

Abstract
The chemokine CCL5 prevents neuronal cell death mediated both by amyloid β, as well as the human immunodeficiency virus (HIV) viral proteins gp120 and Tat. Because CCL5 binds to CCR5, CCR3 and/or CCR1 receptors, it is unclear which of these receptors plays a role in neuroprotection. Indeed, CCL5 also has neuroprotective activity in cells lacking these receptors. CCL5 may bind to a G protein-coupled receptor 75 (GPR75), which encodes for a 540 amino-acid orphan receptor of the Gqα family. In this study, we have used SH-SY5Y human neuroblastoma cells to characterize whether CCL5 could activate a Gq signaling through GPR75. Both qPCR and flow cytometry show that these cells express GPR75 but do not express CCR5, CCR3 or CCR1 receptors. SY-SY5Y cells were then used to examine CCL5-mediated signaling. We report that CCL5 promotes a time- and concentration-dependent phosphorylation of protein kinase B (AKT), glycogen synthase kinase 3β and extracellular signal–regulated kinase (ERK) 1/2. Specific antagonists of CCR5, CCR3 and CCR1 did not prevent CCL5 from increasing phosphorylated AKT or ERK. Moreover, CCL5 promotes a time-dependent internalization of GPR75. Lastly, knocking down GPR75 expression by a CRISPR-Cas9 approach inhibited the ability of CCL5 to activate pERK in SH-SY5Y cells. Therefore, we propose that GPR75 is a novel receptor for CCL5 that could explain some of the pharmacological action of this chemokine. These findings may help in the development of small molecule GPR75 agonists that mimic CCL5.
Keywords: AKT, CCR5, CRISPR-Cas9, CXCR4, ERK, signal transduction
Graphical Abstract
The chemokine CCL5 is known to bind predominantly to chemokine receptor CCR5. However, CCL5 induces extracellular signaling-regulated kinases (Erk) and protein kinase B (Akt) pathways, even in cells that do not express CCR5. In this report we show that CCL5 activates G protein-coupled receptor 75 (GPR75). This receptor is predominantly expressed in neurons and does not belong to the chemokine receptor family. Our results suggest that GPR75 is a novel receptor that could mediate the effect of CCL5 on neurons.
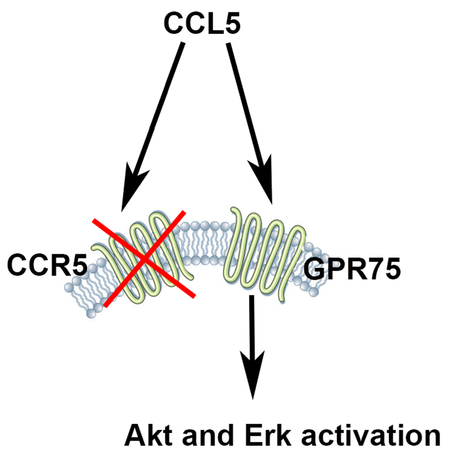
Introduction
Chemokines, which belong to a class of polypeptides that play a role in regulating migration of immune cells, are notoriously promiscuous ligands. CCL5 is not an exception because, in addition to CCR5, it also binds to chemokine receptors CCR1 and CCR3 (Rossi & Zlotnik 2000), members of the G protein-coupled receptor (GPCR) family. These receptors are expressed not only by immune cells but also in the central nervous system (Avdoshina et al. 2011), where they may participate in neuro-inflammation (Kaul & Lipton 1999), neuroprotection (Maung et al. 2014) as well as pain modulation (Szabo et al. 2002).
More recently, it was reported that CCL5 activates GPCR 75 (GPR75), a 540 amino acid receptor that belongs to the GPCR family (Ignatov et al. 2006). GPR75 has been considered an orphan receptor of the Gqα family of G proteins, which was originally characterized for its expression in the human retina (Tarttelin et al. 1999, Sauer et al. 2001). Other studies have indicated high expression of this receptor in the mouse brain and heart when compared to traditional immune organs such as spleen (Ignatov et al. 2006). Comparison of amino sequence between GPR75 and other GPCRs has shown that GPR75 has little homology to classical (e.g. dopamine, serotonin) neurotransmitter/hormone GPCRs, ~25% homology to neuropeptide Y receptor and 12% homology to chemokine receptors (Tarttelin et al. 1999). Importantly, in cells overexpressing GPR75, CCL5, but not other chemokines such as CXCL12, CCL4 or CCL7 (Ignatov et al. 2006), activates phosphatidylinositol 3–kinase (PI3K) and its down-stream effector protein kinase B, or AKT, as well as extracellular signaling-regulated kinases (ERK). Thus, GPR75 could be a missing link to explain physiological properties of CCL5 that do not pertain to its immune function.
The neuronal expression of GPR75 could have an important neuroprotective function. In fact, experimental data have shown that GPR75 activation by CCL5 inhibits (Ignatov et al. 2006) the neurotoxic effect of amyloid-β peptide (Aβ), a hallmark of Alzheimer’s disease (Stokin et al. 2005). Intriguingly, neuroprotection by CCL5 has also been observed in cortical cultures from CCR5 knockout mice (Kaul et al. 2007), suggesting that GPR75 could be a novel receptor whose signal transduction mediates CCL5-induced neuronal survival. However, there are no specific antagonists for GPR75 that can be used to definitely prove that CCL5 activates this receptor. Given the potential importance of GPR75 agonists as neuroprotectants, in this work, we have used a variety of techniques to demonstrate that CCL5 is a cognate ligand for the GPR75 receptor.
MATERIALS AND METHODS
Materials.
Recombinant human CCL5 (cat# 278-RN), CXCL12 (cat# 350-NS) and pertussis toxin (PTX, cat# 3097) were purchased from R&D (Minneapolis, MN). Wortmannin (cat# W1628), 1-[6-[((17β)-3-Methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione (U73122, cat# U6756) and retinoic acid (RA, cat# R2625) were from Sigma-Aldrich (St. Louis, MO).
Cell cultures.
