

Abstract
The aim of this study was to assess and compare the pharmacokinetics (PK) and safety of Enasidenib in healthy adult male Japanese subjects to healthy adult male Caucasian subjects. This was a phase 1, single dose study to evaluate the PK and safety of Enasidenib in healthy adult male Japanese subjects relative to healthy adult male Caucasian subjects. A total of 62 subjects (31 Japanese and 31 Caucasian) were enrolled into three dose cohorts (single doses of 50 mg, 100 mg, or 300 mg Enasidenib). Blood samples for PK assessment were collected up to 672 hours postdose. Safety was evaluated throughout the study. In the present study, we found that PK exposures of Enasidenib and its metabolite AGI‐16903 for Caucasian and Japanese subjects were comparable at the 50, 100, and 300 mg dose levels, demonstrated by that the 90% confidence intervals (CIs) of geometric mean ratios for AUCs and C max between these two populations generally contained 100% from all three treatment cohorts. In conclusion, PK exposures of Enasidenib and its metabolite AGI‐16903 for Caucasians and Japanese subjects were comparable and Enasidenib was safe and well tolerated with no apparent differences between Japanese and Caucasian subjects when administered as single oral doses of 50 mg, 100 mg, and 300 mg.
Keywords: Caucasian, Enasidenib, IDHIFA, Japanese, pharmacokinetics, race
Abbreviation
- AEs
adverse events
- AITL
angioimmunoblastic T‐cell lymphoma
- BSA
body surface area
- CI
confidence intervals
- ECG
electrocardiograms
- ET
early termination
- FDA
food and drug Administration
- GCP
good clinical practice
- HVs
healthy volunteers
- ICF
informed consent form
- IDH2
isocitrate dehydrogenase‐2
- LS
least squares
- MRP
multiple reactive monitoring
- TEAEs
treatment emergent AEs
1. INTRODUCTION
Enasidenib (IDHIFA®, AG‐221) (Figure 1) is a first‐in‐class, selective, potent inhibitor of the isocitrate dehydrogenase‐2 (IDH2) mutant protein, making it a targeted therapeutic drug for the treatment of cancer patients with IDH2 mutation, including those with AML.1, 2, 3, 4, 5 Direct inhibition of the gain‐of‐function activity of the IDH2 mutated protein is intended to inhibit the production of the oncogenic metabolite 2 hydroxyglutarate (2‐HG).6, 7, 8, 9, 10, 11, 12, 13 Enasidenib has been demonstrated to reduce 2‐HG levels by >90% and reverse histone and DNA hypermethylation in vitro, and to induce differentiation in IDH2 mutant positive leukemia cell models.5, 14 Enasidenib is being investigated for the treatment of patients with advanced hematologic malignancies with IDH2 mutation.2, 3, 5 Other indications have been studied, including solid tumors (including glioma) and angioimmunoblastic T‐cell lymphoma (AITL) with IDH2 mutation.15, 16 Enasidenib was approved in the United States (US) for the treatment of adult patients with relapsed or refractory (R/R) AML with an IDH2 mutation (as detected by a Food and Drug Administration [FDA] approved test) at a dose of 100 mg daily until disease progression or unacceptable toxicity.17 Patients treated with Enasidenib may experience symptoms of differentiation syndrome and the most common adverse reactions (≥20%) included nausea, vomiting, diarrhea, elevated bilirubin, and decreased appetite.17, 18, 19
Figure 1.
