

Abstract
We characterize the encapsulation of supercharged green fluorescent protein, GFP(+36), by thermophilic ferritin from Archaeoglobus fulgidus (AfFtn). The AfFtn–GFP(+36) assembly is rapid, nearly stoichiometric, and robust. Using a more stably assembled mutant AfFtn, we show that encapsulation can occur in the presence of mostly assembled cages, in addition to encapsulation starting from AfFtn individual subunits. Assembly and encapsulation do not occur with non‐supercharged GFP or the alternately supercharged GFP(−30), highlighting the role of complementary electrostatic interactions between the cargo and AfFtn cage interior. We also present a method for verifying protein–protein encapsulation, using nickel nitrilotriacetic acid agarose resin. AfFtn‐supercharged protein host‐guest complexes could find applications in enzyme studies, protein separations, and in vivo protein stabilization and targeted delivery.
Keywords: ferritin, encapsulation, protein–protein interactions, supercharged protein, host–guest interactions
Abbreviations
- AfFtn
thermophilic ferritin from Archaeoglobus fulgidus
- GFP(+36)
superpositively charged green fluorescent protein
- DLS
dynamic light scattering
- eGFP
enhanced green fluorescent protein
- GFP(−30)
supernegatively charged green fluorescent protein
- Ni‐NTA
nickel nitrilotriacetic acid
- SEC
size exclusion chromatography
- TEM
transmission electron microscopy
Introduction
Supramolecular host–guest complexes have wide‐ranging applications that include sensing,1 catalysis,2 separations,3 and drug delivery.4 Small‐molecule host–guest chemistry has explored the roles of non‐covalent interactions, shape and size complementarity, and electronic and solvation effects in molecular recognition.5, 6, 7 Protein–ligand interactions have been studied and designed in the context of enzyme active sites as well as protein surfaces.8, 9 In nature, host–guest complexes often involve protein–protein interactions. Chaperone proteins can bind or sequester a misfolded guest protein and help it to refold properly.10 Bacterial microcompartments provide a polyhedral protein shell that encapsulates an enzyme and prevents the release of toxic metabolic intermediates.11 Here, we characterize a small (~10 nm), versatile protein‐cage system capable of encapsulating two‐to‐three, full‐length supercharged proteins in high yield.
Non‐native protein–protein host–guest complexes have been reported previously, where protein cages such as lumazine synthase from Aquifex aeolicus, encapsulin, or viral capsids act as hosts. These large (>25 nm diameter) protein cages have been shown to effectively encapsulate fluorescent proteins,12, 13, 14, 15 HIV protease,16 Cas9,17 Escherichia coli alkaline phosphatase,12 and other enzymes with wide ranging structures and functions.18, 19 Encapsulation of a guest protein within a host virus capsid has enabled kinetic studies of enzymes in confined spaces,20, 21 as well as the development of confined enzyme cascades.22 Protein host–guest assemblies have also been engineered to enable structural studies of a guest peptide.23 These host‐guest assemblies differ from larger scale protein–protein ordered arrays,24, 25, 26, 27 in that their utility arises from the behavior of a single particle, rather than material properties gained from many proteins assembling. Many prior protein–protein host–guest examples have employed extensive directed evolution,16 addition of osmolyte,12 pH modulation,20 or covalent attachment.15 Recent work by Tetter et al.19 presented a host–guest complex featuring thermophilic ferritin from Archaeoglobus fulgidus (AfFtn) (Fig. 1), which required no added reagents and encapsulated a supercharged variant of green fluorescent protein with a theoretical net charge of +36 (GFP(+36)).28
Figure 1.
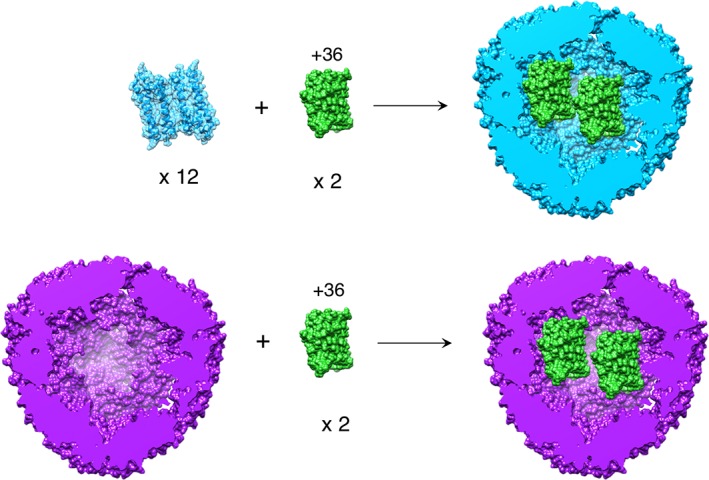
Encapsulation of GFP(+36) by wt‐AfFtn (top) and E65R‐AfFtn (bottom). For wt, 12 AfFtn dimers self‐assemble around positively charged GFP(+36) to form a 1:2 AfFtn 24mer:GFP(+36) host–guest complex. E65R‐AfFtn is predominantly assembled in low ionic strength conditions but also forms a 1:2 complex with GFP(+36), starting with the assembled ferritin cage.
AfFtn exists as dimers of tetrahelical bundles with a short fifth helix at low ionic strength (< 200 mM NaCl). Self‐assembly of 12 dimers occurs in high ionic strength buffer (Fig. S1), yielding a hollow, cage‐like 24mer with tetrahedral symmetry and four large (4.5 nm), triangular pores.29 Cargo smaller than the 24mer inner diameter (8 nm) can be encapsulated within the protein interior, as has been shown with other ferritins.30, 31, 32, 33, 34 Previously, we demonstrated the encapsulation of 6‐nm gold nanoparticles (AuNPs), where the particles nucleate AfFtn assembly via their high surface charge35, 36, 37, 38, 39 without the need to increase the solution ionic strength. The interior of AfFtn is negatively charged,35 and thus an attractive electrostatic interaction with GFP(+36) is expected. The experiments conducted by Tetter et al.19 raised several questions regarding the encapsulation process, such as whether cargo can be encapsulated within fully assembled AfFtn cages or whether the encapsulation process must start with disassembled AfFtn subunits. To address these questions, we use a designed AfFtn mutant which we recently characterized that remains predominantly assembled in low ionic strength solutions.40 We show that AfFtn and its more stably assembled mutant can encapsulate GFP(+36), demonstrating that unlike other ferritins,41 AfFtn is capable of encapsulating large cargo without complete prior disassembly. We also present further evidence that the encapsulation process is dependent on complementary charge interactions, by attempting encapsulation of non‐supercharged eGFP and supernegatively charged GFP(−30).
Results and Discussion
Encapsulation of GFP(+36) in wt‐AfFtn
GFP(+36) and AfFtn were mixed in varying stoichiometries (AfFtn 24mer:GFP(+36)) ranging from 0.75:1 to 1:6. A low ionic strength buffer (“no‐salt buffer,” meaning no added NaCl, 20 mM sodium phosphate, pH 7.6) was used to promote AfFtn disassembly prior to mixing. We have found that using phosphate buffer enables almost complete disassembly into dimers (Fig. S1), whereas Tris buffer gives ~40% 24mer as shown by Tetter et al.19 Thus, our experimental conditions enable us to investigate cleanly the effect of AfFtn assembly state on encapsulation. The solutions were analyzed by native agarose gel electrophoresis [Fig. 2(a)]. A single fluorescent green band was observed to run toward the positive gel terminal, corresponding to the AfFtn–GFP(+36) complex. With GFP(+36) in excess of 2 per AfFtn 24mer, a green band running toward the negative terminal appeared, corresponding to free GFP(+36). Size exclusion chromatography (SEC) was used to verify the association between GFP(+36) and AfFtn 24mer. When monitored using absorbance (A280), the main peak corresponded to the AfFtn 24mer elution volume and overlapped with the GFP(+36) elution peak monitored using GFP(+36) fluorescence at 509 nm (I509), indicating association and assembly of AfFtn:GFP(+36) complex [Fig. 2(b)]. Without AfFtn present, GFP(+36) alone does not elute from the column in no‐salt buffer, presumably due to non‐specific interactions with the column medium (Fig. S2). The encapsulation product fractions were combined, concentrated, and analyzed by dynamic light scattering (DLS) to verify AfFtn assembly [Fig. 2(c)]. The average particle diameter matched that of AfFtn 24mer, 13.9 nm (PDI 0.26) (vs. 13.5 nm previously published),40 suggesting that GFP(+36) was successfully encapsulated within AfFtn, rather than adsorbing to its surface or forming a non‐specific, disordered aggregate with multiple AfFtn dimers. Transmission electron microscopy (TEM) also supports GFP(+36) encapsulation, as negatively stained AfFtn–GFP(+36) samples excluded more stain from the cage interior than empty AfFtn, indicating the cage contains protein [Fig. 2(d), additional micrographs in Fig. S3]. GFP(+36) absorbance and emission spectra collected pre‐ and post‐encapsulation were nearly identical, indicating the fluorescent protein remains folded (Fig. S4). The encapsulation process is quite rapid, as native gel electrophoresis and SEC show association of AfFtn and GFP(+36) within 30 min (the measurement time, Fig. S5).
