

Abstract
Many interactions between proteins are mediated by intrinsically disordered regions (IDRs). Intrinsically disordered proteins (IDPs) do not adopt a stable three‐dimensional structure in their unbound form, but they become more structured upon binding to their partners. In this communication, we study how a bound IDR adapts to mutations, preventing the formation of hydrogen bonds at the binding interface that needs a precise positioning of the interacting residues to be formed. We use as a model the YAP:TEAD interface, where one YAP (IDP) and two TEAD residues form hydrogen bonds via their side chain. Our study shows that the conformational flexibility of bound YAP and the reorganization of water molecules at the interface help to reduce the energetic constraints created by the loss of H‐bonds at the interface. The residual flexibility/dynamic of bound IDRs and water might, therefore, be a key for the adaptation of IDPs to different interface landscapes and to mutations occurring at binding interfaces.
Keywords: intrinsically disordered proteins, protein–protein interactions, YAP, TEAD, water
Introduction
The Lock and Key model was an early attempt to explain the selectivity of the interactions between a protein and its ligand. In this model, the optimal ligand (key) has the right shape and size to fit into the keyhole of the protein (lock). This model, where both interacting partners have a precise/fixed geometry, gives a very static view of the interactions involving proteins. With the availability of more structural data, it became apparent that the flexibility of the binding partners is an important feature in molecular recognition. This led to the development of new models where the conformational flexibility of the interacting partners is integrated into the binding process. Today there are two models that have been extensively discussed in the literature.1, 2, 3, 4 In the conformational selection model, one of the two partners binds to a specific conformation (or a small subset) the other binding partner can adopt in its unbound state. In the induced fit model, the initial interaction between the two partners is followed by a conformational change that leads to the final complex. The frontier between both mechanisms may not be so strict, since both the protein dynamics and the ligand concentration can shift the binding mechanism.5 Furthermore, it is conceivable that binding processes may involve both conformational selection and induced fit.
In the past few years, a new area in molecular recognition has emerged with the study of intrinsically disordered proteins (IDPs) or intrinsically disordered regions (IDRs) of proteins. IDPs/IDRs do not have a fixed three‐dimensional structure in their unbound state, but they become more structured once they are bound to their partner(s). It should be kept in mind that IDPs/IDRs may have some level of structural organization (e.g., transient secondary or molten‐like structures) in their unbound form. The interesting properties of IDPs/IDRs have prompted numerous investigations, and many studies have focused on their binding mechanism (see e.g., Refs. [6, 7, 8, 9). Some of these studies used kinetics methods (φ‐value analysis10) to probe the transition state and thus map the interactions established during the binding process (see e.g., Refs. [11, 12, 13, 14). A – very cautious – interpretation of the data obtained from the limited number of systems studied so far suggests that IDPs/IDRs fold after binding9 and that hydrophobic residues located at the interface are important for this step. However, in view of the diversity of IDPs/IDRs and their binding partners, they may not all follow the same mechanism for binding.
In this manuscript, we are focusing on a different aspect of the interaction between an IDR and its binding counterpart. The aim of this work was to study the effect of mutations that prevent the formation of hydrogen bonds between the side chains of the two interacting partners at the binding interface. As H‐bonds require a precise geometry to be formed, this study should enable us to determine how a bound IDP/IDR adapts to the loss of the structural constrain required for its formation. Furthermore, since water is important for the dynamics of H‐bonds, this analysis should provide an insight into the contribution of water molecules in maintaining/establishing H‐bond networks at the binding interface between an IDP/IDR and its target. As a model, we use the YAP:TEAD interaction. The TEAD binding region of YAP is a bona fide IDR, since it adopts a random coil conformation in solution.15 YAP binds to TEAD, which is a well‐folded and rather a rigid partner, via two main secondary structure elements, an α‐helix and an Ω‐loop.16, 17 The interface at the Ω‐loop binding site is key for the YAP:TEAD interaction16, 17, 18 and, within this region, Ser94YAP makes hydrogen bonds with Glu263TEAD4 and Tyr429TEAD4 [Fig. 1(A)]. The mutation of these residues significantly affects the YAP:TEAD interaction as measured in biochemical or cellular assays.16, 17, 19 This interaction, which requires the formation of two well‐defined hydrogen bonds, therefore, represents a good model for the purpose of this study.
Figure 1.
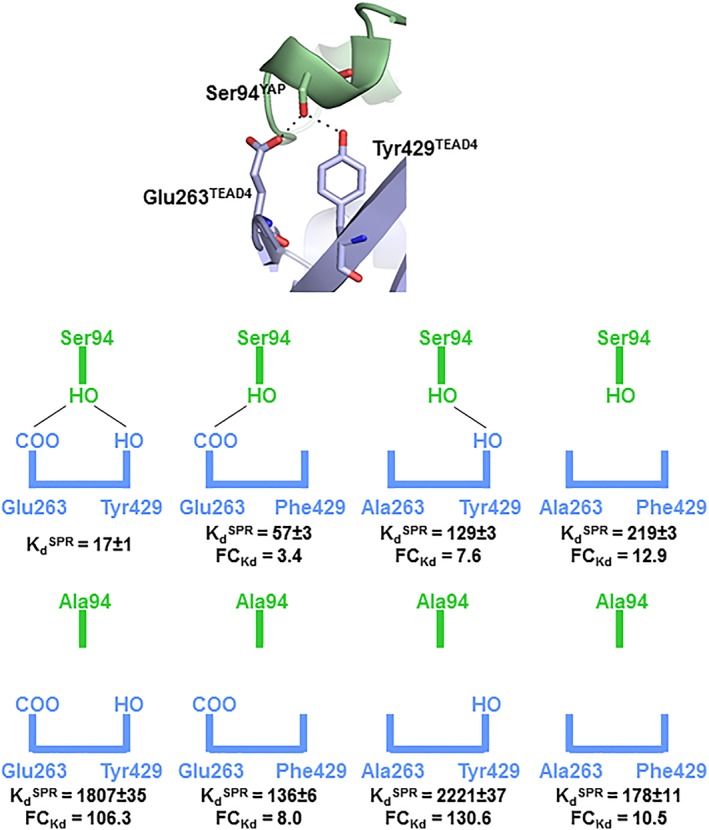
Biochemical study of the YAP:TEAD complexes. (A) Close view of the interaction site between Ser94YAP and Glu263TEAD4/Tyr429TEAD4 in the wtYAP:wtTEAD4 complex. YAP and TEAD4 are in green and blue, respectively. H‐bonds are represented by black dotted lines. (B) Schematic view of the interaction site in the different YAP:TEAD complexes. H‐bonds are represented by black lines. Fold change in (= K dmutant/K dwt) are indicated.
