

Abstract
Adequate supply of blood and structural and functional integrity of blood vessels is key to normal brain functioning. On the other hand, cerebral blood flow (CBF) shortfalls and blood-brain barrier (BBB) dysfunction are early findings in neurodegenerative disorders in humans and animal models. Here, we first examine molecular definition of cerebral blood vessels, and pathways regulating CBF and BBB integrity. Then, we examine the role of CBF and BBB in the pathogenesis of Alzheimer’s disease (AD), Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis and multiple sclerosis. We focus on AD as a platform of our analysis because more is known about neurovascular dysfunction in this disease than in other neurodegenerative disorders. Finally, we propose a hypothetical model of AD biomarkers to include brain vasculature as a factor contributing to the disease onset and progression, and suggest a common pathway linking brain vascular contributions to neurodegeneration in multiple neurodegenerative disorders.
Introduction
Neurons work hard. To keep the 86 billion neurons in the human brain working properly requires an adequate supply of blood, which is accomplished through a vast, well-regulated vascular network of arteries, arterioles, capillaries, venules and veins reaching approximately 400 miles in length1,2. Several cell types work in concert to regulate cerebral blood flow (CBF) and maintain blood-brain barrier (BBB) integrity. This assortment of cells, collectively called the neurovascular unit (NVU), is comprised of endothelial cells forming the inner layer of the vessel walls, mural cells along the vessels that help regulate vascular tone (pericytes and vascular smooth muscle cells; SMCs), astrocytes whose endfeet cover most of the vasculature, and neurons1,2. The NVU cellular composition varies along the vascular tree, with rubber band-like SMCs wrapping vessels at the arterial and arteriole level, pericytes along capillaries, and “stellate” SMCs along venules3 (Figure 1a).
Figure 1. Molecular definition of the blood-brain barrier and cerebral blood vessels.
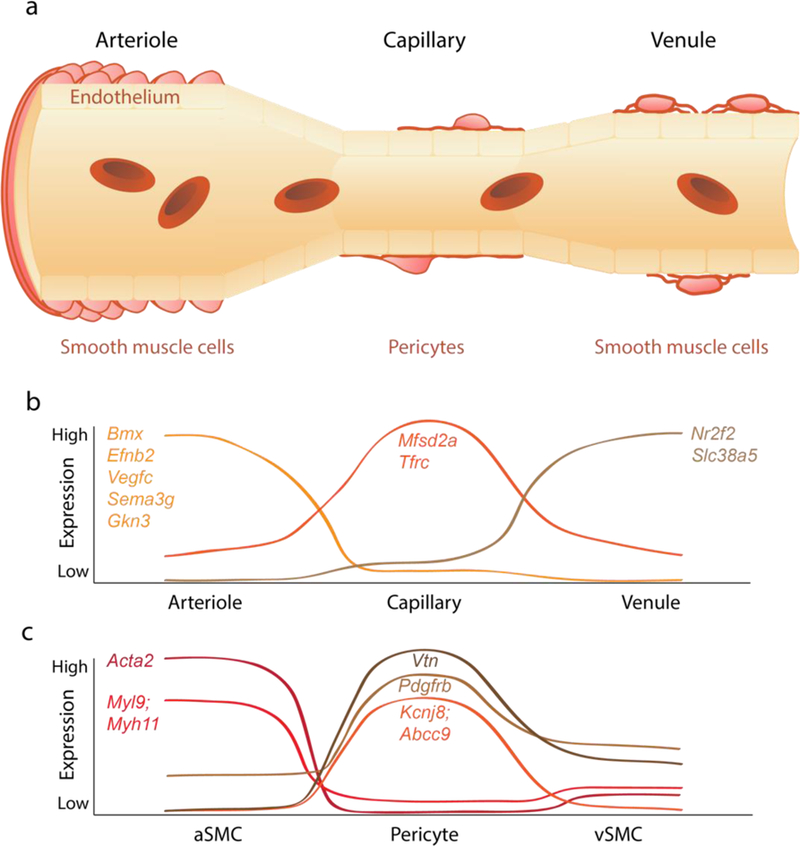
(a) The brain vasculature is a continuum from artery to arteriole to capillary to venule to vein. The blood-brain barrier is formed by a continuous endothelium monolayer surrounded by mural cells. Vascular ‘zonation’ refers to the molecular and phenotypic changes along the vascular endothelial continuum. Molecularly, the endothelium is a gradual continuum enriched with cell-specific markers at the arterial/arteriolar, capillary and venule/veins levels. Mural cells also cluster at different vascular segments: smooth muscle cells (SMCs) at arterioles and venules and pericytes at capillaries. (b) Representative curves showing molecular expression patterns of endothelial cells. The arteriole-specific genes include Bmx, Efnb2, Vegfc, Sema3g, and Gkn3, capillary-specific genes include Mfsd2a and Tfrc, and venule-specific genes include Nr2f2 and Slc38a5. (c) Representative curves showing molecular expression patterns of mural cells. Arteriole SMCs enriched genes include Acta2, Myl9, and Myh11. Capillary pericyte enriched genes include Vtn, Pdgfrβ, Kcnj8, and Abcc9. The molecular characterization is informed from recent single cell RNA-sequencing studies in multiple murine models7. Arteriole markers represent averaged artery and arteriole expression. See main text for details.
A continuous endothelium monolayer forms the BBB, which maintains cerebrovascular integrity through continuous cross-talk between endothelium, mural cells, astrocytes and neurons. The BBB restricts paracellular and transcellular endothelial transport of most blood-derived macromolecules, blood cells (e.g., leukocytes) and microbial pathogens between blood and brain, and is present at all levels of the vascular tree from arteries and arterioles to capillaries to venules and veins4,5. The largest surface area of the BBB (> 85%) is provided by the capillary endothelium6. Numerous transport systems expressed mainly in brain capillary endothelium and venule endothelium7 facilitate or actively shuttle molecules across BBB, as reviewed elsewhere4.
Here, we first examine molecular definition of cerebral blood vessels, cell types and zonation in the brain vasculature. Next, we review key pathways along the vascular tree regulating CBF and BBB integrity. Then, we discuss the role of CBF and BBB in the pathogenesis of neurodegenerative changes in humans in different neurodegenerative disorders concentrating on recent neuroimaging data. We focus on Alzheimer’s disease (AD) because more is known about vascular dysfunction in this disease than in other neurodegenerative disorders. Finally, we propose an updated hypothetical model of AD biomarkers8 to incorporate brain vascular changes as an important factor of the disease onset and progression, and suggest a common pathway linking brain vascular changes to neurodegeneration in multiple neurodegenerative disorders.
Molecular definition of cerebral blood vessels
Endothelial zonation.
The concept of vascular ‘zonation’ landmarks was recently introduced based on single cell RNA-sequencing of brain vascular cell types isolated from different murine models suggesting molecular and functional phenotypic differences along the vasculature7 (Figure 1b). Single endothelial cells isolated from Cldn5-GFP reporter mice with green fluorescent protein (GFP) expression driven by claudin-5, an endothelial tight junction protein, and transcriptional data allowed endothelial cell clustering into groups based on transcriptomic (dis)similarity. For example, arterial vascular zonation could be identified with arterial-specific endothelial markers, Bmx [encoding BMX non-receptor tyrosine kinase], Efnb2 [encoding ephrin B2], Vegfc [encoding vascular endothelial growth factor C], Sema3g [encoding semaphorin 3G], and Gkn3 [encoding gastrokine-3], and venous vascular zonation with venous-specific endothelial markers, Nr2f2 [encoding nuclear receptor subfamily 2 group F member 2] and Slc38a5 [encoding a sodium-dependent amino acid transporter]7. Analysis of the gradual arterial-to-venous (A-V) zonation reveals endothelial genes that peaked in the middle, representing the capillary enriched genes Mfsd2a [major facilitator superfamily domain-containing protein 2a (MFSD2a)] and Tfrc [transferrin receptor]7. MFSD2a is an essential omega-3 fatty acid transporter9 required for BBB formation and omega-3 fatty acid transport function10, and transferrin receptor is important for brain delivery of iron4. For the first time, this novel approach enables studies of transcriptional expression with respect to vascular zonation revealing molecular and functional differences along the brain vascular tree. For example, transcription factors predominated at the arterial endothelium, whereas transporters predominated at the capillary and venous endothelium. Some endothelial cells did not fit the A-V zonation pattern exhibiting high expression of ribosomal protein transcripts, which indicates that protein synthesis occurs throughout the A-V axis7.
Mural cell pattern.
In contrast to the endothelium with gradual zonation, mural cells exhibited a segregated zonation pattern. Isolated from Pdgfrb-GFP; Cspg4-DsRed mice, mural cells transcriptionally separated into two distinct groups including pericytes and venous SMCs (vSMCs), and arterial and arteriolar SMCs (aSMCs)7. Clustering analysis revealed pericyte-enriched genes, Pdgfrβ [encoding platelet-derived growth factor receptor-β], Cspg4 [encoding chondroitin sulfate proteoglycan neuron-glial antigen 2 (NG2)], Anpep [encoding N-aminopeptidase CD13], Rgs5 [encoding regulator of G protein signaling 5], Abcc9 [encoding SUR2 subunit of K+-ATP channel], Kcnj8 [encoding Kir6.1], Vtn [encoding vitronectin], and Ifitm1 [encoding interferon-induced transmembrane protein 1], consistent with reported literature7,11. aSMCs enriched genes included Acta2 [encoding alpha smooth muscle actin, α-SMA], Tagln [encoding transgelin], Myh11 [encoding myosin heavy chain 11], Myl9 [encoding myosin light chain 9], Mylk [encoding myosin light chain kinase], Sncg [encoding synuclein gamma], Cnn1 [encoding calponin-1], and Pln [encoding phospholamban]7 (Figure 1c).
In contrast to aSMCs, pericytes express barely detectable levels of Acta2 encoding α-SMA7, a protein that plays a key role in cell contractile apparatus, which would argue against pericyte role in contractility and CBF regulation. However, using strategies that allow rapid filamentous-actin (F-actin) fixation or prevent F-actin depolymerization, it has been recently shown that pericytes on mouse retinal capillaries, including those in intermediate and deeper plexus, express α-SMA12. Junctional pericytes were more frequently α-SMA-positive compared to pericytes on linear capillary segments. Additionally, short interfering α-SMA-siRNA suppressed α-SMA expression preferentially in high order branch capillary pericytes, confirming the existence of a smaller pool of α-SMA in distal capillary pericytes that is quickly lost by depolymerization12. Recent RNA-seq studies also indicated that pericytes express moderate-to-robust levels of other contractile proteins including Des (desmin) and Cnn2 (calponin-2)7 and Myl9 (myosin light chain 9)11. How and whether these contractile proteins contribute to the contractile apparatus in pericytes remains to be determined. Nevertheless, beyond expression of contractile proteins, more functional studies are needed to determine how exactly pericytes contribute to CBF regulation, as discussed in greater detail below.