Figure 2.
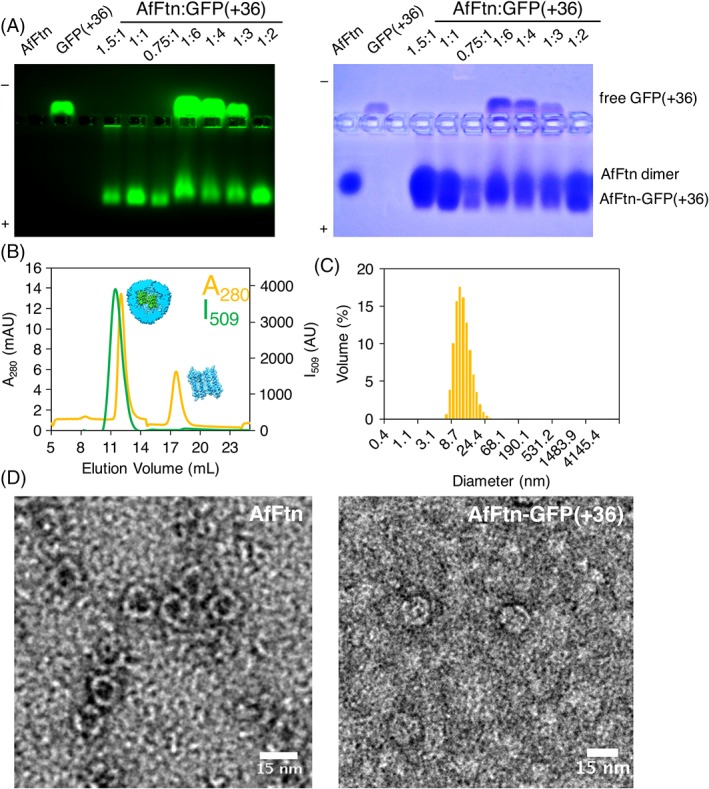
(A) Native gel electrophoresis varying AfFtn 24mer:GFP(+36) stoichiometry. The green band traveling toward the positive terminal can be attributed to the AfFtn–GFP(+36) encapsulation product, while the green band traveling toward the negative terminal corresponds to free or excess GFP(+36). “1” = concentration of 0.6 μM. (B) SEC of AfFtn–GFP(+36) complex, monitored by absorbance at 280 nm (A 280, yellow) and fluorescence at 509 nm (I509, green). Peak at 12.5 mL corresponds to AfFtn–GFP(+36), peak at 17.5 mL corresponds to AfFtn dimer (6 μM AfFtn, 12 μM GFP(+36)). (C) Dynamic light scattering diameter size distribution of sample corresponding to SEC peak at 12.5 mL. Average diameter is 13.9 nm with PDI of 0.26, matching that of assembled AfFtn without GFP(+36) present (6 μM AfFtn, 12 μM GFP(+36)). (D) TEM micrographs of AfFtn without (left) and with (right) GFP(+36), stained with uranyl acetate. Scale bars are 15 nm.
As further confirmation of encapsulation, we incubated AfFtn–GFP(+36) with nickel nitrilotriacetic acid (Ni‐NTA) agarose resin (Fig. 3). Only GFP(+36) has a His‐tag for purification purposes and therefore binds strongly to the nickel agarose resin. If GFP(+36) is encapsulated within the ferritin cage, however, the His‐tag should be less accessible for binding to the resin. Significant fluorescence was seen in solution for the AfFtn–GFP(+36) load sample, indicating that little GFP(+36) bound to the resin. In the positive control, GFP(+36)‐only sample remained bound to resin during loading and wash steps, producing low solution fluorescence until elution with 250 mM imidazole buffer triggered dissociation from Ni‐NTA. Importantly, folded GFP(+36) is unlikely to diffuse through the pores of the wild type (wt) AfFtn 24mer, based on protein size and geometry – assuming that the crystal structures of both proteins are representative of their structures in solution and that these crystal structures can be simplified to a hollow sphere (AfFtn 24mer) and a cylinder (GFP(+36)) (see Methods – Geometric analysis of AfFtn and GFP(+36)). These data confirm that AfFtn encapsulates and protects GFP(+36) within the ~8‐nm ferritin cavity.
Figure 3.
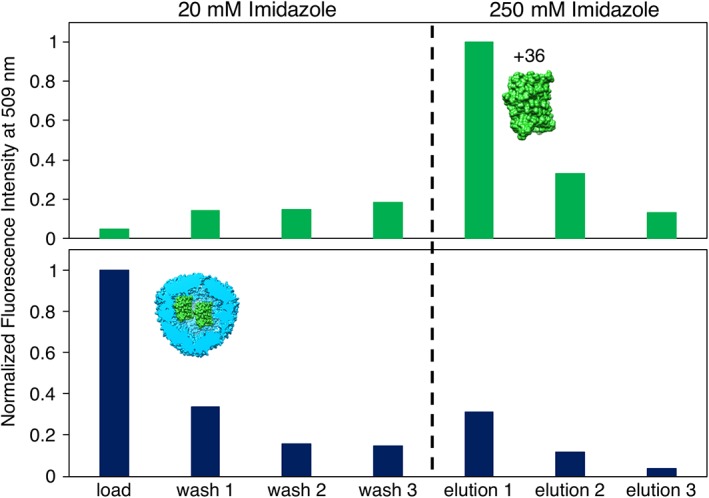
Ni‐NTA assay shows greatly reduced GFP(+36) binding to resin after encapsulation within AfFtn. Samples were incubated at 6 μM AfFtn, 12 μM GFP(+36) for 1 h to enable binding of GFP His‐tag to Ni resin. Samples were then centrifuged and the supernatants were analyzed by fluorescence (“load”), followed by three washings of the resin (“wash 1–3”). Increasing [imidazole] to 250 mM dissociated any protein bound to the resin (“elution 1–3”). Green bars are for GFP(+36) alone, navy bars are for AfFtn–GFP(+36). High fluorescence in the load sample for AfFtn–GFP(+36) indicates a lack of binding, due to encapsulation, while high fluorescence in the Elution 1 sample for free GFP(+36) indicates resin binding and release, as expected for a protein with a His‐tag.