SH-SY5Y neuroblastoma cells (ATCC, Manassas, VA, Cat# CRL-2266, RRID:CVCL_0019) were grown as previously described with minor modifications (Dedoni et al. 2014). The International Cell Line Authentication Committee does not list these cells as a commonly misidentified cell line. In brief, cells used up to the 16th passage, were maintained in Ham’s F12 (cat# 11765054)/MEM (cat# 11095072) (1:1) supplemented with 2 mM L-glutamine (cat# 25030081), 1% non-essential amino acids (cat# 11140050), 10% fetal calf serum and 1% antibiotic-antimycotic (cat# 15240062) (all from Thermo Fisher Scientific, Waltham, MA) at 37°C in a humidified atmosphere of 5% CO2 in air. Cells were differentiated to neurons by exposure for 6–8 days to RA (10 μM) dissolved in dimethyl sulfoxide (Thermo Fisher Scientific, cat# D12345). The medium was renewed every 48 h, and during the RA treatment, care was taken to avoid cell culture exposure to light. A microtubule associated protein 2 (MAP2) antibody (1:5000; Sigma-Aldrich, cat# M4403, RRID:AB_477193) was used to verify the presence of neurons in these cultures before the experiments. Differentiated cells were arbitrarily assigned to be exposed to medium control or containing CCL5 or other compounds.
Human leukemia monocytic cell line THP-1 (ATCC Cat# TIB-202, RRID:CVCL_0006) and human embryonic kidney cell line HEK293 (ATCC Cat # CRL-1573, RRID:CVCL _0045) were used. Cells were grown following ATCC protocol and authenticated before the experiments. The international Cell Line Authentication Committee does not list these cells as a commonly misidentified cell lines.
Preparation of rat primary cultures.
Animal studies were done in strict accordance with the Laboratory Animal Welfare Act, the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and after approval from the Georgetown University Animal Care and Use Committee (#2016–1189).
Rat cortical neurons were prepared from E17 embryos Sprague Dawley rats (Taconic, Hudson, NY, RRID:RGD:1566440) as previously described (Avdoshina et al. 2010). In brief, time-pregnant dams were euthanized by CO2 inhalation. Embryos were removed from the uterus and quickly euthanized by sharp scissors. Their brains were removed and cortices were dissected and cleaned from blood vessels. Cortices were minced and dissociated in trypsin (Sigma Aldrich, cat# T4665). Trypsin was inactivated by the addition of soybean trypsin inhibitor (cat# T6522) and DNase (cat# D4527) all from Sigma Aldrich. Cell were re-suspended in Neurobasal medium (ThermoFisher Scientific, cat# 21103049,) containing 2% B-27 supplement (ThermoFisher Scientific, cat# 17504044), 25 mM glutamate (Sigma Aldrich cat# 496215), 0.5 mM L-glutamine (ThermoFisher Scientific, cat# 25030081), and 1% antibiotic-antimycotic solution (ThermoFisher Scientific). Cells were seeded at a density of 0.5×106 cells per ml onto poly-L-lysine (Sigma Aldrich, cat# P9155,) pre-coated 6 well plates (Corning, Tewksbury, WA, cat# 3335,). Cells were maintained at 37°C in 5% CO2 in air for 7–8 days before adding any compound. At the day of experiment (8 days in culture) 95% of cells were neurons as determined either by βIII-tubulin antibody (1:5000; Covance Research Products Inc., Dedham, MA, cat# PBR-435P-100, RRID:AB_291637) or MAP2 antibody. Neuronal cultures were arbitrary assigned to be exposed to medium alone or containing CCL5.
Rat cortical astrocytes were prepared and grown as previously described (Avdoshina et al. 2010). By the time of experiment (11 days in culture), 98% of cells were positive for the astrocytic marker, glial fibrillary acidic protein (1:5000; Dako, Carpinteria, CA, cat# N1506, RRID:AB_10013482).
Culture of human T lymphocytes.
The collection of blood was done in healthy adult human subjects. Written consent was obtained from all individual participants included in the study. Peripheral blood mononuclear cells were isolated with the use of Histopaque® (Sigma-Aldrich, cat#11191). CD4+ T cells (purity of >95% when analyzed by flow cytometry) were isolated from resting peripheral blood mononuclear cells using naïve CD4+ T cell isolation kit II (Miltenyi Biotec, Auburn, CA, cat# 130-094-131). T cells were rested for 2 h in Roswell Park Memorial Institute 1640 medium with 1% L-glutamine (Thermo Fisher Scientific, cat#11875–085), supplemented with 10% fetal calf serum (Thermo Fisher Scientific, cat# MT35015CV) and containing 1% penicillin/streptomycin 1 μg/mL and phytohemagglutinin (Thermo Fisher Scientific, cat# 1514148 and 10576015, respectively). Cell were then plated and cultured for 4 days in fresh medium as described above.
Flow cytometry analysis.
RA-treated SH-SY5Y cells were stained and analyzed by flow cytometry for the cell surface markers CCR1, CCR3, and CCR5. Cells were washed once with PBS and resuspended at 106 cells/ml. 100 μl of cells were stained in 12×75 tubes for 20 min at room temperature in the dark with 1 μg of antibody against CCR1 (cat# 362903, RRID:AB_2563897), CCR3 (cat# 310707, RRID:AB_1134156), or CCR5 (cat# 359106, RRID:AB_2562335), all from BioLegend, San Diego, CA. THP-1 cells were stained using same method to provide a positive control. Twenty thousand cells were processed on a BDFortessa (Becton Dickinson, San Jose, CA) and analyzed utilizing FCSExpress 5 (DeNovo Software, Glendale, CA).
Real time polymerase chain reaction (qPCR).
Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen, Valencia, CA, cat# 74136) and reverse transcribed using SuperScript® VILO Master Mix with ezDNase™ Enzyme Kit (Thermo Fischer Scientific, cat# 11766050) according to manufacturer’s specifications. The qPCR mix (20 μl volume) contained Platinum SYBR Green qPCR SuperMix (Thermo Fisher Scientific, cat# 11733038), cDNA template or negative controls and primers. Reactions were carried out at 95°C for 10 min and 40 cycles at 95°C for 15 s, 60°C for 30 s and 72°C for 30 s, using 7900HT Fast Real-Time PCR System (Thermo Fisher Scientific). Primers for CCR5 were TTTTCCAGCAAGAGGCTCC (forward) and ATGTGCACAACTCTGACTGG (reverse); for CCR3 AATGACTGTGAGCGGAGCAA (forward) and CCTCTCTCCAACAAAGGCG (reverse); for CCR1 TGCTCTGCTCACACTCATGG (forward) and TCCAAAGCTGTCCGTTTGAT (reverse); for GPR75 GCCACCCGGCAGGCTTATCT (forward) and TTCGGAGAGAAATGTCTCCTTC (reverse). Primers for the housekeeping gene hypoxanthine phosphoribosyltransferase were obtained from RealTimePrimers.com (Elkins Park, PA). Standard curves for each set of primers were plotted using serial dilution of cDNA to verify equal amplification efficiency. Reverse-transcriptase negative controls were used to exclude genomic DNA contamination. Data were analyzed using SDS v2.3 software (Thermo Fisher Scientific).
Western blot analysis.
SH-SY5Y cells were exposed to various compounds in serum-free medium whereas primary neurons were treated in neurobasal medium without B27 supplement, at 37° C in a humidified atmosphere of 5% CO2. Inhibitors were dissolved in DMSO (final concentration <0.5%). Control samples received an equal amount of vehicle. After treatments, cells were lysed in 1X RIPA buffer (Millipore Sigma, Burlington, MA, cat# 20188), with 1X Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, cat# 78440) at 4°C. The samples were sonicated for 10 s on ice and Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, cat# 23225) was used to determine protein concentrations. Proteins were separated by NuPAGE 4–12% Bis-Tris gel (Thermo Fisher Scientific, cat# NP0336BOX,) and transferred to nitrocellulose membranes using the iBlot device (Thermo Fisher Scientific). Staining of the gel with SimplyBlue™ SafeStain (Thermo Fisher Scientific, cat# LC6065) ensured the efficiency of the transfer. Membranes were blocked in 5% skim milk in PBS-Tween 20 (Sigma Aldrich, cat# P2287), washed and incubated overnight at 4° C with anti-GPR75 (1:500; Abcam, Cambridge, MA, cat# ab75581, RRID:AB_1523717). Thereafter, the membranes were washed and incubated with an appropriate horseradish peroxidase-conjugated secondary antibody (Jackson Immuno Research, West Grove, PA). A mouse monoclonal β-actin (1:10,000, Sigma-Aldrich, cat # A2228, RRID:AB_476697) antibody was used for loading control.
For signaling molecules, blots were probed with anti-phospho-(Thr308)-AKT (cat# 9275, RRID:AB_329828), anti-phosho ERK1/2 (cat# 9102, RRID:AB_331646) and anti-phospho glycogen synthase kinase 3 β (GSK3β, 9332s, RRID:AB_2335664), all at 1:1000 dilution, Cell Signaling Technology, Danvers, MA. For assurance that an equal amount of total protein was loaded in each lane, after each experiment the membranes were stripped of the antibodies by using the Restore Western Blot Stripping Buffer (cat# 21059; Thermo Fisher Scientific) and re-probed with AKT (cat# 9272, RRID:AB_329827), ERK1/2 (cat# 5013S, RRID:AB_10693607) or GSK3β (cat# 9332s, RRID:AB_2335664) antibodies (all at 1:1000 dilution, Cell Signaling Technology). Immunoreactive bands were detected by using ECL Plus (Thermo Fisher Scientific, cat# 32132). Band densities were determined using NIH ImageJ software (National Institutes of Health, Bethesda, MD). The optical densities of phosphoproteins were normalized to the density of the corresponding total protein.
Biotinylation of surface proteins.
Surface biotinylation of cell proteins was performed as previously described (Dedoni et al. 2014) with minor modifications. In brief, SH-SY5Y cells, grown in 100 mm dishes at 90% confluence, were exposed to either vehicle (0.1% bovine serum albumin, cat# A2153, Sigma Aldrich) or CCL5 in serum free medium. Cells were then washed with ice-cold PBS (pH 8.0) and incubated for 35 min at 4°C without or with the cell impermeable biotinylating agent sulfosuccinimidyl-6-(biotin-amido)hexanoate (sulpho-NHS-LC-biotin) (0.50 mg/ml; Thermo Fisher Scientific, cat# PG82075). Thereafter, the medium was aspirated and the cells were washed in ice-cold PBS containing 20 mM glycine (Sigma Aldrich, cat# G5417). Cells were then solubilized by incubation for 60 min at 4° C in 1X RIPA lysis buffer (Millipore Sigma) with 1X Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, cat# 78441B) supplemented with 0.1% Triton X 100 (Sigma Aldrich, cat# T8787). Cell extracts were centrifuged at 15,000 x g for 10 min at 4° C and the supernatants incubated overnight with streptavidin-conjugated agarose beads (Thermo Fisher Scientific, cat# 20334) with continuous rotation at 4° C. The samples were then centrifuged to obtain a supernatant and a pellet fraction containing the plasma membrane-associated proteins. The agarose beads were washed three times with ice-cold Tris buffer and 0.1% Triton X 100, followed by one wash with Tris buffer. The pellet was then mixed with sample buffer and incubated 5 min at 100 °C. Proteins were then analyzed by western blot. A pan-cadherin antibody (Thermo Fisher Scientific, cat# MA5–15036) was used to control for protein loading.
CRISPR-Cas9 knockdown.
We utilized the CRISPR-Cas9 design site crispr.mit.edu to identify gRNAs that target the protein-coding region (bp 5278–6091) of the human GPR75 gene. We chose two gRNA’s (gRNA A 5’-CAAGGTAGCCAAGGTGGCAA-3’, gRNA B 5’-CCCATGTCCAGTCTGATTCG-3’) based on efficiency and low frequency of off target sites. The gRNA’s would create a mutation at bp 5865 and 5804, resulting in inhibition of GPR75 transcription. The gRNA’s were ligated into the pSpCas9(BB)-2A-GFP backbone PX458 (Addgene, Cambridge, MA, plasmid #48138) after linearization using BbsI (New England Biolabs, Ipswich, MA, cat# R0539L). This plasmid expresses the Cas9 protein, ligated gRNA, in addition to green fluorescent protein (GFP) simultaneously. Plasmids were then sequenced to confirm proper insertion of gRNA (Eurofins, Hanover, MD).