Pathways regulating CBF along the vascular tree
The NVU of the mammalian brain is functionally integrated to regulate CBF responses to neuronal stimulation in a process called neurovascular coupling or functional hyperemia1,2, which ensures a rapid increase in CBF and oxygen delivery to activated brain regions. Mural cells can depolarize and contract, or hyperpolarize and relax in response to different stimuli, to either constrict or dilate blood vessels, respectively, which in turn decreases or increases blood flow. SMCs regulate arteriolar and arterial vessel diameter1. Most in vivo rodent studies in the brain and retina, as well as studies using cortical and cerebellar slices and retinal explants suggest that pericytes regulate blood flow at the capillary level13–20. However, this has not been confirmed by some in vivo studies in the mouse cortex21,22, leaving this subject still as a matter of controversy. An exact explanation for these conflicting reports remains unclear at present, but could reflect technical challenges or underlying properties of the studied models that should be clarified by future studies. As pointed out in a recent review1, the evidence to date also does not rule out a role of capillaries (and pericytes) in the retrograde propagation of intramural vascular signals from capillary level upstream1,23,24. Given that the vast majority of brain vasculature is composed of capillaries (~90% in mouse cortex)13,25, capillaries and pericytes are well positioned to regulate CBF throughout the brain parenchyma. Figure 2 illustrates key cellular and molecular pathways regulating CBF.
Figure 2. Key cellular and molecular pathways regulating cerebral blood flow.
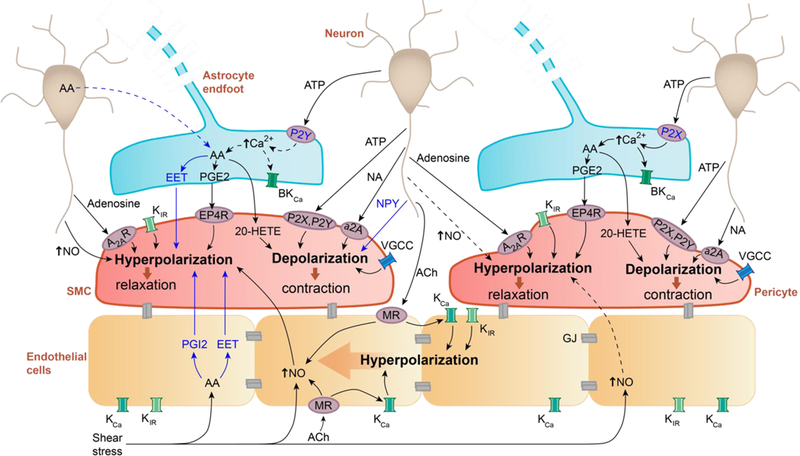
Neuron-mural cells crosstalk. ATP and noradrenaline (NA) released by neurons act on smooth muscle cells (SMCs) and pericytes through adenosine A2A receptors (A2AR) or α2-adrenergic receptors (α2A), respectively, causing cell depolarization and constriction, which reduces blood flow. Adenosine acts via purinergic P2X and P2Y receptors to hyperpolarize SMCs and pericytes, which increases blood flow. Neuropeptide Y (NPY) causes SMCs contraction. In both SMCs and pericytes, nitric oxide (NO) produced by neurons leads to hyperpolarization resulting in blood flow increase. Pericyte response to NO may vary by brain region, indicated by dashed arrows. Extracellular potassium ions (K+) released during neuronal activation can act on K+ (inward rectifier, KIR) and Ca2+ (Voltage-gated, VGCC) channels in SMCs and pericytes to hyperpolarize and relax the cells, or depolarize and contract cells. Astrocyte-mural cells crosstalk. ATP acts on P2X or P2Y receptors on astrocytes, which according to some studies can increase intracellular [Ca2+]. However, the role of arteriolar astrocyte [Ca2+] changes remains debatable (indicated by dashed arrows; see text). [Ca2+] increase triggers production of arachidonic acid (AA) and its metabolites (prostaglandin E2, PGE2, through PGE2 receptor EP4, EP4R; 20-hydroxyeicosatetraenoic acid; 20-HETE; epoxyeicosatetraenoic acids, EETs) that act on SMCs and pericytes to regulate blood flow. Alternatively, neurons may release AA to be further metabolized by astrocytes, indicated by dashed line. Endothelial-mural cells crosstalk. Acetylcholine (ACh) released from neurons or blood-derived ACh act on endothelial muscarinic ACh receptors (MRs) to increase endothelial NO production causing hyperpolarization and relaxation of mural cells, which increases blood flow. Shear stress can also increase NO endothelial production as well as production of AA and metabolites EETs and prostacyclin (PGI2) that hyperpolarize and relax SMCs, increasing arteriolar blood flow. Extracellular [K+] increase or ACh can activate KIR or calcium-activated K+ (KCa) channels on endothelial cells, leading to endothelial hyperpolarization that can propagate via gap junctions (GJs) between endothelial cells in a retrograde direction to increase blood flow. Altogether these findings are informed from various CNS regions and from both in vivo and in vitro studies; see main text for details.
Briefly, neurons signal the vasculature through transmitters adenosine triphosphate (ATP) and adenosine that act on their receptors - purinergic P2X and P2Y for ATP (also expressed on other CNS cell types7,11) and adenosine A2A for adenosine - on SMCs and pericytes causing depolarization and cell contraction that reduces blood flow, or hyperpolarization and cell relaxation that increases blood flow, as shown in vivo in cortex and retina, and ex vivo in cortical slices and whole mount retina2,13,26. Noradrenaline contracts SMCs ex vivo in isolated retina and in vivo in cortical arterioles23 and pericytes ex vivo in rat cerebellar slices and isolated retina14, whereas neuropeptide Y contracts cortical arterioles in vivo27. Neuronal nitric oxide (NO) can hyperpolarize and relax SMCs and pericytes increasing blood flow in vivo and ex vivo2,15,20. Although it has been initially suggested that NO can act by blocking 20-hydroxyeicosatetraenoic acid (20-HETE) production, this mechanism may play a more significant role in autoregulation of vascular tone than neurovascular coupling28. In contrast, a recent study suggested that dilation of arterioles depends on NMDA receptor activation and Ca2+-dependent NO generation by interneurons, whereas the dilation of capillary pericytes in mouse cortex is mediated by local Ca2+ elevations in glial processes and endfeet along capillaries16 although regional differences may exist15–17,20,29. These findings indicate that different signaling cascades regulate CBF at the capillary and arteriole levels. Neuronal activity elevates extracellular potassium ([K+]) directly and/or via release from astrocytes (as an effect of neuronal ATP stimulation, see below), which acts on various K+ and Ca2+ channels to alter pericyte and SMCs tone, leading to alterations in blood flow as shown in brain and retina in vivo2,29 (Figure 2).
Astrocytes also respond to neuronal ATP through P2X or P2Y receptors by increasing intracellular calcium concentration ([Ca2+]) (similar to microglia and oligodendrocytes7,11), as shown in live and whole mount retina26, which triggers production of arachidonic acid (AA) and its vasoactive metabolites (20-HETE; prostaglandin E2, PGE2; and epoxyeicosatetraenoic acids, EETs) as shown in vivo in cortex and retina and ex vivo in cortical and cerebellar slices and retinal explants2,15,16,29, and secretion of K+. Mural cells generate 20-HETE from AA causing cell depolarization and blood flow reduction, while PGE2 hyperpolarizes SMCs and pericytes (via PGE2 receptor EP4, EP4R) increasing vessel diameter, as shown in vivo in cortex and retina and ex vivo in cortical and cerebellar slices and retinal explants2,15,16,29. The role of astrocytes in CBF regulation has been, however, a matter of controversy, stemming from conflicting data as to whether astrocyte [Ca2+] can increase in response to stimulus, and whether if any [Ca2+] increase can occur rapidly enough to influence neurovascular coupling29 (Figure 2). Additional studies are needed to resolve this controversy.
Endothelial cells respond to acetylcholine (ACh) from the blood stream via muscarinic ACh receptors30,31, and blood flow shear stress to generate NO23, which increases blood flow by relaxing pericytes and SMCs, as described above. ACh also causes NO-independent endothelial hyperpolarization23. Shear stress triggers production of AA and AA-derived metabolites (EETs; prostacyclin, PGI2), which act on SMCs to increase blood flow23. Extracellular [K+] elevations can hyperpolarize endothelium at both the capillary and arteriole level, which can propagate in a retrograde direction via gap junctions shared between endothelial cells and endothelial-mural cell junctions, leading to relaxation and dilation of upstream vasculature to increase blood flow1,2,23. These endothelial pathways have been described ex vivo in various rodent aortic and brain arteriole ring preparations, and in vivo in cortex and cerebellum, but further validation studies in brain are needed.
Expression of all major receptors, ion channels and other key proteins in SMCs, pericytes, endothelial cells and astrocytes contributing to CBF regulation, as shown in Figure 2, has been supported by recent single cell RNA-seq analysis of the brain vasculature in murine models7.
Pathways regulating BBB integrity along the vascular tree
Formation and maintenance of the BBB is accomplished through expression of tight junctions (e.g., claudins, occludin, zona occludens) and adherens junctions (e.g., vascular endothelial (VE)-cadherin, platelet endothelial cell adhesion molecule (PECAM-1), connexins) connecting neighboring endothelial cells, as well as the paucity of trans-endothelial bulk flow transcytosis4. Gap junctions between endothelial cells, pericytes and astrocytes contribute to cerebrovascular integrity. Upholding endothelial barrier is essential for specialized transport properties and functions of BBB, as reviewed elsewhere4. Figure 3 summarizes key cellular and molecular pathways underlying BBB establishment and maintenance.
Figure 3. Key cellular and molecular pathways regulating blood-brain barrier integrity.