By measuring UV–vis spectra of SEC‐purified AfFtn–GFP(+36), we calculated N GFP, the average number of GFP(+36) molecules per AfFtn 24mer (see Methods – Determination of GFP(+36) loading for details). For wt‐AfFtn, N GFP = 2.6 ±0.3, reflecting a mixture of AfFtn cages with 2 or 3 GFP(+36) encapsulated. The value for wt falls within the expected range N GFP = 0–4, where 4 is the maximum based on approximate geometry of the AfFtn cavity and GFP(+36) (see Methods – Geometric analysis of AfFtn and GFP(+36)). This UV–vis result is also consistent with the approximately 2 encapsulated GFP(+36) molecules observed by native agarose gel electrophoresis [Fig. 2(a)]. The high charge of GFP(+36) likely disfavors close packing within the AfFtn cage, leading to the observed 50% loading in wild‐type for N GFP = 2. N GFP = 2 corresponds to a confinement molarity of ~12 mM, and N GFP = 3 is nearly 20 mM. For an encapsulated enzyme, such high confinement could significantly increase the catalytic rate.42 Similar levels of encapsulation were observed by Tetter et al. for GFP(+36) encapsulation with AfFtn, where the addition of 1.5 GFP(+36) per 24mer (adjusted for our BCA assay‐determined GFP(+36) extinction coefficient value) led to encapsulation of 1.5 GFP(+36) per 24mer (mixture of 1 or 2 GFP(+36) per cage) when incubated in 100 mM NaCl, 50 mM Tris buffer.19 Increasing the ratio of GFP(+36) to 24mer led to a linear increase in encapsulated GFP(+36), up to a reported value of 4 GFP(+36)/24mer, at that ionic strength in Tris buffer. Our loading is also similar to that reported for teal fluorescent protein within virus (CCMV) capsids.15 Attempting to increase the loading by adding higher ratios of GFP(+36) (1:4 or 1:6 AfFtn:GFP(+36)) was unsuccessful and led to significant protein precipitation (Fig. S6). Tetter et al. noted similar observations of precipitation at low ionic strength for high GFP(+36) loading.19
Encapsulation occurs with assembled cage E65R
We recently reported a new AfFtn mutant with enhanced cage stability at low ionic strengths: E65R,40 which differs from other ferritin mutants where cage assembly is destabilized.43, 44, 45 The crystal structure of E65R (PDB ID 5V5K) reveals octahedral symmetry and lacks the large triangular pores of wt‐AfFtn (Fig. 1). Shown in Figure 4(a), we saw evidence of GFP(+36) encapsulation by AfFtn E65R using native gel electrophoresis, with overlapping GFP(+36) fluorescent bands and E65R Coomassie staining. E65R AfFtn–GFP(+36) 1:2 samples were also analyzed by SEC, DLS, and TEM. GFP(+36) fluorescence was observed primarily in SEC peaks corresponding to the AfFtn 24mer, indicating an association between E65R and GFP(+36) [Fig. 4(b)]. SEC fractions containing the highest GFP(+36) fluorescence were further analyzed by DLS [Fig. 4(c)] and TEM [Fig. 4(d), see Fig. S7 for additional micrographs]. DLS results show an average particle size similar to that of native E65R: 13.1 nm (PDI 0.08) versus 12.9 (PDI 0.06).40 The TEM images are consistent with the ferritin 24mer, further indicating the AfFtn E65R assembled with GFP(+36) inside. Based on gel assays and absorbance measurements post‐FPLC, there appear to be approximately 2 GFP(+36) per E65R 24mer, similar to wt results. However, because E65R remains mostly assembled (and elutes as 24mer) at low ionic strength whether or not encapsulation has occurred, SEC absorbance (A280) measurements are less informative about the number of GFP(+36) molecules per cage.
Figure 4.
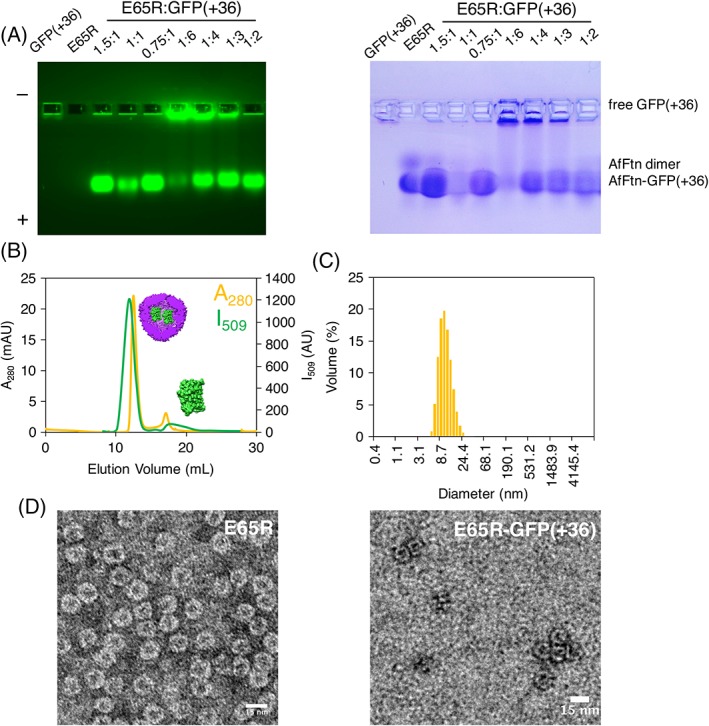
E65R‐GFP(+36) characterization. (A) Native gel electrophoresis with varying stoichiometry. “1” = concentration of 0.6 μM. (B) Size exclusion chromatography showing overlap of absorbance at 280 nm (yellow) and fluorescence at 509 nm (green), suggesting encapsulation of GFP(+36) within the E65R‐AfFtn cage (6 μM AfFtn, 12 μM GFP(+36)). (C) Volume size distribution from dynamic light scattering measurements. Average diameter is 13.1 nm (PDI 0.08), matching that of E65R without GFP(+36) present (6 μM AfFtn, 12 μM GFP(+36)). (D) Transmission electron micrographs of E65R (left) and E65R‐GFP(+36) (right), stained with uranyl acetate. Scale bars are 15 nm.
E65R AfFtn–GFP(+36) samples were also seen to have little immobilized protein in the Ni‐NTA binding assay, again supporting encapsulation (Fig. S8). Interestingly, E65R‐GFP(+36) had even less binding to Ni‐NTA resin compared with wt‐AfFtn, likely due to its closed form40 rendering the encapsulated His‐tag more inaccessible. AfFtn assembly occurs at low ionic strength with only two equivalents of GFP(+36) present, and the His‐tag of GFP(+36) appears sequestered in the presence of AfFtn. These observations are consistent with GFP(+36) residing within the cage interior.
It is striking that E65R is able to efficiently encapsulate GFP(+36), given its assembled, closed‐pore conformation (Fig. 1). This differs from previous experiments, where E65R was not observed to encapsulate a 6‐nm gold nanoparticle (AuNP).40 Encapsulation of large protein cargo without first disassembling into small subunits is not without precedent, however. Lumazine synthase was found to encapsulate supercharged ferritin starting from either assembled capsids or capsid subunits, albeit with lower yields for assembled capsids.46 The cage dynamics were such that protein cargo significantly larger than the 4‐nm pores47 could still enter the interior cavity. Förster resonance energy transfer (FRET) experiments exploring the dynamics of lumazine synthase confirmed that the assembled capsid can rapidly uptake and exchange cargo.14 In addition, coarse‐grained simulations of HIV‐1 capsid assembly have suggested individual subunits can come on and off of an assembled virion.48 Similar dynamics may be at play for E65R, enabling GFP(+36) encapsulation. GFP(+36) is also smaller than a 6‐nm AuNP and potentially more flexible, compared with the inorganic AuNP.
Investigation of electrostatic effects
To confirm that encapsulation is triggered by the high charge of GFP(+36), we repeated native gel and size exclusion experiments with enhanced GFP (eGFP), a non‐supercharged variant with enhanced fluorescence.49 At pH 7.6, eGFP has an estimated net charge of −6.4, based on individual amino acid pK a values. As seen in Figure 5(a), we observed no overlapping eGFP‐AfFtn bands by native gel, nor did we see AfFtn 24mer overlapping with eGFP fluorescence by SEC [Fig. 5(b)]. Thus, the encapsulation observed with GFP(+36) is dependent on its high charge. E65R also does not show encapsulation of eGFP, via SEC (Fig. S9). We suggest that similar to studies with AuNP encapsulation, the high charge density of GFP(+36) mitigates the need for increased ionic strength and allows encapsulation to occur, even if the bulk solution ionic strength is low. We also attempted encapsulation using supernegatively charged GFP,50 GFP(−30), to determine whether the sign of the charge was important for encapsulation. GFP(−30) did not induce encapsulation and AfFtn 24mer assembly as determined by SEC [Fig. 5(c)], indicating that charge complementarity between the cargo and AfFtn interior is necessary for supercharged protein loading. Using SEC, E65R also showed no encapsulation with GFP(−30) (Fig. S10). These results are similar to those presented by Deshpande et al., where an engineered AfFtn with different overall interior cage charges was best able to stabilize the folding of guest proteins that had charge complementarity between host and guest.51
Figure 5.