Structures of enasidenib and its metabolite AGI‐16903
Enasidenib PK has been well characterized both in healthy subjects and in subjects with relapsed and refractory AML.14, 17, 20 The absolute bioavailability of Enasidenib after 100 mg oral dose was approximately 57%. After a single oral dose, the median time to C max (t max) was 4 hours. Food intake had no clinically relevant impact on Enasidenib plasma exposures. Human plasma protein binding of Enasidenib was 98.5% and the mean volume of distribution (V d) of Enasidenib after IV administration was 55.8 L indicating an extensive extravascular distribution. Enasidenib had a terminal half‐life (t 1/2) of approximately 25 hours and a plasma clearance (CL/F) of approximately 2.5 L/hour in healthy subjects, while a much longer t 1/2 of approximately 137 hours and a smaller CL/F of 0.74 L/hour in subjects with relapsed and refractory AML. Enasidenib was extensively metabolized prior to excretion and in vitro studies suggested that metabolism of Enasidenib was mediated by multiple CYP enzymes (eg, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4), and multiple UGTs (eg, UGT1A1, UGT1A3, UGT1A4, UGT1A9, UGT2B7, and UGT2B15). The [14C] ADME study in healthy volunteers showed that AGI‐16903 (Figure 1), the N‐dealkylation product, was the most abundant metabolite of Enasidenib but was <10% of Enasidenib exposure at steady state after multiple doses in AML patients. Enasidenib accounted for 89% of the radioactivity in circulation and AGI‐16903 represented 10% of the circulating radioactivity. Eighty‐nine percent of Enasidenib was eliminated in feces and 11% in the urine. Excretion of unchanged Enasidenib accounts for 34% of the radiolabeled drug in the feces and 0.4% in the urine.
A population pharmacokinetic analysis pooling data from 428 healthy subjects and subjects with advanced hematologic malignancies that harbor an IDH2 mutation has been conducted to assess the influence of various intrinsic and extrinsic covariates on Enasidenib PK (data on file). Although it was concluded that all tested covariates of interest such as age, body weight, body surface area (BSA), sex, hepatic function markers, renal function marker, IDH2 mutation type, tumor type, bone marrow blasts burden (%), formulation, and more specifically, race and ethnicity didn't contribute to the interindividual variations observed in Enasidenib PK, no definite conclusion can be made specifically on the covariate of race considering the dataset is not balanced on the covariate of race (70% Caucasians vs 12% Asians). Therefore, a dedicated Japanese‐Caucasian PK study is still necessary. In addition, the Pharmaceuticals and Medical Devices Agency (PMDA) of Japan requires that at least one single‐dose phase 1 study to be conducted to confirm the safety and PK of an investigational product in Japanese healthy volunteers (HVs); results should be compared with those obtained in similar studies with non‐Japanese individuals; and the possible ethnic differences should be assessed prior to Japanese people joining a global clinical trial.21, 22
Therefore, conducting a phase 1 study to evaluate the PK and safety of Enasidenib in healthy male Japanese subjects relative to healthy male Caucasian subjects was necessary to support the inclusion of Japanese sites in the Enasidenib global phase 3 clinical trial. The primary objective of the present study was to assess and compare the PK of Enasidenib in healthy adult male Japanese subjects to healthy adult male Caucasian subjects after single, oral doses of 50 mg, 100 mg, or 300 mg Enasidenib and the second objectives of the present study were to assess the safety of Enasidenib in Caucasian and Japanese healthy adult male subjects and to evaluate the PK of its metabolite AGI‐16903 in a Japanese population. A total of 62 healthy adult male subjects from 20 to 50 years of age, inclusive, were enrolled (31 Japanese and 31Caucasian). Blood samples for PK assessment were collected up to 672 hours postdose and safety was evaluated throughout the study.
2. MATERIALS AND METHODS
This study was conducted and monitored in accordance with Celgene procedures and the study protocol. These procedures complied with the ethical principles of the International Conference on Harmonization (ICH) harmonized tripartite guideline E6 (R1): Good Clinical Practice (GCP), as required by the major regulatory authorities. The conduct also complied with the Declaration of Helsinki, Title 21 of the US Code of Federal Regulations, Parts 50 and 56 concerning informed consent and IRB regulations and applicable national, state, and local laws or regulations. This study was conducted by California Clinical Trials (Glendale, California, USA) and was approved by the Aspire independent review board (Santee, California, US). The investigator obtained written informed consent from the subject prior to any study‐related procedures.