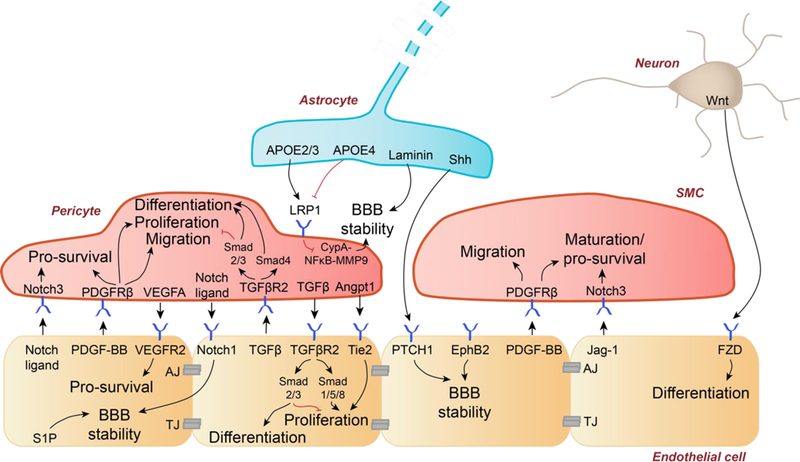
BBB integrity is maintained by tight junction (TJ) and adherens junction (AJ) proteins between endothelial cells and low-level bulk flow transcytosis. Pericyte-endothelial cells crosstalk: Notch ligands-Notch3 receptor signaling promotes pericyte survival. Platelet-derived growth factor-BB (PDGF-BB) binds to PDGFRβ on pericytes causing pericyte survival, proliferation, and migration. Vascular endothelial growth factor-A (VEGFA) binds to endothelial VEGF receptor-2 (VEGFR2) mediating endothelial survival. Pericyte-derived notch ligands bind to endothelial Notch1 receptor which mediates BBB stability, as does endothelial sphingosine-1 phosphate (S1P). Transforming growth factor-β (TGFβ) and TGFβ receptor-2 (TGFβR2) signaling occurs bi-directionally between pericytes and endothelial cells. Pericyte-secreted angiopoietin-1 (Angpt1) binds Tie2 receptor on endothelial cells to promote proliferation. Astrocyte-endothelial cells crosstalk: Astrocyte-secreted APOE2 and APOE3, in contrast to APOE4, suppresses the pro-inflammatory signaling cyclophilin A-NFkB-matrix metalloproteinase-9 (MMP9) pathway in pericytes to maintain BBB stability. Similarly, astrocyte-produced laminin maintains BBB stability. Astrocyte-secreted sonic hedgehog (Shh) interacts with patched-1 (PTCH1) at the endothelium to further promote BBB stability. Smooth muscle cell (SMC)-endothelial cells crosstalk: Ephrin B2 (EphB2) on the endothelium promotes BBB stability. PDGF-BB binds PDGFRβ on SMCs to promote survival and migration. Endothelial-secreted jagged-1 (Jag-1) binds Notch3 to promote SMC maturation and survival. Neuron-endothelial cells crosstalk. Neuron secreted Wnt is a ligand of frizzled (FZD) at the endothelium that promotes endothelial cell differentiation.
Studies using pericyte-deficient murine models with reduced endothelial-derived platelet-derived growth factor-BB (PDGF-BB) bioavailability or deficient PDGF receptor-β (PDGFRβ) signaling in pericytes, have suggested that pericytes regulate BBB formation and maintenance at the level of brain capillaries5,32–35. PDGF-BB is a ligand for PDGFRβ on pericytes, which controls pericyte survival, proliferation, and migration32–36 (Figure 3, left). Endothelial-secreted Notch ligands signal via Notch3 receptor in pericytes to promote pericyte survival5. Similarly, pericyte-derived Notch ligands bind to endothelial Notch1 receptor, which promotes N-cadherin synthesis stabilizing BBB5. Vascular endothelial growth factor-A (VEGF-A) signals VEGF receptor-2 (VEGFR2) in capillary endothelium reinforcing cell survival5. Transforming growth factor-β (TGFβ) and TGFβ receptor-2 (TGFβR2) signaling between pericytes and endothelial cells regulates endothelial proliferation and differentiation and pericytes proliferation and migration5 (Figure 3, left).
Ephrin type B receptor 2 (EphB2) expressed by arterial endothelial cells regulates SMC recruitment to the developing vessels contributing to cerebrovascular integrity, which is indirectly indicative of BBB stability4,37. Loss of EphB2 disrupts mural cell coverage causing BBB breakdown37. Ablation of astrocytic laminin leads to BBB breakdown38. Apolipoprotein E (APOE) isoforms, secreted by astrocytes regulate BBB integrity by signaling the low-density lipoprotein receptor-related protein-1 (LRP1) on pericytes39. Additionally, neuron-secreted Wnt, a ligand of frizzled (FZD) receptor on endothelium, promotes endothelial cell differentiation during brain vasculogenesis40,41.
Expression of all major receptors and key proteins in endothelial cells, pericytes, SMCs and astrocytes regulating BBB integrity, as shown in Figure 3, has been supported by recent single cell RNA-seq analysis of the brain vasculature in murine models7. Besides contributing to BBB formation, these pathways continue to play an important role in BBB maintenance in the adult and aging brain.
Gap between basic, translational and human studies of the brain vasculature
The molecular and cellular mechanisms regulating CBF and BBB integrity, as discussed in the first half of this Review, have been extensively examined in animal models generating important basic physiological data. However, these mechanisms have not been studied in great detail in models of neurodegenerative diseases that show early neurovascular dysfunction6, nor in humans with early CBF and BBB deficits that are consistently observed in different neurodegenerative diseases2,4,6,42–45. Whether the pathways and molecular makeup of the human brain vasculature can reproduce findings in animal models remains unclear, illustrating a major gap between animal models and human studies. Nevertheless, knowledge from animal studies offers a roadmap to examine the link between neurovascular dysfunction and neurodegeneration in humans.
Neurovascular dysfunction is increasingly recognized in AD1,2,4,42–48, besides classical pathological hallmarks of the disease including amyloid-β (Aβ) plaques, hyperphosphorylated tau neurofibrillary tangles, and neuronal loss8. Neuropathological studies show that cerebrovascular pathology contributes to dementia and clinically diagnosed AD46 and lowers the threshold for dementia due to AD47. Similarly, vascular risk factors also lower the threshold and act synergistically with Aβ burden to promote cognitive decline49. Neurovascular dysfunction is also seen as an early event in AD, influenced by genetic, lifestyle and environmental factors, as demonstrated by vascular biomarkers studies42,44,45,50, and discussed in sections to follow.
In addition to AD, the brain vasculature has been implicated in the pathogenesis of frontotemporal dementia, Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS) and other neurodegenerative conditions such as HIV-induced neurocognitive disorder, and chronic traumatic encephalopathy1,5,45. Here, we focus on AD because more is known about neurovascular dysfunction in this disease than in other neurodegenerative disorders, but also discuss the emerging role of neurovascular dysfunction in PD, HD, ALS and MS. We concentrate on CBF and BBB neuroimaging biomarkers that have advanced the field in recent years establishing early role of brain vasculature in multiple neurodegenerative disorders.
Cerebral blood flow in Alzheimer’s disease
CBF reductions, impaired cerebrovascular reactivity and impaired hemodynamic responses are increasingly recognized in early stages of AD and across normal aging-to-mild cognitive impairment (MCI)-to-AD spectrum (Table 1). However, no evidence to date shows that CBF deficits are causal to AD. Below, we examine studies showing that CBF is a useful biomarker in preclinical AD.
Table 1.
Cerebral blood flow deficits in the normal aging-to-mild cognitive impairment-to-Alzheimer’s disease spectrum studied with neuroimaging.
| Affected CNS regions | Disease Stages |
Methods | References | CBF biomarker prior to |
References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NCI | MCI | Early AD |
AD | Brain atrophy |
Amyloid | Tau | |||||
| CBF reductions | |||||||||||
| Middle cerebral artery territory | C | TCD | 51 | Yes | Yes | — | 2,42,43,51,53,65,66 | ||||
| Precuneus, posterior cingulate, parietotemporal, frontal and occipital cortices, parahippocampal gyrus, hippocampus, caudate nucleus, thalamus |
C | C | ASL-MRI | 56 | |||||||
| C | C | 55 | |||||||||
| C | 59 | ||||||||||
| C | 57 | ||||||||||
| C | C | 58 | |||||||||
| C | 52 | ||||||||||
| Posterior DMN | C | 62 | |||||||||
| Whole brain | C | 61 | |||||||||
| C | 66 | ||||||||||
| → → → | 42 | ||||||||||
| Parietotemporal cortices and basal ganglia | C | DSC-MRI | 65 | ||||||||
| Internal carotids and vertebral arteries | C | C | PC-MRI | 63 | |||||||
| Whole brain | C | C | 64 | ||||||||
| Orbitofrontal, middle and inferiorfrontal, middle and superior temporal, inferior temporal, and superior parietal cortices |
→ | [15O]–PET | 67 | ||||||||
| Impaired cerebrovascular reactivity | |||||||||||
| Hippocampus | C | BOLD-fMRI (CO2- challenge) |
71 | — | — | — | 2,43 | ||||
| Prefrontal, cingulate and insular cortices | C | 69 | |||||||||
| Middle cerebral artery territory | C | TCD (CO2- challenge) |
68 | ||||||||
| C | 72 | ||||||||||
| Impaired neurovascular coupling | |||||||||||
| Hippocampal formation | C | C | task-based BOLD-fMRI |
73 | Yes | — | — | 2,43 | |||
| Visual cortex | C | 75 | |||||||||
Abbreviations: C, cross-sectional study; →, longitudinal study; —, not studied; CNS, central nervous system; NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; CBF, cerebral blood flow; ASL, arterial spin labelling; MRI, magnetic resonance imaging; fMRI, functional MRI; BOLD, blood-oxygenation-level-dependent; DSC, dynamic susceptibility-contrast; PC, phase-contrast; TCD, transcranial Doppler; SPECT, single-photon emission-computed tomography ; PET, positron emission tomography
CBF reductions.
Early studies found that individuals with greater CBF velocity, as shown by transcranial Doppler measurements in the middle cerebral artery, are less likely to develop dementia or hippocampal and amygdalar atrophy51. Similar, individuals with MCI or early AD develop early CBF reductions in the posterior cingulate gyrus and precuneus, as shown by arterial spin labelling (ASL)-magnetic resonance imaging (MRI) 2,43,52–54. Progression of MCI to AD is associated with more global55–60 and severe, up to 40%, CBF reductions61,62, which includes other CNS regions besides the posterior cingulate gyrus and precuneus, such as bilateral parietotemporal, frontal and occipital cortex, parahippocampal gyrus, hippocampus and entorhinal cortex (Table 1). Studies using two-dimensional phase contrast MRI confirmed CBF reductions in MCI63 and full blown AD64.
A large cohort longitudinal study from the Alzheimer’s Disease Neuroimaging Initiative database suggested that changes in vascular biomarkers (e.g., cerebrospinal fluid (CSF) heart-type fatty acid-binding protein, cortisol, and apolipoprotein A that are all associated with cardiovascular disorders42) and decreased CBF by 2D ASL occur before detectable changes in classical Aβ and tau CSF AD biomarkers42. Ideally, the CBF findings in this study42 should be confirmed using more sensitive methods such as those that use contrast agents (e.g., dynamic susceptibility contrast (DSC), DCE perfusion) to achieve better signal to noise ratio compared to ASL, which is less optimal and noisy. Reduced CBF in parietotemporal cortex and basal ganglia in MCI was also shown to occur before brain atrophy by DSC-MRI studies65. ASL imaging and Pittsburgh compound B (PiB) amyloid positron emission tomography (PET) imaging revealed widespread and Aβ-independent CBF reductions in cognitively normal APOE4 carriers with strongest genetic risk for AD66. [15O]-labeled water-PET longitudinal study showed widespread decline of CBF occurs over time in cognitively normal APOE4 carriers67. More longitudinal studies are needed to better understand the relationship between CBF and Aβ and tau pathological changes.
