

Abstract
This report describes a cost‐effective experimental method for determining an intrinsically disordered protein (IDP) region in a given protein sample. In this area, the most popular (and conventional) means is using the amide (1HN) NMR signal chemical shift distributed in the range of 7.5–8.5 ppm. For this study, we applied an additional step: analysis of 1HN chemical shift temperature coefficients (1HN‐CSTCs) of the signals. We measured 1H–15N two‐dimensional NMR spectra of model IDP samples and ordered samples at four temperatures (288, 293, 298, and 303 K). We derived the 1HN‐CSTC threshold deviation, which gives the best correlation of ordered and disordered regions among the proteins examined (below −3.6 ppb/K). By combining these criteria with the newly optimized chemical shift range (7.8–8.5 ppm), the ratios of both true positive and true negative were improved by approximately 19% (62–81%) compared with the conventional “chemical shift‐only” method.
Keywords: intrinsically disordered protein, chemical shift temperature coefficient, nuclear magnetic resonance, hydrogen bond
Abbreviations
- CS
chemical shift
- CSTC
chemical shift temperature coefficient
- heteroNOE
heteronuclear Overhauser effect
- HSQC
heteronuclear single quantum coherence
- IDP
intrinsically disordered protein
- IDR
intrinsically disordered region
- MCC
Matthews's correlation coefficient
- NMR
nuclear magnetic resonance
- PDB
Protein Data Bank
- RDC
residual dipolar coupling
- ROC
receiver operating characteristic
- SOFAST‐HMQC
band‐selective optimized flip angle short transient‐heteronuclear multi quantum coherence
Introduction
Intrinsically disordered protein (IDP)1 is an emerging key idea for the protein sequence–function relationship.2, 3, 4, 5, 6 Even under physiological conditions, IDPs (and intrinsically disordered regions, IDRs) do not adopt unique and compact three‐dimensional (3D) structures overall or in the part of the polypeptide chain. Growing interest in IDPs has led to an increasing number of reports describing that they possess various physiological functions. Such functions include transcription, translation, and signaling cascades. A unique biological feature of IDPs is believed to be structural polymorphism among the free and multiple target‐bound states with their biological partner proteins. It is particularly interesting that IDP–IDP interaction happens to exhibit ultrahigh picomolar affinity between linker histone H1 and prothymosin α, both of which remain in a full disordered state, even in complex.7 We built a database, IDEAL, in which the protein segments (ProS) in IDPs, which are important for interaction with their targets, were annotated extensively.8, 9 Recent reports describe that IDPs from tardigrades contribute to resistance against drought by IDP's unique vitrification mechanism.10, 11 More recently, we have discovered a sequence‐independent function of human‐genome derived IDPs as a cryoprotectant against other enzymes and non‐enzymatic proteins.12 Consequently, methods for experimental discrimination of IDPs from non‐IDPs are expected to become increasingly important.
To date, two major experimental methods have been used to confirm the presence of disordered regions in a given polypeptide chain: X‐ray crystallography and solution NMR. Because substantial difficulty underlies X‐ray crystallography of IDPs in theory, solution NMR methodology becomes indispensable for physicochemical studies of IDPs.13, 14, 15 The conventional NMR method to ascertain whether the residue of interest belongs to either an IDP region (IDR) or a structured domain (non‐IDR), and probably the simplest, is to acquire 1H‐15N HSQC spectrum of the sample and to measure the dispersion of amide 1H chemical shift (1HN‐CS). Because 1HN‐CS is sensitive to its local environment of the amide groups in the 3D structure, a typical HSQC of a folded protein sample shows a widely dispersed spectrum in the range of 6–11 ppm on the 1H axis. By contrast, a sample consisting only of an IDR might show a very narrow range of 1HN‐CS: 7.5–8.5 ppm. However, this tendency does not suggest a theory. As a result, some amide protons of the residues within the structured domain region might eventually give a chemical shift at 7.5–8.5 ppm. For example, ubiquitin and the mouse ZO1 PDZ1 domain, both of which we selected as folded protein examples, respectively possess 13 (out of 70) and 16 (out of 90) amide signals (Table 1).16 Consequently, although 1HN‐CS is a good indicator of protein residues for IDR/non‐IDR discrimination, room for improvement remains.
Table 1.
Amino‐Acid Sequences of Proteins Used for this Study. Residues Showing Well‐Separated Signals Used for the CSTC Analysis Are Underlined
| Protein name | Total residues | Used signals | |
|---|---|---|---|
| Training dataset (IDP) | |||
| B3 (thymosin) | 44 | 38a | MADKPDMGEIASFDKAKLKKTETQEKNTLPTKETIEQEKRSEIS |
| C1 (WWOX) | 36 | 33 | MAALRYAGLDDTDSEDELPPGWEERTTKDGWVYYAK |
| Training dataset (structured) | |||
| hUb | 76 | 64 | MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDLEGIPPDQQRLIFAGLQL |
| EDGRTLSDYNIQKESTLHLVLRLRGG | |||
| mZO1‐PDZ1 | 100 | 84 | GPLGSDHIWEQHTVTLHRAPGFGFGIAISGGRDNPHFQSGETSIVISDVL |
| KGGPAEGQLQENDRVAMVNGVSMDNVEHAFAVQQLRKSGKNAKITIRRKK | |||
| hVps4b‐MIT | 81 | 68 | GSDHMSSTSPNLQKAIDLASKAAQEDKAGNYEEALQLYQHAVQYFLHVVK |
| YEAQGDKAKQSIRAKCTEYLDRAEKLKEYLK | |||
| Evaluation dataset | |||
| hSUMO1 | 97 | 93 | MSDQEAKPSTEDLGDKKEGEYIKLKVIGQDSSEIHFKVKMTTHLKKLKES |
| YCQRQGVPMNSLRFLFEGQRIADNHTPKELGMEEEDVIEVYQEQTGC | |||
| hSUMO2 | 93 | 88 | MADEKPKEGVKTENNDHINLKVAGQDGSVVQFKIKRHTPLSKLMKAYCER |
| QGLSMRQIRFRFDGQPINETDTPAQLEMEDEDTIDVFQQQTGG | |||
Chemical shifts were not assigned. Thirty‐eight well‐separated signals were used for the analysis.
