in the process of nanomedicinal product translation (Tables 1and2), this article intends to help researchers and clinicians better understand the development hurdles and regulatory requirements concerning new (nano)medicines, highlighting the tension that exists between these complexities on the one hand and the feasibility and affordability desired by the cardiovascular clinical arena on the other hand. As many early-stage innovative nanomedicine development efforts take place within academia, beyond the large R&D budgets of the pharmaceutical industry, there can only be hope that more funding options become available in the field of CVD to perform systematic basic studies concerning the mechanisms of nanoparticle transport, their interactions with cells and disease targeting efficacy that should in the future guide their improved design. Large scale funding and/or pharmaceutical industry investments will be necessary to help promising new nanomedicinal drug products reach the clinical stage in which proof of efficacy and added therapeutic benefit can really be shown in patients.
Table 1.
Translation checklist
| Development step | Performed (Y/N) | Qualified (Y/N) |
|---|---|---|
| Characterization and physicochemical evaluation | ||
| Size and charge analysis | ||
| Dispersibility analysis in complex media | ||
| Degradation products analysis | ||
| Stability and shelf life | ||
| Costs analysis | ||
| Manufacturability and scale-up | ||
| Sterisability, pyrogen content check | ||
| In vitro safety | ||
| Target cell response analysis | ||
| First-contact cell response | ||
| Haemolytic response | ||
| Immune response | ||
| Thrombogenicity analysis | ||
| In vivo evaluation | ||
| In vivo efficacy in appropriate animal model | ||
| CARPA | ||
| Biodistribution | ||
| Regulatory toxicology and PK | ||
| GMP-compliant production | ||
| Scale-up | ||
| GMP synthesis | ||
| Approval for clinical use | ||
| IMPD, preparation of regulatory dossiers | ||
| Ethical approval | ||
| Evaluation of clinical adoption readiness | ||
CARPA, complement activation-related pseudoallergy; GMP, good manufacturing practice; IMPD, investigational medicinal product dossier; PK, pharmacokinetics..
Table 2.
Barriers to translation and possible mitigation steps
| Barrier to translation | Mitigation steps |
|---|---|
| Approval for novel nanomaterials/nanoparticle use in humans is challenging compared with small molecules |
|
| Regulatory approval for diagnostic imaging agents has, to date, been limited to trace amounts |
|
| Potential toxicity of nanomaterials |
|
| Insufficient standardization between pre-clinical studies |
|
| Cost of manufacturing/upscaling production |
|
| Nanoparticles do not maintain original physiochemical/biological properties over time precluding clinical use |
|
| Prolonged bioaccumulation in organs associated with nanoparticle elimination such as liver, spleen, or kidney |
|
| Agents do not meet expectations of efficacy |
|
| Lack of clinical adoption/market value |
|
eIND, exploratory investigative new drug application.
Acknowledgements
The authors thank Dr Jasmin Matuszak for help with clinical trial search.
Conflict of interest: none declared.
Funding
This work was supported by the EU (‘NanoAthero’ project FP7-NMP-2012-LARGE-6-309820), the DFG (German Research Foundation, grant CI 162/2-2), Manfred Roth Foundation (Fürth, Germany), the British Heart Foundation grant RE/13/5/30177; the European Commission Marie Skłodowska-Curie Individual Fellowships 661369; and the Engineering and Physical Sciences Research Council (EPSRC) grant EP/L014165/1. LETI/DTBS is part of the Arcane Labex programme, funded by the French National Research Agency (ARCANE project no ANR-12-LABX-003).