Impaired cerebrovascular reactivity.
Impaired cerebrovascular reactivity, reflecting diminished vasodilation of cerebral vessels in response to a CO2 inhalation challenge, was found early in AD compared to cognitively normal controls using transcranial Doppler68 and the blood oxygenation level dependent (BOLD)-functional MRI (fMRI)69. However, the evidence that this is etiologic is lacking, and while it is possible that these effects could also be downstream to Aβ-mediated vasculopathy, neither of these two studies investigated the presence of cerebral amyloid angiopathy (CAA). CAA is characterized by deposition of Aβ in the wall of small and mid-sized cerebral and leptomeningeal arteries, veins and cerebral capillaries, and is related to AD70. Impaired cerebral blood vessels reactivity to CO2 has also been shown in the hippocampus of young, non-demented APOE4 carriers compared to non-carriers by BOLD-fMRI in response to a memory task71, and by reduced CBF velocity in the middle cerebral artery using transcranial Doppler72 (Table 1).
Impaired neurovascular coupling.
BOLD-fMRI provides a measure of neurovascular coupling by detecting an increase in blood oxygen delivery and CBF in response to neuronal stimulation43. The physiological basis of the BOLD signal has not been fully elucidated and current models include contributions from different hemodynamic and blood parameters43. Most fMRI studies treat the BOLD response as an indirect qualitative measure of neuronal activity and interpret BOLD signal differences as changes in neuronal activity. However, the BOLD signal reflects local changes in deoxyhemoglobin content, which in turn exhibits an intricate dependence on changes in CBF, cerebral blood volume, and the cerebral metabolic rate of oxygen consumption (CMRO2). Factors that disturb the connection between CBF and CMRO2 (e.g., altered cerebrovascular structure, reduced blood vessel elasticity, atherosclerosis, reduced resting state CBF, decreased resting CMRO2, reduced vascular reactivity to chemical modulators) may therefore significantly change the BOLD signal during aging and AD, even when neuronal activity is unchanged, which should be taken into account in data interpretation43. Reduced CBF response to visual stimuli and memory encoding tasks were detected in the hippocampus, parahippocampal gyrus, precuneus and posterior cingulate cortex in MCI patients73, and in the hippocampus in AD patients73,74. Changes in the hemodynamic response in the visual cortex were also found in patients with CAA75 that associate with cerebrovascular dysfunction45. The alterations in BOLD signal activity during AD progression appear to be regionally specific, and depend on the nature of cognitive tasks, indicating that they indeed may reflect the pathophysiological changes in neurovascular coupling (Table 1). However, two important caveats have limited the application and interpretation of BOLD studies in AD including head motion artifacts and often limited ability of subjects with neurodegenerative diseases to perform tasks.
Animal studies.
Most animal CBF studies have been done in transgenic AD APP models of Aβ amyloidosis. These studies have shown that Aβ has vasoactive and vasculotoxic effects on brain vessels, as recently reviewed2. Briefly, Aβ vasoactive effects were shown on cerebral blood vessels both in vitro and in vivo in transgenic mouse models overexpressing human APP mutations1,2, including impaired CBF responses to vasodilators76, and dysfunctional neurovascular coupling77. Thus, preventing Aβ deposition may clearly abolish Aβ-dependent effects on CBF reductions and dysregulation. A recent study in APP mice expressing human Swedish, Dutch and Iowa mutations has demonstrated, however, that counteracting the harmful effects of Aβ after vascular depositions is not effective in reversing the neurovascular dysfunction owing to the mural cell damage that is caused by aging and massive Aβ deposition78.
Several studies reported that CBF dysregulation develops early in experimental models of mural cell or endothelial dysfunction, which can lead to neuronal dysfunction and loss, independent of Aβ, as recently reviewed2. For instance, pericyte-deficient mutant mice develop early CBF reductions in grey33 and white79 matter in the absence of Aβ pathology, and aberrant CBF responses in the presence of initially normal neuronal activity, endothelial vasodilation, and astrocyte coverage of the blood vessels13. Thus, CBF reductions and dysregulation in mouse models can develop independent of Aβ and precede neuronal dysfunction, similar as suggested by human studies (Table 1). In humanized APOE4 transgenic mice, early CBF reductions precede neuronal dysfunction independently of Aβ39,80 similar as in human APOE4 carriers, as discussed above.
Blood-brain barrier in Alzheimer’s disease
In parallel to early CBF changes, BBB breakdown and dysfunction have been shown in early stages during AD pathophysiological progression (Table 2). Below, we examine studies indicating that BBB is a useful biomarker in preclinical AD.
Table 2.
Blood-brain barrier dysfunction in the normal aging-to-mild cognitive impairment-to-Alzheimer’s disease spectrum studied with neuroimaging.
| Affected CNS region(s) | Disease Stage | Accelerated with risk factors |
Method | Ref. | BBB biomarker prior to | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NCI | MCI | Early AD |
AD | Age | VRFs | APOE4 | Brain atrophy |
Amyloid | Tau | |||||
| Increased BBB permeability to Gadolinium (Ktrans) | ||||||||||||||
| Hippocampus and its subfields | C | Yes | — | — | DCE-MRI | 43,44 | Yes | Yes | Yes | 43,44 | ||||
| Gray matter, normal-appearing white matter, deep gray matter, hippocampus and cortex |
C | — | No | — | 59,81,82 | |||||||||
| Microbleeds | ||||||||||||||
| Deep infratentorial regions | C | Yes | Yes | Yes | T2*/SWI-MRI | 87 | Yes | Yes | Yes |
83– 85,87,89 |
||||
| C | Yes | — | No | 84 | ||||||||||
| C | C | — | No | — | 89 | |||||||||
| Lobar and deep infratentorial regions |
C | C | Yes | — | — | 83 | ||||||||
| Lobar regions | C | — | — | — | 90 | |||||||||
| → | Yes | No | Yes | 86,88 | ||||||||||
| C | C | C | Yes | Yes | — | 85 | ||||||||
| Diminished glucose transport | ||||||||||||||
| Parietal, frontal and/or temporal cortex |
C | — | — | — | Dynamic FDG-PET |
106–108 | — | — | — | NA | ||||
| Diminished glucose transport and/or metabolism | ||||||||||||||
| Posterior cingulate, parieto- temporal, and entorhinal cortices, hippocampus, whole brain |
C* | — | — | — | FDG-PET | 99,100 | Yes | Yes | Yes | 93–98,100 | ||||
| Posterior cingulate and parieto- temporal cortex |
C | — | — | Yes | 96 | |||||||||
| C# | — | — | — | 94 | ||||||||||
| Posterior cingulate, parieto- temporal, occipital and frontal cortices, precuneus, and/or hippocampus |
C | C | — | — | — | 98 | ||||||||
| C | C | C | — | — | — | 95 | ||||||||
| C | — | — | — | 52 | ||||||||||
| Posterior cingulate, parieto- temporal and frontal cortices, hippocampus |
— | — | — | 103 | ||||||||||
| — | — | — | 97,101 | |||||||||||
| Yes | — | — | 102 | |||||||||||
| — | — | Yes | 93 | |||||||||||
| Diminished P-glycoprotein function | ||||||||||||||
| Parieto-temporal, frontal, occipital and cingulate cortices, hippocampus |
C | — | — | — | Verapamil- PET |
115,116 | Yes | — | — | 116 | ||||
Abbreviations: C, cross-sectional study; →, longitudinal study; —, not studied; CNS, central nervous system; AD, Alzheimer’s disease; VRFs, vascular risk factors; APOE4, apolipoprotein E4; NCI, no cognitive impairment; MCI, mild cognitive impairment; DCE, dynamic contrast-enhanced; MRI, magnetic resonance imaging; FDG, fluorodeoxyglucose; PET, positron emission tomography.
Footnotes:
Accelerated in asymptomatic PSEN1 mutation carriers compared to non-carriers
Accelerated in individuals with positive maternal history of AD.
BBB permeability.
Blood-to-brain leakage of an intravenously administered gadolinium-based contrast agent, as measured with dynamic contrast-enhanced (DCE)-MRI and quantified with the Patlak analysis, has recently enabled detection of subtle, regional changes in BBB permeability during the normal aging-to-MCI-to-AD spectrum43,44. These studies have shown an increase in BBB permeability in the hippocampus and its CA1 and dentate gyrus subfields during normal aging44, which is further accelerated in individuals with MCI prior to brain atrophy or detectable changes in Aβ and tau CSF biomarkers44. Individuals with MCI and early AD develop, with disease progression, increased BBB permeability in other brain regions including cortex, deep gray matter and white matter, all prior to dementia81,59,82. During MCI, the increased BBB permeability correlated with pericyte injury, as measured via CSF PDGFRβ pericyte biomarker, further supporting the presence of early BBB breakdown and capillary damage44. Interestingly, vascular risk factors such as atherosclerosis, cardiac arrhythmia, and coronary disease did not influence BBB permeability59, suggesting that BBB breakdown during preclinical and early AD is not related to concomitant vascular conditions, but is rather an early independent biomarker contributing to AD pathophysiology. Future longitudinal studies are needed to precisely define the role of BBB breakdown in cognitive impairment and loss of structural and functional connectivity during AD progression and elucidate the cellular and molecular mechanisms through which it promotes AD pathology.
Microbleeds.
Microbleeds caused by blood leakage into brain parenchyma from damaged blood vessels due to loss of BBB integrity can be visualized by T2*-MRI sequences as hyposignals reflecting perivascular accumulation of blood-derived hemosiderin deposits43. Several studies indicate appearance of microbleeds during normal aging83–88, which is further accelerated in individuals with MCI83–85,89, early AD83,85,86,88–90, and AD85. Microbleeds reflect cerebral small vessel disease, which is observed in approximately 50% of all dementia cases worldwide48,91, and is associated with worse cognitive performance85 and white matter hyperintensities87,88. APOE4 status accelerates microbleed prevalence in a majority of studies86–88, but not all84. However, at present there is no evidence that microbleeds are etiologically important in causing the AD pathology of plaques and tangles, and/or that they cause AD.