2.1. Study design
This was a phase 1, single dose study to evaluate the PK and safety of Enasidenib in healthy adult male Japanese subjects (defined as must have been born in Japan to both a Japanese mother and father and also have maternal and paternal Japanese grandparents) relative to healthy adult male Caucasian subjects (defined as being of European or Latin American descent, ie, White). A total of 62 subjects (31 Japanese subjects and 31 Caucasian subjects) were enrolled into three different dose cohorts. Subjects entered the study center prior to the evening meal on the day before dosing (Day ‐1). On the morning of Day 1, subjects enrolled in each of the following dose cohorts received an oral dose of Enasidenib under fasted condition:
50 mg Enasidenib (1 × 50‐mg tablet)
100 mg Enasidenib (1 × 100‐mg tablet)
300 mg Enasidenib (3 × 100‐mg tablets)
Food and beverages were withheld from subjects for at least 10 hours prior to dosing until at least 2 hours post dosing. All doses were administered with approximately 240 mL of noncarbonated, room temperature water. During fasting periods, water was allowed ad libitum except from between 1 hour prior to dosing and 2 hours post dosing. After the dose was administered, subjects were observed for at least 7 days to assess the acute safety profile before the next protocol specified dose cohorts enrolled.
Subjects were confined to the study center from Day ‐1 to Day 3 and discharged from the study center upon completion of the 48‐hour PK blood draw. Subjects returned to the study center for additional PK blood draws on Days 5, 8, 11, 15, 22, and 29. Should a subject discontinued from the study, an early termination visit was performed.
Safety parameters including adverse events (AEs), physical examinations (PEs), vital signs, 12‐lead electrocardiograms (ECGs), clinical laboratory safety tests, and concomitant medications/procedures, were reviewed prior to initiating the next dose cohort.
2.2. Blood collection for pharmacokinetic analysis
Blood samples (approximately 5 mL each) for measurement of Enasidenib and its metabolite AGI‐16903 concentrations were collected at the following time points: prior to the dose (0 hour) and 1, 2, 3, 4, 6, 9, 12, 18, 24, 48, 96, 168, 240, 336, 504, and 672 hours postdose. PK blood draws were performed at the nominal time(s) specified in this clinical protocol. All actual PK blood sample collection times were recorded in the source documents and case report form (CRF). An explanation was provided in the source documents and CRF for missed or mishandled samples and for samples collected outside the following time windows: Within 60 minutes prior to dosing for predose PK samples; ±5 minutes for 0.5 to ≤3 hours postdose PK samples; ±10 minutes for >3 hours to <24 hours postdose PK samples; ±60 minutes for 24‐48 hours post dose PK samples; and ±6 hours for ≥48 hours PK samples.
2.3. Safety assessment
Safety was evaluated throughout the study by monitoring of AEs, ECGs, PEs, clinical laboratory tests, and pregnancy tests for female subjects, vital signs, and recording of concomitant medications and procedures. All AEs were monitored and recorded from the time the informed consent form (ICF) was signed until study completion, and when made known to the Investigator within 28 days after the last dose of Enasidenib (and those serious AEs [SAEs] made known to the Investigator at any time thereafter that are suspected of being related to Enasidenib).
2.4. Bioanalytical methodology
Enasidenib and its metabolite AGI‐16903 were extracted from 100 μL human plasma by protein precipitation extraction using 100 μL of internal standards (stable isotope labeled Enasidenib and AGI‐16903) solution in methanol then 300 μL methanol. Samples were centrifuged and a 300 μL aliquot of supernatant was transferred, evaporated to dryness and reconstituted with 300 μL of acetonitrile: water: formic acid (50:50:0.1, v: v: v). Validated liquid chromatography–tandem mass spectrometry (LC‐MS/MS) assays were used to assess Enasidenib and its metabolite AGI‐16903 concentrations in the plasma samples. Chromatographic separation of Enasidenib and AGI‐16903 was achieved on a Fortis C18 column (2.1 × 50 mm, 5 μm, Fortis F18‐020305). The mobile phases were 0.1% formic acid in water (MPA), 0.1% formic acid in acetonitrile (MPB), and 10 mmol/L ammonium acetate in methanol (MPC) at a total flow rate of 0.3 mL/min. The ternary gradient for analytes separation started with 20% MPB and 40% MPC for 0.5 min, then MPB was increased to 60% over 2.5 min. At 3.01 min, MPB was reduced to 0% while MPC was adjusted to 90%. MPC then increased to 95% over 0.5 min. At 4.01 min, original condition was restored (20% MPB and 40% MPC) and equilibrated for 1 min (total run time was 5 min). MRM (multiple reactive monitoring) transitions in ESI positive mode for both Enasidenib and its metabolite AGI‐16903 were optimized and detected on the AB Sciex 4000 mass spectrometer coupled with CTC autosampler system. The lower limit of quantification was 1.0 ng/mL for both Enasidenib and its metabolite AGI‐16903.