Results
Biochemical study of the YAP:TEAD complexes
The affinity (K d) of wtYAP for the different TEAD4 proteins was measured by surface plasmon resonance [SPR; Fig. 1(B) and Table 1]. In agreement with earlier results,19 the K d of wtYAP for wtTEAD4 is in the low double‐digit nanomolar range. The Tyr429PheTEAD4 mutation has a smaller effect (ΔΔG = ΔG mutant – ΔG wt =0.71 kcal/mol) than the Glu263AlaTEAD4 mutation (ΔΔG = 1.19 kcal/mol) and, in approximate terms, both mutations have an additive effect on the interaction with wtYAP (ΔΔG double mutant = 1.51 kcal/mol similar to 1.9 kcal/mol). The Ser94AlaYAP mutation has a major destabilizing effect on wtTEAD4 binding (ΔΔG = 2.76 kcal/mol), which is significantly greater (by 0.86 kcal/mol) than the cumulated effect of two TEAD4 mutations. There is, therefore, an asymmetry in the impact of the mutations, the loss of Ser94‐OHYAP being more detrimental for binding than the loss of both Glu263‐COOTEAD4 and Tyr429‐OHTEAD4. This asymmetry is also observed when comparing the Ser94AlaYAP:wtTEAD4 and wtYAP:Glu263Ala–Tyr429PheTEAD4 complexes. Since the Ser94AlaYAP and Glu263Ala–Tyr429PheTEAD4 mutations prevent the formation of the same interactions at the YAP:TEAD interface, the complexes would be expected to have the same affinity. But this is not the case. Their affinity differs by 1.25 kcal/mol. When Tyr429TEAD4 is mutated, the Ser94AlaYAP mutation, which suppresses only the formation of an H‐bond with Glu263TEAD4, has a moderate effect with the wtYAP:Tyr429PheTEAD4 and Ser94AlaYAP:Tyr429PheTEAD4 complexes having a similar affinity (ΔΔG = 0.52 kcal/mol). However, when Glu263TEAD4 is mutated, the Ser94AlaYAP mutation, which suppresses only the formation of an H‐bond with Tyr429TEAD4, has a large impact on the interaction with the wtYAP:Glu263AlaTEAD4 and Ser94AlaYAP:Glu263AlaTEAD4 complexes showing significantly different affinities (ΔΔG = 1.69 kcal/mol). The structure of the wtYAP:wtTEAD4 complex suggests that the Glu263AlaTEAD4 and Tyr429PheTEAD4 mutations should have little impact on the binding of Ser94AlaYAP since the side chain with which they form an H‐bond in the wt complex is not present in this YAP mutant. A similar prediction can also be made for the interaction of Ser94AlaYAP with Glu263Ala–Tyr429PheTEAD4. These predictions hold true both for the Glu263AlaTEAD4 mutation, which has little impact (ΔΔG = 0.12 kcal/mol, comparison between Ser94AlaYAP:Glu263AlaTEAD4 and Ser94AlaYAP:wtTEAD4) on Ser94AlaYAP binding, and for the Ser94AlaYAP mutation, which has a negligible effect on the binding of Glu263Ala–Tyr429PheTEAD4 (ΔΔG = −0.13 kcal/mol, comparison between Ser94AlaYAP:Glu263Ala–Tyr429PheTEAD4 and wtYAP:Glu263Ala–Tyr429PheTEAD4). However, a very different result is obtained with the Tyr429PheTEAD4 mutation, which enhances the affinity of Ser94AlaYAP by 1.53 kcal/mol when compared to the Ser94AlaYAP:wtTEAD4 complex.
Table 1.
Biochemical characterization of the different complexes. The dissociation constants (K d) were measured at equilibrium (295°K) by Surface Plasmon Resonance. Average values and standard errors are indicated (n ≥ 3). The binding energies (ΔG) are calculated from the K d values
| wtYAP | Ser94AlaYAP | |||
|---|---|---|---|---|
| TEAD4 | K d (nM) | ΔG (kcal/mol) | K d (nM) | ΔG (kcal/mol) |
| wt | 17 ± 1 | −10.58 ± 0.04 | 1807 ± 35 | −7.83 ± 0.04 |
| Tyr429Phe | 57 ± 3 | −9.88 ± 0.03 | 136 ± 6 | −9.36 ± 0.01 |
| Glu263Ala | 129 ± 3 | −9.39 ± 0.01 | 2221 ± 37 | −7.70 ± 0.01 |
| Tyr429Phe‐Glu263Ala | 219 ± 2 | −9.08 ± 0.01 | 178 ± 11 | −9.20 ± 0.01 |
Overall, this analysis reveals that the mutations of Ser94YAP and Glu263/Tyr429TEAD4 have a clear impact on the YAP:TEAD interaction. However, some of the observed effects seem to involve more complex mechanisms than the simple loss of H‐bonds at the interface, and the structure of the wtYAP:wtTEAD4 complex is not sufficient for them to be appreciated.
Structural study of the YAP:TEAD complexes
Synthetic peptides mimicking the region 60–100 of wtYAP and Ser94AlaYAP were co‐crystallized with the different TEAD4 proteins and the structures of these eight complexes were determined by X‐ray crystallography (Table S1).
In the wtYAP:wtTEAD4 complex, Glu263‐COOTEAD4 and Tyr429‐OHTEAD4 make an H‐bond with Ser94‐OHYAP [Fig. 2(A)]. A water molecule (W1) is present between Glu263‐COOTEAD4 and Ser94‐NHYAP. Glu263TEAD4, Ile428TEAD4, and His427TEAD4 are engaged in an H‐bond network involving the two water molecules, W2 and W3. The imidazole ring of His427TEAD4 is oriented toward Lys97YAP.
Figure 2.
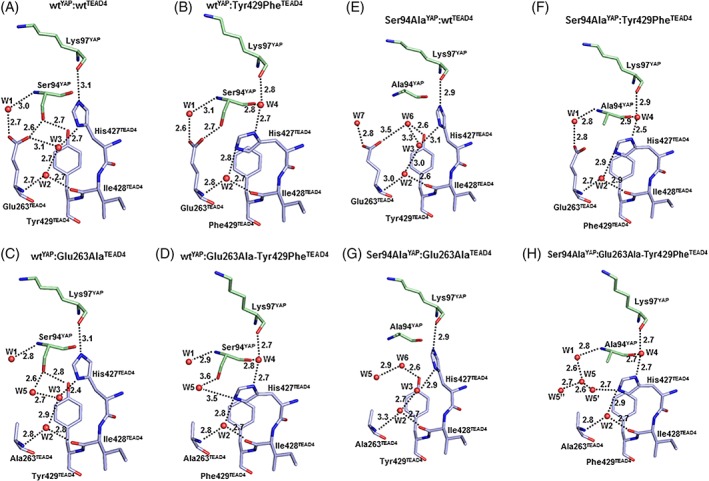
(A–D). Structure of the different YAP:TEAD complexes in the region surrounding the mutation sites. The figure represents the residues and water molecules discussed in the text. YAP and TEAD4 residues are in green and blue, respectively. Water molecules are indicated by red spheres. The distance (Å) between atoms is indicated (dotted lines).
The Tyr429PheTEAD4 mutation induces several changes at the interface [Fig. 2(B)]. The small cavity (~5 Å3) created by the mutation allows YAP to move toward TEAD [Fig. 3(A)]. This movement, which brings Ser94‐CYAP closer to Phe429‐CzTEAD4 (4.1 and 4.7 Å in the wtYAP:Tyr429PheTEAD4 and the wtYAP:wtTEAD4 complex, respectively), preserves the interaction between Glu263‐COOTEAD4 and Ser94‐OHYAP that remains within H‐bond distance. The side chain of His427TEAD4 is rotated versus the wt complex and is oriented toward Glu263TEAD4. This movement affects the water molecules at the binding interface. W3 in the wt complex is displaced by the rotation of His427TEAD4, and the space left empty between this residue and Lys97YAP becomes occupied by a new water molecule (W4). W1 and W2 occupy a position similar to that in the wt complex.