Microbleed etiology and topography are related, with CAA causing lobar microbleeds in AD and hypertensive arteriopathy causing deep infratentorial microbleeds91. Hypertension positively associates with microbleed size and prevalence85,87 particularly in infratentorial regions, whereas other vascular risk factors such as diabetes and hyperlipidemia do not associate with microbleeds86. Some studies indicate that microbleeds predominate in deep infratentorial regions during early preclinical AD and MCI stages84,87 supporting the view that they may precede amyloid pathology, determined by no difference in CSF Aβ42 in MCI subjects with and without microbleeds84, but are later seen in lobar regions85,86,88,90 reflecting CAA etiology during AD progression (Table 2). CAA further contributes to BBB breakdown, infarcts, white matter changes, and cognitive impairment leading the detection of dementia earlier92. Of note, studies using different magnetic field strengths indicate that the 7T magnet has approximately 3 times greater ability to detect smaller microbleeds compared to 3T magnet89; this has implications for future research to determine the true quantity and size of microbleeds that appear throughout the normal aging-MCI-AD spectrum.
Glucose transport.
Numerous 18F-fluoro-2-deoxyglucose (FDG)-PET studies show decreased glucose brain uptake in early stages of AD prior to amyloid and tau changes detected by amyloid-PET and/or Aβ42 and phosphorylated-tau CSF analysis, and/or brain atrophy93–98. Cognitively normal individuals with increased genetic risk for AD such as PSEN1 mutation carriers99,100 and APOE4 carriers96 or positive AD maternal history94 also display decreased FDG brain uptake as an initial pathophysiological event. Longitudinal studies indicate that FDG brain uptake is further diminished in AD in individuals that converted into AD either from cognitively unimpaired stage or MCI93,97,101–103. These reductions in brain uptake of glucose in AD patients are thought to reflect pathological hypometabolism resulting from neurodegeneration8,104. However, there is evidence from animal models that brain uptake of 2-deoxyglucose (2DG) depends on the BBB glucose transporter-1 (GLUT1). In particular, deletion of a single Slc2a1 allele encoding GLUT1 at the BBB in mice diminishes 14C-2DG brain uptake followed by BBB breakdown, CBF reductions and dysregulation, capillary rarefaction, and eventually neuronal dysfunction and loss105. In humans, a few kinetic FDG-PET studies demonstrated reductions in FDG BBB transport in AD106–108. These reductions in BBB transport may be explained by the fact that the reduced expression of GLUT1 was found in the brain capillaries of AD patients in post-mortem studies109–112 (Table 2). Thus, in our opinion reduced FDG-PET does not reflect only glucose neuronal hypometabolism but can also reflect reduced transport of glucose across the BBB. Future studies in animal models and humans using glucose, FDG and glucose analogs that track only BBB transport (e.g., 3-O-[11C]-methyl glucose)113, and in addition to PET more sensitive analytical methods such as nuclear magnetic resonance spectroscopy should further explore brain glucose transport and metabolism in AD and the therapeutic potential of GLUT1 BBB transporter.
Diminished P-glycoprotein function.
P-glycoprotein is an active efflux transporter expressed at the BBB luminal endothelium4. Laboratory studies indicate that Aβ is cleared from brain-to-blood via abluminal LRP1-dependent efflux followed by luminal P-glycoprotein transport4,114. Individuals with early AD develop widespread reductions in P-glycoprotein BBB function in the parieto-temporal, frontal, occipital and cingulate cortices, and hippocampus, as shown using PET with the radiolabeled P-glycoprotein substrate, 11C-verapamil115,116. Diminished P-glycoprotein BBB function occurred independently of reduced CBF115 and prior to brain atrophy116. In addition to P-glycoprotein, other BBB transporter changes including increased levels of the receptor for advanced glycation endproducts (RAGE), a major Aβ influx transporter at the BBB, and decreased levels of LRP1, a major Aβ efflux transporter at the BBB, were found in cerebral blood vessels of AD patients45.
Animal studies.
Consistent with human studies, studies in different models relevant to AD pathophysiology also indicate the presence of BBB breakdown and dysfunction. This topic has been comprehensively reviewed recently6, and will not be examined in great detail here. Briefly, animal studies using transgenic models with various mutations in human APP gene, have demonstrated perivascular accumulation of blood-derived proteins (e.g., fibrinogen, immunoglobulin G (IgG), albumin), vascular leakage of circulating exogenous tracers, loss of tight junction proteins, loss of pericyte coverage, pericyte and endothelial degeneration, and microbleeds, altogether indicating BBB breakdown6,117. Although most studies did not examine the time course of BBB changes in relation to other brain pathologies, those that did indicated that BBB breakdown occurs early prior to amyloid accumulation, behavioral deficits or brain degenerative changes6,117,118. Simultaneously, BBB transporter dysfunction occurs, including decreased luminal P-glycoprotein function, decreased LRP1-mediated Aβ clearance, increased RAGE-mediated Aβ influx, which all accelerates Aβ accumulation in APP models114,119–121. Decreased GLUT1 BBB expression also contributes to BBB breakdown and Aβ pathology by transcriptionally downregulating LRP1105, thus corroborating evidence from human BBB studies.
Different PSEN1 models (e.g., PSEN1 knockouts and PSEN1 mutations driven by neuronal promoters) exhibit microbleeds and endothelial degeneration indicative of BBB breakdown122,123, occurring prior to brain Aβ and CAA pathology122. AD models with tau mutations show also BBB leakage of exogenous tracers, IgG deposits, microbleeds, and leukocyte infiltration despite no evidence of brain Aβ or CAA accumulation124. Finally, APOE knockout mice and mice with targeted replacement of human APOE4 gene show BBB breakdown and dysfunction (e.g., loss of BBB GLUT1 and increased RAGE)39,125 prior to development of behavioral deficits, synaptic changes and neuronal dysfunction39.
Cerebral blood flow and blood-brain barrier in other neurodegenerative disorders
Early CBF and BBB changes are also found in other neurodegenerative disorders including PD, HD, MS and ALS (Table 3).
Table 3.
Cerebral blood flow deficits and blood-brain barrier dysfunction in other neurodegenerative disorders studied with neuroimaging.
| Affected CNS regions | Disease | Study design |
Methods | References | Biomarker prior to | References | ||
|---|---|---|---|---|---|---|---|---|
| Motor symptoms |
Cognitive decline |
|||||||
| CBF reductions | ||||||||
| Posterior parieto-occipital cortex, superior temporal gyrus, posterior cingulate, precuneus and cuneus, and middle frontal gyri |
PD | C | ASL-MRI | 126,127 | — | Yes | 127 | |
| Sensorimotor, paracentral, inferior temporal and lateral occipital cortices, postcentral gyrus, insula and medial occipital areas |
HD | C | ASL-MRI | 128 | — | Yes | 128 | |
| White matter including lesions | MS | C | DCE-MRI | 129 | — | — | NA | |
| White matter including lesions, gray matter | C | DSC-MRI | 130 | — | Yes | 130 | ||
| Frontal and parietal cortices | ALS | C | ASL-MRI | 132 | — | Yes | 131,132 | |
| All cortical lobes, and subcortical grey and white matter | → | CT | 131 | |||||
| Impaired cerebrovascular reactivity | ||||||||
| Whole brain | PD | C | ASL-MRI | 134 | — | — | NA | |
| Middle cerebral artery territory | C | TCD | 133 | — | — | NA | ||
| Default mode, frontoparietal, somatomotor, visual, limbic, dorsal and ventral attention networks |
MS | C | ASL-MRI | 135 | — | — | NA | |
| Increased BBB permeability | ||||||||
| Basal ganglia | PD | C | DCE-MRI | 136 | — | Yes | 136 | |
| Caudate nucleus | HD | C | 137 | — | Yes | 137 | ||
| White matter | MS | C | 44,129,138–140 | Yes | Yes | 44,139 | ||
| Microbleeds | ||||||||
| Cortical gray and white matter | PD | C | T2*/SWI-MRI | 141–143 | — | Yes | 141 | |
| Deep layers of motor cortex | ALS | C | 144 | — | Yes | 144 | ||
| Diminished P-glycoprotein function | ||||||||
| Mid-brain | PD | C | Verapamil- PET |
145 | — | Yes | 145 | |
| CNS leukocyte infiltration | ||||||||
| White matter | MS | → | MMP inhibitor-PET |
146 | — | Yes | 146 | |
Abbreviations: C, cross-sectional; →, longitudinal study; —, not studied; PD, Parkinson’s disease; HD, Huntington’s disease; MS, multiple sclerosis; ALS, amyotrophic lateral sclerosis.
CBF reductions.
Decreased CBF was found throughout the cortical mantles and precuneus in PD patients with a range of motor and cognitive impairments using ASL-MRI126. A recent study showed that CBF reductions were also detectable in non-demented patients with PD127, suggesting that perfusion abnormalities may be an early biomarker upstream of cognitive impairment and neurodegeneration. Similarly, perfusion deficits were found in the cortex in early HD prior to cognitive dysfunction128. Using contrast MRI, early CBF reductions were detected in white matter lesions and in normally appearing white matter in relapsing-remitting MS patients with and without cognitive impairment129,130. ALS patients also develop early perfusion deficits in fronto-parietal cortex, subcortical grey matter as well as white matter in the absence of cognitive decline131, which is independent of brain atrophy132.
Impaired cerebrovascular reactivity.
Global impaired vasodilation of cerebral vessels in response to CO2 has been shown in PD using transcranial Doppler133 and BOLD-fMRI134. Decreased cerebrovascular reactivity was also reported in MS135. Whether altered cerebrovascular reactivity occurs prior to motor and cognitive impairments remains, however, unknown.
BBB permeability.
DCE-MRI studies indicated increased regional BBB permeability (i.e., Ktrans values) in the basal ganglia in PD136, caudate nucleus in HD137, and white matter in MS44,129,138–140 prior to cognitive decline44,136,137,139. Increased Ktrans BBB permeability values indicated perivascular lesion growth in MS with minimal motor symptoms139, suggesting loss of BBB integrity could be an early event driving MS pathophysiology.
Microbleeds.
Brain microbleeds are found in cortical gray matter, basal ganglia, corpus callosum, and internal and external capsules in PD141–143, as well as in deep layers of motor cortex in ALS144, occurring prior to cognitive decline141,144. However, it remains unclear whether microbleeds precede motor symptoms in PD and ALS. Moreover, the prevalence and regional distribution of microbleeds in HD and MS is presently unknown.
Diminished P-glycoprotein function.
[11C]-verapamil-PET scans indicated diminished P-glycoprotein efflux transport function at the BBB in the mid-brain in PD subjects with no cognitive impairment145. Whether or not P-glycoprotein or other BBB transporters play a role in HD, ALS, or MS, is currently elusive.
CNS leukocyte infiltration.
PET studies using a radiolabeled matrix metalloproteinase (MMP) inhibitor ligand indicated increased MMP activity in MS lesions suggesting enhanced leukocyte infiltration across the BBB into the brain of cognitively normal MS patients146.
Animal studies.
CBF and BBB dysfunctions have also been shown in animal models of PD, HD, ALS, and MS, as recently examined elsewhere147,148.