We have specifically examined the parameter 1HN chemical shift temperature coefficients (1HN‐CSTCs).16, 17 Actually, 1HN‐CSTC can be readily estimated from a small number of additional HSQC measurements at different temperatures (at least two). Strong correlation between 1HN‐CSTC and formation of intramolecular hydrogen bonds has been reported in the relevant literature.18, 19, 20, 21 Consequently, it seems promising that the additional use of 1HN‐CSTC for IDR/non‐IDR discrimination can improve accuracy, although the border line between IDR and non‐IDR definition is not equivalent to the absence or presence of intramolecular hydrogen bonds. To evaluate this idea, we designed the following experiments: 1 preparation of a dataset of 1HN‐CSTC of proteins for method development, for which residues had been classified as either IDR or non‐IDR; 2 defining a single 1HN‐CSTC threshold value that gives the best Matthew's correlation coefficient (MCC) upon IDR/non‐IDR discrimination; 3 evaluating the method by its application to sample realistic IDR/non‐IDR discrimination problems, human SUMO1 and SUMO2 cases (the method was also evaluated by comparing the orthodox 1HN‐CS criteria); 4 evaluating the combinatorial use of the 1HN‐CSTC method and the conventional 1HN‐CS criterion and comparing it with the single use of each two criteria.
Results
Experimental design
For this study, we started to obtain an amide chemical shift temperature coefficient (1HN‐CSTC) data for statistical analysis. We analyzed the amide temperature coefficients of two IDP samples with 30–50 residues: IDP‐B3 and IDP‐C1 [Fig. 1(a,b), S1, and Table 1]. Among them, backbone signals of 33 out of 36 residues (92%) were assigned for IDP‐C1, whereas 38 well‐separated backbone signals out of 44 residues (86%) of IDP‐B3 were used for this analysis without assignment. We were unable to assign IDP‐B3 because of a poor signal‐to‐noise ratio of 3D experiments. These samples were confirmed as unstructured in solution by our membrane fusion‐based systematic IDP assessment22 as well as CD (Supporting Information Fig. S1). Subsequently, we analyzed 1HN‐CSTC of hUb, mZO1‐PDZ1, and hVps4bMIT [Fig. 1(c–e)] as a control of the structured protein domains. For the IDP samples, all residues were classified as disordered. For the structured proteins, the residues were classified into two classes (structured regions and disordered regions) using Ota's criteria to identify IDP regions from the NMR structure ensemble23 (Table 1). For all these residues, 1H–15N SOFAST‐HMQC spectra,24 instead of HSQC, were recorded at four temperatures to calculate 1HN‐CSTC: 288, 293, 298, and 303 K. These data were then used for statistical analyses (histogram and Matthew's correlation). Finally, two proteins including both the structured and disordered parts of the polypeptides (hSUMO1 and hSUMO2) were used to evaluate the proposed 1HN‐CSTC method [Fig. 1(f, g)].
Figure 1.
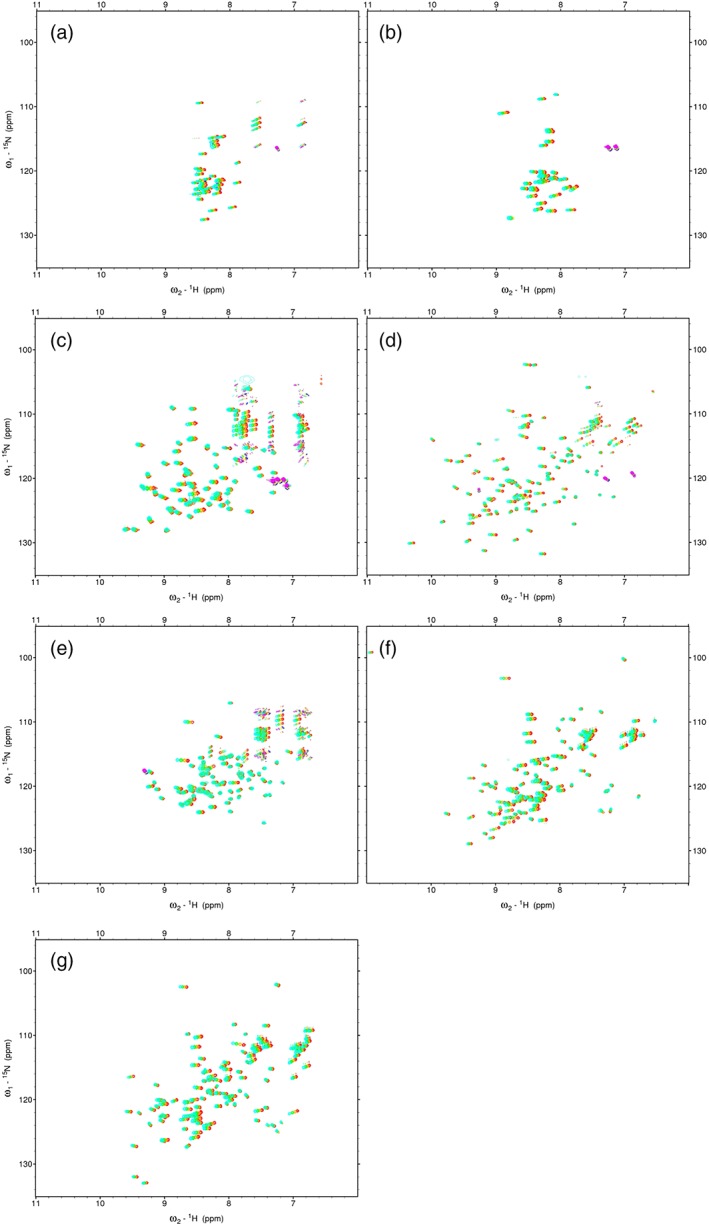
SOFAST‐HMQC spectra of the analyzed proteins recorded at four temperatures: (a) IDP‐B3, (b) IDP‐C1, (c) hUb, (d) mZO1‐PDZ1, (e) hVps4bMIT, (f) hSUMO1, and (g) hSUMO2. For all spectra, signals of 288, 293, 298, and 303 K are shown, respectively, as cyan, green, orange, and red.