Figure 3.
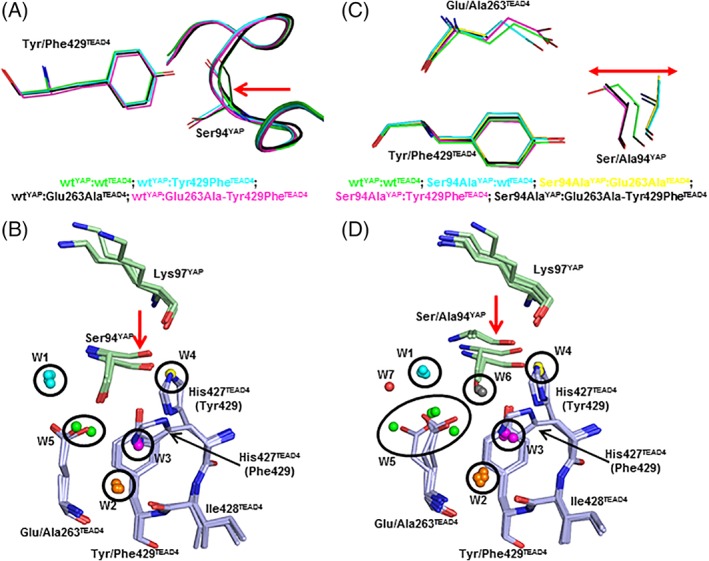
Conformational and solvation changes induced by the mutations. (A) The structures of wtYAP in complex with the four TEAD4 proteins have been superimposed. The four complexes are each represented in a different color and the main chain of YAP is pictured as a ribbon. The red arrow shows the region where the movement of YAP toward TEAD is observed. B. YAP and TEAD4 residues are in green and blue, respectively. Water molecules are indicated by spheres (see text for more details). The position of His427TEAD4 in the complexes with Tyr429TEAD4 or Phe429TEAD4 is indicated. The red arrow highlights the movement of YAP observed in the wtYAP:Tyr429PheTEAD4 and wtYAP:Glu263Ala–Tyr429PheTEAD4 complexes. (C) The structures of the different complexes, pictured in different colors, have been superimposed. The residues at position‐94 (YAP) and position‐263/429 (TEAD4) are indicated. The red arrow represents the movement of YAP at Position‐94. D. YAP and TEAD4 residues are in green and blue, respectively. Water molecules are indicated by spheres (see text for more details). The position of His427TEAD4 in the complexes with Tyr429TEAD4 or Phe429TEAD4 is indicated. The red arrow highlights the movement of YAP.
The shift of YAP toward TEAD observed in the wtYAP:Tyr429PheTEAD4 complex does not take place in the wtYAP:Glu263AlaTEAD4 complex [Fig. 3(A)]. As in the wt complex, Tyr429‐OHTEAD4 is within H‐bond distance of Ser94‐OHYAP, His427TEAD4 is oriented toward Lys97YAP and its imidazole ring is also engaged in an H‐bond network, via W2 and W3, with Ala263TEAD4 and Ile428TEAD4 [Fig. 2(C)]. A water molecule, W5, occupies a position similar to Glu263‐COOTEAD4 in the wt complex, and it is within H‐bond distance of Ser94‐OHYAP and W3. W1 is in the same position as in the wt complex.
The movement of YAP detected in the wtYAP:Tyr429PheTEAD4 complex is also observed in the wtYAP:Glu263Ala–Tyr429PheTEAD4 complex and is of similar magnitude [Fig. 3(A)]. The side chain of His427TEAD4 is rotated toward Ala423TEAD4 and the two water molecules W2 and W4 occupy positions similar to those in the wtYAP:Tyr429PheTEAD4 complex [Fig. 2(D)]. As in the wtYAP:Glu263AlaTEAD4 complex, W5 is present at the interface, but it is not in exactly the same position as in the wtYAP:Glu263AlaTEAD4 complex probably because of the absence of W3 and the different orientation of His427TEAD4 in the wtYAP:Glu263Ala–Tyr429PheTEAD4 complex. W1 has the same position as in the wt complex.
The four previous structures were solved from crystals belonging to the same space group (P41212), and the possibility cannot be ruled out that the movement of YAP observed in the wtYAP:Tyr429PheTEAD4 and wtYAP:Glu263Ala–Tyr429PheTEAD4 complexes might be triggered because they crystallize in this specific crystal form. To test this hypothesis, new crystallization conditions were developed, crystals from a different space group (P212121) were obtained with the wtYAP:Tyr429PheTEAD4 complex and a new structure was solved by X‐ray crystallography (Table S1). The same movement of YAP around position‐94 is observed in this additional structure (Fig. S1) indicating that it is the presence of Phe429TEAD4 at the interface and not the crystallization conditions which induce the shift of YAP toward TEAD in the wtYAP:Tyr429PheTEAD4 complex. This movement of YAP toward TEAD probably triggers the rotation of the side chain of His427TEAD4 and consequently a change in the position of interfacial water molecules (loss of W3 and presence of W4).
Figure 3(B) represents the region surrounding the mutation sites when the four structures obtained with wtYAP are superimposed. W1 (cyan) and W2 (orange) have the same position in all the complexes. W3 (magenta), which is present in the wtYAP:wtTEAD4 and wtYAP:Glu263AlaTEAD4 complexes, is at the position occupied by His427‐Nε2TEAD4 in the wtYAP:Tyr429PheTEAD4 and wtYAP:Glu263Ala–Tyr429PheTEAD4 complexes. W4 (yellow), which is found in the wtYAP:Tyr429PheTEAD4 and wtYAP:Glu263Ala–Tyr429PheTEAD4 complexes, is at the position of His427‐Nε2EAD4 in the wtYAP:wtTEAD4 and wtYAP:Glu263AlaTEAD4 complexes. Finally, W5 (green), which is present in the wtYAP:Glu263AlaTEAD4 and wtYAP:Glu263Ala–Tyr429PheTEAD4 complexes, is located in the region occupied by Glu263‐COOTEAD4 in the wtYAP:wtTEAD4 and wtYAP:Tyr429PheTEAD4 complexes.
We shall now study the structures of the YAP:TEAD complexes when Ser94YAP is mutated to alanine. The structure of the Ser94AlaYAP:wtTEAD4 complex again highlights the adaptation of bound YAP to mutations [Fig. 2(E)]. Glu263TEAD4 and Tyr429TEAD4 are in the same position as in the wt complex. His427TEAD4 is oriented toward Lys97YAP, and the network of H‐bonds created by W2 and W3 is maintained. However, changes are observed around the mutation site. If Ala94YAP kept the position occupied by Ser94YAP in the wt complex, its hydrophobic Cβ atom would be in the vicinity of the polar Glu263‐COOTEAD4 and Tyr429‐OHTEAD4, which in addition would have limited ability to form H‐bonds with water. The residual flexibility of bound YAP allows such highly unstable conformations to be avoided and it moves away from TEAD4, separating Ala94‐CβYAP from these polar groups [Fig. 3(C)]. This movement allows a water molecule, W6, to be located at the interface close to Glu263‐COOTEAD4 and Tyr429‐OHTEAD4 [Fig. 2(E)]. This displacement of YAP also shifts Ala94‐NHYAP away from Glu263‐COOTEAD4 (6.3 Å vs. 5 Å measured between Ser94‐NHYAP and Glu263‐COOTEAD4 in the wt complex). This distance is too long for W1 to efficiently bridge Glu263‐COOTEAD4 and Ala94‐NHYAP, and in the Ser94AlaYAP:wtTEAD4 complex, W7 connects Glu263‐COOTEAD4 to Asp93‐NHYAP [not shown in Fig. 2(E)].