References
- 1. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, AlMazroa MA, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Abdulhak AB, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FGR, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo J-P, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Memish ZA, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KMV, Nasseri K, Norman P, O'Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Pope CA, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De León FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh P-H, Yip P, Zabetian A, Zheng Z-J, Lopez AD, Murray CJL.. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin KY, Kwong GA, Warren AD, Wood DK, Bhatia SN.. Nanoparticles that sense thrombin activity as synthetic urinary biomarkers of thrombosis. ACS Nano 2013;7:9001–9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cowles CL, Zhu X.. Dual signal amplification for bioassays using ion release from nanolabels and ion-activated enzyme kinetics. Analyst 2012;137:4815–4821. [DOI] [PubMed] [Google Scholar]
- 4. Tang TY, Muller KH, Graves MJ, Li ZY, Walsh SR, Young V, Sadat U, Howarth SP, Gillard JH.. Iron oxide particles for atheroma imaging. Arterioscler Thromb Vasc Biol 2009;29:1001–1008. [DOI] [PubMed] [Google Scholar]
- 5. Richards JM, Semple SI, MacGillivray TJ, Gray C, Langrish JP, Williams M, Dweck M, Wallace W, McKillop G, Chalmers RT, Garden OJ, Newby DE.. Abdominal aortic aneurysm growth predicted by uptake of ultrasmall superparamagnetic particles of iron oxide: a pilot study. Circ Cardiovasc Imaging 2011;4:274–281. [DOI] [PubMed] [Google Scholar]
- 6. Kim DE, Kim JY, Sun IC, Schellingerhout D, Lee SK, Ahn CH, Kwon IC, Kim K.. Hyperacute direct thrombus imaging using computed tomography and gold nanoparticles. Ann Neurol 2013;73:617–625. [DOI] [PubMed] [Google Scholar]
- 7. Yilmaz A. Visualising inflammation after myocardial infarction with the use of iron oxide nanoparticles. Heart 2017;103:1479–1480. [DOI] [PubMed] [Google Scholar]
- 8. Juenet M, Aid-Launais R, Li B, Berger A, Aerts J, Ollivier V, Nicoletti A, Letourneur D, Chauvierre C.. Thrombolytic therapy based on fucoidan-functionalized polymer nanoparticles targeting P-selectin. Biomaterials 2018;156:204–216. [DOI] [PubMed] [Google Scholar]
- 9. Zhang M, He J, Jiang C, Zhang W, Yang Y, Wang Z, Liu J.. Plaque-hyaluronidase-responsive high-density-lipoprotein-mimetic nanoparticles for multistage intimal-macrophage-targeted drug delivery and enhanced anti-atherosclerotic therapy. Int J Nanomedicine 2017;12:533–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. MacRitchie N, Grassia G, Noonan J, Garside P, Graham D, Maffia P.. Molecular imaging of atherosclerosis: spotlight on Raman spectroscopy and surface-enhanced Raman scattering. Heart 2018;104:460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blázquez R, Sánchez-Margallo FM, Crisóstomo V, Báez C, Maestre J, García-Lindo M, Usón O, Álvarez V, Casado JG. Intrapericardial administration of mesenchymal stem cells in a large animal model: a bio-distribution analysis. PLoS One 2015;10:e0122377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu C, Zhang Y, Li Z, Li C, Wang Q. A novel photoacoustic nanoprobe of ICG@PEG-Ag2S for atherosclerosis targeting and imaging in vivo. Nanoscale 2016;8:12531–9. [DOI] [PubMed] [Google Scholar]
- 13. Yilmaz A, Dengler MA, van der Kuip H, Yildiz H, Rosch S, Klumpp S, Klingel K, Kandolf R, Helluy X, Hiller KH, Jakob PM, Sechtem U. Imaging of myocardial infarction using ultrasmall superparamagnetic iron oxide nanoparticles: a human study using a multi-parametric cardiovascular magnetic resonance imaging approach. Eur Heart J 2013;34:462–475. [DOI] [PubMed] [Google Scholar]