Histogram of 1HN‐CSTC but not 15N‐CSTC showed deviation between ordered and disordered regions
We selected 211 and 76 residues, respectively, as structured and IDR residue datasets. We then measured SOFAST‐HMQC spectra of the corresponding NMR samples at four temperatures and calculated 1HN‐CSTCs and 15NH‐CSTCs of the structured and IDR residues. The average and the standard deviation for 1HN‐CSTCs of the IDR and non‐IDR (structured) residues were, respectively, −5.62 ±1.56 and − 3.19 ±2.58 ppb/K. Similarly, the average and the standard deviation for 15NH‐CSTCs of the IDR and non‐IDR residues were, respectively, −7.09 ±9.52 and 3.25 ±12.63 ppb/K. Figure 2(a, b) represents histograms of 1HN‐CSTC and 15NH‐CSTC for the residues classified in both the structured and IDR residues. The histogram of 1HN‐CSTC showed clear deviation between the two datasets, in which 1HN of the ordered region showed large (small negative) CSTCs, whereas that of the disordered region showed small (large negative) CSTCs. This tendency was readily confirmed from the SOFAST‐HMQC spectra of hSUMO1. Actually, hSUMO1 includes 16 IDR residues at its N‐terminus, whereas hSUMO2 has 12 IDR residues. Typical chemical shift changes of the amide signals upon temperature shift taken from 2D NMR spectra of hSUMO1 are shown [Fig. 1(f)]. Although a similar tendency of the histogram of 15NH‐CSTC was obtained, we abandoned the use of 15NH‐CSTC for IDR/non‐IDR discrimination because the standard deviation for non‐IDR residues was too large [12.63 ppb/K, Fig. 2(b)].
Figure 2.
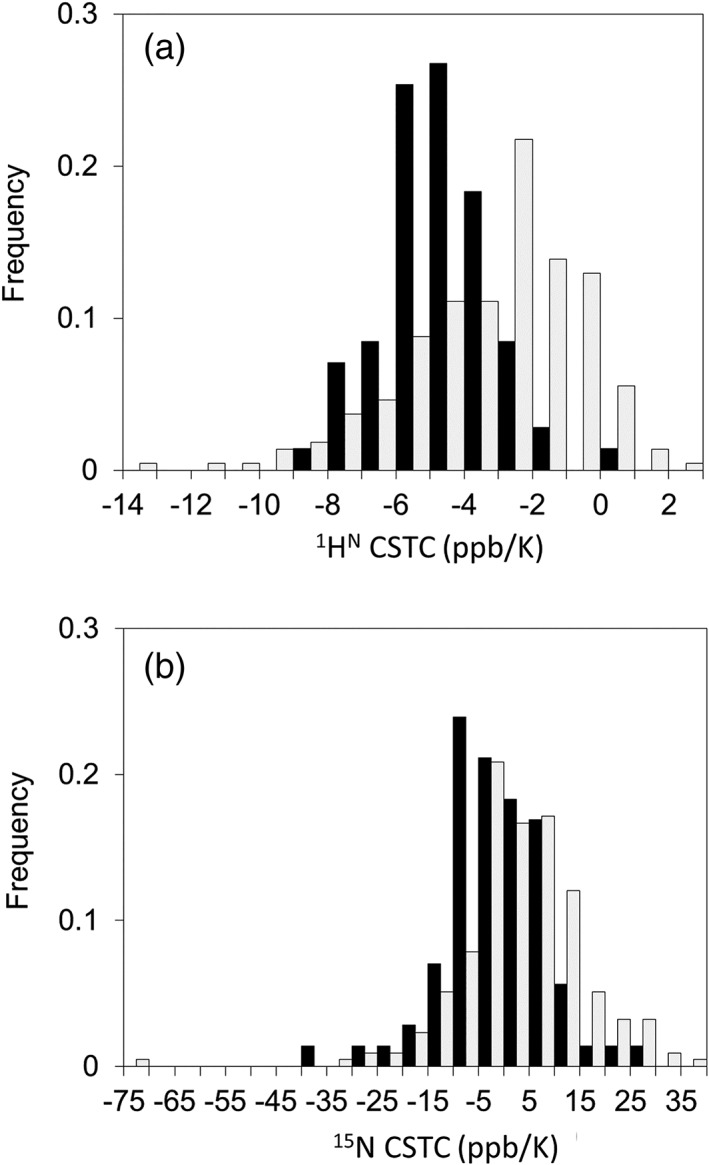
Histograms of (a) 1HN‐CSTC and (b) 15NH‐CSTC for residues classified in both structured (light gray) and IDR (black) residues.
Defining the threshold of 1HN‐CSTC for order/disorder discrimination and redefinition of 1HN‐CS range
We calculated the MCC26 only for any given 1HN‐CSTCs [Eq. (3)]. By changing the threshold value in 0.1 ppb/K increments, we sought the threshold that produced the maximum MCC for discriminating the structured and IDR states in the evaluation dataset of 1HN‐CSTCs. We chose the threshold value with care. For instance, had we chosen a small (large negative) threshold, the denominator would have been zero. All residues in the NMR structure would have been labeled as being ordered. Consequently, N(*, D) will be zero. For all 287 residues, we were able to calculate all the MCCs (i.e., denominators were non‐zero) when we adjusted the threshold value within −4.5 to −3.0 ppb/K. Results demonstrated that threshold values between −3.6 and − 3.9 ppb/K gave almost maximum MCC values (greater than 0.55) [Fig. 3(a)]. The calculated MCC at −3.6 ppb/K was the highest (0.626). Therefore, we set −3.6 ppb/K as the 1HN‐CSTC threshold (CSTCTH) used to label structured and IDR residues based on NMR spectra. We also calculated the receiver operating characteristic (ROC) curve of this defined CSTCTH against the training dataset [Fig. 3(b)].