2.5. Pharmacokinetic analyses
Noncompartmental PK parameters, such as C max, t max, AUC0‐t, AUC0‐inf, t 1/2, CL/F, Vz/F, and AUC%extrap (percentage of AUC0‐inf due to extrapolation from the time for the last quantifiable concentration to infinity) were calculated from the plasma concentration‐time data with Phoenix™ WinNonlin® Professional Version 6.3 (Pharsight®, a Certara™ company, St. Louis, Missouri). Actual sampling times were used in the calculations.
2.6. Statistical analyses
Sixty‐two eligible healthy adult male subjects (31 Japanese subjects and 31 Caucasian subjects) were enrolled. This sample size was chosen as a suitable number based on empirical considerations, literature, and previous experience. No formal sample size calculation was performed.
To compare Enasidenib PK between Japanese subjects with Caucasian subjects, an analysis of variance (ANOVA) was performed on the natural log‐transformed C max, AUC0‐t, and AUC0‐inf for Enasidenib and its metabolite AGI‐16903. SAS procedure PROC MIXED will be used. The ANOVA model included ethnic group, dose, and ethnic group by dose interaction as fixed effects, subject as a random effect, along with ethnic group and subject as the repeated group and subject terms, respectively. The geometric least squares (LS) means, percent ratios of the geometric LS mean between ethnic groups, and 90% CI for the geometric LS mean percent ratios was estimated for each cohort. The parameter t max was analyzed using the Wilcoxon rank sum test. Hodges‐Lehmann estimate and its 90% CI were calculated for the median difference between ethnic groups (Japanese vs Caucasian) within each cohort.
All safety assessments, including AEs, vital sign measurements, clinical laboratory information, concomitant medications, physical exams, and ECG interpretations, were tabulated and summarized as appropriate. Adverse events were recorded and classified using the Medical Dictionary for Drug Regulatory Activities (MedDRA) classification system. Treatment emergent AEs (TEAEs) were summarized by frequency, severity, and relatedness to study drug. The frequency (the number of TEAEs and the number of subjects experiencing a TEAE) of TEAEs were tabulated by system organ class and preferred term.
3. RESULTS
3.1. Demographic and other baseline characteristics
A total of 62 subjects enrolled in the study and two subjects discontinued the study early but completed Enasidenib administration (one subject discontinued due to visa complications and the other subject discontinued due to a family emergency). Every effort was made to ensure that all end of study procedures were performed at the early termination (ET) visit. A summary of demographics and baseline characteristics is presented in Table 1. The mean and range of age were similar between Japanese and Caucasian subjects. Although in all cohorts, mean BMI and body weight were lower in Japanese subjects compared to Caucasian subjects by approximately 2.1‐2.4 kg/m2 and 7.68‐10.33 kg for BMI and body weight, respectively, a previous PK analysis showed that neither BMI nor body weight had a significant impact on Enasidenib plasma exposure. All enrolled subjects satisfied the inclusion and exclusion criteria, with no clinically significant abnormalities prior to dose administration, and the Investigator approved all the subjects for study participation.
Table 1.