- 14. Guo RM, Cao N, Zhang F, Wang YR, Wen XH, Shen J, Shuai XT.. Controllable labelling of stem cells with a novel superparamagnetic iron oxide-loaded cationic nanovesicle for MR imaging. Eur Radiol 2012;22:2328–2337. [DOI] [PubMed] [Google Scholar]
- 15. Riegler J, Liew A, Hynes SO, Ortega D, O’Brien T, Day RM, Richards T, Sharif F, Pankhurst QA, Lythgoe MF.. Superparamagnetic iron oxide nanoparticle targeting of MSCs in vascular injury. Biomaterials 2013;34:1987–1994. [DOI] [PubMed] [Google Scholar]
- 16. Polyak B, Medved M, Lazareva N, Steele L, Patel T, Rai A, Rotenberg MY, Wasko K, Kohut AR, Sensenig R, Friedman G.. Magnetic nanoparticle-mediated targeting of cell therapy reduces in-stent stenosis in injured arteries. ACS Nano 2016;10:9559–9569. [DOI] [PubMed] [Google Scholar]
- 17. Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR.. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res 2016;33:2373–2387. [DOI] [PubMed] [Google Scholar]
- 18. Kelly B. Nanomedicines: regulatory challenges and risks ahead. Regulatory Affairs Pharma 2010;10:14–17. [Google Scholar]
- 19. Bleeker EA, de Jong WH, Geertsma RE, Groenewold M, Heugens EH, Koers-Jacquemijns M, van de Meent D, Popma JR, Rietveld AG, Wijnhoven SW, Cassee FR, Oomen AG.. Considerations on the EU definition of a nanomaterial: science to support policy making. Regul Toxicol Pharmacol 2013;65:119–125. [DOI] [PubMed] [Google Scholar]
- 20. Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J.. The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine 2013;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anselmo AC, Mitragotri S.. Nanoparticles in the clinic. Bioeng Transl Med 2016;1:10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bayarri L. Drug-device combination products: regulatory landscape and market growth. Drugs Today 2015;51:505–513. [DOI] [PubMed] [Google Scholar]
- 23. Sanna V, Pala N, Sechi M.. Targeted therapy using nanotechnology: focus on cancer. Int J Nanomedicine 2014;9:467–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chew SA, Danti S.. Based implantable devices for cancer therapy. Adv Healthcare Mater 2017;6:1600766. [DOI] [PubMed] [Google Scholar]
- 25. Chan CKW, Zhang L, Cheng CK, Yang H, Huang Y, Tian XY, Choi CHJ.. Recent advances in managing atherosclerosis via nanomedicine. Small 2018;14:1702793.. [DOI] [PubMed] [Google Scholar]
- 26. Varna M, Juenet M, Bayles R, Mazighi M, Chauvierre C, Letourneur D.. Nanomedicine as a strategy to fight thrombotic diseases. Future Sci OA 2015;1:FSO46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang J, Zu Y, Dhanasekara CS, Li J, Wu D, Fan Z, Wang S.. Detection and treatment of atherosclerosis using nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2017;9:e1412.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alaarg A, Perez-Medina C, Metselaar JM, Nahrendorf M, Fayad ZA, Storm G, Mulder WJM.. Applying nanomedicine in maladaptive inflammation and angiogenesis. Adv Drug Deliv Rev 2017;119:143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trivedi RA, Mallawarachi C, U-King-Im JM, Graves MJ, Horsley J, Goddard MJ, Brown A, Wang L, Kirkpatrick PJ, Brown J, Gillard JH.. Identifying inflamed carotid plaques using in vivo USPIO-enhanced MR imaging to label plaque macrophages. Arterioscler Thromb Vasc Biol 2006;26:1601–1606. [DOI] [PubMed] [Google Scholar]
- 30. Howarth SPS, Tang TY, Trivedi R, Weerakkody R, U-King-Im J, Gaunt ME, Boyle JR, Li ZY, Miller SR, Graves MJ, Gillard JH.. Utility of USPIO-enhanced MR imaging to identify inflammation and the fibrous cap: a comparison of symptomatic and asymptomatic individuals. Eur J Radiol 2009;70:555–560. [DOI] [PubMed] [Google Scholar]