Cell transfection.
SH-SY5Y cells were transfected with plasmid containing Cas9+gRNA A or Cas9+gRNA B using Lipofectamine 2000 (Thermo Fisher Scientific, cat# 11668027). Cells were also transfected with plasmid containing GFP only to produce wild type controls (WT). GFP expression was assessed to determine successful transfection. Cells were then split, re-plated, and transfected again to enhance the presence of mutation. This process was repeated up to four cell passages/transfections before experimentation. Site-specific mutations were assayed using the T7 endonuclease I (T7E1) enzyme. In brief, genomic DNA from SH-SY5Y cells was purified using the NucleoSpin Tissue kit (Macherey-Nagel, Bethlehem, PA, cat# 740952.10). The coding region for GPR75 was then amplified using the following primers (Forward: 5’-CAATGCCACCTCGCTCCATG-3’, Reverse: 5’-TGACCAGTTGTTGGGACTC-3’). The PCR product was gel purified and 100 ng of purified product was denatured for 10 min at 95o and subsequently exposed to a cooling gradient. One microliter of T7E1 (New England Biolabs, cat# M0302S) was added to the newly annealed product and incubated at 37o for 1 h. The resulting products were then run on a 1% agarose gel to determine mutation/cleavage of PCR product.
Statistical analysis.
The study was not preregistered. No sample calculation was performed. No blinding was performed but data on signaling were confirmed by two different investigators. No outliers were excluded. Data were analyzed using one-way ANOVA followed by Tuckey’s post hoc test (GraphPad Prism, San Diego, CA, USA). Control values are set arbitrarily either to 100 or one.
RESULTS
GPR75 expression in neurons.
GPR75 levels in the brain appear to correlate with those of CCL5. Indeed, CCL5 is highly abundant in the cerebellum and cerebral cortex (Avdoshina et al. 2011), two areas that exhibit the highest GPR75 expression (Sauer et al. 2001). To examine whether the expression of GPR75 is cell specific, we used western blot to analyze lysates from primary rat cortical neurons, astrocytes, or human neuroblastoma SH-SY5Y cells. SH-SY5Y cells were exposed to RA to promote differentiation into a neuronal phenotype (Ammer & Schulz 1994). Lysates from the thymus and spleen were used to provide an assessment of the expression of GPR75 in peripheral tissues. The antibody against GPR75 detected an immunoreactive band with a molecular weight of approximately 59 kDa in rat cortical neurons and differentiated SH-SY5Y cells (Fig. 1A). Astrocytes, undifferentiated SH-SY5Y cells, thymus, and spleen all exhibited a band with a lower intensity than neurons (Fig. 1B) and a slightly higher molecular weight (Fig. 1A), which may be due to posttranslational modifications.
Figure 1. GPR75 is expressed in neurons and astrocytes.

Western blot was used to analyze lysates from rat primary cortical neurons, astrocytes, undifferentiated, or RA differentiated SH-SY5Y cells. Differentiation of SH-SY5Y by RA was confirmed by immunocytochemistry using a MAP2 antibody (not shown). Rat thymus and spleen were used as representatives of immune tissues. A. Representative western blot probed with an antibody against GPR75. Blot was stripped and reprobed with an antibody against β-actin. B. Relative levels of GPR75 expressed in arbitrary units (AU) were determined by optical density of GPR75 immunoreactivity and corrected by β-actin density. Data are the mean + SEM of three independent samples for each tissue/cell line.
qPCR was then used to confirm expression of GPR75 in SH-SY5Y cells. Table 1 shows that these cells expressed GPR75 mRNA; however, mRNA for CCR1, CCR3, and CCR5 receptors was undetectable even by 40 cycles of qPCR (Table 1). The validity of the qPCR method was assured by measuring GPR75, CCR1, CCR3, and CCR5 mRNA in human CD4+ cells. These cells expressed all four receptors tested (Table 1). To further confirm that CCR1, CCR3, and CCR5 are not expressed on the cell surface, SH-SY5Y ceIls were analyzed by flow cytometry. As a control we used THP-1 cells, a human leukemic cell line which expresses all these receptors (Schecter et al. 1997, Giri et al. 2004). While all three receptors were detected in THP-1 cells, none were present on SH-SY5Y cells (Table 1). Thus, for the continuation of this study we used RA-treated SH-SY5Y cells to determine whether CCL5 activates GPR75 signal transduction.
Table 1.
SH-SY5Y cells express GPR75 but not other CCL5 receptors.
| GPR75 | CCR1 | CCR3 | CCR5 | |||||
|---|---|---|---|---|---|---|---|---|
| qPCR | flow cytometry | qPCR | flow cytometry | qPCR | flow cytometry | qPCR | flow cytometry | |
| SH-SY5Y | yes | yes | no | no | no | no | no | no |
| Human CD4+ | yes | yes | yes | yes | ||||
| THP-1 | yes | yes | yes | yes | ||||
qPCR and flow cytometry were used to detect expression of the indicated mRNAs and membrane receptors, respectively, in SH-SY5Y cells. CD4 and TH-1 cells were used as positive controls for qPCR and flow cytometry, respectively. Details of the methods are described in Materials and Methods. For qPCR, yes indicates amplification of mRNA of the indicated genes after 40 cycles, no indicates undetectable levels of mRNA for CCR1, CCR3, and CCR5 after 40 cycles. Hypoxanthine phosphoribosyltransferase was used as housekeeping gene. For flow cytometry, no indicates undetectable expression of indicated chemokine receptors on cell membranes.
CCL5 signal transduction.
Signaling kinases that are phosphorylated when Gqα are activated include AKT, GSK3β and ERK1/2. Therefore, to test whether CCL5 induces GPR75-mediated signaling, we exposed SH-SY5Y cells to CCL5 for various time points and examined phospho (p)-AKT (Figs. 2A and B), pGSK3β (Figs. 2C and D) and pERK1/2 (Figs. 2E and F). CCL5 (100 nM) promoted a rapid and time-dependent phosphorylation of all signaling molecules with a maximal activation for all three phosphoproteins by ~15 min. Total levels of AKT, GSK3β, and ERK were used to determine whether CCL5 affects the expression of these kinases. No changes in their total levels were observed (Fig. 2) suggesting that CCL5 activates signaling rather than expression. Since the time and relative net activation of pGSK3β and pERK by CCL5 were equivalent (Figs. 2D and F), we decided for the continuation of this study to narrow the focus to only pAKT and pERK.