Figure 3.
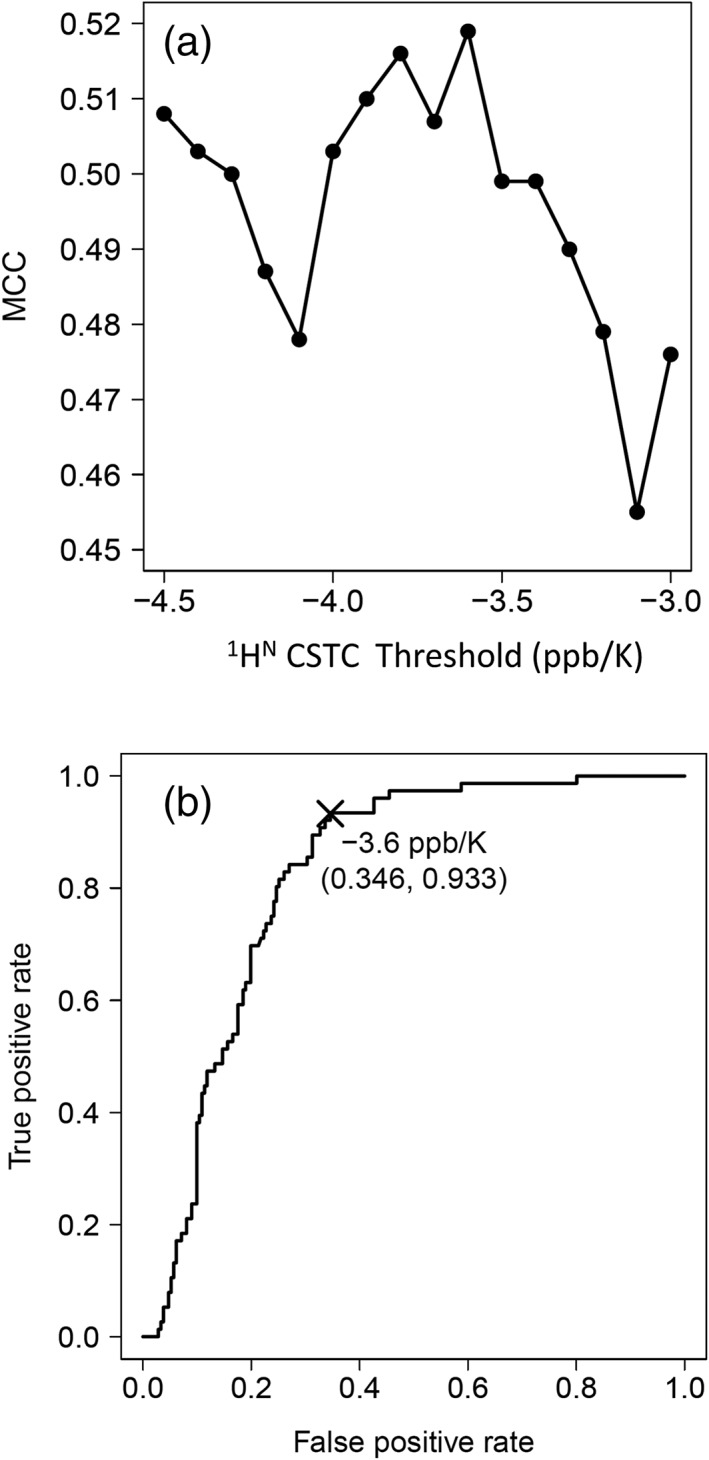
(a) MCC curve of IDP discrimination by change of the 1HN‐CSTC threshold. (b) ROC curve of IDP discrimination by 1HN‐CSTC < −3.6 ppb/K as IDR.
Evaluation of the new IDP/non‐IDP discrimination method against naturally occurring examples, hSUMO1 and hSUMO2
We then evaluated the usefulness the new IDR discrimination method with the 1HN‐CSTC criterion (1HN‐CSTC < −3.6 ppb/K as IDR) against the evaluation dataset of mixture of IDR and non‐IDR in naturally occurring proteins (hSUMO1 and hSUMO2). Both proteins contain N‐terminal disordered regions followed by compactly folded ubiquitin‐like domains (Ubl). We compared the new method with the orthodox IDR criterion of 1HN‐chemical shift distribution range (1HN‐CS), which is in the range of 7.5–8.5 ppm. When applying the 1HN‐CS criterion alone, the discrimination method accuracy was 62% (Table 2); the score closely resembled that of using only 1HN‐CSTC (59%). In detail, the numerous false positive residues that were judged as IDR residues, although they were non‐IDR, impaired the accuracy. For example, 67 false‐positive IDR residues were judged by the 1HN‐CS criterion, whereas 72 false positives were judged by 1HN‐CSTC. These results demonstrate that the 1HN‐CSTC criterion alone is not useful.
Table 2.
Results of the Two Naturally Occurring, Mixed Examples of IDP/Non‐IDP Residues, hSUMO1 and hSUMO2, by the Indicated IDP Discrimination Methods. Numbers of the Predicted Residues Are Shown. CS Shows the IDP Discrimination Only by the 1HN‐CS Range of 7.5–8.5 ppm, Whereas CSTC Shows IDP Discrimination Only by 1HN‐CSTC with −3.6 ppb/K as the CSTC Threshold
| CS | CSTC | CS†/CSTC | |
|---|---|---|---|
| True | |||
| Positive (a) | 28 | 30 | 28 |
| Negative (b) | 84 | 77 | 118 |
| Total (%) | 112 (62) | 107 (59) | 146 (81) |
| False | |||
| Positive (c) | 67 | 74 | 33 |
| Negative (d) | 2 | 0 | 2 |
| Total | 69 (38%) | 74 (41%) | 35 (19%) |
| Positive total (a + c) | 95 | 104 | 61 |
| Negative total (b + d) | 86 | 77 | 120 |
1HN chemical shift range of IDP is defined between 7.5 ppm and 8.5 ppm.