- 31. Sadat U, Howarth SP, Usman A, Tang TY, Graves MJ, Gillard JH.. Sequential imaging of asymptomatic carotid atheroma using ultrasmall superparamagnetic iron oxide-enhanced magnetic resonance imaging: a feasibility study. J Stroke Cerebrovasc Dis 2013;22:e271–e276. [DOI] [PubMed] [Google Scholar]
- 32. Florian A, Ludwig A, Rosch S, Yildiz H, Klumpp S, Sechtem U, Yilmaz A.. Positive effect of intravenous iron-oxide administration on left ventricular remodelling in patients with acute ST-elevation myocardial infarction—a cardiovascular magnetic resonance (CMR) study. Int J Cardiol 2014;173:184–189. [DOI] [PubMed] [Google Scholar]
- 33. Alam SR, Shah AS, Richards J, Lang NN, Barnes G, Joshi N, MacGillivray T, McKillop G, Mirsadraee S, Payne J, Fox KA, Henriksen P, Newby DE, Semple SI.. Ultrasmall superparamagnetic particles of iron oxide in patients with acute myocardial infarction: early clinical experience. Circ Cardiovasc Imaging 2012;5:559–565. [DOI] [PubMed] [Google Scholar]
- 34. Smits LP, Tiessens F, Zheng KH, Stroes ES, Nederveen AJ, Coolen BF.. Evaluation of ultrasmall superparamagnetic iron-oxide (USPIO) enhanced MRI with ferumoxytol to quantify arterial wall inflammation. Atherosclerosis 2017;263:211–218. [DOI] [PubMed] [Google Scholar]
- 35. Banai S, Finkelstein A, Almagor Y, Assali A, Hasin Y, Rosenschein U, Apruzzese P, Lansky AJ, Kume T, Edelman ER.. Targeted anti-inflammatory systemic therapy for restenosis: the Biorest Liposomal Alendronate with Stenting sTudy (BLAST)-a double blind, randomized clinical trial. Am Heart J 2013;165:234–240.e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kharlamov AN, Tyurnina AE, Veselova VS, Kovtun OP, Shur VY, Gabinsky JL.. Silica-gold nanoparticles for atheroprotective management of plaques: results of the NANOM-FIM trial. Nanoscale 2015;7:8003–8015. [DOI] [PubMed] [Google Scholar]
- 37. Lobatto ME, Calcagno C, Otten MJ, Millon A, Ramachandran S, Paridaans MP, van der Valk FM, Storm G, Stroes ES, Fayad ZA, Mulder WJ, Metselaar JM.. Pharmaceutical development and preclinical evaluation of a GMP-grade anti-inflammatory nanotherapy. Nanomedicine 2015;11:1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van der Valk FM, van Wijk DF, Lobatto ME, Verberne HJ, Storm G, Willems MC, Legemate DA, Nederveen AJ, Calcagno C, Mani V, Ramachandran S, Paridaans MP, Otten MJ, Dallinga-Thie GM, Fayad ZA, Nieuwdorp M, Schulte DM, Metselaar JM, Mulder WJ, Stroes ES.. Prednisolone-containing liposomes accumulate in human atherosclerotic macrophages upon intravenous administration. Nanomedicine 2015;11:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van der Valk FM, Schulte DM, Meiler S, Tang J, Zheng KH, Van den Bossche J, Seijkens T, Laudes M, de Winther M, Lutgens E, Alaarg A, Metselaar JM, Dallinga-Thie GM, Mulder WJ, Stroes ES, Hamers AA.. Liposomal prednisolone promotes macrophage lipotoxicity in experimental atherosclerosis. Nanomedicine 2016;12:1463–1470. [DOI] [PubMed] [Google Scholar]
- 40. Almer G, Frascione D, Pali-Scholl I, Vonach C, Lukschal A, Stremnitzer C, Diesner SC, Jensen-Jarolim E, Prassl R, Mangge H.. Interleukin-10: an anti-inflammatory marker to target atherosclerotic lesions via PEGylated liposomes. Mol Pharmaceutics 2013;10:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alaarg A, Senders ML, Varela-Moreira A, Perez-Medina C, Zhao Y, Tang J, Fay F, Reiner T, Fayad ZA, Hennink WE, Metselaar JM, Mulder WJM, Storm G.. A systematic comparison of clinically viable nanomedicines targeting HMG-CoA reductase in inflammatory atherosclerosis. J Control Release 2017;262:47–57. [DOI] [PubMed] [Google Scholar]
- 42. Suzuki M, Bachelet-Violette L, Rouzet F, Beilvert A, Autret G, Maire M, Menager C, Louedec L, Choqueux C, Saboural P, Haddad O, Chauvierre C, Chaubet F, Michel JB, Serfaty JM, Letourneur D.. Ultrasmall superparamagnetic iron oxide nanoparticles coated with fucoidan for molecular MRI of intraluminal thrombus. Nanomedicine (Lond) 2015;10:73–87. [DOI] [PubMed] [Google Scholar]