Figure 2. CCL5 increases phosphorylation of signaling proteins.

Differentiated SH-SY5Y cells were exposed to CCL5 (100 nM) for the indicated time points and lysates were prepared. An equal amount of proteins was loaded on a gel, transferred and immunoblotted with an antibody against (A) pAKT, (C) pGSK3β or (E) pERK1/2. Blots were stripped and reprobed with antibodies against AKT, GSK3β or ERK1/2. B, D and F: relative levels of phosphoproteins were calculated by optical density as described in Materials and Methods. Data are the mean + SEM of three independent samples for each time point.
Chemokine receptor antagonists do not block CCL5-mediated increase in pAKT.
CCL5 binds to CCR5, CCR3, and CCR1 (Rossi & Zlotnik 2000). To further confirm that these receptors do not mediate CCL5 signaling, SH-SY5Y cells were preincubated for 15 min with selective antagonists of various chemokine receptors, alone or in combination. These include D-ala-peptide T-amide (DAPTA), an inhibitor of CCR5 (Polianova et al. 2005), BX471, an inhibitor of CCR1 (Liang et al. 2000), and SB328437, an inhibitor of CCR3 (White et al. 2000). CCL5-induced pAKT was observed in cells despite pre-treatment with inhibitors alone or in combination (Fig. 3), confirming that CCL5 signaling is not mediated by CCR5, CCR3, and CCR1 in SH-SY5Y cells.
Figure 3. CCL5-induced signaling is not blocked by chemokine receptor antagonists.

RA-treated SH-SY5Y cells were exposed to DAPTA (20 nM), BX471 (25 nM), SB328437 (80 nM) 15 min prior to CCL5 (100 nM). Cells were lysed and induction of pAKT was determined by western blot analysis (A) as described in Materials and Methods. B. Data are the mean + SD of four samples (two separate samples for each compound, from two independent experiments). **p<0.01, ***p<0.001 vs control or respective antagonists.
We then carried out concentration-dependent studies to establish the relative potency of CCL5. We observed a concentration-dependent and saturable effect of CCL5 on pAKT (Figs. 4A and B) and pERK1/2 (Figs. 4C and D) with an EC50 ~50 nM. Our data support previous studies obtained in mouse hippocampal cell line HT22 (Ignatov et al. 2006).
Figure 4. CCL5 elicits a concentration dependent increase in pAKT and pERK1/2.

Differentiated SH-SY5Y cells were exposed to the indicated concentrations of CCL5. Lysates were prepared and analyzed for pAKT (A and B) or pERK1/2 (C and D) by western blot. B and D. Data are presented as mean + SEM of four independent samples for each CCL5 concentration.
Ligands of GPCRs activate PI3K, which can be blocked by wortmannin (Wort). They can also induce protein kinase C, leading to downstream phosphorylation of ERK1/2. To further test whether CCL5 activation of GPR75 occurs through a GPCR, we exposed SH-SY5Y cells to Wort or U73122, a compound that uncouples Gqα-phospholipase C complex. Wort (Figs. 5A and B) and U73122 (Figs. 5C and D) strongly inhibited CCL5-induced pAKT and pERK1/2 respectively, further suggesting an involvement of Gq proteins in CCL5 activation of GPR75.
Figure 5. Inhibitors of signaling pathways block the effect of CCL5.

Differentiated SH-SY5Y cells were pretreated with 0.1 mM PI3K inhibitor wortmannin (Wort) for 1 h, or 5 μM PLC inhibitor U73122 for 15 min before adding CCL5 (100 nM). Cell extracts were prepared 15 min later and analyzed for pAKT and total AKT (A and B) or pERK1/2 and total ERK1/2 (C and D). Values are the mean ± SEM of three independent samples for each compound. *p<0.05, **p< 0.01 vs control; #p<0.05, ###p<0.001 vs CCL5.
CCL5 signaling is pertussis toxin (PTX)-insensitive.
Chemokine receptors are known to signal preferentially through PTX-sensitive Gi/G0 proteins (Cartier et al. 2005). To investigate the nature of the G protein involved in CCL5 activation of GPR75, SH-SY5Y cells or rat primary cortical neurons were exposed for 24 h to PTX (200 ng/ml), which uncouples the receptor from Gi/Go proteins by ADP-ribosylating their α subunit. PTX failed to abolish pAKT induction by CCL5 in both SH-SY5Y cells (Figs. 6A and B) and rat primary cortical neurons (Figs. 6C and D). To establish whether the concentration of PTX used in this experiment ADP-ribosylates Gi/Go, neurons were exposed to PTX and CXCL12, a ligand that activates Gi/Go CXCR4 receptor (Busillo & Benovic 2007). PTX blocked CXCL12- mediated activation of pAKT (not shown), suggesting that CCL5 activation of pAKT occurs most likely through a Gq protein.
Figure 6. CCL5 activation of signaling is PTX independent.

RA-treated SH-SY5Y cells (A and B) and primary rat cortical neurons (C and D) were exposed to vehicle alone (control) or containing 200 ng/ml PTX for 24 h. Cells were then incubated with either vehicle alone or containing 100 nM CCL5 for 15 min. Values are the mean ± SEM of three independent samples for each treatment. *p<0.05 vs control, #p<0.05 vs PTX.
CCL5 promotes internalization of GPR75.
GPCRs are rapidly internalized upon ligand activation (Ferguson et al. 1996), which can be measured by cell-surface biotinylation techniques using sulfo-NHS-biotin. Therefore, to further examine whether CCL5 acts as a ligand for GPR75 we determined whether CCL5 alters membrane-associated GPR75. Differentiated SH–SY5Y cells were exposed to CCL5 and membrane-impermeant NHS-LC-biotin, and then biotinylated cell surface proteins were precipitated with immobilized streptavidin and subjected to GPR75 immunoblotting. CCL5 induced a time-dependent internalization of GPR75, which began by 30 min and continued up to 1 h (Fig. 7A).