Subsequently, we combined the two criteria: 1HN‐CS and 1HN‐CSTC. Only when a residue fulfills both the criteria is the residue judged as IDR, otherwise non‐IDR. This combinatorial use of CS and CSTC drastically improved the accuracy to 81%, which was 19% higher than that of the orthodox method alone (7.5–8.5 ppm criterion). This improvement was driven by marked reduction of false positives (67–33). It is noteworthy that the number of the true‐positives was unchanged. Finally, we inferred that the combinatorial use of 1HN‐CSTC and 1HN‐CS was helpful for discriminating IDR residues from the NMR HSQC spectra of naturally occurring IDR/non‐IDR mixed case of proteins.
Re‐assessment of 1HN‐chemical shift range for IDP/non‐IDP discrimination
Next, we reassessed the threshold values (upper and lower limits of 1HN‐CS) for discriminating structured and IDR/non‐IDR residues. Historically, the chemical shift ranges of 7.5–8.5 ppm or 7.6–8.6 ppm have been used to confirm that residues with 1HN chemical shift in the range belong to IDR, when the NMR samples are entirely disordered. However, if the protein sample of interest includes both ordered and IDRs in one polypeptide, then the signals raised from the ordered region might eventually appear in the chemical shift range. Therefore, we calculated the ratio of true positive residues by changing the upper and lower threshold values of the chemical shift in 0.1 ppm when this criterion was used in combination with the previously determined 1HN‐CSTC threshold (−3.6 ppb/K). Results demonstrate that 1HN‐CS of 7.8–8.5 ppm gave the best result for discriminating ordered/IDR residues, providing accuracy of 83%, which was 2% higher than that of the previous CS/CSTC combination.
Discussion
For this study, we introduced 1HN‐CSTC as another experimental indicator for IDR/non‐IDR discrimination. We defined the threshold value CSTCTH as −3.6 ppb/K. Our approach was fundamentally based on earlier findings about the relation between 1HN‐CSTC and hydrogen bond formation inside a protein molecule. First, weak correlation between 1HN‐CSTC and the secondary structures of peptides or proteins had been reported respectively by Andersen et al.27 and by Baxter et al.16 Cierpicki et al. subsequently reported strong correlation between 1HN‐CSTC and intramolecular hydrogen bonding N–H···O=C.19, 20 This fact was rediscovered and analyzed further by Bouvignies et al.18 and by Tomlinson and Williamson.17 An advanced study by Hong et al. combining NMR and parallel MD simulations demonstrated that 1HN‐CSTC originates from thermal expansions of intramolecular N–H···O=C hydrogen bonding distances.25 However, thermal expansion of hydrogen bonding distances more likely occurs between protein backbone amide groups and solvent water rather than intramolecular hydrogen bonds. Based on this knowledge, we attempted to use 1HN‐CSTC as an indication of IDRs.
The threshold value of 1HN‐CSTC for discriminating hydrogen‐bonded and solvent‐exposed 1HN was found to be as −4.6 ppb/K.19, 20 The value was slightly smaller than our discrimination threshold for IDR/non‐IDR residues, CSTCTH as −3.6 ppb/K, because most IDR residues are assumed to be fully exposed to the solvent, whereas non‐IDR residues might still contain solvent‐exposed residues at the surface of compactly folded domains. Accordingly, we decided not to use 1HN‐CSTC alone as the criterion for IDR, but to use a combination of two criteria: the 1HN‐CS range and 1HN‐CSTC. This approach improved the discrimination accuracy. Nevertheless, our determined 1HN‐CSTC threshold of −3.6 ppb/K might still estimate non‐IDR residues erroneously in solvent‐exposed β‐strands as IDR. Therefore, we tested the application of an additional rule further as follows:
If one residue of “state D” is sandwiched by two residues of “state O”, then the “state D” residue is changed to “state O”.[Rule α]
The use of this additional Rule α improved IDR/non‐IDR discrimination further, producing results as high as 92% for our evaluation datasets hSUMO1/hSUMO2 (Table 3). This Rule α was only applicable to limited cases in which all the amide signals have been assigned sequentially. By contrast, the combinatorial use of 1HN‐CS range (7.8–8.5 ppm) and 1HN‐CSTC was easier because the method did not require signal assignment.
Table 3.
Results of the Two Naturally Occurring, Mixed Examples of IDP/Non‐IDP Residues, hSUMO1 and hSUMO2, Obtained Using the Indicated IDP Discrimination Methods. Numbers of Predicted Residues Are Shown. CS Shows the IDP Discrimination Only by the Refined 1HN‐CS Range of 7.8–8.5 ppm, Whereas CSTC Shows the IDP Discrimination Only by 1HN‐CSTC with −3.6 ppb/K as the CSTC Threshold; α Denotes That the Rule α (see the text) was Additionally Applied
| CS† | CS†/CSTC | CS†/CSTC/α‡ | |
|---|---|---|---|
| True | |||
| Positive (a) | 28 | 28 | 28 |
| Negative (b) | 97 | 123 | 139 |
| Total | 125 (69%) | 151 (83%) | 167 (92%) |
| False | |||
| Positive (c) | 54 | 28 | 12 |
| Negative (d) | 2 | 2 | 2 |
| Total | 56 (31%) | 30 (17%) | 14 (8%) |
| Positive total (a + c) | 82 | 56 | 40 |
| Negative Total (b + d) | 99 | 125 | 141 |
1HN chemical shift range of IDP is defined between 7.8 ppm and 8.5 ppm.
The additional Rule α was applied.