Zhang et al. recently designed synthetic peptides mimicking the Ω‐loop of YAP and found that the Glu93AlaYAP mutation leads to a two‐fold enhancement in the affinity of YAP for TEAD.20 Their interpretation of this finding is that the mutation enhances the stability of the one‐turn α‐helix present in this region of the Ω‐loop because of the α‐helix stabilizing nature of alanine. We measured the Φ and Ψ dihedral angles of YAP Residues 93, 94, and 95 in the wtYAP:wtTEAD4 and Ser94AlaYAP:wtTEAD4 complexes (Table S2). The Ser94AlaYAP mutation has little effect on the dihedral angles of the two residues surrounding the mutation site, but it significantly alters those of the residue at position‐94. The Φ and Ψ dihedral angles of Ala94YAP depart more from the canonical angles measured in α‐helices (clustered around Φ = −57° and Ψ = −47°) than those measured for Ser94YAP. Ala94YAP adopts a conformation which is closer to that found in 310 helices (clustered around Φ = −74° and Ψ = −4°). The presence of a residue with such a conformation in this single turn α‐helix may have a destabilizing effect on bound YAP.
The Ser94AlaYAP:Tyr429PheTEAD4 [Fig. 2(F)] and wtYAP:Tyr429PheTEAD4 [Fig. 2(B)] complexes are structurally related. His427TEAD4 is oriented toward Glu263TEAD4, and the three water molecules W1, W2, and W4 occupy the same position in both complexes. As observed in the wtYAP:Tyr429PheTEAD4 complex, there is also a movement of YAP in the direction of TEAD, bringing Ser94‐CβYAP within van der Waals distance from Phe429‐CzTEAD4 and potentially creating a new interaction at the interface [Fig. 3(C)]. The main difference between the two complexes is the absence of an H‐bond between Glu263TEAD4 and Ser94YAP in the Ser94AlaYAP:Tyr429PheTEAD4 complex.
In the Ser94AlaYAP:Glu263AlaTEAD4 complex, His427TEAD4, W2 and W3 occupy the same position as in the wt complex [Fig. 2(G)]. As observed in the Ser94AlaYAP:wtTEAD4 complex, Ala94YAP moves away from TEAD, and the magnitude of this movement is similar in both complexes [Fig. 3(C)]. A water molecule, W6, is also located at the interface close to Tyr429‐OHTEAD4, and the movement of YAP also modifies the Φ, Ψ dihedral angles of the residue at Position‐94 without affecting those of the surrounding amino acids (Table S2). This movement away from TEAD is, therefore, only observed in the presence of Tyr429TEAD4 at the interface. W5, which occupies the position of Glu263‐COOTEAD4, is in the vicinity of W6.
The structure of the Ser94AlaYAP:Glu263Ala–Tyr429PheTEAD4 complex has several features in common with the complexes already described [Fig. 2(H)]. His427TEAD4, W2, and W4 occupy a position similar to that observed in every complex formed with Phe429TEAD4. Ala94YAP has moved toward TEAD and is at a position similar to that in the Ser94AlaYAP:Tyr429PheTEAD4 complex [Fig. 3(C)]. W1 is at the same location as in the wt complex. Several molecules, W5–W5’–W5”, are located in the area occupied by Glu263‐COOTEAD4 in the wt complex.
The superimposition of the structures obtained with Ser94AlaYAP with that of the wt complex shows the interplay between conformational changes and the presence of water molecules at the interface [Fig. 3(D)]. The observations made for W1, W2, W3, W4, and W5 in the structures obtained with wtYAP also apply to Ser94AlaYAP [see above and compare Fig. 3(D) and (B)]. However, in the structures obtained with this Ser94AlaYAP, a new water molecule, W6 [grey in Fig. 3(D)] occupies the position of Ser94‐OHYAP when Tyr429TEAD4 is present at the interface. This shows that as soon as an H‐bond donor/acceptor atom is absent at the interface, its position is occupied by a water molecule which helps maintaining the cohesiveness of the H‐bond network between the two interacting proteins. These changes in interfacial water are not only passive events, where water comes to fill empty cavities, but, as observed with W6, they can require readjustments at the expense of conformational constraints (e.g., effect on the dihedral angles of Ala94YAP) in the final complex.
Thermodynamic study of the YAP:TEAD complexes
The effect of the different mutations on the YAP:TEAD interaction was studied by Isothermal Titration Calorimetry (ITC). ITC measures the change in heat signal which results from the multiple events taking place during a binding reaction (e.g., breaking/formation of interactions, conformational changes, exchange of protons with the buffer, etc.). The precise interpretation of ITC data is, therefore, often challenging.21 The two thermodynamic parameters enthalpy (ΔH) and entropy (ΔS) derived from ITC experiments measure the overall difference between the unbound and bound states of a system. Even if YAP is an IDR, it may adopt transient structures in its unbound form and the Ser94AlaYAP mutation could then affect the dynamics between them and consequently the ground state of the system. Therefore ΔH and ΔS would also reflect this effect, which cannot be appreciated from the analysis of the structures of the YAP:TEAD complexes. Keeping this in mind, we voluntarily limited our analysis to avoid any overinterpretation of the data.
In line with the structural data, the stoichiometry of the different complexes is 1 (0.93 ≤ n ≤ 1.1, Table 2). The K d values obtained by ITC (Table 2) and those measured by SPR (Table II) are in good agreement, with less than a 2.2‐fold difference. The ΔH obs and –TΔS values for the different complexes are given in Table 2 and schematically represented in Figure 4. The interaction between wtYAP and wtTEAD4 is driven by a favorable enthalpic contribution (ΔH obs = −10.15 kcal/mol) which is partially offset by an unfavorable entropic component (–TΔS = 2.8 kcal/mol). The wtYAP:Tyr429PheTEAD4 and wtYAP:Glu263AlaTEAD4 complexes have a ΔH obs similar to that of the wt complex [Fig. 4(A)], while it is reduced by about 2 kcal/mol for the other interactions. The wtYAP:Tyr429PheTEAD4, wtYAP:Glu263AlaTEAD4, Ser94AlaYAP:wtTEAD4, and Ser94AlaYAP:Glu263AlaTEAD4 complexes show an increased entropic penalty (between 0.7 and 1.3 kcal/mol) but TΔS is more favorable by 0.8–1.1 kcal/mol for the wtYAP:Glu263Ala–Tyr429PheTEAD4, Ser94AlaYAP:Tyr429PheTEAD4, and Ser94AlaYAP:Glu263Ala–Tyr429PheTEAD4 complexes [Fig. 4(B)].
Table 2.