Vascular pathways to neurodegeneration
The first hypothetical model of in vivo AD dynamic biomarkers, often called the Jack model, was proposed in 2010 and updated in 2013 to include interim evidence and present inter-subject variability in cognitive impairments8. This model was intended to summarize literature showing temporal evolution of in vivo AD biomarkers relative to each other and to the onset and progression of clinical symptoms. This initial model, however, did not include currently available evidence that vascular dysfunction contributes to AD pathophysiology in a significant way1. Therefore, it is time now to update the hypothetical biomarkers model of AD and include the impact of cerebral blood vessels on AD pathophysiology, as previously suggested42,149. Figure 4 presents an updated revised model of AD biomarkers to show the role of brain vasculature and early changes in CBF and BBB biomarkers in AD that, according to some studies (see Table 1 and Table 2), are altered prior to cognitive decline, brain atrophy, neurodegeneration, and/or amyloid and tau biomarker abnormalities. The sigmoid shape of the vascular curve reflects growing evidence obtained from imaging, biofluid and post-mortem tissue studies indicating an initial acceleration followed by deceleration of brain vascular changes, which do not plateau, similar as shown for the other biomarkers. However, future longitudinal studies may warrant amendment of the proposed trajectories of the vascular curve as well as of the other biomarkers curves and/or their order of their respective appearance.
Figure 4. Hypothetical updated Jack model of Alzheimer’s disease biomarkers to include the role of brain vasculature.
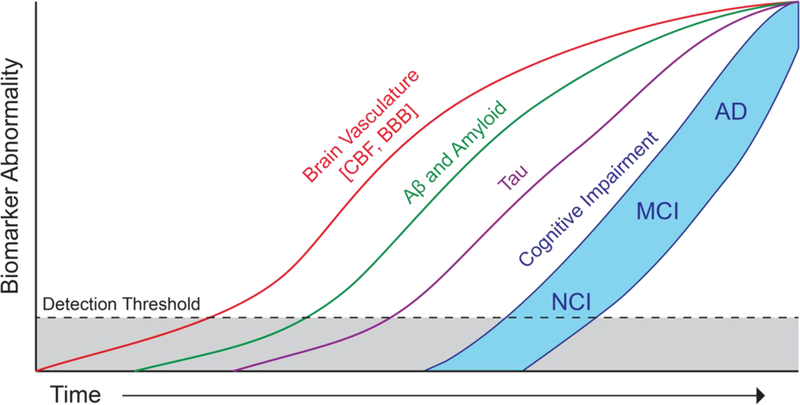
Hypothetical model of Alzheimer’s disease (AD) biomarker changes illustrating that early cerebral blood flow (CBF) and blood-brain barrier (BBB) biomarkers and vascular dysfunction may contribute to initial stages of AD pathophysiological progression from no cognitive impairment (NCI) to mild cognitive impairment (MCI) to AD, which is followed by cerebrospinal fluid and brain changes in Aβ and amyloid, and tau biomarkers. All biomarker curves converge at the top right-hand corner of the plot, that is the point of maximum abnormality. The horizontal axis of disease progression is expressed as time. Cognitive response is illustrated as a zone (blue filled area) with low and high-risk borders. Subjects with high risk of AD-related cognitive impairment are shown with a cognitive response curve that is shifted to the left. In contrast, the cognitive response curve is shifted to the right in subjects with a protective genetic profile, high cognitive reserve and the absence of comorbid brain pathologies.
As discussed above, AD can be viewed as a model for other neurodegenerative diseases that are beginning to reveal notable vascular contributions to disease pathophysiology such as PD, HD, ALS and MS (Table 3). Figure 5 illustrates the commonalities of vascular dysfunction across neurodegenerative diseases, focusing on specific regions where CBF shortfalls and BBB dysfunction are evident during early stages of each disease.
Figure 5. Commonality of an early involvement of brain vasculature in different neurodegenerative disorders.
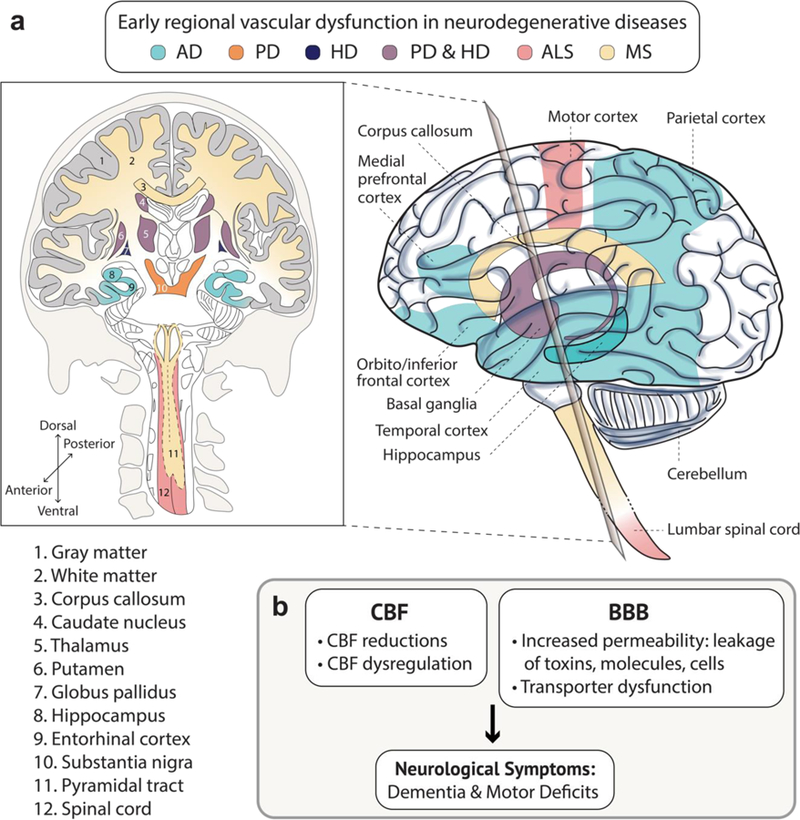
(a–b) Region-specific brain vascular dysfunction including cerebral blood flow (CBF) shortfalls (reductions and dysregulation) and/or blood-brain barrier (BBB) breakdown (increased vascular permeability and transporter dysfunction) is a common pathway seen early in multiple neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS), as illustrated schematically in a and b. See Tables 1, 2 and 3 for details. Specifically, some studies suggest that vascular dysfunction (CBF and/or BBB) in the hippocampus, gray matter and entorhinal cortex in AD may precede dementia, brain atrophy and/or detectable Aβ and tau biomarker changes. Similar, vascular dysfunction in the white matter and corpus callosum in MS, basal ganglia (the caudate nucleus, thalamus, putamen, globus pallidus and substantia nigra) in PD and HD, the spinal cord white matter pyramidal tract in MS, and motor cortex and spinal cord in ALS is found by some studies in early stages of these disorders prior to progression of neurological symptoms including motor deficits.
Conclusions and future directions
An emerging role of brain vasculature in the pathogenesis of human neurodegenerative diseases, particularly AD, has led to increasingly recognized importance of healthy blood vessels for normal brain functioning. A recent single cell transcriptomics approach to examine vascular zonation along the A-V axis in mouse models is a huge conceptual advancement7, providing important insights to biological processes at different levels of the vascular tree and BBB. The single cell RNA-seq approach holds great promise to help us understand the cellular and molecular basis of CBF and BBB dysfunction in models of neurodegenerative disorders and AD, and by extension in humans with these disorders. While the molecular atlas of cerebral blood vessels and BBB is being elucidated in animal models, we lack the molecular definition of human brain vasculature, BBB, and perhaps NVU, to generate an atlas of blood vessels in the human brain during health and disease. Comparative knowledge of molecular makeup between humans and animals is essential to bridge this translational gap, and for future studies to take advantage of discoveries in animal models. This has the potential to reveal why regional changes in the brain vasculature may lead to disease-specific neurological phenotypes in different neurodegenerative diseases and inform gene networks and upstream regulators driving the link between cerebrovascular dysfunction and neurodegeneration.
Neuroimaging holds the potential to further examine the regional vascular pathophysiology in the living human brain. Further advances are needed such as development of novel neuroimaging modalities for biomarkers of different vascular cell types including SMCs, pericytes and endothelial cells, markers of vascular zonation, and/or molecular pathways in blood vessels. For example, PET imaging with radiolabeled matrix metalloproteinase inhibitors examines in vivo leukocyte infiltration across the human BBB in multiple sclerosis, which can be applied to AD and other neurodegenerative disorders45. Similarly, MRI imaging of endothelial adhesion molecules detects the inflammatory phenotype of brain vascular endothelium in the rodent brain and models of different brain diseases150, which could be extended and developed for the living human brain. Similar types of probes could be developed to interrogate RAGE-mediated Aβ vascular influx, and/or LRP1-mediated Aβ vascular efflux capacity. Longitudinal studies with high field strength 7T (or greater) magnets will provide improved sensitivity to detect early, subtle, regional changes in cerebrovascular dysfunction, which will confirm or amend the temporal course of biomarkers abnormality in AD and other neurodegenerative disorders. The advances in imaging will continue to establish new early vascular biomarkers in the living human brain, hopefully revealing untapped novel targets of disease-modifying therapeutics for multiple neurodegenerative disorders.
Acknowledgements
The work of B.V.Z. is supported by the National Institutes of Health grants R01AG023084, R01NS090904, R01NS034467, R01AG039452, 1R01NS100459, 5P01AG052350, and 5P50AG005142 in addition to the Alzheimer’s Association, Cure Alzheimer’s Fund, and the Foundation Leducq Transatlantic Network of Excellence for the Study of Perivascular Spaces in Small Vessel Disease reference no. 16 CVD 05. We apologize to those authors whose excellent original papers we were not able to cite due to the journal’s limit of 150 references. Instead, we sometimes cited recent reviews by leading authorities so that the reader can access all key primary papers in the field of our review.
References
- 1.Iadecola C The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 96, 17–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kisler K, Nelson AR, Montagne A & Zlokovic BV Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci (2017). [DOI] [PMC free article] [PubMed]
- 3.Hartmann DA et al. Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics 2, 041402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Z, Nelson AR, Betsholtz C & Zlokovic BV Establishment and Dysfunction of the Blood-Brain Barrier. Cell 163, 1064–1078 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sweeney MD, Ayyadurai S & Zlokovic BV Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci 19, 771–783 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montagne A, Zhao Z & Zlokovic BV Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med 214, 3151–3169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanlandewijck M et al. A molecular atlas of cell types and zonation landmarks in the brain vasculature. Nature 554, 475–480 (2018).This is the first study to examine molecular landmarks and heterogeneity of cerebrovascular cells in the adult mouse brain and investigate the concept of zonation along the arterio-capillary-venous axis.