- 43. Li B, Juenet M, Aid-Launais R, Maire M, Ollivier V, Letourneur D, Chauvierre C.. Development of polymer microcapsules functionalized with fucoidan to target p-selectin overexpressed in cardiovascular diseases. Adv Healthcare Mater 2017;6:1601200. [DOI] [PubMed] [Google Scholar]
- 44. Unterweger H, Janko C, Schwarz M, Dézsi L, Urbanics R, Matuszak J, Őrfi E, Fülöp T, Bäuerle T, Szebeni J, Journé C, Boccaccini AR, Alexiou C, Lyer S, Cicha I.. Non-immunogenic dextran-coated superparamagnetic iron oxide nanoparticles: a biocompatible, size-tunable contrast agent for magnetic resonance imaging. Int J Nanomedicine 2017;12:5223–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Merian J, Boisgard R, Decleves X, Theze B, Texier I, Tavitian B.. Synthetic lipid nanoparticles targeting steroid organs. J Nucl Med 2013;54:1996–2003. [DOI] [PubMed] [Google Scholar]
- 46. Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J 2012;14:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crist RM, Grossman JH, Patri AK, Stern ST, Dobrovolskaia MA, Adiseshaiah PP, Clogston JD, McNeil SE.. Common pitfalls in nanotechnology: lessons learned from NCI's Nanotechnology Characterization Laboratory. Integr Biol (Camb) 2013;5:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Coty JB, Vauthier C.. Characterization of nanomedicines: a reflection on a field under construction needed for clinical translation success. J Control Release 2018;275:254–268. [DOI] [PubMed] [Google Scholar]
- 49. Asharani PV, Lianwu Y, Gong Z, Valiyaveettil S.. Comparison of the toxicity of silver, gold and platinum nanoparticles in developing zebrafish embryos. Nanotoxicology 2011;5:43–54. [DOI] [PubMed] [Google Scholar]
- 50. Bar-Ilan O, Albrecht RM, Fako VE, Furgeson DY.. Toxicity assessments of multisized gold and silver nanoparticles in zebrafish embryos. Small 2009;5:1897–1910. [DOI] [PubMed] [Google Scholar]
- 51. Dokka S, Toledo D, Shi X, Castranova V, Rojanasakul Y.. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm Res 2000;17:521–525. [DOI] [PubMed] [Google Scholar]
- 52. McNeil SE. Nanoparticle therapeutics: a personal perspective. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2009;1:264–271. [DOI] [PubMed] [Google Scholar]
- 53. Hall JB, Dobrovolskaia MA, Patri AK, McNeil SE.. Characterization of nanoparticles for therapeutics. Nanomedicine (Lond) 2007;2:789–803. [DOI] [PubMed] [Google Scholar]
- 54. Cicha I. Strategies to enhance nanoparticle-endothelial interactions under flow. J Cell Biotechnol 2016;1:191–208. [Google Scholar]
- 55. Ta HT, Truong NP, Whittaker AK, Davis TP, Peter K.. The effects of particle size, shape, density and flow characteristics on particle margination to vascular walls in cardiovascular diseases. Expert Opin Drug Deliv 2018;15:33–45. [DOI] [PubMed] [Google Scholar]
- 56. Charoenphol P, Huang RB, Eniola-Adefeso O.. Potential role of size and hemodynamics in the efficacy of vascular-targeted spherical drug carriers. Biomaterials 2010;31:1392–1402. [DOI] [PubMed] [Google Scholar]
- 57. Thompson AJ, Mastria EM, Eniola-Adefeso O.. The margination propensity of ellipsoidal micro/nanoparticles to the endothelium in human blood flow. Biomaterials 2013;34:5863–5871. [DOI] [PubMed] [Google Scholar]
- 58. Keene AM, Peters D, Rouse R, Stewart S, Rosen ET, Tyner KM.. Tissue and cellular distribution of gold nanoparticles varies based on aggregation/agglomeration status. Nanomedicine 2012;7:199–209. [DOI] [PubMed] [Google Scholar]
- 59. Oberdörster G. Safety assessment for nanotechnology and nanomedicine: concepts of nanotoxicology. J Intern Med 2010;267:89–105. [DOI] [PubMed] [Google Scholar]