Some alternative NMR techniques are useful for discriminating IDR/non‐IDR residues: 13C secondary chemical shifts,26, 27, 28, 29 the 1H–15N heteronuclear Overhauser effect (heteroNOE30), and residual dipolar coupling (RDC14, 31, 32). The 13C secondary chemical shifts of Cα, Cβ, and CO, which are differences of the chemical shifts from those of a random coil state, were shown to be sensitive to its backbone φ and ψ torsion angles, thereby being useful for ascertaining the secondary structure of proteins.26, 28, 29 This technique is also useful for discriminating IDR residues from non‐IDR residues.27 The major shortcoming of this approach, however, is to complete sequential assignments of backbone signals; otherwise the secondary chemical shifts cannot be calculated. For this purpose, long NMR measuring time (at least two 3D datasets, HNCACB, and HN(CA)CO) with a 13C/15N‐labeled protein sample, and an effort to assign the signals, are required. Accordingly, neither the use of 1H–15N heteroNOE nor of RDC requires signal assignment. Both are 2D‐based experiments. The measurement time is shorter than that of 3D experiments. Although no comprehensive threshold of 1H–15N heteroNOE exists for discriminating IDR residues, we found in our earlier study that 1H–15N heteroNOE <0.5 showed a reasonable confidence.23 However, 1H–15N heteroNOE experiments require a long measuring time: typically 1 day (with a reference spectrum). Furthermore, measurement of RDC requires a special medium for the NMR sample preparation, as reviewed in an earlier report.33 In contrast, our proposed method is simpler, requiring only additional measurements of HSQC spectra at other temperatures. We evaluated it in naturally occurring protein cases, with or without amide signal assignment.
Conclusion
In summary, we introduced amide chemical shift temperature coefficients as an additional simple parameter to assess whether the protein region of interest fits the criteria of IDPs. In addition to a narrow dispersion of overall 1H‐15N HSQC (and SOFAST‐HMQC) spectra in the 1H axis, large negative temperature dependency (large up‐field change) of its chemical shift was shown to be a good indication of IDPs. If the region is intrinsically disordered, then it merely shows a state transition from ordered to disordered upon temperature increase. It remains disordered because it must be intrinsically disordered by definition. Results show that most of the amide chemical shifts might also exhibit linear change upon the temperature shift. This tendency suggests that only two HSQC spectra at the different temperature points are sufficient to drive 1HN‐CSTC, thereby assessing the disordered region.
Materials and Methods
Protein sample preparation
We selected three proteins (including protein domains), human ubiquitin (hUb, 76 aa), MIT domain from human Vps4B (Residues 1–77, hVps4bMIT, 81 aa), and the first PDZ domain of mouse zonula occludens 1 (Residues 18–110, mZO1‐PDZ1, 100 aa) as mainly folded protein examples. We then selected two IDPs from our in‐house human genome derived IDP library,22 IDP‐B3 (thymosin β10, 44 aa) and IDP‐C1 (WWOX Isoform 3, 33 aa) as the well‐characterized IDPs. All five of these proteins were used as the training dataset for method development. Finally, we chose human SUMO1 (hSUMO1, 97 aa) and SUMO2 (hSUMO2, 93 aa) as the validation set for the method developed in this study. Among them, chemical shift assignments of the main‐chain amide group (NH) signals for hUb20, 21 and hSUMO134, 35 were obtained from the literature. The NH signals of hVps4bMIT 36 and mZO1‐PDZ137 were assigned and reported by us. The NH signals of IDP‐C1 and hSUMO2 were found (this study, see below). The NH signals of IDP‐B3 were used without assignment.
Isotopically labeled proteins for NMR study were generated in Escherichia coli BL21(DE3) grown in 1 or 2 L M9 minimal medium culture, respectively, in the presence of [13C]‐glucose and [15N]‐NH4Cl as the sole carbon and nitrogen sources. Recombinant expression and purification of hUb, mZO1‐PDZ1, and hVps4bMIT were done according to the protocols.37, 38 15N‐labeled proteins of IDP‐B3 and IDP‐C1 were expressed using an NPRO(EDDIE)‐fusion protein expression system39 and were purified according to our slightly modified protocol.40 13C/15N‐labeled IDP‐C1 was also prepared and used for NH signal assignment.
For hSUMO1 and hSUMO2, the plasmids harboring the genes of N‐terminally GST‐tagged SUMOs, pET‐GST‐SUMO1/2, were transformed to BL21(DE3). After induction of protein expression by IPTG, the cells from 2 L M9 medium were harvested. Then the cells were suspended in lysis buffer (50 mM Tris–HCl, pH 7.4, 200 mM NaCl, 2 mM DTT) and were disrupted by sonication. The supernatant was applied to a DEAE–Sepharose (GE Healthcare, Little Chalfont, UK) column. The pass‐through was used for glutathione affinity column chromatography (3 mL bed/volumes, GST‐Accept®, Nacalai Tesque Inc., Tokyo, Japan). The GST‐tag was removed by addition of thrombin on beads. The crude protein was passed through benzamidine sepharose (GE Healthcare, Little Chalfont, UK) and was subsequently concentrated to a small volume. Finally, the sample was purified by gel filtration (Superdex® 75 pg; GE Healthcare, Little Chalfont, UK), concentrated to approximately 0.2 mM, and was dialyzed against 25 mM phosphate buffer (pH 6.25) containing 75 mM NaCl and 2 mM DTT.
NMR experiments
NMR experiments were conducted using an NMR spectrometer (600 MHz, Avance III; Bruker AXS GmbH, Karlsruhe, Germany) equipped with a cryogenic triple‐resonance probe. To determine 1HN‐CSTCs, approximately 100 μM protein samples were dissolved in the NMR measurement buffer (25 mM phosphate buffer (pH 6.25) containing 75 mM NaCl and 2 mM DTT). Then, to shorten the experimental time, the 1H–15N SOFAST‐HMQC spectra instead of HSQC to shorten experimental time were measured at 288, 293, 298, and 303 K. For all the well‐dispersed NH signals, 1HN‐CSTCs were found following the equation of
| (1) |
where δH is the 1HN chemical shift and T is the temperature. 15NH‐CSTC was ascertained similarly as
| (2) |
where δN represents the 15NH chemical shift.