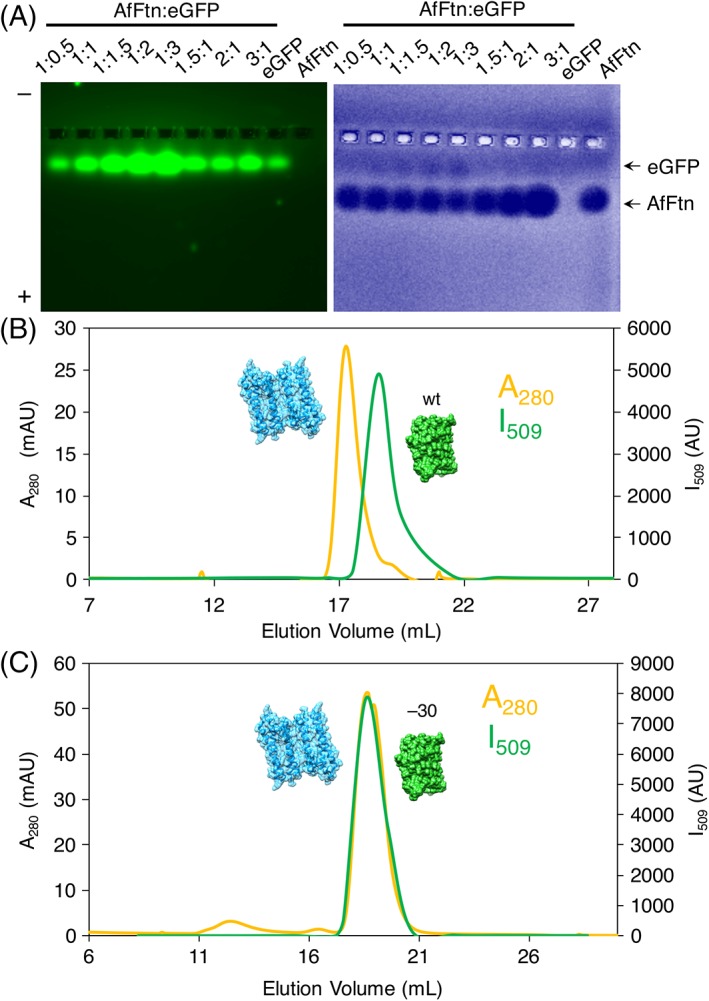
Non‐superpositively charged GFP cargo is not encapsulated. (A) Native gel electrophoresis of wt‐AfFtn mixed with eGFP at varying stoichiometries. “1” = concentration of 0.6 μM. (B) SEC of wt‐AfFtn–eGFP shows no peak corresponding to encapsulation product, indicating that high charge is needed to induce encapsulation (6 μM AfFtn, 12 μM GFP(+36)). (C) SEC of wt‐AfFtn–GFP(−30) shows no peak corresponding to encapsulation product, indicating that charge complementarity between the AfFtn cage interior and the cargo protein is important for encapsulation (6 μM AfFtn, 12 μM GFP(+36)).
To disentangle any effect of pre‐assembled cages, we investigated the effect of ionic strength on encapsulation using E65R, which mostly retains the 24mer structure across a wide range of NaCl concentrations (>90% 24mer, 0–800 mM NaCl).40 We mixed E65R and GFP(+36) in solutions of increasing NaCl concentration (Fig. 6). With increasing ionic strength, the fluorescent band in the native gel corresponding to encapsulated product grew dimmer, confirming that the extent of encapsulation decreases as electrostatic interactions are attenuated. Similar decreases in encapsulation with increasing ionic strength from 100 mM to 300 mM and 600 mM NaCl were also observed by Tetter et al. with AfFtn encapsulation of GFP(+36), although, again, the buffer conditions differed from ours with the use of Tris rather than phosphate.19
Figure 6.
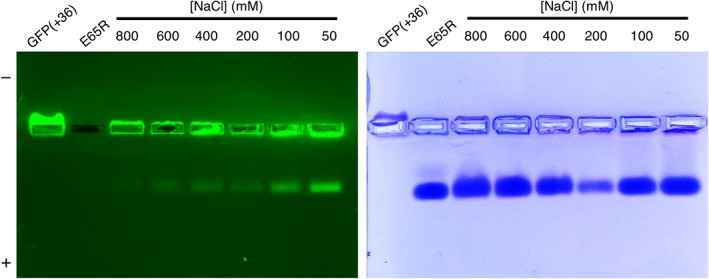
E65R‐GFP(+36) native gel with increasing [NaCl]. 0.6 μM AfFtn, 1.2 μM GFP(+36). 0.7% agarose gel run at 100 V, 20 min, on ice, covered in foil. At increasing ionic strength less encapsulation was observed, as indicated by dimmer fluorescent band traveling towards the positive terminal.
The role of ionic strength in the GFP(+36) encapsulation process was also explored with wt‐AfFtn. Notably, after the formation of the protein–protein host–guest complex, raising the ionic strength of the solution did not entirely disrupt the assembled complex. After incubating wt‐AfFtn with GFP(+36) 1:2 overnight in no‐salt buffer (20 mM sodium phosphate, pH 7.6), the NaCl concentration was increased from 0 mM to 800 mM, and the sample was incubated overnight to ensure equilibrium was reached. The sample was run on a size exclusion column equilibrated in high‐salt buffer (20 mM sodium phosphate, 800 mM NaCl, pH 7.6), as seen in Figure 7. While a free GFP(+36) peak was observed (matching the elution volume of a GFP(+36) alone sample in the high‐salt buffer, see Fig. S11), most GFP(+36) fluorescence overlapped with the AfFtn 24mer peak, indicating the majority remained encapsulated. High salt concentrations should stabilize the AfFtn 24mer but also screen electrostatic interaction between AfFtn and GFP(+36). When wt‐AfFtn and GFP(+36) were initially incubated in high‐salt buffer overnight and then analyzed by SEC, encapsulation was not observed. Instead, AfFtn 24mer was seen eluting with minimal GFP(+36) fluorescence, while a separate free GFP(+36) peak eluted later (Fig. 8). This result differs somewhat from Tetter's observation of roughly 1 GFP loaded per AfFtn 24mer in 600 mM NaCl solution,19 but these experiments serve to confirm that encapsulation of GFP(+36) by wt‐AfFtn is driven by electrostatic interactions. Control experiments incubating wt‐AfFtn with either eGFP or GFP(−30) in the high‐salt buffer also did not show encapsulation (Fig. S12).
Figure 7.
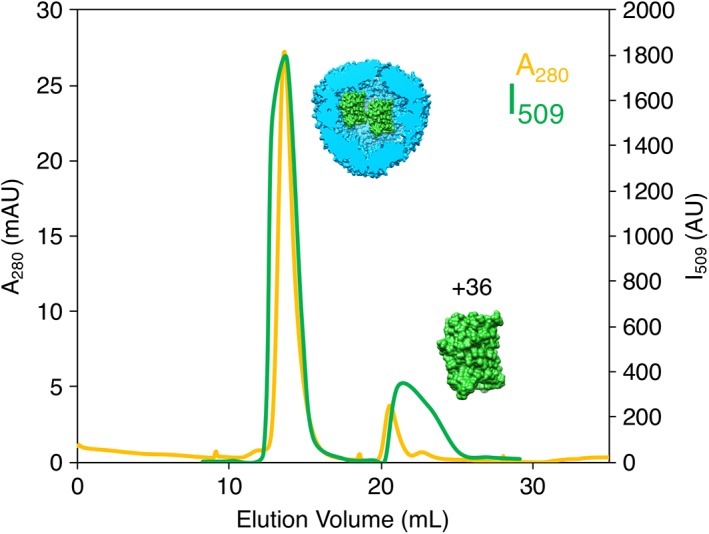
SEC of AfFtn–GFP(+36) in high‐salt buffer after overnight incubation in no‐salt buffer mixed at 1:2 AfFtn:GFP(+36) (6 μM AfFtn, 12 μM GFP(+36)), demonstrating the effect of increasing the ionic strength after initial encapsulation. Free GFP(+36) was observed, indicating that increasing ionic strength partially disrupted the assembly. However, the majority remained encapsulated, indicating that post‐encapsulation, the complex is fairly stable.
Figure 8.
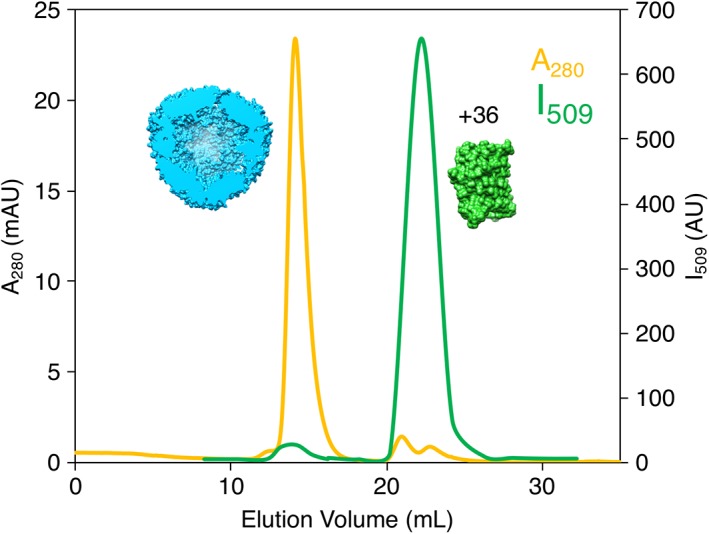
SEC of AfFtn and GFP(+36) in high‐salt buffer after overnight incubation in high‐salt buffer mixed at 1:2 AfFtn:GFP(+36) (6 μM AfFtn, 12 μM GFP(+36)). Minimal encapsulation was observed, indicating that high ionic strength inhibits the encapsulation process.