Figure 7. CCL5 promotes internalization of GPR75.

RA-treated SH-SY5Y cells were exposed to medium control (0.1% BSA) or medium containing 100 nM of CCL5 (A) or 100 nM of CXCL12 (B) for the indicated time points. Cells were then washed and treated with 0.5 mg/ml NHS-LC-biotin for 35 min at 4°C to biotinylate cell surface proteins. Biotinylated proteins were precipitated with immobilized streptavidin. Precipitates were then electrophoresed on SDS-polyacrylamide gels and subjected to GPR75 immunoblotting. Pan-cadherin was used to control for protein loading. The experiment was repeated twice with comparable data.
To confirm the specificity of GPR75 internalization by CCL5, SH-SY5Y cells were exposed to the CXCR4 ligand CXCL12, and internalization of GPR75 determined as described above. CXCL12 failed to promote internalization of GPR75 (Fig. 7B) suggesting that endocytosis of GPR75 is induced by selected chemokines.
Knockdown of GPR75 by mutagenesis.
To further support the hypothesis that CCL5 activates GPR75, we used CRISPR-Cas9 mutagenesis to knockdown this receptor. Two guide RNA’s (gRNA- A, B) were utilized to create target mutations within the GPR75 gene (Fig. 8A). T7E1 assay was then used to determine mutation in the genomic DNA of transfected SH-SY5Y and wild type SH-SY5Y (WT) cells. HEK293 cells were used as a comparison. After PCR amplification of the GPR75 coding region, the T7E1 assay showed specific cleavage of the band resulting in the expected 494bp + 363bp products for gRNA A and 555bp + 302bp products for gRNA (Fig. 8B). WT control cells displayed no T7E1 digestion indicating no heterogeneity in genomic DNA sequence of GPR75 in the target region.
Figure 8. CRISPR-Cas9 knockdown of GPR75.

A. Schematic of CRISPR-Cas9 targeting of human GPR75. Two guide RNA’s (gRNA- A, B) were used to create target mutations within the GPR75 gene. B. Analysis of genomic DNA of WT and transfected SH-SY5Y cells. HEK293 cells were used as a comparison. After PCR amplification of the GPR75 coding region (as described in Materials and Methods), the T7E1 assay of mutated cells shows specific cleavage of the band resulting in the expected 555bp + 302bp products for gRNA A and 494bp + 363bp products for gRNA B but not in WT cells.
WT and SH-SY5Y cells transfected with GPR75 gRNA A or B were then exposed for various time points to CCL5 to activate pERK. Western blot analysis of cell lysates revealed that the ability of CCL5 to induce pERK was significantly reduced in transfected cells when compared to WT cells (Figs. 9A and B). Western blot analysis was also used to measure levels of GPR75 in WT and gRNA transfected cells. A significant decrease (~70%) in GPR75 levels was observed in the transfected cells (Figs. 9C and D). Additionally, both gRNAs failed to change the amount of ERK or β-Actin (Figs. 9A and C), suggesting that GPR75 mutagenesis does not induce a generalized reduction in protein levels. Together these data suggest that CCL5 signals through GPR75.
Figure 9. Knockdown of GPR75 decreases CCL5-induced pERK.

A. SH-SY5Y cells were transfected with CRISPR/Cas9 components once per cell passage (4 cell passages total) to enrich GPR75 knockdown within the population. After confirmation of GPR75 knockdown, cells were split and plated onto a 12 well plate. After 24 hours cells were stimulated with CCL5 (50 ng/ml) for the specified time-points. Cell lysates were separated on a gel for western blot analysis of pERK1/2 and total ERK1/2. B. Densitometric analysis of the ratio of pERK1/2-total ERK1/2. Data are the mean + SD of three independent samples for each time-point. *p<0.05 GPR75 gRNA A vs. WT, #p<0.05 GPR75 gRNA B vs. WT. C. An example of a western blot stripped and re-probed for GPR75 and β-Actin to confirm protein knockdown. D. Densitometric analysis shows significant decrease of GPR75 expression in mutant cell lines. *p<0.05 GPR75 gRNA A vs. WT, #p<0.05 GPR75 gRNA B vs. WT. Data, expressed in AU, are the mean + SD of three independent samples for each treatment.
DISCUSSION
In the present report, we have explored the interaction of CCL5 with GPR75, an orphan receptor of the Gqα family of GPCRs, which appears to be expressed more abundantly in neuron-like cells than astrocytes. We initially characterized GPR75 signaling in SH-SY5Y cells because, according to qPCR and flow cytometry, this human neuronal cell line is GPR75 positive but does not express CCR1, CCR3, or CCR5. These receptors, which belong to the pertussis toxin-sensitive Gi subfamily of G proteins (Griffith et al. 2014), bind to and are activated by CCL5. Our data show that CCL5 triggers a signal cascade including pAKT, pERK1/2 and pGSKβ in a PTX insensitive manner, which is characteristic of Gqα receptors. Moreover, we show that CCL5 activates these signaling molecules even in the presence of selective antagonists of CCR1, CCR3, or CCR5 receptors. We obtained additional evidence confirming that CCL5 binds to and activates GPR75. First, CCL5 promoted a selective time-dependent internalization of GPR75, and second, CCL5 failed to significantly induce phosphorylation of AKT and ERK1/2 in neurons in which GPR75 was knockdown by CRISPR-Cas9 approach. Overall, our findings suggest that GPR75 is a novel CCL5 receptor that could mediate an important physiological function of this chemokine related to neuronal activity.