Demographic and other baseline characteristics
| 50 mg | 100 mg | 300 mg | Overall | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Japanese | Caucasian | Total | Japanese | Caucasian | Total | Japanese | Caucasian | Total | ||
| N | 10 | 10 | 20 | 11 | 11 | 22 | 10 | 10 | 20 | 62 |
| Mean age (range) in years | 33.0 (27‐47) | 33.3 (22‐46) | 33.2 (22‐47) | 31.2 (21‐46) | 36.1 (24‐50) | 33.6 (21‐50) | 31.8 (23‐45) | 30.7 (20‐49) | 31.3 (20‐49) | 32.7 (20‐50) |
| Mean height (range) in cm | 171.50 (162.0‐184.0) | 175.50 (164.0‐186.0) | 173.50 (162.0‐186.0) | 173.55 (167.0‐181.0) | 176.00 (166.0‐188.0) | 174.77 (166.0‐188.0) | 174.20 (163.0‐180.0) | 176.00 (165.0‐186.0) | 175.10 (163.0‐186.0) | 174.47 (162.0‐188.0) |
| Mean weight (range) in kg | 67.19 (55.6‐83.4) | 77.52 (64.6‐94.2) | 72.36 (55.6‐94.2) | 68.87 (54.2‐82.0) | 78.25 (66.7‐103.4) | 73.56 (54.2‐103.4) | 68.49 (56.8‐82.7) | 76.17 (65.9‐90.2) | 72.33 (56.8‐90.2) | 72.77 (54.2‐103.4) |
| Mean BMI (range) in kg/m2 | 22.79 (19.4‐25.5) | 25.13 (21.3‐29.9) | 23.96 (19.4‐29.9) | 22.83 (19.4‐26.8) | 25.24 (22.1‐29.3) | 24.03 (19.4‐29.3) | 22.52 (19.8‐25.5) | 24.62 (20.8‐29.5) | 23.57 (19.8‐29.5) | 23.86 (19.4‐29.9) |
BMI, body mass index; N, number of subjects in category.
3.2. Enasidenib plasma exposures: Japanese subjects vs caucasian subjects
Mean (±SD) Enasidenib plasma concentration vs time profiles by race (Japanese and Caucasian) and treatment (50 mg, 100 mg and 300 mg) are summarized in Figure 2. Mean Enasidenib plasma concentration time profiles were well characterized over the 672‐hour postdose sampling interval and the AUC% extrap was less than 10% (ranging from 0.03% to 8.16%), suggesting an adequate PK sampling schedule for both Japanese and Caucasian subjects and all treatment cohorts. Figure 2 showed that Enasidenib was readily absorbed and reached the maximum plasma concentration at approximately 3‐13 hours and declined in a monoexponential manner in both Japanese and Caucasian subjects. The Enasidenib concentration‐time profiles were similar between Japanese and Caucasian subjects across all three dose levels.
Figure 2.
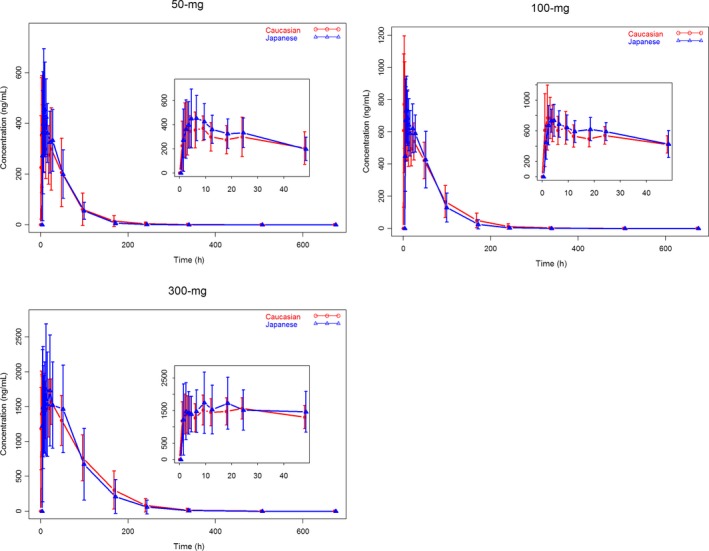
Mean (±SD) Enasidenib Plasma Concentration – Time Profiles by Race and Dose Level (red lines and symbols represent data from Caucasian subjects and blue lines and symbols represent data from Japanese subjects)
The plasma pharmacokinetic parameters of Enasidenib by race and treatment are summarized in Table 2. Metrics of the rate of absorption, as reflected by C max and the extent of absorption, as reflected by AUC0‐inf and AUC0‐t were either higher in Japanese subjects as compared to that in Caucasian subjects from 50 mg and 300 mg treatment cohorts (Table 2), or lower in Japanese subjects as compared to that in Caucasian subjects from 100 mg treatment cohort (Table 2). The lack of a universal trend of Enasidenib plasma exposures between Japanese and Caucasian subjects suggested that there was no race difference in Enasidenib plasma exposures.