Conclusions
In conclusion, we have successfully tailored a non‐native, stoichiometric protein–protein host–guest complex using archaeal thermostable ferritin and supercharged GFP(+36). Careful characterization of the protein–protein host–guest complexes was achieved by working in phosphate buffer where the wt‐AfFtn protein is normally disassembled. Encapsulation was confirmed by several characterization methods, including Ni‐NTA assay, which showed little‐to‐no binding of the encapsulated His‐tagged GFP(+36) protein. The stoichiometry of encapsulated protein was determined through careful measurement of both AfFtn and GFP extinction coefficients, after rigorous protein purification. We demonstrated that the encapsulation process does not require pre‐disassembled cages, as we encapsulated GFP(+36) within a mutant AfFtn (E65R), which forms highly stable cages that lack large pores.40 The cage dynamics may allow for the encapsulation of the significantly smaller GFP(+36). Charge complementarity between cargo protein and the negatively charged AfFtn interior is necessary for encapsulation, as eGFP and supernegatively charged GFP(−30) were not encapsulated. Encapsulating native supercharged proteins52 or GFP(+36) fusion proteins within ferritins could be a useful delivery method, for example, by rendering therapeutic cargo less vulnerable to proteolysis, unfolding, and immune response. Such a system could also harbor toxic cargo or deliver cargo to cells presenting specific cell‐surface receptors. More fundamentally, we provide an example of a small 1:2 protein–protein host–guest complex, achieving significantly high confinement molarity due to the small cavity volume, which opens new possibilities for studying protein biophysical phenomena such as crowding, folding, molecular recognition, protein threading, and enzymology within discrete protein nanocapsules.
Materials and Methods
AfFtn protein expression and purification
AfFtn wt and AfFtn E65R mutant were expressed and purified as previously published.40 Briefly, a plasmid containing AfFtn wt or E65R genes were transformed in BL21(DE3)CodonPlus‐RP cells. Cells were grown overnight at 30°C in LB medium. Cultures were transferred to 1 L Terrific Broth and grown at 37°C until OD600 ~0.8. Expression was induced with 1 mM IPTG for 4 h at 37°C. Cells were pelleted by centrifugation (10 min, 4 krpm, 4°C) and stored at −20°C. Cells were resuspended in buffer (20 mM sodium phosphate, 20 mM NaCl, pH 7.6) and lysed by treatment with lysozyme (~1 mg/mL final concentration), DNAse, and sonication (amplitude of 30, 1 s on, 1 s off, 15 min total processing time). Cellular debris was removed by centrifugation (30 min, 6 krpm, 4°C) and the soluble fraction was treated with benzonase nuclease for 15 min at rt after addition of MgCl2 to a final concentration of 2 mM. The solution was heat shocked for 10 min at 80°C to precipitate most E. coli proteins and centrifuged (9 krpm, 60 min, 4°C). The supernatant was concentrated, buffer exchanged (20 mM sodium phosphate, 2.5 M NaCl, 2 mM EDTA, pH 7.6) and injected onto a HiLoad 16/60 Superdex 200 size exclusion column equilibrated with high‐salt buffer (20 mM sodium phosphate, 800 mM NaCl, pH 7.6). Fractions corresponding to 24mer (~60 mL elution volume) were collected and concentrated. Protein concentration was determined by the extinction coefficient at 280 nm calculated using ProtParam53 (1.67 mL mg−1 cm−1 for both wt and E65R), and purity was confirmed using SDS‐PAGE (Fig. S13). Protein solutions were stored at 4°C until needed for experiments. Experiments described below were repeated with at least three different preparations of each protein with similar results.
GFP protein expression and purification
GFP(+36)‐His6 plasmid was purchased from DNA 2.0 (now ATUM), based on the sequence published by Lawrence et al.28 Plasmid containing eGFP‐His6 (pUCBB‐ntH6‐eGFP) was a gift from Claudia Schmidt‐Dannert (Addgene plasmid #32557).54 Plasmid containing GFP(−30) (pET‐6xHis‐(−30)GFP) was a gift from David Liu (Addgene plasmid #62936).50 Plasmids were transformed in E. coli BL21CodonPlus(DE3)‐RP cells. Cells were grown at 30°C overnight in LB broth, transferred to 1 L LB, and grown at 37°C until OD600 ~0.6. Induction was done with 1 mM IPTG for 4 h at 37°C. Cells were harvested by centrifugation and stored at −20°C. Cells were resuspended in lysis buffer (phosphate buffered saline with 2 M NaCl) and lysed by treatment with lysozyme (~1 mg/mL final concentration) and DNase with stirring at 4°C for 30 min, followed by sonication on ice (amplitude of 30, 1 s on, 2 s off, 10 min processing time). Cellular debris was removed by centrifugation (6 krpm, 30 min, 4°C). Initial purification was performed using Ni‐NTA spin columns, washing three times with lysis buffer containing 20 mM imidazole, followed by elution with lysis buffer containing 500 mM imidazole. Green fractions were collected and buffer exchanged to PBS. Benzonase nuclease and MgCl2 (2 mM final concentration) were added and the solution was incubated at rt for 15 min to further remove nucleic acids. Using a HiTrap SP HP column, cation exchange was performed by running a gradient 0–100% lysis buffer over 25 mL using lysis buffer and PBS. The sample was then concentrated and injected into a HiLoad 16/60 Superdex200 size exclusion column equilibrated with lysis buffer at a flow rate of 0.7 mL/min. Green fractions were collected and concentrated. Purity was verified by SDS‐PAGE (Fig. S14) and concentration was determined using extinction coefficients at 488 nm: 4.7 × 104 M−1 cm−1 (GFP(+36) and GFP(−30)), 5.3 × 104 M−1 cm−1 (eGFP).55 Protein solutions were stored at 4°C in the dark until needed for experiments. GFP(+36) samples from which nucleic acids were rigorously removed during purification gave a ratio of absorbances at 280 nm (A 280) and 260 nm (A 260) of A 280/A 260 = 1.6–1.8. The extinction coefficient of GFP(+36) was determined experimentally by preparing a standard curve, using concentrations determined via the Bicinchoninic Acid Assay (BCA Assay, Pierce) using bovine serum albumin (Pierce) as a standard. Absorbance at 488 nm was measured with samples of varying concentration (3, 4.5, 6, 9, 18 μM) (Fig. S15). The experiment was performed in triplicate. Importantly, this value differs by nearly 30% from the value calculated from the extinction coefficients of individual amino acids (36,600 M−1 cm−1),18 which has significant consequence for the determination of GFP(+36) loading. The extinction coefficient at 488 nm for GFP(−30) was assumed to be within error of that for GFP(+36), due to the similar chromophore environments in the two proteins.28
AfFtn–GFP(+36) complex formation
AfFtn–GFP(+36) complexes were formed by mixing AfFtn and GFP(+36) in a 1:2 ratio in no‐salt buffer (20 mM sodium phosphate, pH 7.6) at a concentration of 0.6 μM (based on AfFtn 24mer) and 1.2 μM, respectively, and equilibrating overnight at 4°C.
Native gel electrophoresis
Native gels (0.7% agarose) were prepared to present the solution conditions 5 mM NaCl, 20 mM sodium phosphate, and pH 7.6. Samples were mixed with glycerol (final concentration 16% v/v). Gels were run at 100 V for 20 min on ice, covered with foil. Gels were imaged using a Typhoon FLA7000 imager using an excitation wavelength of 472 nm and PMT setting of 500 V. Following fluorescence imaging, gels were stained with Coomassie Brilliant Blue R‐250. For the varying ionic strength gel (Fig. 6), E65R and GFP(+36) were mixed in increasing ionic strength buffers ([NaCl] = 50, 100, 200, 400, 600, 800 mM) and incubated overnight at 4°C. The samples were run on the gel as described above.
Analytical SEC
Analytical SEC was performed with an AKTA FPLC system using a Superdex200 Increase 10/300 GL column equilibrated with no‐salt buffer (20 mM sodium phosphate, pH 7.6). Samples were prepared as above (“AfFtn–GFP(+36) complex formation”), but at concentrations of [AfFtn] = 6 μM and [GFP(+36)] = 12 μM for greater signal. Sample (200 μL) was injected and A 280 was monitored. For experiments to determine the average number of GFP(+36) molecules per AfFtn 24mer, N GFP, 2 mL of sample was injected to ensure adequate signal for subsequent UV–vis measurements. The sample was eluted at 4°C using a flow rate of 0.9 mL/min. Fluorescence of individual fractions was measured using a microplate reader as described below.