- 60. Owens DE 3rd, Peppas NA.. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm 2006;307:93–102. [DOI] [PubMed] [Google Scholar]
- 61. Zaloga J, Pottler M, Leitinger G, Friedrich RP, Almer G, Lyer S, Baum E, Tietze R, Heimke-Brinck R, Mangge H, Dorje F, Lee G, Alexiou C.. Pharmaceutical formulation of HSA hybrid coated iron oxide nanoparticles for magnetic drug targeting. Eur J Pharm Biopharm 2016;101:152–162. [DOI] [PubMed] [Google Scholar]
- 62. Corbo C, Molinaro R, Parodi A, Toledano Furman NE, Salvatore F, Tasciotti E.. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine (Lond) 2016;11:81–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barrán-Berdón AL, Pozzi D, Caracciolo G, Capriotti AL, Caruso G, Cavaliere C, Riccioli A, Palchetti S, Laganà A.. Time evolution of nanoparticle-protein corona in human plasma: relevance for targeted drug delivery. Langmuir 2013;29:6485–6494. [DOI] [PubMed] [Google Scholar]
- 64. Varenne F, Botton J, Merlet C, Beck-Broichsitter M, Legrand FX, Vauthier C.. Standardization and validation of a protocol of size measurements by dynamic light scattering for monodispersed stable nanomaterial characterization. Colloid Surface A 2015;486:124–138. [Google Scholar]
- 65. Varenne F, Botton J, Merlet C, Vachon JJ, Geiger S, Infante IC, Chehimi MM, Vauthier C.. Standardization and validation of a protocol of zeta potential measurements by electrophoretic light scattering for nanomaterial characterization. Colloid Surface A 2015;486:218–231. [Google Scholar]
- 66. Matuszak J, Baumgartner J, Zaloga J, Juenet M, da Silva AE, Franke D, Almer G, Texier I, Faivre D, Metselaar JM, Navarro FP, Chauvierre C, Prassl R, Dezsi L, Urbanics R, Alexiou C, Mangge H, Szebeni J, Letourneur D, Cicha I.. Nanoparticles for intravascular applications: physicochemical characterization and cytotoxicity testing. Nanomedicine (Lond) 2016;11:597–616. [DOI] [PubMed] [Google Scholar]
- 67. Dobrovolskaia MA, Neun BW, Clogston JD, Grossman JH, McNeil SE.. Choice of method for endotoxin detection depends on nanoformulation. Nanomedicine 2014;9:1847–1856. [DOI] [PubMed] [Google Scholar]
- 68. Kucki M, Cavelius C, Kraegeloh A.. Interference of silica nanoparticles with the traditional Limulus amebocyte lysate gel clot assay. Innate Immun 2014;20:327.. [DOI] [PubMed] [Google Scholar]
- 69. Paliwal R, Babu RJ, Palakurthi S.. Nanomedicine scale-up technologies: feasibilities and challenges. AAPS Pharmscitech 2014;15:1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Muller RH, Shegokar R, Keck CM.. 20 years of lipid nanoparticles (SLN and NLC): present state of development and industrial applications. Curr Drug Discov Technol 2011;8:207–227. [DOI] [PubMed] [Google Scholar]
- 71. Friedrich RP, Janko C, Poettler M, Tripal P, Zaloga J, Cicha I, Durr S, Nowak J, Odenbach S, Slabu I, Liebl M, Trahms L, Stapf M, Hilger I, Lyer S, Alexiou C.. Flow cytometry for intracellular SPION quantification: specificity and sensitivity in comparison with spectroscopic methods. Int J Nanomedicine 2015;10:4185–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Unterweger H, Dezsi L, Matuszak J, Janko C, Poettler M, Jordan J, Bauerle T, Szebeni J, Fey T, Boccaccini AR, Alexiou C, Cicha I.. Dextran-coated superparamagnetic iron oxide nanoparticles for magnetic resonance imaging: evaluation of size-dependent imaging properties, storage stability and safety. Int J Nanomedicine 2018;13:1899–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Beer C. Nanotoxicology and regulatory affairs In: Howard KA, Vorup-Jensen T, Peer D (eds). Nanomedicine. New York: Springer; 2016. pp. 279–310. [Google Scholar]
- 74. Alaarg A, Zheng KH, van der Valk FM, da Silva AE, Versloot M, van Ufford LC, Schulte DM, Storm G, Metselaar JM, Stroes ES, Hamers AA.. Multiple pathway assessment to predict anti-atherogenic efficacy of drugs targeting macrophages in atherosclerotic plaques. Vascul Pharmacol 2016;82:51–59. [DOI] [PubMed] [Google Scholar]