Thermodynamic analysis of the interaction between the different TEAD4 variants and wt/Ser94AlaYAP. The different parameters were measured by Isothermal Titration Calorimetry at 295°K. Average values and standard errors are indicated (n ≥ 2)
| wtYAP | Ser94AlaYAP | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TEAD4 | n a | K d (nM) | ΔG (kcal/mol) | ΔH obs (kcal/mol) | −TΔS
(kcal/mol) |
n | K d (nM) | ΔG (kcal/mol) | ΔH obs (kcal/mol) | −TΔS (kcal/mol) |
| wt | 1.05 ± 0.02 | 37 ± 5 | −10.15 ± 0.05 | −13.0 ± 0.1 | 2.8 ± 0.1 | 1.1 ± 0.00 | 3300 ± 400 | −7.5 ± 0.1 | −11.0 ± 0.1 | 3.52 ± 0.01 |
| Tyr429Phe | 0.96 ± 0.01 | 92 ± 3 | −9.61 ± 0.02 | −13.3 ± 0.3 | 3.7 ± 0.4 | 0.93 ± 0.02 | 210 ± 20 | −9.1 ± 0.1 | −11.10 ± 0.04 | 2.0 ± 0.1 |
| Glu263Ala | 0.99 ± 0.03 | 212 ± 2 | −9.11 ± 0.00 | −13.2 ± 0.2 | 4.1 ± 0.2 | 0.98 ± 0.03 | 4160 ± 300 | −7.35 ± 0.04 | −11.4 ± 0.1 | 4.1 ± 0.2 |
| Glu263Ala‐Tyr429Phe | 1.06 ± 0.04 | 289 ± 11 | −8.93 ± 0.02 | −10.85 ± 0.05 | 1.93 ± 0.01 | 1.05 ± 0.03 | 291 ± 40 | −8.9 ± 0.1 | −10.60 ± 0.00 | 1.7 ± 0.1 |
n: stoichiometry.
Figure 4.
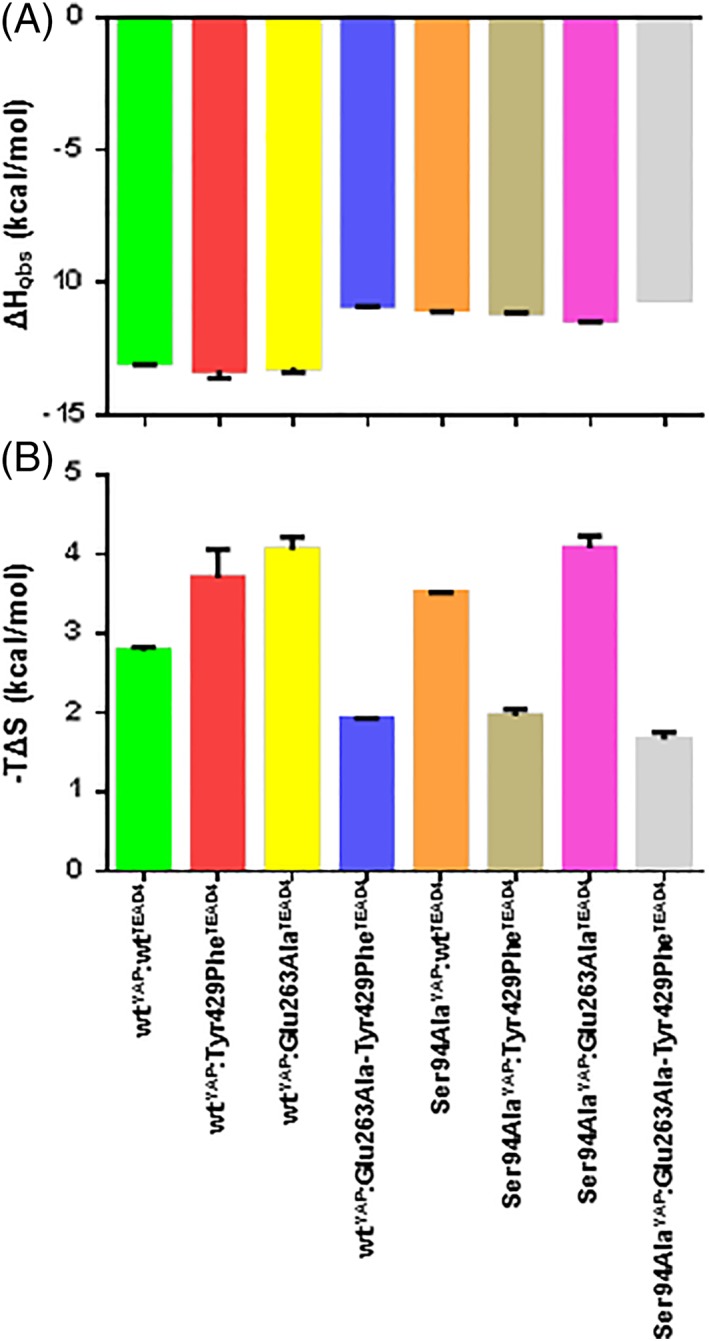
Thermodynamic characterization of the YAP:TEAD complexes. The thermodynamic data were obtained by Isothermal Titration Calorimetry (Table 2 and Fig. S5). A. Measured ΔH obs . (B) Calculated –TΔS. Average values and standard errors are represented.
The two single mutations Glu263AlaTEAD4 and Tyr429PheTEAD4 have a similar impact on ΔH and ΔS (Table 2). The minor change in ΔH (ΔΔH obs = −0.2 kcal/mol) measured with the wtYAP:Glu263AlaTEAD4 complex suggests that the loss of the interaction with Ser94YAP might be minimized by the formation of compensating H‐bonds at the interface via W5 [Fig. 2(D)]. However, the immobilization of W5 may trigger the observed increase in the entropic penalty when compared to the wt complex (Δ‐TΔS = 1.3 kcal/mol). It is difficult to come up with an unequivocal explanation regarding the data obtained for the wtYAP:Tyr429PheTEAD4 complex, because the findings probably result from multiple changes at the interface: loss of H‐bond with Ser94YAP, conformational changes (movement of YAP and His427TEAD4) and changes in solvation (displacement of W3 and binding of W4). The wtYAP:Tyr429PheTEAD4 and wtYAP:Glu263Ala–Tyr429PheTEAD4 complexes have a similar structure [Fig. 2(B) and (D)] and the later complex has a less favorable ΔH (ΔΔH obs = 2.45 kcal/mol) but a more favorable ΔS (Δ‐TΔS = −1.77 kcal/mol). The better ΔH for the wtYAP:Tyr429PheTEAD4 could be explained by the presence of the H‐bonds which are formed by Glu263TEAD4 in this complex. The desolvation of Glu263TEAD4 and/or Ser94YAP upon the formation of the wtYAP:Tyr429PheTEAD4 complex may explain the greater entropic penalty measured with this complex. Changes in conformation and solvation are also observed between the wt and Ser94AlaYAP:wt TEAD4 complexes, making it difficult to interpret the thermodynamic data. In contrast to the two other single mutations, which affect only ΔS, the Ser94AlaYAP mutation significantly alters both ΔH (ΔΔH obs = 2 kcal/mol) and ΔS (Δ‐TΔS = 0.72 kcal/mol) (Table 2). We also mentioned that the wtYAP:Glu263Ala–Tyr429PheTEAD4 and Ser94AlaYAP:wtTEAD4 complexes have a very different affinity, whereas they might be expected to be similar in this respect. The ITC data show that this difference has an entropic origin, since both complexes have a similar ΔH (ΔΔH obs = −0.15 kcal/mol) but a significantly different TΔS (Δ‐TΔS = 1.59 kcal/mol). The Ser94AlaYAP:wtTEAD4 and Ser94AlaYAP:Glu263AlaTEAD4 complexes, which have a similar structure [Fig. 2(E) and (G)], also have much the same thermodynamic properties (Table 2). This also applies to the Ser94AlaYAP:Tyr429PheTEAD4 and Ser94AlaYAP:Glu263Ala–Tyr429PheTEAD4 complexes [Fig. 2(F) and (H) and Table II]. These two groups of complexes have a similar ΔH (ΔH obs ~ −11 kcal/mol), but the Ser94AlaYAP:wtTEAD4 and Ser94AlaYAP:Glu263AlaTEAD4 complexes have a less favorable ΔS, which is reduced by more than 1.5 kcal/mol. A tentative explanation is that the immobilization of W6 and the constraints created on the dihedral angles of Ala94YAP lead to this more unfavorable ΔS. The wtYAP:Glu263Ala–Tyr429PheTEAD4 and Ser94AlaYAP:Glu263Ala–Tyr429PheTEAD4 complexes are structurally related [Fig. 2(D) and (H)] and have similar thermodynamic parameters (Table 2). The wtYAP:Tyr429PheTEAD4 and Ser94AlaYAP:Tyr429PheTEAD4 complexes, which are structurally related [Fig. 2(B) and (F)], have significantly different ΔH and ΔS (Table II). The loss of one H‐bond in the Ser94AlaYAP:Glu263Ala–Tyr429PheTEAD4 complex may explain its less favorable ΔH. The higher ΔS obtained for the wtYAP: Tyr429PheTEAD4 complex might be triggered by the desolvation of both Glu263TEAD4 and Ser94YAP.