Signal assignment
For the assignment of backbone 1H, 13C, and 15N resonances of IDP‐C1 and hSUMO2, 1H‐15N HSQC, HNCACB, CBCA(CO)NH, HNCO, and HN(CA)CO spectra were recorded. All 2D and 3D spectra were processed using NMRPipe41 and were analyzed with the Sparky program.42
Dataset preparation
For all 1HN‐CS, 15NH‐CS, 1HN‐CSTC, and 15NH‐CSTC data, we regarded that each residue belongs to either IDRs (D state, representing “disordered” state) or non‐IDRs (O state, representing “ordered” state). The states of respective residues were assigned based on results obtained from our earlier study.23 Briefly, it was defined from their Cα’s atomic co‐ordinate deviations as RMSD in the corresponding solution structures, which were retrieved from the Protein Data Bank (PDB). The following structures were used for the corresponding NMR data, PDB ID: 1D3Z (hUb), 2RRM (mZO1‐PDZ1), 1WR0 (hVps4bMIT), 1A5R (hSUMO1), and 2AWT (hSUMO2). If the deviation was larger than Ota's threshold, 3.2 Å, then the residue was classified as disordered, or part of an IDR (d), and as ordered (O) otherwise.23 Before analyzing the method, each coordinate entry of 1A5R and 2AWT was pre‐fitted to the first entry of each ensemble, particularly addressing their structured ubiquitin‐like regions. All the states for the residues from IDP‐B3 and IDP‐C1 were designated as D because they were confirmed by two experiments: CD spectra (manuscript in preparation, Supporting Information Fig. S1) and indirect systematic NMR assessment.22 Simultaneously, the O/D states for the same residues were determined by 1HN‐CSTC (and 15NH‐CSTC). If the CSTC was smaller (larger negative value) than a given threshold of CSTC (CSTCTH), then the residue was designated as D, and otherwise as O.
We attempted to optimize the value for CSTSTH as described below. We compared the two O/D states of each residue using two methods: one found using Ota's method based on PDB data and another defined by the new CSTC method (this study). When the state of a residue defined by PDB was A, and when that of CSTC was B, then we designated the state of the residue as (A, B), where A and B represent one of O or D. We evaluated the agreement of the residues’ states between the two ways by PDB and by CSTC using Matthews's correlation coefficient (MCC),43 defined as
| (3) |
where N(A, B) represents the number of residues in state (A, B) in a protein, and
| (4) |
Conflict of Interest
The authors declare that they have no conflict of interest in relation to this study or contents of this report describing it.
Supporting information
Appendix S1: CD spectra of each IDP.
Supplementary Figure S1. CD spectra of the selected human genome‐derived IDP samples. Spectra were measured at 278K (circle), 293K (triangle), and 308K (square).
Acknowledgments
This work was supported by JSPS KAKENHI Grant nos. 21113007 and 16 K14707. This work was also partially supported by a grant from the Salt Science Research Foundation and by the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japan Agency for Medical Research and Development (AMED).
References
- 1. Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re‐assessing the protein structure–function paradigm. J Mol Biol 293:321–331. [DOI] [PubMed] [Google Scholar]
- 2. Tompa P, Fersht A. Structure and function of intrinsically disordered proteins. CRC Press Book, Chapman and Hall, Boca Raton, FL, 2009. [Google Scholar]
- 3. Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 6:197–208. [DOI] [PubMed] [Google Scholar]
- 4. Dyson HJ, Wright PE (2002) Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol 12:54–60. [DOI] [PubMed] [Google Scholar]
- 5. Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z (2001) Intrinsically disordered protein. J Mol Graph Model 19:26–59. [DOI] [PubMed] [Google Scholar]
- 6. Uversky VN (2002) Natively unfolded proteins: a point where biology waits for physics. Protein Sci 11:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, Kragelund BB, Best RB, Schuler B (2018) Extreme disorder in an ultrahigh‐affinity protein complex. Nature 555:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukuchi S, Sakamoto S, Nobe Y, Murakami SD, Amemiya T, Hosoda K, Koike R, Hiroaki H, Ota M (2012) IDEAL: intrinsically disordered proteins with extensive annotations and literature. Nucleic Acids Res 40:507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fukuchi S, Amemiya T, Sakamoto S, Nobe Y, Hosoda K, Kado Y, Murakami SD, Koike R, Hiroaki H, Ota M (2014) IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res 42:D320–D325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boothby TC, Tapia H, Brozena AH, Piszkiewicz S, Smith AE, Giovannini I, Rebecchi L, Pielak GJ, Koshland D, Goldstein B (2017) Tardigrades use intrinsically disordered proteins to survive desiccation. Mol Cell 65:975–984.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boothby TC, Pielak GJ (2017) Intrinsically disordered proteins and desiccation tolerance: elucidating functional and mechanistic underpinnings of anhydrobiosis. BioEssays 39:1700119. [DOI] [PubMed] [Google Scholar]
- 12. Matsuo N, Goda N, Shimizu K, Fukuchi S, Ota M, Hiroaki H (2018) Discovery of cryoprotective activity in human genome‐derived intrinsically disordered proteins. Int J Mol Sci 19:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Konrat R (2014) NMR contributions to structural dynamics studies of intrinsically disordered proteins. J Magn Reson 241:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kosol S, Contreras‐Martos S, Cedeño C, Tompa P (2013) Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules 18:10802–10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jensen MR, Ruigrok RWH, Blackledge M (2013) Describing intrinsically disordered proteins at atomic resolution by NMR. Curr Opin Struct Biol 23:426–435. [DOI] [PubMed] [Google Scholar]