DLS
Sample (100 μL) was pipetted into a disposable micro cuvette. DLS was performed on a Malvern ZetaSizer Nano ZS with a scattering angle of 173° at 25°C (1 min equilibration time).
Fluorescence measurements
For GFP(+36) fluorescence spectrum, a Varian Cary Eclipse spectrophotometer was used, exciting at 488 nm and scanning 490–550 nm at a rate of 30 nm/min at 25°C. For fluorescence analysis of size exclusion fractions and Ni‐NTA assay samples, a Tecan M1000 microplate reader was used. Sample (100 μL) was pipetted into a black 96‐well plate. Samples were excited at 488 nm and fluorescence was measured at 509 nm.
Ni‐NTA assay
Fractions from SEC analysis containing encapsulation product were concentrated using a 10 k Centricon to a volume of ~100 μL. Concentrated sample (100 μL) was mixed with 100 μL of Ni‐NTA resin that had been pre‐washed with no‐salt buffer (20 mM sodium phosphate, pH 7.6). Samples were incubated for 1 h at 4°C on an end‐over rocker, covered in foil. Post‐incubation, samples were centrifuged for 2 min at 13.2 krpm to pellet the resin. The supernatant was moved to a clean tube for later analysis (“load”). The resin was resuspended in lysis buffer containing 20 mM imidazole, centrifuged, and again the supernatant was removed (“wash 1”). This was repeated twice more (“wash 2,” “wash 3”). After washing, the resin was resuspended in lysis buffer containing 250 mM imidazole, centrifuged, and the supernatant was removed (“elution 1”). Again, this was repeated twice more (“elution 2,” “elution 3”). The fluorescence of all supernatant samples was measured using a microplate reader as described above.
TEM
Grids were prepared by floating a copper‐coated carbon grid on 5 μL of the sample and staining with 2% (w/v) uranyl acetate. Grids were imaged on a Tecnai T‐12 microscope operating at 120 keV.
Determination of GFP(+36) loading
1:2 AfFtn:GFP(+36) samples were purified by SEC, and the fraction containing the highest fluorescence intensity was selected for loading analysis. The UV–visible spectrum of the sample was measured using an Agilent 8453 UV–visible spectrometer. A 280 and A 488 were used to calculate the number of GFP(+36) per AfFtn 24mer. The measured A 280 has contributions from both AfFtn and GFP(+36), while A 488 is only due to GFP(+36). An average ratio of A 280/A 488 for GFP(+36) was determined to be 0.46. This was used to calculate the A 280 that can be attributed to AfFtn:
The concentration of AfFtn was then calculated using the extinction coefficient above, while the concentration of GFP(+36) was calculated based on A 488 and the extinction coefficient listed above. The loading, [GFP(+36)] / [AfFtn], was then calculated.
Geometric analysis of AfFtn and GFP(+36)
The pores of wt‐AfFtn are ~4.5 nm from base to vertex, forming an approximately equilateral triangle with sides of ~5 nm. GFP(+36) can be approximated to be a cylinder56 of a length of 4 nm and a diameter of 3 nm. To fit GFP(+36) lengthwise through the pore, its diameter must be smaller than the largest circle that can be inscribed in an equilateral triangle of side length s:
For s = 5 nm, d = 2.9 nm, just smaller than the diameter of an idealized GFP(+36) cylinder. To fit GFP(+36) height‐wise through the pore, its cross‐sectional area if flattened to a rectangle must be smaller than 50% of the area of the triangle pore. This results in an area for a GFP(+36) rectangle of 12 nm2, while the largest possible rectangle to fit through the pores has an area of 11.25 nm2. Thus, GFP(+36) should be slightly too large to be encapsulated or escape only via movement through pores of the 24mer.
The theoretical maximum N GFP is based on the same simplified geometry. Four circles of diameter 3 nm can fit within an 8 nm diameter circle, approximating 4 GFP(+36) cylinders all with the same orientation inside the AfFtn cage. The dimensions of the cage will not fit another 3 × 4 nm cylinder lying perpendicular to the four upright cylinders, and N GFP is, therefore, 4. Thus, two GFP(+36) molecules encapsulated inside ferritin corresponds to ~50% loading.
Supporting information
The following file is available free of charge: Protein stock solution characterization, determination of GFP(+36) extinction coefficient, GFP(+36) optical properties upon encapsulation, encapsulation kinetics, precipitation with excess GFP(+36) per AfFtn 24mer, Ni‐NTA assay for E65R‐GFP(+36) (PDF).
Conflict of Interest
The authors declare no competing financial interest.
Supporting information
Figure S1. Size exclusion chromatography (SEC) trace showing assembled 24mer in 800 mM NaCl, 20 mM sodium phosphate solution (dashed line) and disassembled dimer in 0 mM NaCl, 20 mM sodium phosphate solution (solid line). Both samples are 6 μM protein. This is in contrast to when 50 mM Tris buffer is used, where ~40% of the population remains assembled in low‐salt conditions.1
Figure S2. SEC trace of GFP(+36) in no‐salt buffer shows no protein elution, as evidenced by lack of peak in the A280 curve (yellow) or I509 curve (green). [GFP(+36)] = 30 μM.
Figure S3. Additional TEM micrographs of AfFtn‐GFP(+36), post‐FPLC. Grids were stained with 2% (w/v) uranyl acetate. Scale bars are 50 μm.
Figure S4. Optical properties of GFP(+36) pre‐ and post‐encapsulation. (a) Absorbance spectrum with and without AfFtn. (b) Fluorescence spectrum with and without AfFtn.
Figure S5. (a) Native gel time course for GFP(+36) encapsulation by AfFtn. Fluorescent band that appears within the analysis time corresponds to encapsulation product. Samples are 0.6 μM AfFtn, 0.6 μM GFP(+36). (b) Size exclusion chromatograms show the fast overlap of GFP(+36) fluorescence (green) and A280 peak corresponding to AfFtn 24mer (yellow).
Figure S6. Excess GFP(+36) per AfFtn 24mer leads to precipitation of both AfFtn and GFP(+36). (a) Green precipitate was seen for 1:4 and 1:6 samples, but not 1:2. (b) SDS‐PAGE shows precipitate contains both AfFtn and GFP(+36).
Figure S7. Additional TEM micrographs of AfFtn E65R‐GFP(+36), post‐FPLC. Grids were stained with 2% (w/v) uranyl acetate. Scale bars are 50 μm.
Figure S8. Ni‐NTA assay shows lower His‐tag binding for E65R GFP(+36) compared with wt AfFtn GFP(+36) or GFP(+36) alone.
Figure S9. SEC trace of E65R–eGFP shows no encapsulation occurring. The sample was incubated overnight and eluted in the no‐salt buffer.
Figure S10. SEC trace of E65R–GFP(−30) shows no encapsulation occurring. The sample was incubated overnight and eluted in the no‐salt buffer.
Figure S11. SEC trace of GFP(+36) in the high‐salt buffer. [GFP(+36)] = 30 μM.
Figure S12. SEC traces show no encapsulation of eGFP or GFP(−30) by wt‐AfFtn after overnight incubation and column elution in high‐salt conditions. (a) SEC trace of wt‐AfFtn–eGP and (b) SEC trace of wt‐AfFtn–GFP(−30).
Figure S13. SDS‐PAGE showing pure protein for AfFtn wt and E65R used.
Figure S14. SDS‐PAGE showing pure protein for GFP(+36), GFP(−30), eGFP used. The two samples per protein for GFP(+36) and eGFP correspond to two different concentrations used.
Figure S15. Standard curve used to determine the extinction coefficient of GFP(+36) at 488 nm. Three replicate measurements were performed, with very small standard error bars.
Acknowledgments
This work was supported by the U.S. National Science Foundation (NSF) (PD 09‐6885). I. J. D. and J. G. S. acknowledge support from NSF CHE‐1508318 and partial support from NSF DMR‐1120901 (MRSEC). AfFtn mutants were designed with the aid of XSEDE resources (NSF ACI‐1053575, TG‐CHE110041). J. A. V. acknowledges partial support from an NIH Chemistry/Biology Interface training grant (T32 GM071339). We thank the University of Pennsylvania Electron Microscopy Resource Laboratory for use of TEM. We are grateful to Andrew Tsourkas for use of DLS.