Discussion
IDPs/IDRs are flexible entities in their unbound form, and they fold when they bind to their interacting partners. However, the fact that IDPs/IDRs become structured in their bound state does not necessarily mean they are completely rigid and have lost all their flexibility. Indeed, it has been shown that IDPs/IDRs retain some dynamic properties once they are bound to their target (e.g., Refs. [22, 23, 24]). The ability to preserve some flexibility once they are part of a complex may allow IDPs/IDRs to bind to different binding interfaces and/or to minimize the entropic cost associated with folding upon binding. This also suggests that the residual flexibility of bound IDPs/IDRs could allow them to adapt to mutations at binding interfaces to some extent. In this study, we looked at the ability of a bound IDR to adapt to mutations that delete H‐bonds which it engages with its interacting partner. H‐bonds require a precise positioning of the interacting residues, and we were interested to determine how a bound IDR can adapt to the loss of these geometrically constrained interactions. Furthermore, residues that form H‐bonds can also interact with water molecules. This study should, therefore, provide an insight into the contribution of water to the binding of IDRs. We used the interaction between Ser94YAP and Glu263/Tyr429TEAD4, which form two well‐defined H‐bonds at the interface of the YAP:TEAD complex [Fig. 1(A)], as a model system. Ser94YAP is in a region of YAP that folds in an Ω‐loop conformation upon binding to TEAD, and this binding interface is key to the formation of the YAP:TEAD complex interaction.16, 17, 18
Our results show that none of the tested mutations of these three residues induces significant changes in the main chain of the rigid partner, TEAD4. In contrast, some of the mutations affect the position of the main chain of YAP, and we observed its movement in two opposite directions. In the four complexes formed with Phe429TEAD4, YAP moves toward TEAD, but in the two complexes where both Tyr429TEAD4 and Ala94YAP are present, YAP moves away from TEAD. In this case we also noticed a change in the dihedral angles of Ala94YAP when compared to Ser94YAP in the wt complex. Ala94YAP adopts a conformation which is closer to that found in 310 helices than the α‐helix conformation observed for Ser94YAP in the wt complex. This movement shows that an IDR can adapt to different interfaces by assuming different conformations, even if some of them create constrains in its bound structure. These conformational changes do not come in isolation, and water plays an active role in facilitating them. In the different mutant complexes, water molecules occupy the position of the mutated residues or of those that have moved (e.g., His427TEAD4). Water is, therefore, instrumental in maintaining the hydrogen bond network between both proteins and minimizing any binding energy penalties resulting from desolvation. These findings highlight the importance of including the contribution of water when studying the interaction between IDPs/IDRs and their targets. As IDPs/IDRs are largely solvated in their unbound conformation, water might also be crucial at this stage, playing an important role in establishing/maintaining the dynamics between the different forms they can adopt in solution. The “teamwork” between YAP/TEAD and the water molecules we describe in this study is essential for finding new energy minima to compensate for the loss of these key interactions at the interface.
The conformational and solvation changes induced by the mutations lead to complex thermodynamic signatures. The measured effects on ΔH and ΔS are, therefore, composite values reflecting the different readjustments taking place at the binding interface. We have formulated different hypotheses to interpret these data, but the use of computational methods may allow greater insight to be gained into the individual energetic contributions that lead to these thermodynamic profiles.
Overall, this study reveals the adaptability of bound IDR to the loss of geometrically constrained bonds at a binding interface. Our work was conducted using the YAP:TEAD interface as a model, and it is, of course, difficult to extrapolate our findings to other IDPs/IDRs. Nevertheless, it is reasonable to speculate that the residual flexibility of bound IDRs/IDPs may help them adapt not only to different binding interfaces but also to mutations at these interfaces. This could be an evolutional advantage to maintain existing or create new interactions in cells. The concepts of “frustrated interfaces” and of “fuzzy complexes” have been used to describe complexes involving IDPs/IDRs25, 26; our findings continue along these lines and exemplify the exquisite structural malleability of these proteins or protein fragments.
Material and methods
Proteins
Wild‐type N‐Avi‐tagged human TEAD4217–434 (referred to hereinafter as wtTEAD4) and wild‐type human YAP51–171 (referred to hereinafter as wtYAP) were cloned as previously described.18, 19 Mutations were introduced in TEAD and YAP with the QuikChange II Lightning site‐directed mutagenesis kit (Agilent Technologies, Berlin, Germany) according to the manufacturer's instructions. The DNA sequence of the constructs was confirmed by Sanger sequencing. The YAP and TEAD4 proteins were expressed, purified and analyzed as already described (19 and Scheme C27). All the TEAD4 proteins are acylated (see for more details, Ref. [27). Representative LC–MS analyses of the proteins are shown in Figure S3.
SPR
The experimental conditions used in SPR have been previously described.19 The dissociation constants, K d, measured by SPR were obtained from equilibrium data. Representative sensorgrams are presented in Figure S4.