- 8.Jack CR et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12, 207–216 (2013).Proposed and recognized hypothetical model of biomarker changes during Alzheimer’s disease pathophysiological progression.
- 9.Nguyen LN et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509, 503–506 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Ben-Zvi A et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509, 507–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeisel A et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Alarcon-Martinez L et al. Capillary pericytes express α-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. eLife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kisler K et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci 20, 406–416 (2017).This was the first study to demonstrate that pericyte degeneration in a pericyte loss‑of‑function model leads to a loss of neurovascular coupling and diminished oxygen delivery to brain.
- 14.Peppiatt CM, Howarth C, Mobbs P & Attwell D Bidirectional control of CNS capillary diameter by pericytes. Nature 443, 700–704 (2006).This important work in brain slices and retina demonstrated pericyte contractility and control of capillary diameter.
- 15.Hall CN et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60 (2014).This was the first study to show that pericytes have an active role in cerebral blood flow regulation in vivo and that capillaries can dilate ahead of arterioles. In ischemic conditions, pericytes rapidly constrict capillaries and die, consistent with the no‑reflow phenomenon observed in stroke.
- 16.Mishra A et al. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci 19, 1619–1627 (2016).This was the first study to show that astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles in brain, which involves a rise of calcium in astrocytes caused by entry through adenosine triphosphate-gated channels.
- 17.Biesecker KR et al. Glial Cell Calcium Signaling Mediates Capillary Regulation of Blood Flow in the Retina. J. Neurosci. Off. J. Soc. Neurosci 36, 9435–9445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernández-Klett F, Offenhauser N, Dirnagl U, Priller J & Lindauer U Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc. Natl. Acad. Sci. U. S. A 107, 22290–22295 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shih A & Hartmann D In vivo Optical Imaging and Manipulation of Pericytes in the Mouse Brain. in Optics and the Brain BrW3B-1 (2017).
- 20.Mapelli L et al. Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO-Dependent System. J. Neurosci. Off. J. Soc. Neurosci 37, 1340–1351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill RA et al. Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron 87, 95–110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei HS et al. Erythrocytes Are Oxygen-Sensing Regulators of the Cerebral Microcirculation. Neuron 91, 851–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hillman EMC Coupling mechanism and significance of the BOLD signal: a status report. Annu. Rev. Neurosci 37, 161–181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Longden TA et al. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci 20, 717–726 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blinder P et al. The cortical angiome: an interconnected vascular network with noncolumnar patterns of blood flow. Nat. Neurosci 16, 889–897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kur J & Newman EA Purinergic control of vascular tone in the retina. J. Physiol 592, 491–504 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uhlirova H et al. Cell type specificity of neurovascular coupling in cerebral cortex. eLife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gebremedhin D et al. Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ. Res 87, 60–65 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Nippert AR, Biesecker KR & Newman EA Mechanisms Mediating Functional Hyperemia in the Brain. The Neuroscientist 107385841770303 (2017). [DOI] [PMC free article] [PubMed]
- 30.Bény J-L, Nguyen MN, Marino M & Matsui M Muscarinic receptor knockout mice confirm involvement of M3 receptor in endothelium-dependent vasodilatation in mouse arteries. J. Cardiovasc. Pharmacol 51, 505–512 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Yamada M et al. Cholinergic dilation of cerebral blood vessels is abolished in M(5) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U. S. A 98, 14096–14101 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daneman R, Zhou L, Kebede AA & Barres BA Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell RD et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427 (2010).This study not only describes the role of pericytes in maintaining in vivo blood-brain barrier integrity, microvascular density and functional hyperemia during adulthood and brain aging but also shows that a primary loss of pericytes may lead to two parallel pathways of neurodegeneration, blood-brain barrier breakdown and hypoperfusion, which lead to secondary neurodegenerative changes.
- 34.Armulik A et al. Pericytes regulate the blood-brain barrier. Nature 468, 557–561 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Winkler EA, Bell RD & Zlokovic BV Central nervous system pericytes in health and disease. Nat. Neurosci 14, 1398–1405 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikolakopoulou AM, Zhao Z, Montagne A & Zlokovic BV Regional early and progressive loss of brain pericytes but not vascular smooth muscle cells in adult mice with disrupted platelet-derived growth factor receptor-β signaling. PloS One 12, e0176225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foo SS et al. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 124, 161–173 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Yao Y, Chen Z-L, Norris EH & Strickland S Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat. Commun 5, 3413 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bell RD et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516 (2012).Important study demonstrating APOE4-dependent activation of a pro-inflammatory signaling pathway in pericytes involving cyclophilin A – nuclear factor-kappa B – matrix metalloproteinase-9-mediated degradation of blood-brain barrier basement membrane and tight junction proteins.
- 40.Wang Y, Chang H, Rattner A & Nathans J Frizzled Receptors in Development and Disease. Curr. Top. Dev. Biol 117, 113–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liebner S et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol 183, 409–417 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iturria-Medina Y et al. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun 7, 11934 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montagne A et al. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol. (Berl.) 131, 687–707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montagne A et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302 (2015).This study was the first to demonstrate age-associated blood-brain barrier breakdown in the hippocampus in the living human brain and accelerated breakdown in humans with mild cognitive impairment.
- 45.Sweeney M, Sagare A & Zlokovic B Blood-brain barrier breakdown in Alzheimer’s disease and other neurodegenerative disorders. Nat. Rev. Neurol 14, 133–150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA & Schneider JA Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol 15, 934–943 (2016).This large cross-sectional neuropathological study showed that cerebral vessel disease plays a part in dementia that is typically attributed to Alzheimer’s disease during life.
- 47.Toledo JB et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain J. Neurol 136, 2697–2706 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iadecola C The pathobiology of vascular dementia. Neuron 80, 844–866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabin JS et al. Interactive Associations of Vascular Risk and β-Amyloid Burden With Cognitive Decline in Clinically Normal Elderly Individuals Findings From the Harvard Aging Brain Study. JAMA Neurol (2018). [DOI] [PMC free article] [PubMed]
- 50.Sweeney MD, Sagare AP & Zlokovic BV Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer’s disease. J. Cereb. Blood Flow Metab 35, 1055–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruitenberg A et al. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann. Neurol 57, 789–794 (2005).This large population-based study showed that diminished cerebral blood flow precedes cognitive decline and hippocampal atrophy.
- 52.Chen Y et al. Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology 77, 1977–1985 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hays CC, Zlatar ZZ & Wierenga CE The Utility of Cerebral Blood Flow as a Biomarker of Preclinical Alzheimer’s Disease. Cell. Mol. Neurobiol 36, 167–179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wierenga CE et al. Effect of mild cognitive impairment and APOE genotype on resting cerebral blood flow and its association with cognition. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 32, 1589–1599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alexopoulos P et al. Perfusion abnormalities in mild cognitive impairment and mild dementia in Alzheimer’s disease measured by pulsed arterial spin labeling MRI. Eur. Arch. Psychiatry Clin. Neurosci 262, 69–77 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Dai W et al. Mild cognitive impairment and alzheimer disease: patterns of altered cerebral blood flow at MR imaging. Radiology 250, 856–866 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu WT et al. Distinct cerebral perfusion patterns in FTLD and AD. Neurology 75, 881–888 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nation DA et al. Cortical and subcortical cerebrovascular resistance index in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. JAD 36, 689–698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van de Haar HJ et al. Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol. Aging 45, 190–196 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Yoshiura T et al. Simultaneous measurement of arterial transit time, arterial blood volume, and cerebral blood flow using arterial spin-labeling in patients with Alzheimer disease. AJNR Am. J. Neuroradiol 30, 1388–1393 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leeuwis AE et al. Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc 13, 531–540 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Ma HR et al. Aberrant pattern of regional cerebral blood flow in Alzheimer’s disease: a voxel-wise meta-analysis of arterial spin labeling MR imaging studies. Oncotarget 8, 93196–93208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Eulate RG et al. Reduced Cerebral Blood Flow in Mild Cognitive Impairment Assessed Using Phase-Contrast MRI. J. Alzheimers Dis. JAD 58, 585–595 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Leijenaar JF et al. Lower cerebral blood flow in subjects with Alzheimer’s dementia, mild cognitive impairment, and subjective cognitive decline using two-dimensional phase-contrast magnetic resonance imaging. Alzheimers Dement. Amst. Neth 9, 76–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wirth M et al. Divergent regional patterns of cerebral hypoperfusion and gray matter atrophy in mild cognitive impairment patients. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab (2016). [DOI] [PMC free article] [PubMed]
- 66.Michels L et al. Arterial spin labeling imaging reveals widespread and Aβ-independent reductions in cerebral blood flow in elderly apolipoprotein epsilon-4 carriers. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 36, 581–595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thambisetty M, Beason-Held L, An Y, Kraut MA & Resnick SM APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch. Neurol 67, 93–98 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.den Abeelen ASSM, Lagro J, van Beek AHEA & Claassen JAHR Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s disease. Curr. Alzheimer Res 11, 11–17 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Yezhuvath US et al. Forebrain-dominant deficit in cerebrovascular reactivity in Alzheimer’s disease. Neurobiol. Aging 33, 75–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hecht M, Krämer LM, von Arnim CAF, Otto M & Thal DR Capillary cerebral amyloid angiopathy in Alzheimer’s disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol. (Berl.) 135, 681–694 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Suri S et al. Reduced cerebrovascular reactivity in young adults carrying the APOE ε4 allele. Alzheimers Dement. J. Alzheimers Assoc 11, 648–657. e1 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Hajjar I, Sorond F & Lipsitz LA Apolipoprotein E, carbon dioxide vasoreactivity, and cognition in older adults: effect of hypertension. J. Am. Geriatr. Soc 63, 276–281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Small SA, Perera GM, DeLaPaz R, Mayeux R & Stern Y Differential regional dysfunction of the hippocampal formation among elderly with memory decline and Alzheimer’s disease. Ann. Neurol 45, 466–472 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Rombouts SA et al. Functional MR imaging in Alzheimer’s disease during memory encoding. AJNR Am. J. Neuroradiol 21, 1869–1875 (2000). [PMC free article] [PubMed] [Google Scholar]
- 75.Dumas A et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann. Neurol 72, 76–81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Iadecola C et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci 2, 157–161 (1999). [DOI] [PubMed] [Google Scholar]
- 77.Niwa K et al. Abeta 1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. U. S. A 97, 9735–9740 (2000).This paper was the first to demonstrate altered neurovascular coupling by amyloid-β.