Broad audience statement: Encapsulating proteins within protein cages has potential use in drug delivery, protein separations, and furthering fundamental understanding of enzyme behavior in confined spaces. We demonstrate electrostatic‐based encapsulation of superpositively charged green fluorescent protein within thermophilic ferritin. Using a more stably assembled mutant ferritin, we show that, surprisingly, encapsulation can occur starting either from disassembled ferritin dimers or a fully assembled ferritin cage. More generally, we also present a new method for verifying protein–protein encapsulation, using nickel agarose resin.
References
- 1. Sun X, James TD (2015) Glucose sensing in supramolecular chemistry. Chem Rev 115:8001–8037. [DOI] [PubMed] [Google Scholar]
- 2. Wang ZJ, Clary KN, Bergman RG, Raymond KN, Toste FD (2013) A supramolecular approach to combining enzymatic and transition metal catalysis. Nat Chem 5:100–103. [DOI] [PubMed] [Google Scholar]
- 3. Harada A, Hashidzume A, Yamaguchi H, Takashima Y (2009) Polymeric rotaxanes. Chem Rev 109:5974–6023. [DOI] [PubMed] [Google Scholar]
- 4. Da Hu Q, Tang GP, Chu PK (2014) Cyclodextrin‐based host‐guest supramolecular nanoparticles for delivery: from design to applications. Acc Chem Res 47:2017–2025. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y, Dmochowski IJ (2016) An expanded palette of Xenon‐129 NMR biosensors. Acc Chem Res 49:2179–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biros SM, Rebek J (2007) Structure and binding properties of water‐soluble cavitands and capsules. Chem Soc Rev 36:93–104. [DOI] [PubMed] [Google Scholar]
- 7. Pluth MD, Raymond KN (2007) Reversible guest exchange mechanisms in supramolecular host–guest assemblies. Chem Soc Rev 36:161–171. [DOI] [PubMed] [Google Scholar]
- 8. Babine RE, Bender SL (1997) Molecular recognition of protein–ligand complexes: applications to drug design. Chem Rev 97:1359–1472. [DOI] [PubMed] [Google Scholar]
- 9. Pérot S, Sperandio O, Miteva MA, Camproux AC, Villoutreix BO (2010) Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov Today 15:656–667. [DOI] [PubMed] [Google Scholar]
- 10. Hendrick JP, Hartl F‐U (1995) The role of molecular chaperones in protein folding. FASEB J 9:1559–1569. [DOI] [PubMed] [Google Scholar]
- 11. Corchero JL, Cedano J (2011) Self‐assembling, protein‐based intracellular bacterial organelles: emerging vehicles for encapsulating, targeting and delivering therapeutical cargoes. Microb Cell Fact 10:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glasgow JE, Capehart SL, Francis MB, Tullman‐Ercek D (2012) Osmolyte‐mediated encapsulation of proteins inside MS2 viral capsids. ACS Nano 6:8658–8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seebeck FP, Woycechowsky KJ, Zhuang W, Rabe JP, Hilvert D (2006) A simple tagging system for protein encapsulation. J Am Chem Soc 128:4516–4517. [DOI] [PubMed] [Google Scholar]
- 14. Zschoche R, Hilvert D (2015) Diffusion‐limited cargo loading of an engineered protein container. J Am Chem Soc 137:16121–16132. [DOI] [PubMed] [Google Scholar]
- 15. Rurup WF, Verbij F, Koay MST, Blum C, Subramaniam V, Cornelissen JJLM (2014) Predicting the loading of virus‐like particles with fluorescent proteins. Biomacromolecules 15:558–563. [DOI] [PubMed] [Google Scholar]
- 16. Worsdorfer B, Woycechowsky KJ, Hilvert D (2011) Directed evolution of a protein container. Science 331:589–592. [DOI] [PubMed] [Google Scholar]
- 17. Qazi S, Miettinen HM, Wilkinson RA, McCoy K, Douglas T, Wiedenheft B (2016) Programmed self‐assembly of an active P22‐Cas9 nanocarrier system. Mol Pharm 13:1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Azuma Y, Zschoche R, Tinzl M, Hilvert D (2016) Quantitative packaging of active enzymes into a protein cage. Angew Chem Int Ed 55:1531–1534. [DOI] [PubMed] [Google Scholar]
- 19. Tetter S, Hilvert D (2017) Enzyme encapsulation by a ferritin cage. Angew Chem Int Ed 129:15129–15132. [DOI] [PubMed] [Google Scholar]
- 20. Comellas‐Aragonès M, Engelkamp H, Claessen VI, Sommerdijk NAJM, Rowan AE, Christianen PCM, Maan JC, Verduin BJM, Cornelissen JJLM, Nolte RJM (2007) A virus‐based single‐enzyme nanoreactor. Nat Nanotechnol 2:635–639. [DOI] [PubMed] [Google Scholar]
- 21. Patterson DP, Prevelige PE, Douglas T (2012) Nanoreactors by programmed enzyme encapsulation inside the capsid of the bacteriophage P22. ACS Nano 6:5000–5009. [DOI] [PubMed] [Google Scholar]
- 22. Patterson DP, Schwarz B, Waters RS, Gedeon T, Douglas T (2014) Encapsulation of an enzyme cascade within the bacteriophage P22 virus‐like particle. ACS Chem Biol 9:359–365. [DOI] [PubMed] [Google Scholar]
- 23. Ni TW, Tezcan FA (2010) Structural characterization of a microperoxidase inside a metal‐directed protein cage. Angew Chem Int Ed 49:7014–7018. [DOI] [PubMed] [Google Scholar]
- 24. Bellapadrona G, Sinkar S, Sabanay H, Liljeström V, Kostiainen M, Elbaum M (2015) Supramolecular assembly and coalescence of ferritin cages driven by designed protein‐protein interactions. Biomacromolecules 16:2006–2011. [DOI] [PubMed] [Google Scholar]
- 25. Yang R, Chen L, Zhang T, Yang S, Leng X, Zhao G (2014) Self‐assembly of ferritin nanocages into linear chains induced by poly(α, L‐lysine). Chem Commun 50:481–483. [DOI] [PubMed] [Google Scholar]
- 26. Kostiainen M a, Hiekkataipale P, Laiho A, Lemieux V, Seitsonen J, Ruokolainen J, Ceci P (2013) Electrostatic assembly of binary nanoparticle superlattices using protein cages. Nat Nanotechnol 8:52–56. [DOI] [PubMed] [Google Scholar]
- 27. Men D, Zhang OT, Hou OL, Zhou OJ, Zhang Z, Shi Y (2015) Self‐assembly of ferritin nanoparticles into an enzyme nanocomposite with tunable size for ultrasensitive. ACS Nano 9:10852–10860. [DOI] [PubMed] [Google Scholar]
- 28. Lawrence MS, Phillips KJ, Liu DR (2007) Supercharging proteins can impart unusual resilience. J Am Chem Soc 129:10110–10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson E, Cascio D, Sawaya MR, Gingery M, Schröder I (2005) Crystal structures of a tetrahedral open pore ferritin from the hyperthermophilic archaeon Archaeoglobus fulgidus . Structure 13:637–648. [DOI] [PubMed] [Google Scholar]
- 30. Cornell TA, Orner BP (2018) Medium throughput cage state stability screen of conditions for the generation of gold nanoparticles encapsulated within a mini‐ferritin. Bioorg Med Chem in press. https://www.ncbi.nlm.nih.gov/pubmed/29615283 [DOI] [PubMed] [Google Scholar]
- 31. Falvo E, Tremante E, Arcovito A, Papi M, Elad N, Boffi A, Morea V, Conti G, Toffoli G, Fracasso G, Giacomini P, Ceci P (2016) Improved doxorubicin encapsulation and pharmacokinetics of ferritin‐fusion protein nanocarriers bearing proline, serine, and alanine elements. Biomacromolecules 17:514–522. [DOI] [PubMed] [Google Scholar]
- 32. Ferraro G, Monti DM, Amoresano A, Pontillo N, Petruk G, Pane F, Cinellu MA, Merlino A (2016) Gold‐based drug encapsulation within a ferritin nanocage: X‐ray structure and biological evaluation as a potential anticancer agent of the Auoxo3‐loaded protein. Chem Commun 52:9518–9521. [DOI] [PubMed] [Google Scholar]
- 33. Zhen Z, Tang W, Chen H, Lin X, Todd T, Wang G, Cowger T, Chen X, Xie J (2013) RGD‐modified apoferritin nanoparticles for efficient drug delivery to tumors. ACS Nano 7:4830–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yan F, Zhang Y, Kim KS, Yuan H‐K, Vo‐Dinh T (2010) Cellular uptake and photodynamic activity of protein nanocages containing methylene blue photosensitizing drug. Photochem Photobiol 86:662–666. [DOI] [PubMed] [Google Scholar]