- 16. Baxter NJ, Williamson MP (1997) Temperature dependence of 1H chemical shifts in proteins. J Biomol NMR 9:359–369. [DOI] [PubMed] [Google Scholar]
- 17. Tomlinson JH, Williamson MP (2012) Amide temperature coefficients in the protein G B1 domain. J Biomol NMR 52:57–64. [DOI] [PubMed] [Google Scholar]
- 18. Bouvignies G, Vallurupalli P, Cordes MHJ, Hansen DF, Kay LE (2011) Measuring 1HN temperature coefficients in invisible protein states by relaxation dispersion NMR spectroscopy. J Biomol NMR 50:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cierpicki T, Otlewski J (2001) Amide proton temperature coefficients as hydrogen bond indicators in proteins. J Biomol NMR 21:249–261. [DOI] [PubMed] [Google Scholar]
- 20. Cierpicki T, Zhukov I, Byrd RA, Otlewski J (2002) Hydrogen bonds in human ubiquitin reflected in temperature coefficients of amide protons. J Magn Reson 157:178–180. [DOI] [PubMed] [Google Scholar]
- 21. Cordier F, Grzesiek S (2002) Temperature‐dependence of protein hydrogen bond properties as studied by high‐resolution NMR. J Mol Biol 317:739–752. [DOI] [PubMed] [Google Scholar]
- 22. Goda N, Shimizu K, Kuwahara Y, Tenno T, Noguchi T, Ikegami T, Ota M, Hiroaki H (2015) A method for systematic assessment of intrinsically disordered protein regions by NMR. Int J Mol Sci 16:15743–15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin C, Shiyanova T, Shen Z, Liao X (2001) Heteronuclear nuclear magnetic resonance assignments, structure and dynamics of SUMO‐1, a human ubiquitin‐like protein. Int J Biol Macromol 28:227–234. [DOI] [PubMed] [Google Scholar]
- 24. Kumar A, Srivastava S, Hosur RV (2007) NMR characterization of the energy landscape of SUMO‐1 in the native‐state ensemble. J Mol Biol 367:1480–1493. [DOI] [PubMed] [Google Scholar]
- 25. Achmüller C, Kaar W, Ahrer K, Wechner P, Hahn R, Werther F, Schmidinger H, Cserjan‐Puschmann M, Clementschitsch F, Striedner G, Bayer K, Jungbauer A, Auer B (2007) N(pro) fusion technology to produce proteins with authentic N termini in E. coli . Nat Methods 4:1037–1043. [DOI] [PubMed] [Google Scholar]
- 26. Goda N, Matsuo N, Tenno T, Ishino S, Ishino Y, Fukuchi S, Ota M, Hiroaki H (2015) An optimized N(pro)‐based method for the expression and purification of intrinsically disordered proteins for an NMR study. Intrinsically Disord Proteins 3:e1011004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293. [DOI] [PubMed] [Google Scholar]
- 28. Goddard TD, Kneller DG. Sparky 3. San Francisco: University of California, 2004. [Google Scholar]
- 29. Ota M, Koike R, Amemiya T, Tenno T, Romero PR, Hiroaki H, Dunker AK, Fukuchi S (2013) An assignment of intrinsically disordered regions of proteins based on NMR structures. J Struct Biol 181:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matthews BW (1975) Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta 405:442–451. [DOI] [PubMed] [Google Scholar]
- 31. Schanda P, Kupče Ē, Brutscher B (2005) SOFAST‐HMQC experiments for recording two‐dimensional heteronuclear correlation spectra of proteins within a few seconds. J Biomol NMR 33:199–211. [DOI] [PubMed] [Google Scholar]
- 32. Andersen NH, Neidigh JW, Harris SM, Lee GM, Liu Z, Tong H (1997) Extracting information from the temperature gradients of polypeptide NH chemical shifts. 1. The importance of conformational averaging. J Am Chem Soc 119:8547–8561. [Google Scholar]
- 33. Hong J, Jing Q, Yao L (2013) The protein amide 1H(N) chemical shift temperature coefficient reflects thermal expansion of the N‐H···O=C hydrogen bond. J Biomol NMR 55:71–78. [DOI] [PubMed] [Google Scholar]
- 34. Wishart DS, Sykes BD (1994) The 13C chemical‐shift index: a simple method for the identification of protein secondary structure using 13C chemical‐shift data. J Biomol NMR 4:171–180. [DOI] [PubMed] [Google Scholar]
- 35. Tamiola K, Acar B, Mulder FAA (2010) Sequence‐specific random coil chemical shifts of intrinsically disordered proteins. J Am Chem Soc 132:18000–18003. [DOI] [PubMed] [Google Scholar]
- 36. Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13:289–302. [DOI] [PubMed] [Google Scholar]
- 37. Umetsu Y, Goda N, Taniguchi R, Satomura K, Ikegami T, Furuse M, Hiroaki H (2011) 1H, 13C, and 15N resonance assignment of the first PDZ domain of mouse ZO‐1. Biomol NMR Assign 5:207–210. [DOI] [PubMed] [Google Scholar]
- 38. Shen Y, Bax A (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR 56:227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kay LE, Torchia DA, Bax A (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28:8972–8979. [DOI] [PubMed] [Google Scholar]
- 40. Salmon L, Nodet G, Ozenne V, Yin G, Jensen MR, Zweckstetter M, Blackledge M (2010) NMR characterization of long‐range order in intrinsically disordered proteins. J Am Chem Soc 132:8407–8418. [DOI] [PubMed] [Google Scholar]
- 41. Ullman O, Fisher CK, Stultz CM (2011) Explaining the structural plasticity of α‐synuclein. J Am Chem Soc 133:19536–19546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen K, Tjandra N (2012) The use of residual dipolar coupling in studying proteins by NMR. Top Curr Chem 326:47–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takasu H, Jee JG, Ohno A, Goda N, Fujiwara K, Tochio H, Shirakawa M, Hiroaki H (2005) Structural characterization of the MIT domain from human Vps4b. Biochem Biophys Res Commun 334:460–465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: CD spectra of each IDP.
Supplementary Figure S1. CD spectra of the selected human genome‐derived IDP samples. Spectra were measured at 278K (circle), 293K (triangle), and 308K (square).