ITC
The proteins were dialyzed overnight at 277°K in the HEPES ITC assay solution: 50 mM HEPES, 100 mM KCl, 0.25 mM TCEP, 1 mM EDTA, 2% DMSO, pH 7.4. The dialyzed proteins were centrifuged at 14,000 rpm for 30 min at 277°K and their concentration determined by HPLC as previously described.27 ITC measurements were made in a MicroCal PEAQ‐ITC (Malvern, UK). The calorimeter was checked regularly over the entire course of the experiments using the EDTA‐CaCl2 test kit provided by the manufacturer (Malvern Instruments, Malvern, UK). Before each experiment, the solutions were filtered and degassed. In all the experiments, YAP (~150 μM) was in the cell and TEAD4 (~15 μM) in the syringe. For the experiments carried out in different buffers, TRIS and PIPES were chosen because their pK a is within 1 pH unit of the HEPES ITC assay solution pH and because the ionization enthalpy of these buffers differs from that of HEPES (see legend of Fig. S2). The assay solutions containing TRIS and PIPES were tested using a conductivity meter (Mettler Toledo, Columbus, OH) and their [KCl] was adjusted such that their conductivity was equal to that of the HEPES ITC assay solution (13.1 ±0.2 mS/cm at 298°K; final [KCl] for the TRIS and PIPES buffer, 80 and 65 mM, respectively). Aliquots of 2 μL (first injection 0.5 μL) were injected into the cell for 4 s at 150 s intervals with constant stirring (750 rpm). A titration was completed after 19 injections. The heat of dilution was measured by injecting TEAD4 into the cell containing only the assay solution. These values were subtracted from the heat of reaction generated during the titration of YAP. The baseline of the thermograms was eventually adjusted manually and the corrected heat of reaction values fitted using the MicroCal PEAQ‐ITC Analysing Software (Malvern Instruments, Malvern, UK) using a single site binding model. The c‐values (c = [YAP]n/K d, where [YAP] is the concentration of YAP in the cell, n is the stoichiometry, and K d the dissociation constant28) were calculated for each YAP:TEAD pair (experiments done in HEPES): 456 ≤ c ≤ 474 for wtYAP:wtTEAD4; 134 ≤ c ≤ 190 for wtYAP:Tyr429PheTEAD4; 71 ≤ c ≤ 73 for wtYAP:Glu263AlaTEAD4; 16 ≤ c ≤ 58 for wtYAP:Glu263Ala–Tyr429PheTEAD4; 4 ≤ c ≤ 6 for Ser94Ala YAP:wtTEAD4; 62 ≤ c ≤ 82 for Ser94Ala YAP:Tyr429PheTEAD4; 4 ≤ c ≤ 5 for Ser94Ala YAP:Glu263AlaTEAD4; 40 ≤ c ≤ 48 for Ser94Ala YAP:Glu263Ala–Tyr429PheTEAD4. Representative thermograms are presented in Figure S5.
To determine the influence of protonation/deprotonation processes on the measurement of binding enthalpy (ΔH obs ),29 plots of ΔH obs versus ΔH ioni (ionization enthalpy of the buffer) were constructed (Fig. S2). This analysis reveals that no significant exchange of protons takes place with the buffer during the formation of the wtYAP:wtTEAD4 complex (nH+ = −0.11 ±0.06). The enthalpy in the absence of a buffer effect, ΔH nbe (−13.0 ±0.5 kcal/mol), is similar to ΔH obs measured in HEPES buffer (−13.0 kcal/mol, Table 2). The same experiments were conducted with each complex bearing a single mutation: wtYAP:Tyr429PheTEAD4 (nH+ = 0.01 ±0.05 and ΔH nbe = −13.2 ±0.4 kcal/mol), wtYAP:Glu263AlaTEAD4 (nH+ = −0.02 ±0.04 and ΔH nbe = −13.4 ±0.4 kcal/mol) and Ser94YAP:wtTEAD4 (nH+ = −0.10 ±0.03 and ΔH nbe = −10.7 ±0.2 kcal/mol). These additional data confirm the absence of proton exchange with buffer upon complex formation when YAP and TEAD4 are mutated at each of the mutation sites. On the basis of these results, the measured ΔH obs values were used without any correction.
X‐ray structural studies
The TEAD4217–434 proteins used for crystallization were obtained as previously described.27 Synthetic peptides mimicking the region 60–100 of hYAP, wtYAP, or Ser94AlaYAP were used to obtain co‐crystals with the TEAD4 proteins. All the crystallization experiments were carried out at 293°K using the sitting drop vapor diffusion method. The drops were made up of 0.2 μL of protein solution, 0.16 μL of reservoir solution and 0.04 μL of seeding solution (using the automated microseed matrix‐seeding method30). The reservoir solutions comprised the following: wtYAP:wtTEAD4 complex (space group P41212): 16% (w/v) PEG3350, 2% (v/v) Tacsimate pH 5.0, 0.1 M sodium citrate pH 5.6; wtYAP:Glu263AlaTEAD4 complex (space group P41212): 20% (w/v) PEG3350, 0.2 M Mg nitrate hexahydrate; wtYAP:Tyr429PheTEAD4 complex (space group P41212): 20% (w/v) PEG3350, 8% (v/v) Tacsimate pH 4.0; wtYAP:Tyr429PheTEAD4 complex (space group P212121): 25% (w/v) PEG3350, 0.2 M MgCl2, 0.1 M BIS–TRIS pH 5.5; wtYAP:Glu263–Tyr429PheTEAD4 complex (space group P41212): 2 M ammonium sulfate, 0.1 M BIS–TRIS pH 5.5; Ser94AlaYAP:wtTEAD4 complex (space group P41212): 20% (w/v) PEG3350, 0.2 M Na tartrate dibasic dehydrate; Ser94AlaYAP:Glu263AlaTEAD4 complex (space group P41212): 17% (w/v) PEG10000, 0.1 M ammonium acetate, 0.1 M BIS–TRIS pH 5.5; Ser94AlaYAP:Tyr429PheTEAD4 complex (space group P41212): 20% (w/v) PEG3350, 0.2 M Na Tartrate dibasic dehydrate; and Ser94AlaYAP:Glu263Ala–Tyr429PheTEAD4 complex (space group P41212): 20% (w/v) PEG3350, 0.2 M sodium acetate trihydrate pH 7.0.
All crystals were cryo‐protected in reservoir solution supplemented with 20% ethylene glycol and flash‐frozen in liquid nitrogen. X‐ray data sets were collected at the Swiss Light Source Facility (SLS, Villigen, Switzerland) on beamline X10SA. The data were processed with XDS.31 The structures were determined by molecular replacement (PHASER32) using various previous in‐house TEAD4 X‐ray structures as search models. Programs REFMAC33 and COOT34 were used for refinement and model (re)building. The final refined structures have R (R free) values of 0.218–0.243 (0.261–0.350) and showed excellent geometry in the Ramachandran plots. The details of data collection and structure refinement are given in Table S1. The crystallographic data have been deposited at the RSCB Protein Data Bank (PDB, http://www.pdb.org) with the access codes: 6GE3 (wtYAP:wtTEAD4), 6GE4 (wtYAP:Glu263AlaTEAD4), 6GE5 (wtYAP:Tyr429PheTEAD4, space group P41212), 6GEK (wtYAP:Tyr429PheTEAD4, space group P212121), 6GE6 (wtYAP:Glu263–Tyr429PheTEAD4), 6GEC (Ser94AlaYAP:wtTEAD4), 6GEE (Ser94AlaYAP:Glu263AlaTEAD4), 6GEG (Ser94AlaYAP:Tyr429PheTEAD4), 6GEI (Ser94AlaYAP:Glu263Ala–Tyr429PheTEAD4).
Supplementary Material
File name: Supplementary Material. Table S1: X‐ray data collection and refinement statistics. Table S2: Φ and Ψ dihedral angles of the YAP Residues 93, 94, and 95 in the different YAP:TEAD complexes. Figure S1: Structures of the wtYAP:Tyr429PheTEAD4 complex solved from different crystal forms. Figure S2: Effect of proton exchange with buffer on ΔH obs . Figure S3: LC–MS analyses of the YAP and TEAD4 proteins. Figure S4: Surface Plasmon Resonance. Figure S5: Isothermal Titration Calorimetry.