- 35. Swift J, Butts CA, Cheung‐Lau J, Yerubandi V, Dmochowski IJ (2009) Efficient self‐assembly of Archaeoglobus fulgidus ferritin around metallic cores. Langmuir 25:5219–5225. [DOI] [PubMed] [Google Scholar]
- 36. Cheung‐Lau JC, Liu D, Pulsipher KW, Liu W, Dmochowski IJ (2014) Engineering a well‐ordered, functional protein–gold nanoparticle assembly. J Inorg Biochem 130:59–68. [DOI] [PubMed] [Google Scholar]
- 37. Pulsipher KW, Dmochowski IJ. Ferritin encapsulation and templated synthesis of inorganic nanoparticles In: Orner BP, Ed. , 2014, 2014. Methods in molecular biology. New York, NY: Springer New York; p. 27–37. [DOI] [PubMed] [Google Scholar]
- 38. Pulsipher KW, Honig S, Deng S, Dmochowski IJ (2017) Controlling gold nanoparticle seeded growth in thermophilic ferritin protein templates. J Inorg Biochem 174:169–176. [DOI] [PubMed] [Google Scholar]
- 39. Ping J, Pulsipher KW, Vishnubhotla R, Villegas J, Hicks TL, Honig S, Saven JG, Dmochowski IJ, Johnson ATC (2017) Structural–functional analysis of engineered protein–nanoparticle assemblies using graphene microelectrodes. Chem Sci 8:5329–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pulsipher KW, Villegas JA, Roose BW, Hicks TL, Yoon J, Saven JG, Dmochowski IJ (2017) Thermophilic ferritin 24mer assembly and nanoparticle encapsulation modulated by interdimer electrostatic repulsion. Biochemistry 56:3596–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Webb B, Frame J, Zhao Z, Lee ML, Watt GD (1994) Molecular entrapment of small molecules within the interior of horse spleen ferritin. Arch Biochem Biophys 309:178–183. [DOI] [PubMed] [Google Scholar]
- 42. Minten IJ, Claessen VI, Blank K, Rowan AE, Nolte RJM, Cornelissen JJLM (2011) Catalytic capsids: the art of confinement. Chem Sci 2:358–362. [Google Scholar]
- 43. Zhang Y, Fu J, Chee SY, Ang EXW, Orner BP (2011) Rational disruption of the oligomerization of the mini‐ferritin E. coli DPS through protein–protein interface mutation. Protein Sci 20:1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cornell TA, Ardejani MS, Fu J, Newland SH, Zhang Y, Orner BP (2018) A structure‐based assembly screen of protein cage libraries in living cells: experimentally repacking a protein–protein interface to recover cage formation in an assembly‐frustrated mutant. Biochemistry 57:604–613. [DOI] [PubMed] [Google Scholar]
- 45. Zhang Y, Wang L, Ardejani MS, Aris NF, Li X, Orner BP, Wang F (2015) Mutagenesis study to disrupt electrostatic interactions on the twofold symmetry interface of Escherichia coli bacterioferritin. J Biochem 158:505–512. [DOI] [PubMed] [Google Scholar]
- 46. Beck T, Tetter S, Künzle M, Hilvert D (2015) Construction of matryoshka‐type structures from supercharged protein nanocages. Angew Chem Int Ed 127:951–954. [DOI] [PubMed] [Google Scholar]
- 47. Sasaki E, Böhringer D, van de Waterbeemd M, Leibundgut M, Zschoche R, Heck AJR, Ban N, Hilvert D (2017) Structure and assembly of scalable porous protein cages. Nat Commun 8:14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grime JMA, Dama JF, Ganser‐Pornillos BK, Woodward CL, Jensen GJ, Yeager M, Voth GA (2016) Coarse‐grained simulation reveals key features of HIV‐1 capsid self‐assembly. Nat Commun 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang G, Gurtu V, Kain SR (1996) An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem Biophys Res Commun 227:707–711. [DOI] [PubMed] [Google Scholar]
- 50. Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen Z‐Y, Liu DR (2014) Cationic lipid‐mediated delivery of proteins enables efficient protein‐based genome editing in vitro and in vivo . Nat Biotechnol 33:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Deshpande S, Masurkar ND, Girish VM, Desai M, Chakraborty G, Chan JM, Drum CL (2017) Thermostable exoshells fold and stabilize recombinant proteins. Nat Commun 8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cronican JJ, Beier KT, Davis TN, Tseng JC, Li W, Thompson DB, Shih AF, May EM, Cepko CL, Kung AL, Zhou Q, Liu DR (2011) A class of human proteins that deliver functional proteins into mammalian cells in vitro and in vivo . Chem Biol 18:833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy Server, 2005, 2005. The proteomics protocols handbook. Totowa, NJ: Humana Press; p. 571–607. [Google Scholar]
- 54. Vick JE, Johnson ET, Choudhary S, Bloch SE, Lopez‐Gallego F, Srivastava P, Tikh IB, Wawrzyn GT, Schmidt‐Dannert C (2011) Optimized compatible set of BioBrick vectors for metabolic pathway engineering. Appl Microbiol Biotechnol 92:1275–1286. [DOI] [PubMed] [Google Scholar]
- 55. Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW (1997) Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J 73:2782–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wörsdörfer B, Pianowski Z, Hilvert D (2012) Efficient in vitro encapsulation of protein cargo by an engineered protein container. J Am Chem Soc 134:909–911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Size exclusion chromatography (SEC) trace showing assembled 24mer in 800 mM NaCl, 20 mM sodium phosphate solution (dashed line) and disassembled dimer in 0 mM NaCl, 20 mM sodium phosphate solution (solid line). Both samples are 6 μM protein. This is in contrast to when 50 mM Tris buffer is used, where ~40% of the population remains assembled in low‐salt conditions.1
Figure S2. SEC trace of GFP(+36) in no‐salt buffer shows no protein elution, as evidenced by lack of peak in the A280 curve (yellow) or I509 curve (green). [GFP(+36)] = 30 μM.
Figure S3. Additional TEM micrographs of AfFtn‐GFP(+36), post‐FPLC. Grids were stained with 2% (w/v) uranyl acetate. Scale bars are 50 μm.
Figure S4. Optical properties of GFP(+36) pre‐ and post‐encapsulation. (a) Absorbance spectrum with and without AfFtn. (b) Fluorescence spectrum with and without AfFtn.
Figure S5. (a) Native gel time course for GFP(+36) encapsulation by AfFtn. Fluorescent band that appears within the analysis time corresponds to encapsulation product. Samples are 0.6 μM AfFtn, 0.6 μM GFP(+36). (b) Size exclusion chromatograms show the fast overlap of GFP(+36) fluorescence (green) and A280 peak corresponding to AfFtn 24mer (yellow).
Figure S6. Excess GFP(+36) per AfFtn 24mer leads to precipitation of both AfFtn and GFP(+36). (a) Green precipitate was seen for 1:4 and 1:6 samples, but not 1:2. (b) SDS‐PAGE shows precipitate contains both AfFtn and GFP(+36).
Figure S7. Additional TEM micrographs of AfFtn E65R‐GFP(+36), post‐FPLC. Grids were stained with 2% (w/v) uranyl acetate. Scale bars are 50 μm.
Figure S8. Ni‐NTA assay shows lower His‐tag binding for E65R GFP(+36) compared with wt AfFtn GFP(+36) or GFP(+36) alone.
Figure S9. SEC trace of E65R–eGFP shows no encapsulation occurring. The sample was incubated overnight and eluted in the no‐salt buffer.
Figure S10. SEC trace of E65R–GFP(−30) shows no encapsulation occurring. The sample was incubated overnight and eluted in the no‐salt buffer.
Figure S11. SEC trace of GFP(+36) in the high‐salt buffer. [GFP(+36)] = 30 μM.
Figure S12. SEC traces show no encapsulation of eGFP or GFP(−30) by wt‐AfFtn after overnight incubation and column elution in high‐salt conditions. (a) SEC trace of wt‐AfFtn–eGP and (b) SEC trace of wt‐AfFtn–GFP(−30).
Figure S13. SDS‐PAGE showing pure protein for AfFtn wt and E65R used.
Figure S14. SDS‐PAGE showing pure protein for GFP(+36), GFP(−30), eGFP used. The two samples per protein for GFP(+36) and eGFP correspond to two different concentrations used.
Figure S15. Standard curve used to determine the extinction coefficient of GFP(+36) at 488 nm. Three replicate measurements were performed, with very small standard error bars.