- 78.Park L et al. Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathy. Stroke J. Cereb. Circ 45, 1815–1821 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Montagne A et al. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat. Med 24, 326–337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80.Alata W, Ye Y, St-Amour I, Vandal M & Calon F Human apolipoprotein E ɛ4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 35, 86–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van de Haar HJ et al. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 281, 527–535 (2016). [DOI] [PubMed] [Google Scholar]
- 82.van de Haar HJ et al. Subtle blood-brain barrier leakage rate and spatial extent: considerations for dynamic contrast-enhanced MRI. Med. Phys (2017). [DOI] [PubMed]
- 83.Heringa SM et al. Multiple microbleeds are related to cerebral network disruptions in patients with early Alzheimer’s disease. J. Alzheimers Dis. JAD 38, 211–221 (2014). [DOI] [PubMed] [Google Scholar]
- 84.Poliakova T, Levin O, Arablinskiy A, Vasenina E & Zerr I Cerebral microbleeds in early Alzheimer’s disease. J. Neurol 263, 1961–1968 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Shams S et al. Cerebral microbleeds: different prevalence, topography, and risk factors depending on dementia diagnosis—the Karolinska Imaging Dementia Study. AJNR Am. J. Neuroradiol 36, 661–666 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shams S & Wahlund L-O Cerebral microbleeds as a biomarker in Alzheimer’s disease? A review in the field. Biomark. Med 10, 9–18 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Vernooij MW et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology 70, 1208–1214 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Yates PA et al. Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology 82, 1266–1273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brundel M et al. High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer’s disease. J. Alzheimers Dis. JAD 31, 259–263 (2012).A high prevalence of cerebral microbleeds are detected in mild cognitive impairment and early Alzheimer’s disease brains (78% of subjects) using state-of-the-art 7-Tesla magnetic resonance imaging.
- 90.Uetani H et al. Prevalence and topography of small hypointense foci suggesting microbleeds on 3T susceptibility-weighted imaging in various types of dementia. AJNR Am. J. Neuroradiol 34, 984–989 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wardlaw JM et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12, 822–838 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saito S & Ihara M Interaction between cerebrovascular disease and Alzheimer pathology. Curr. Opin. Psychiatry 29, 168–173 (2016). [DOI] [PubMed] [Google Scholar]
- 93.Iaccarino L et al. A Cross-Validation of FDG- and Amyloid-PET Biomarkers in Mild Cognitive Impairment for the Risk Prediction to Dementia due to Alzheimer’s Disease in a Clinical Setting. J. Alzheimers Dis. JAD 59, 603–614 (2017). [DOI] [PubMed] [Google Scholar]
- 94.Mosconi L et al. Amyloid and metabolic positron emission tomography imaging of cognitively normal adults with Alzheimer’s parents. Neurobiol. Aging 34, 22–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mosconi L et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J. Nucl. Med. Off. Publ. Soc. Nucl. Med 49, 390–398 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Protas HD et al. Posterior cingulate glucose metabolism, hippocampal glucose metabolism, and hippocampal volume in cognitively normal, late-middle-aged persons at 3 levels of genetic risk for Alzheimer disease. JAMA Neurol 70, 320–325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mosconi L Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 32, 486–510 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Bailly M et al. Precuneus and Cingulate Cortex Atrophy and Hypometabolism in Patients with Alzheimer’s Disease and Mild Cognitive Impairment: MRI and (18)F-FDG PET Quantitative Analysis Using FreeSurfer. BioMed Res. Int 2015, 583931 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bateman RJ et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mosconi L et al. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med 47, 1778–1786 (2006). [PubMed] [Google Scholar]
- 101.Landau SM et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 32, 1207–1218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Landau SM et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75, 230–238 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mosconi L et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 36, 811–822 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jack CR et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Winkler EA et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci 18, 521–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gejl M et al. Blood-Brain Glucose Transfer in Alzheimer’s disease: Effect of GLP-1 Analog Treatment. Sci. Rep 7, 17490 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jagust WJ et al. Diminished glucose transport in Alzheimer’s disease: dynamic PET studies. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 11, 323–330 (1991). [DOI] [PubMed] [Google Scholar]
- 108.Piert M, Koeppe RA, Giordani B, Berent S & Kuhl DE Diminished glucose transport and phosphorylation in Alzheimer’s disease determined by dynamic FDG-PET. J. Nucl. Med. Off. Publ. Soc. Nucl. Med 37, 201–208 (1996). [PubMed] [Google Scholar]
- 109.Mooradian AD, Chung HC & Shah GN GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol. Aging 18, 469–474 (1997). [DOI] [PubMed] [Google Scholar]
- 110.Kalaria RN & Harik SI Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J. Neurochem 53, 1083–1088 (1989). [DOI] [PubMed] [Google Scholar]
- 111.Simpson IA, Chundu KR, Davies-Hill T, Honer WG & Davies P Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol 35, 546–551 (1994). [DOI] [PubMed] [Google Scholar]
- 112.Horwood N & Davies DC Immunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer’s disease. Virchows Arch. Int. J. Pathol 425, 69–72 (1994). [DOI] [PubMed] [Google Scholar]
- 113.Kilbourn MR Small Molecule PET Tracers for Transporter Imaging. Semin. Nucl. Med 47, 536–552 (2017). [DOI] [PubMed] [Google Scholar]
- 114.Cirrito JR et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Invest 115, 3285–3290 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deo AK et al. Activity of P-Glycoprotein, a β-Amyloid Transporter at the Blood-Brain Barrier, Is Compromised in Patients with Mild Alzheimer Disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med 55, 1106–1111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.van Assema DME et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain J. Neurol 135, 181–189 (2012). [DOI] [PubMed] [Google Scholar]
- 117.Sagare AP et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun 4, 2932 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 118.Paul J, Strickland S & Melchor JP Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J. Exp. Med 204, 1999–2008 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Deane R et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med 9, 907–913 (2003). [DOI] [PubMed] [Google Scholar]
- 120.Deane R et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43, 333–344 (2004). [DOI] [PubMed] [Google Scholar]
- 121.Deane R et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Invest 122, 1377–1392 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gama Sosa MA et al. Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer’s disease mutations. Am. J. Pathol 176, 353–368 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wen PH et al. Selective expression of presenilin 1 in neural progenitor cells rescues the cerebral hemorrhages and cortical lamination defects in presenilin 1-null mutant mice. Dev. Camb. Engl 132, 3873–3883 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Blair LJ et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun 3, 8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cacciottolo M et al. The APOE4 allele shows opposite sex bias in microbleeds and Alzheimer’s disease of humans and mice. Neurobiol. Aging 37, 47–57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Melzer TR et al. Arterial spin labelling reveals an abnormal cerebral perfusion pattern in Parkinson’s disease. Brain J. Neurol 134, 845–855 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Syrimi ZJ et al. Arterial spin labelling detects posterior cortical hypoperfusion in non-demented patients with Parkinson’s disease. J. Neural Transm. Vienna Austria 1996 124, 551–557 (2017). [DOI] [PubMed] [Google Scholar]
- 128.Chen JJ, Salat DH & Rosas HD Complex relationships between cerebral blood flow and brain atrophy in early Huntington’s disease. NeuroImage 59, 1043–1051 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ingrisch M et al. Quantification of perfusion and permeability in multiple sclerosis: dynamic contrast-enhanced MRI in 3D at 3T. Invest. Radiol 47, 252–258 (2012). [DOI] [PubMed] [Google Scholar]
- 130.Hojjat S-P et al. Cortical Perfusion Alteration in Normal-Appearing Gray Matter Is Most Sensitive to Disease Progression in Relapsing-Remitting Multiple Sclerosis. AJNR Am. J. Neuroradiol 37, 1454–1461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Murphy MJ et al. Widespread cerebral haemodynamics disturbances occur early in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis 13, 202–209 (2012). [DOI] [PubMed] [Google Scholar]
- 132.Rule RR, Schuff N, Miller RG & Weiner MW Gray matter perfusion correlates with disease severity in ALS. Neurology 74, 821–827 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Camargo CHF et al. Abnormal Cerebrovascular Reactivity in Patients with Parkinson’s Disease. Park. Dis 2015, 523041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Al-Bachari S, Vidyasagar R, Emsley HC & Parkes LM Structural and physiological neurovascular changes in idiopathic Parkinson’s disease and its clinical phenotypes. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 37, 3409–3421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Marshall O, Chawla S, Lu H, Pape L & Ge Y Cerebral blood flow modulation insufficiency in brain networks in multiple sclerosis: A hypercapnia MRI study. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 36, 2087–2095 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Al-Bachari S MRI assessment of neurovascular changes in idiopathic Parkinson’s disease Thesis, University of Manchester (2016). [Google Scholar]
- 137.Drouin-Ouellet J et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol 78, 160–177 (2015).Blood-brain barrier leakage and cerebrovascular dysfunction in Huntington’s disease (HD) investigated by neuroimaging and post-mortem tissue analysis in human subjects and further corroborated in a mouse model of HD.
- 138.Taheri S, Gasparovic C, Shah NJ & Rosenberg GA Quantitative measurement of blood-brain barrier permeability in human using dynamic contrast-enhanced MRI with fast T1 mapping. Magn. Reson. Med. Off. J. Soc. Magn. Reson. Med. Soc. Magn. Reson. Med 65, 1036–1042 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cramer SP, Simonsen H, Frederiksen JL, Rostrup E & Larsson HBW Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clin 4, 182–189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gaitán MI et al. Evolution of the blood-brain barrier in newly forming multiple sclerosis lesions. Ann. Neurol 70, 22–29 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ham JH et al. Cerebral microbleeds in patients with Parkinson’s disease. J. Neurol 261, 1628–1635 (2014). [DOI] [PubMed] [Google Scholar]
- 142.Kim J-H, Park J, Kim Y-H, Ma H-I & Kim YJ Characterization of cerebral microbleeds in idiopathic Parkinson’s disease. Eur. J. Neurol 22, 377–383 (2015). [DOI] [PubMed] [Google Scholar]
- 143.Yamashiro K et al. The prevalence and risk factors of cerebral microbleeds in patients with Parkinson’s disease. Parkinsonism Relat. Disord 21, 1076–1081 (2015). [DOI] [PubMed] [Google Scholar]
- 144.Kwan JY et al. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PloS One 7, e35241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kortekaas R et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol 57, 176–179 (2005). [DOI] [PubMed] [Google Scholar]
- 146.Gerwien H et al. Imaging matrix metalloproteinase activity in multiple sclerosis as a specific marker of leukocyte penetration of the blood-brain barrier. Sci. Transl. Med 8, 364ra152 (2016).Developed an important novel approach to image initial leukocyte penetration of the blood-brain barrier in multiple sclerosis patients and animal models.
- 147.Zlokovic BV Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci 12, 723–738 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nelson AR, Sweeney MD, Sagare AP & Zlokovic BV Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 1862, 887–900 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Iadecola C Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci 5, 347–360 (2004). [DOI] [PubMed] [Google Scholar]
- 150.Montagne A et al. Ultra-sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. NeuroImage 63, 760–770 (2012). [DOI] [PubMed] [Google Scholar]