- 75. Janikowska A, Matuszak J, Lyer S, Schreiber E, Unterweger H, Zaloga J, Groll J, Alexiou C, Cicha I.. A novel human artery model to assess the magnetic accumulation of SPIONs under flow conditions. Sci Rep 2017;7:42314.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Saboural P, Chaubet F, Rouzet F, Al-Shoukr F, Azzouna RB, Bouchemal N, Picton L, Louedec L, Maire M, Rolland L, Potier G, Guludec DL, Letourneur D, Chauvierre C.. Purification of a low molecular weight fucoidan for SPECT molecular imaging of myocardial infarction. Mar Drugs 2014;12:4851–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dingley KH, Roberts ML, Velsko CA, Turteltaub KW.. Attomole detection of 3H in biological samples using accelerator mass spectrometry: application in low-dose, dual-isotope tracer studies in conjunction with 14C accelerator mass spectrometry. Chem Res Toxicol 1998;11:1217–1222. [DOI] [PubMed] [Google Scholar]
- 78. Hakimzadeh N, Pinas VA, Molenaar G, de Waard V, Lutgens E, van Eck-Smit BLF, de Bruin K, Piek JJ, Eersels JLH, Booij J, Verberne HJ, Windhorst AD.. Novel molecular imaging ligands targeting matrix metalloproteinases 2 and 9 for imaging of unstable atherosclerotic plaques. PLoS One 2017;12:e0187767.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pérez-Medina C, Binderup T, Lobatto ME, Tang J, Calcagno C, Giesen L, Wessel CH, Witjes J, Ishino S, Baxter S, Zhao Y, Ramachandran S, Eldib M, Sánchez-Gaytán BL, Robson PM, Bini J, Granada JF, Fish KM, Stroes ESG, Duivenvoorden R, Tsimikas S, Lewis JS, Reiner T, Fuster V, Kjær A, Fisher EA, Fayad ZA, Mulder WJM.. In vivo PET imaging of HDL in multiple atherosclerosis models. JACC Cardiovasc Imaging 2016;9:950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Williams J, Sayles HR, Meza JL, Sayre P, Sandkovsky U, Gendelman HE, Flexner C, Swindells S.. Long-acting parenteral nanoformulated antiretroviral therapy: interest and attitudes of HIV-infected patients. Nanomedicine (Lond) 2013;8:1807–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mei L, Zhang Z, Zhao L, Huang L, Yang XL, Tang J, Feng SS.. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv Drug Deliv Rev 2013;65:880–890. [DOI] [PubMed] [Google Scholar]
- 82. Ong KJ, MacCormack TJ, Clark RJ, Ede JD, Ortega VA, Felix LC, Dang MKM, Ma GB, Fenniri H, Veinot JGC, Goss GG.. Widespread nanoparticle-assay interference: implications for nanotoxicity testing. PLoS One 2014;9:e90650.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kroll A, Pillukat MH, Hahn D, Schnekenburger J.. Interference of engineered nanoparticles with in vitro toxicity assays. Arch Toxicol 2012;86:1123–1136. [DOI] [PubMed] [Google Scholar]
- 84. Millon A, Canet-Soulas E, Boussel L, Fayad Z, Douek P.. Animal models of atherosclerosis and magnetic resonance imaging for monitoring plaque progression. Vascular 2014;22:221–237. [DOI] [PubMed] [Google Scholar]
- 85. Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; and Council on Basic Cardiovascular Sciences. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 86. Decano JL, Viereck JC, McKee AC, Hamilton JA, Ruiz-Opazo N, Herrera VL.. Early-life sodium exposure unmasks susceptibility to stroke in hyperlipidemic, hypertensive heterozygous Tg25 rats transgenic for human cholesteryl ester transfer protein. Circulation 2009;119:1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yuan F, Guo L, Park KH, Woollard JR, Taek-Geun K, Jiang K, Melkamu T, Zang B, Smith SL, Fahrenkrug SC, Kolodgie FD, Lerman A, Virmani R, Lerman LO, Carlson DF.. Ossabaw pigs with a PCSK9 gain-of-function mutation develop accelerated coronary atherosclerotic lesions: a novel model for preclinical studies. J Am Heart Assoc 2018;7:e006207.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bentzon JF, Falk E.. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol 2010;21:434–440. [DOI] [PubMed] [Google Scholar]