GPR75 was considered an orphan receptor until 2006 because no natural ligands for this receptor were identified (Civelli 2012). However, using a library of ligands on CHO-K1 cells transfected with GPR75, it was reported that CCL5 promotes inositol 3-phosphate formation, and increases pERK1/2 and pAKT (Ignatov et al. 2006), three representative signaling molecules associated with GPCR activation. No effect was obtained with other chemokines such as CCL4 and CCL7 or in mock transfected cells (Ignatov et al. 2006), suggesting specificity of ligand-receptor interaction. Our data show that CCL5 activates both ERK1/2 and AKT in cells that express physiological levels of GPR75, including rat primary neurons. In addition, we were able to show that CCL5, but not CXCL12 that binds only to CXCR4 (Signoret et al. 1997), promotes a time-dependent endocytosis of GPR75; This is in line with the known property of agonist-mediated GPCR internalization (Ferguson 2001). Taken together, all these data further support the notion that CCL5 is a cognate ligand for GPR75.
GPR75 has no selective agonist and/or antagonist ligands. Thus, to prove the interaction between CCL5 and GPR75, we have used CRISPR-Cas9 technology to reduce the expression of GPR75 in human neuroblastoma cells. We have used two gRNAs that target 5278–6091 sequence in exon 1 of the human GPR75 gene to generate a defined deletion. These two gRNA’s were chosen based on efficiency and low frequency of off target sites to create a mutation at bp 5865 and 5804. This strategy elicited a robust decrease of GPR75 protein as revealed by western blot analysis. Consequently, in cells transfected with CRISPR-Cas9, CCL5-mediated activation of pErk was significantly reduced. Thus, the CRISPR experiments further confirm that CCL5 activates GPR75. It is important to note that CRISPR-Cas9 targeting should result in a mutation of the receptor at amino acid 195 rather than a complete knockdown. At present, we cannot confirm these two possibilities. Indeed, the antibody utilized to detect GPR75 is against amino acid residues 395–425, which would be absent if there was truncation. Moreover, a specific antibody against the amino terminal sequence (upstream of the mutation site) is not available and must be developed. Thus, we cannot confirm or rule out that the observed decrease in GPR75 protein production is due to truncation of the receptor.
Alterations in the levels of Gq protein and associated signaling pathways are linked to impaired cellular functions in several pathological states. GPR75 is not an exception because this receptor has been shown to play a role in human neurodegenerative diseases. For instance, polymorphisms in the GPR75 gene are associated with the development of macular degeneration (Sauer et al. 2001). Experimental data have shown that GPR75 activation (Ignatov et al. 2006) inhibits the neurotoxic effect of Aβ peptide, whose levels have been linked to the neuropathology of Alzheimer’s disease (Murphy & LeVine 2010). Moreover, CCL5 has been shown to prevent the neurotoxic action of the human immunodeficiency virus (HIV) protein Tat (Rozzi et al. 2014) which does not bind to CCR5 or any other chemokine receptors, Likewise, a neuroprotective effect of CCL5 has been described for T-tropic gp120 (Kaul & Lipton 1999, Meucci et al. 1998), the HIV envelope protein that induce neuronal apoptosis by binding to CXCR4 receptor (Hesselgesser et al. 1998, Zheng et al. 1999). Our experiments were not designed to confirm or rule out that activation of GPR75 by CCL5 is the main mechanism of neuroprotection. Experiments in neurons in which GPR75 expression is decreased or abolished must be carried out to definitely prove the role that GPR75 plays in neuroprotection. Nevertheless, our findings provide a rationale for further studies on GPR75 as a valid target for the development of small ligands to prevent neurodegeneration.
GPR75 is a Gq receptor whose activation promotes PLC activity leading to diacylglycerol and inositol-triphosphate formation and the release of Ca2+ from intracellular stores. These intracellular signaling molecules have been shown to mediate the neuroprotective activity of various Gq receptors, including glutamate metabotropic receptor 5 (Doria et al. 2013). In addition, GPR75 activates the ERK and AKT pathways, which have been shown to mediate the neuroprotective effects of various compounds, including neurotrophins (Reichardt 2006), which also prevent gp120-mediated neurodegeneration (Bachis et al. 2003). Therefore, binding of CCL5 to GPR75 could explain why this chemokine exhibits a neuroprotective property against viral proteins. The data presented here and previously (Ignatov et al. 2006) suggest that GPR75 could be an alternative therapeutic target because it has no known interaction with the immune system and is expressed by neurons. Indeed, the neuronal expression of GPR75, linked to the activation of ERK and other neuroprotective signaling molecules, makes it particularly appealing for the development of small-molecule therapeutics against neurodegenerative diseases.
In summary, we report that CCL5 activates GPR75, a neuronal receptor that does not belong to the chemokine receptor family. This finding offers a unique approach for unraveling the molecular mechanisms of CCL5 neuroprotection and GPR75 role in brain function. Overall, our study underlies the importance of better understanding CCL5 action through the activation of GPR75.
Open Science Badges.
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. The complete Open Science Disclosure form for this article can be found at the end of the article. More information about the Open Practices badges can be found at https://cos.io/our-services/open-science-badges/.
Acknowledgements.
This work was supported by National Institute of Health Grant R21 NS089446 and Intramural Research Program at the National Institute on Drug Abuse. The authors declare that they have no conflicts of interest.