Conflict of interest
The authors declare no conflict of interest.
Placeholder Text
Supporting information
Supporting Information
Statement: YAP is an intrinsically disordered protein which becomes structured upon binding to TEAD. YAP and TEAD form two key hydrogen bonds at their binding interface. The study of the abrogation of these interactions by mutations reveals that bound YAP keeps some flexibility, which together with the contribution of interfacial water molecules, permits to mitigate the loss of these hydrogen bonds.
Supplementary Material. File name: Supplementary Material; Description: additional information/experimental results (tables and figures with legend).
References
- 1. Zhou HX (2010) From induced fit to conformational selection: a continuum of binding mechanism controlled by the timescale of conformational transitions. Biophys J 98:L15–L17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vogt AD, Di Cera E (2012) Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 51:5894–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gianni S, Dogan J, Jemth P (2014) Distinguishing induced fit from conformational selection. Biophys Chem 189:33–39. [DOI] [PubMed] [Google Scholar]
- 4. Chakraborty P, Di Cera E (2017) Induced fit is a special case of conformational selection. Biochemistry 56:2853–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greives N, Zhou HX (2014) Both protein dynamics and ligand concentration can shift the binding mechanism between conformational selection and induced fit. Proc Natl Acad Sci USA 111:10197–10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kiefhaber T, Bachmann A, Jensen KS (2012) Dynamics and mechanisms of coupled protein folding and binding reactions. Curr Opin Struct Biol 22:21–29. [DOI] [PubMed] [Google Scholar]
- 7. Dogan J, Gianni S, Jemth P (2014) The binding mechanisms of intrinsically disordered proteins. Phys Chem Chem Phys 16:6323–6331. [DOI] [PubMed] [Google Scholar]
- 8. Berlow RB, Dyson HJ, Wright PE (2015) Functional advantages of dynamic protein disorder. FEBS Lett 589:2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shammas SL, Crabtree MD, Dahal L, Wicky BI, Clarke J (2016) Insights into coupled folding and binding mechanisms from kinetic studies. J Biol Chem 291:6689–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matouschek A, Kellis JT Jr, Serrano L, Fersht AR (1989) Mapping the transition state and pathway of protein folding by protein engineering. Nature 340:122–126. [DOI] [PubMed] [Google Scholar]
- 11. Bachmann A, Wildemann D, Praetorius F, Fischer G, Kiefhaber T (2011) Mapping backbone and side‐chain interactions in the transition state of a coupled protein folding and binding reaction. Proc Natl Acad Sci USA 108:3952–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karlsson OA, Chi CN, Engstrom A, Jemth P (2012) The transition state of coupled folding and binding for a flexible beta‐finger. J Mol Biol 417:253–261. [DOI] [PubMed] [Google Scholar]
- 13. Giri R, Morrone A, Toto A, Brunori M, Gianni S (2013) Structure of the transition state for the binding of c‐Myb and KIX highlights an unexpected order for a disordered system. Proc Natl Acad Sci USA 110:14942–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rogers JM, Oleinikovas V, Shammas SL, Wong CT, De Sancho D, Baker CM, Clarke J (2014) Interplay between partner and ligand facilitates the folding and binding of an intrinsically disordered protein. Proc Natl Acad Sci USA 111:15420–15425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tian W, Yu J, Tomchick DR, Pan D, Luo X (2010) Structural and functional analysis of the YAP‐binding domain of human TEAD2. Proc Natl Acad Sci USA 107:7293–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen L, Chan SW, Zhang XQ, Walsh M, Lim CJ, Hong W, Song H (2010) Structural basis of YAP recognition by TEAD4 in the Hippo pathway. Genes Dev 24:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan KL, Xu Y (2010) Structural insights into the YAP and TEAD complex. Genes Dev 24:235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hau JC, Erdmann D, Mesrouze Y, Furet P, Fontana P, Zimmermann C, Schmelzle T, Hofmann F, Chène P (2013) The TEAD4‐YAP/TAZ protein‐protein interaction: expected similarities and unexpected differences. ChemBioChem 14:1218–1225. [DOI] [PubMed] [Google Scholar]
- 19. Mesrouze Y, Bokhovchuk F, Meyerhofer M, Fontana P, Zimmermann C, Martin T, Delaunay C, Erdmann D, Schmelzle T, Chene P (2017) Dissection of the interaction between the intrinsically disordered YAP protein and the transcription factor TEAD. Elife 6:e25068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Z, Lin Z, Zhou Z, Shen HC, Yan SF, Mayweg AV, Xu Z, Qin N, Wong JC, Zhang Z, Rong Y, Fry DC, Hu T (2014) Structure‐based design and synthesis of potent cyclic peptides inhibiting the YAP‐TEAD protein–protein interaction. ACS Med Chem Lett 5:993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klebe G (2015) Applying thermodynamic profiling in lead finding and optimization. Nat Rev Drug Discov 14:95–110. [DOI] [PubMed] [Google Scholar]
- 22. Brzovic PS, Heikaus CC, Kisselev L, Vernon R, Herbig E, Pacheco D, Warfield L, Littlefield P, Baker D, Klevit RE, Hahn S (2011) The acidic transcription activator Gcn4 binds the mediator subunit Gal11/Med15 using a simple protein interface forming a fuzzy complex. Mol Cell 44:942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, Kragelund BB, Best RB, Schuler B (2018) Extreme disorder in an ultrahigh‐affinity protein complex. Nature 555:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delaforge E, Kragelj J, Tengo L, Palencia A, Milles S, Bouvignies G, Salvi N, Blackledge M, Jensen MR (2018) Deciphering the dynamic interaction profile of an intrinsically disordered protein by NMR exchange spectroscopy. J Am Chem Soc 140:1148–1158. [DOI] [PubMed] [Google Scholar]
- 25. Tompa P, Fuxreiter M (2007) Fuzzy complexes: polymorphism and structural disorder in protein‐protein interactions. Trends Biochem Sci 33:2–8. [DOI] [PubMed] [Google Scholar]
- 26. Jemth P, Mu X, Engstrom A, Dogan J (2014) A frustrated binding interface for intrinsically disordered proteins. J Biol Chem 289:5528–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mesrouze Y, Meyerhofer M, Bokhovchuk F, Fontana P, Zimmermann C, Martin T, Delaunay C, Izaac A, Kallen J, Schmelzle T, Erdmann D, Chène P (2017) Effect of the acylation of TEAD4 on its interaction with co‐activators YAP and TAZ. Protein Sci 26:2399–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Turnbull WB, Daranas AH (2003) On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc 125:14859–14866. [DOI] [PubMed] [Google Scholar]
- 29. Baker BM, Murphy KP (1996) Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry. Biophys J 71:2049–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. D'Arcy A, Villard F, Marsh M (2007) An automated microseed matrix‐screening method for protein crystallization. Acta Cryst D63:550–554. [DOI] [PubMed] [Google Scholar]
- 31. Kabsch W (2010) XDS. Acta Cryst D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Project CC (1994) The CCP4 suite: programs for protein crystallography. Acta Cryst D50:760–763. [DOI] [PubMed] [Google Scholar]
- 33. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 34. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information