Table 2.
Summary of enasidenib plasma pharmacokinetic parameters by race and dose level
| Dose (mg) | Race | AUC0‐t (ng h/mL) | AUC0‐inf (ng h/mL) | C max (ng/mL) | t max a(h) | t 1/2 (h) | CL/F (mL/h) | Vz/F (mL) |
|---|---|---|---|---|---|---|---|---|
| 50 | Japanese (n = 10) | 21800 (46.9) | 21900 (46.7) | 533 (41.0) | 3.98 (0.98, 8.95) | 21.1 (31.6) | 2290 (46.7) | 69700 (40.9) |
| Caucasian (n = 9) | 17800 (55.0) | 18000 (55.0) | 406 (34.9) | 4.00 (2.02, 24.0) | 23.0 (47.4) | 2780 (55.0) | 92300 (44.5) | |
| 100 | Japanese (n = 11) | 40500 (47.2) | 40700 (47.3) | 786 (27.1) | 4.00 (1.97, 24.0) | 19.5 (45.6) | 2460 (47.3) | 69000 (25.1) |
| Caucasian (n = 10) | 49200 (27.5) | 49500 (27.8) | 822 (38.5) | 2.99 (1.03, 9.08) | 25.5 (51.4) | 2020 (27.8) | 74400 (41.2) | |
| 300 | Japanese (n = 10) | 168000 (44.7) | 170000 (44.6) | 2030 (34.3) | 13.4 (1.00, 48.0) | 27.7 (35.6) | 1760 (44.6) | 70300 (15.3) |
| Caucasian (n = 10) | 163000 (44.8) | 163000 (44.9) | 1780 (27.8) | 10.5 (1.97, 24.0) | 28.3 (32.0) | 1840 (44.9) | 74900 (26.9) |
AUC0‐t, AUC from time zero to time t, where t is the last measurable time point; AUC0‐inf,AUC from time zero extrapolated to infinity; CL/F, apparent total plasma clearance; C max, maximum observed plasma concentration; CV%, percent coefficient of variation; t 1/2, estimate of the terminal elimination half‐life; t max, time to C max; V z/F = apparent total volume of distribution.
Geometric mean (Geometric CV%) data are presented.
Median (min, max) data are presented.
Statistical analyses of the plasma exposure parameters were conducted to assess the impact of race on Enasidenib plasma exposures (Table 3). The PK parameters for Enasidenib, including C max, AUC0‐t, AUC0‐inf, and t max, were similar between the Japanese and Caucasians from all three dose levels. Except for the C max at 50 mg dose level, the 90% CI of ratio of geometric LS means for C max, AUC0‐t, and AUC0‐inf from all three treatment cohorts included 100% (Table 3). In addition, the 90% CI of median difference for t max included 0 and all p values from the Wilcoxon rank sum test were larger than 0.05 which further confirmed that there was no race difference in the Enasidenib rate of absorption (Table 3).
Table 3.
Statistical comparison of enasidenib plasma pharmacokinetic parameters between japanese and caucasian subjects: AUC, C max and t max
| Dose (mg) | Pharmacokinetic parameter (unit) | Ratio (%) of geometric LS means (Japanese versus Caucasian) | 90% CI of ratio of geometric LS means |
|---|---|---|---|
| 50 | AUC0‐t (ng h/mL) | 122.0 | (87.9, 169.3) |
| AUC0‐inf (ng h/mL) | 121.5 | (87.6, 168.7) | |
| C max (ng/mL) | 131.1 | (101.6, 169.2) | |
| t max (h) | −0.14a | (−5.02, 1.00)a | |
| 100 | AUC0‐t (ng h/mL) | 82.3 | (60.3, 112.4) |
| AUC0‐inf (ng h/mL) | 82.3 | (60.3, 112.4) | |
| Cmax (ng/mL) | 95.6 | (75.0, 121.8) | |
| t max (h) | 1.00a | (−0.03, 1.98)a | |
| 300 | AUC0‐t (ng h/mL) | 103.2 | (75.0, 142.0) |
| AUC0‐inf (ng h/mL) | 104.4 | (75.9, 143.8) | |
| C max (ng/mL) | 113.5 | (88.6, 145.5) | |
| t max (h) | 0.02a | (−6.87, 15.15)a |
AUC0‐t, AUC from time zero to time t, where t is the last measurable time point; AUC0‐inf, AUC from time zero extrapolated to infinity; CI, confidence interval; C max, maximum observed plasma concentration; LS, least squares; n is the number of observations for each race in each cohort used in the model; t max = time to maximum observed plasma concentration.