Abbreviations used are:
- AKT
protein kinase B
- Aβ
amyloid-β peptide
- CRISPR
clustered regularly interspaced short palindromic repeats
- DAPTA
D-ala-peptide T-amide
- ERK
extracellular signaling-regulated kinases
- GPCR
G protein-coupled receptor
- GFP
green fluorescent protein
- GPR75 CRISPR
lentiCRISPR gRNA B carrying vector
- GSK3β
glycogen synthase kinase 3 β
- PTX
pertussis toxin
- DTSSP
3,3’-dithiobis(sulfosuccinimidylpropionate)
- HIV
human immunodeficiency virus
- HAND
HIV-associated neurocognitive disorders
- MAP2
microtubule associated protein-2
- qPCR
real time-polymerase chain reaction
- PI3K
phosphatidylinositol 3–kinase
- RA
retinoic acid
- RRID
Research Resource Identifier
- sulpho-NHS-LC-biotin
sulfosuccinimidyl-6-(biotin-amido)hexanoate
- U73122
1-[6-[((17β)-3-Methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione
- T7E1
T7 endonuclease I
- Wort
wortmannin
- WT
wild type
REFERENCES
- Ammer H and Schulz R (1994) Retinoic acid-induced differentiation of human neuroblastoma SH-SY5Y cells is associated with changes in the abundance of G proteins. J Neurochem, 62, 1310–1318. [DOI] [PubMed] [Google Scholar]
- Avdoshina V, Becker J, Campbell L, Parsadanian M, Mhyre T, Tessarollo L and Mocchetti I (2011) Neurotrophins modulate the expression of chemokine receptors in the brain. J NeuroVirol, 17, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdoshina V, Biggio F, Palchik G, Campbell LA and Mocchetti I (2010) Morphine induces the release of CCL5 from astrocytes: potential neuroprotective mechanism against the HIV protein gp120. Glia, 58, 1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachis A, Major EO and Mocchetti I (2003) Brain-derived neurotrophic factor inhibits human immunodeficiency virus-1/gp120-mediated cerebellar granule cell death by preventing gp120 internalization. J Neurosci, 23, 5715–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busillo JM and Benovic JL (2007) Regulation of CXCR4 signaling. Biochim Biophys Acta, 1768, 952–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier L, Hartley O, Dubois-Dauphin M and Krause KH (2005) Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev, 48, 16–42. [DOI] [PubMed] [Google Scholar]
- Civelli O (2012) Orphan GPCRs and neuromodulation. Neuron, 76, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedoni S, Olianas MC, Ingianni A and Onali P (2014) Type I interferons up-regulate the expression and signalling of p75 NTR/TrkA receptor complex in differentiated human SH-SY5Y neuroblastoma cells. Neuropharmacology, 79, 321–334. [DOI] [PubMed] [Google Scholar]
- Doria JG, Silva FR, de Souza JM, Vieira LB, Carvalho TG, Reis HJ, Pereira GS, Dobransky T and Ribeiro FM (2013) Metabotropic glutamate receptor 5 positive allosteric modulators are neuroprotective in a mouse model of Huntington’s disease. Br J Pharmacol, 169, 909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SS (2001) Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev, 53, 1–24. [PubMed] [Google Scholar]
- Ferguson SS, Downey WE 3rd, Colapietro AM, Barak LS, Menard L and Caron MG (1996) Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science, 271, 363–366. [DOI] [PubMed] [Google Scholar]
- Giri RK, Rajagopal V and Kalra VK (2004) Curcumin, the active constituent of turmeric, inhibits amyloid peptide-induced cytochemokine gene expression and CCR5-mediated chemotaxis of THP-1 monocytes by modulating early growth response-1 transcription factor. J Neurochem, 91, 1199–1210. [DOI] [PubMed] [Google Scholar]
- Griffith JW, Sokol CL and Luster AD (2014) Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol, 32, 659–702. [DOI] [PubMed] [Google Scholar]
- Hesselgesser J, Taub D, Baskar P, Greenberg M, Hoxie J, Kolson DL and Horuk R (1998) Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol, 8, 595–598. [DOI] [PubMed] [Google Scholar]
- Ignatov A, Robert J, Gregory-Evans C and Schaller HC (2006) RANTES stimulates Ca2+ mobilization and inositol trisphosphate (IP3) formation in cells transfected with G protein-coupled receptor 75. Br J Pharmacol, 149, 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M and Lipton SA (1999) Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A, 96, 8212–8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Ma Q, Medders KE, Desai MK and Lipton SA (2007) HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ, 14, 296–305. [DOI] [PubMed] [Google Scholar]
- Liang M, Mallari C, Rosser M et al. (2000) Identification and characterization of a potent, selective, and orally active antagonist of the CC chemokine receptor-1. J Biol Chem, 275, 19000–19008. [DOI] [PubMed] [Google Scholar]
- Maung R, Hoefer MM, Sanchez AB et al. (2014) CCR5 knockout prevents neuronal injury and behavioral impairment induced in a transgenic mouse model by a CXCR4-using HIV-1 glycoprotein 120. J Immunol, 193, 1895–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW and Miller RJ (1998) Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A, 95, 14500–14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP and LeVine H 3rd (2010) Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis, 19, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polianova MT, Ruscetti FW, Pert CB and Ruff MR (2005) Chemokine receptor-5 (CCR5) is a receptor for the HIV entry inhibitor peptide T (DAPTA). Antiviral Res, 67, 83–92. [DOI] [PubMed] [Google Scholar]
- Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci, 361, 1545–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D and Zlotnik A (2000) The biology of chemokines and their receptors. Annu Rev Immunol, 18, 217–242. [DOI] [PubMed] [Google Scholar]
- Rozzi SJ, Borelli G, Ryan K, Steiner JP, Reglodi D, Mocchetti I and Avdoshina V (2014) PACAP27 is protective against tat-induced neurotoxicity. J Mol Neurosci, 54, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer CG, White K, Stohr H et al. (2001) Evaluation of the G protein coupled receptor-75 (GPR75) in age related macular degeneration. Br J Ophthalmol, 85, 969–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M, Giesen PL, Nemerson Y and Taubman MB (1997) Tissue factor is induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. J Biol Chem, 272, 28568–28573. [DOI] [PubMed] [Google Scholar]
- Signoret N, Oldridge J, Pelchen-Matthews A et al. (1997) Phorbol esters and SDF-1 induce rapid endocytosis and down modulation of the chemokine receptor CXCR4. J Cell Biol, 139, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL et al. (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science, 307, 1282–1288. [DOI] [PubMed] [Google Scholar]
- Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ and Rogers TJ (2002) Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci U S A, 99, 10276–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarttelin EE, Kirschner LS, Bellingham J, Baffi J, Taymans SE, Gregory-Evans K, Csaky K, Stratakis CA and Gregory-Evans CY (1999) Cloning and characterization of a novel orphan G-protein-coupled receptor localized to human chromosome 2p16. Biochem Biophys Res Commun, 260, 174–180. [DOI] [PubMed] [Google Scholar]
- White JR, Lee JM, Dede K et al. (2000) Identification of potent, selective non-peptide CC chemokine receptor-3 antagonist that inhibits eotaxin-, eotaxin-2-, and monocyte chemotactic protein-4-induced eosinophil migration. J Biol Chem, 275, 36626–36631. [DOI] [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Ghorpade A et al. (1999) Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol, 98, 185–200. [DOI] [PubMed] [Google Scholar]