- 89. van der Tuin SJ, Kuhnast S, Berbee JF, Verschuren L, Pieterman EJ, Havekes LM, van der Hoorn JW, Rensen PC, Jukema JW, Princen HM, Willems van Dijk K, Wang Y.. Anacetrapib reduces (V)LDL cholesterol by inhibition of CETP activity and reduction of plasma PCSK9. J Lipid Res 2015;56:2085–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Beere PA, Glagov S, Zarins CK.. Experimental atherosclerosis at the carotid bifurcation of the cynomolgus monkey. Localization, compensatory enlargement, and the sparing effect of lowered heart rate. Arterioscler Thromb 1992;12:1245–1253. [DOI] [PubMed] [Google Scholar]
- 91. Granada JF, Kaluza GL, Wilensky RL, Biedermann BC, Schwartz RS, Falk E.. Porcine models of coronary atherosclerosis and vulnerable plaque for imaging and interventional research. EuroIntervention 2009;5:140–148. [DOI] [PubMed] [Google Scholar]
- 92. Sukhova GK, Williams JK, Libby P.. Statins reduce inflammation in atheroma of nonhuman primates independent of effects on serum cholesterol. Arterioscler Thromb Vasc Biol 2002;22:1452–1458. [DOI] [PubMed] [Google Scholar]
- 93. Szebeni J. Complement activation-related pseudoallergy: a stress reaction in blood triggered by nanomedicines and biologicals. Mol Immunol 2014;61:163–173. [DOI] [PubMed] [Google Scholar]
- 94. Szebeni J, Bedőcs P, Csukás D, Rosivall L, Bünger R, Urbanics R.. A porcine model of complement-mediated infusion reactions to drug carrier nanosystems and other medicines. Adv Drug Deliv Rev 2012;64:1706–1716. [DOI] [PubMed] [Google Scholar]
- 95. Armstrong JK. The occurrence, induction, specificity and potential effect of antibodies against poly(ethylene glycol) In: Veronese FM (ed). PEGylated Protein Drugs: Basic Science and Clinical Applications. Cham, Switzerland: Springer Nature; 2009. pp. 147–168. [Google Scholar]
- 96. Ishida T, Kiwada H.. Anti-polyethyleneglycol antibody response to PEGylated substances. Biol Pharm Bull 2013;36:889–891. [DOI] [PubMed] [Google Scholar]
- 97. Gardner J, Webster A, Barry J.. Anticipating the clinical adoption of regenerative medicine: building institutional readiness in the UK. Regen Med 2018;13:29–39. [DOI] [PubMed] [Google Scholar]
- 98. Sabatine MS, Leiter LA, Wiviott SD, Giugliano RP, Deedwania P, De Ferrari GM, Murphy SA, Kuder JF, Gouni-Berthold I, Lewis BS, Handelsman Y, Pineda AL, Honarpour N, Keech AC, Sever PS, Pedersen TR.. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol 2017;5:941–950. [DOI] [PubMed] [Google Scholar]
- 99. Bottini M, Rosato N, Gloria F, Adanti S, Corradino N, Bergamaschi A, Magrini A.. Public optimism towards nanomedicine. Int J Nanomedicine 2011;6:3473–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bruce D. Ethical implications of nanomedicine: implications of imaging new futures for medicine In: Ge Y, Li S, Wang S, Moore R (eds). Nanomedicine: Principles and Perspectives. New York: Springer; 2014. pp. 251–269. [Google Scholar]
- 101. Bruce D. Ethical and social issues in nanobiotechnologies: nano2Life provides a European ethical ‘think tank’ for research in biology at the nanoscale. EMBO Rep 2006;7:754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kahan DM, Slovic P, Braman D, Gastil J, Cohen GL.. Affect, Values, and Nanotechnology Risk Perceptions: An Experimental Investigation. SSRN Electronic Journal 2007; doi:10.2139/ssrn.968652. [Google Scholar]
- 103. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW.. Analysis of nanoparticle delivery to tumours. Nat Rev Mater 2016;1:16014. [Google Scholar]