The median difference and 90% CI of the median difference were from Hodges‐Lehmann estimates.
Taken together, those results indicate that the Enasidenib plasma exposures were comparable for Japanese and Caucasian subjects at the 50, 100, and 300 mg dose levels.
3.3. AGI‐16903 plasma exposures: Japanese subjects vs caucasian subjects
Mean (±SD) AGI‐16903 plasma concentration vs time profiles by race (Japanese and Caucasian) and treatment (50 mg, 100 mg, and 300 mg) are summarized in Figure 3. Mean AGI‐16903 plasma concentration‐time profiles were well characterized over the 672‐hour postdose sampling interval and the AUC%extrap was less than 10% (ranging from 0.29% to 9.25%), suggesting an adequate PK sampling schedule for both Japanese and Caucasian subjects and all treatment cohorts. AGI‐16903 concentration‐time profiles were similar between Japanese and Caucasian subjects across all three dose levels.
Figure 3.
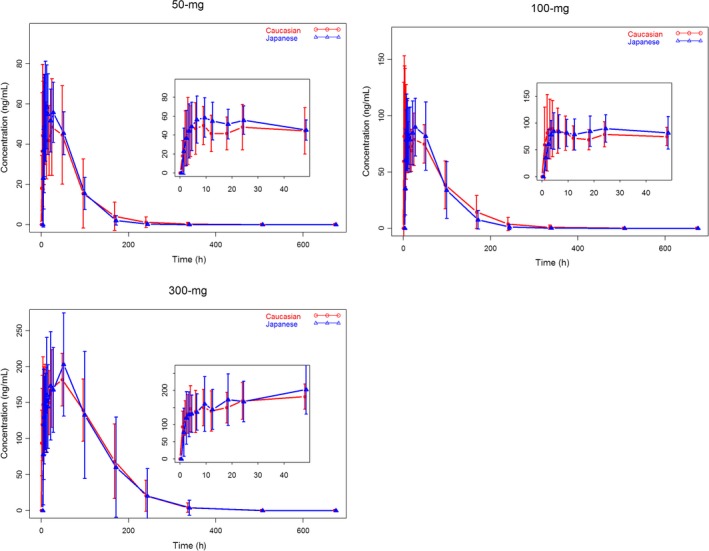
Mean (±SD) AGI‐16903 Plasma Concentration – Time Profiles by Race and Dose Level (red lines and symbols represent data from Caucasian subjects and blue lines and symbols represent data from Japanese subjects)
Consistent with the parent drug of Enasidenib, metrics of the rate of production, as reflected by C max and the extent of production, as reflected by AUC0‐inf and AUC0‐t were either higher in Japanese subjects as compared to that in Caucasian subjects from 50 mg and 300 mg treatment cohorts, or lower in Japanese subjects as compared to that in Caucasian subjects from 100 mg treatment cohort (data not shown). The lack of a universal trend of AGI‐16903 plasma exposure between Japanese and Caucasian subjects suggested that there was no race difference in AGI‐16903 plasma exposures.
Similar statistical analyses of the systemic exposure parameters were conducted to assess the impact of race on AGI‐16903 plasma exposures (data not shown). Consistent with the parent drug of Enasidenib, the PK parameters of AGI‐16903, including C max, AUC0‐t, AUC0‐inf, and t max, were similar between the Japanese and Caucasians from all three dose levels, demonstrated by that the 90% CI of ratio of geometric LS means for C max, AUC0‐t, and AUC0‐inf included 100% from all three dose levels. In addition, the 90% CI of median difference for t max included zero. These comparisons indicate that the PK exposure of AGI‐16903 for Japanese and Caucasian subjects were comparable for the 50, 100, and 300 mg dose levels.
Figure 4 showed the AGI‐16903 to Enasidenib plasma concentration ratio (M/P ratio) vs time profiles by race and treatment cohort. The M/P ratios increased with time from all three treatment cohorts, and Japanese and Caucasian subjects showed similar M/P ratio time course pattern, that is, the median M/P ratios were 12.5% and 13.2% from 50‐mg cohort, 12.5% and 12.2% from 100‐mg cohort, and 9.5% and 9.9% from 300‐mg cohort, for Caucasian and Japanese subjects, respectively, indicating similar in vivo AGI‐16903 production pattern in both populations.
Figure 4.
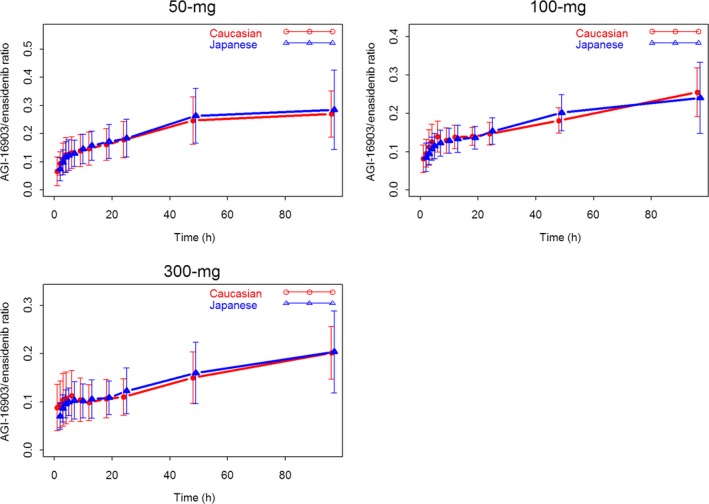
Mean (±SD) AGI‐16903 to Enasidenib Plasma Concentration Ratio – Time Profiles by Race and Dose Level (red lines and symbols represent data from Caucasian subjects and blue lines and symbols represent data from Japanese subjects)
3.4. Safety and tolerability
All 62 enrolled subjects were included in the safety population. Enasidenib was well tolerated following single oral doses of 50, 100, and 300 mg. The incidence of TEAEs was similar between Japanese and Caucasian subjects across the three dose levels. The incidence of total TEAEs (both races) was higher following the 100‐mg dose of Enasidenib (18.2%) compared to 50 mg Enasidenib (5.0%). Following the 300‐mg dose of Enasidenib, the incidence of TEAEs increased to 80.0%. Headache was the most common TEAE, occurring in 5.0%, 9.1%, and 70.0% of subjects following Enasidenib doses of 50 mg, 100 mg, and 300 mg, respectively. Overall, 27.4% of subjects experienced a TEAE of headache. The next most common TEAE (nausea) had an overall incidence of 4.8% and was only reported by Caucasian subjects. The majority of TEAEs were considered by the Investigator to be related to Enasidenib. Overall, 21 subjects (33.9%) reported 32 TEAEs. The majority of TEAEs were mild in severity. There were no deaths, SAEs, or severe AEs in the study. No subjects withdrew from the study due to a TEAE.
Sporadic out‐of‐range values occurred in multiple subjects on various clinical laboratory measurements. There were no clinically significant laboratory abnormalities as per the Investigator, and no clinical laboratory values were reported as a TEAE. Mean values for each parameter were above the reference range, between approximately 1.1 and 1.3 × upper limit of normal. The Day 3 mean total, direct, and indirect bilirubin values for overall subjects were between 1.4‐ and 1.8‐fold higher than baseline values and the increase was greater in Japanese subjects (approximately, 1.7‐ to 2.0‐fold baseline). Mean values for total, direct, and indirect bilirubin had returned to baseline on Day 8. Similar increases were not seen for other markers of liver function including alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase. Bilirubin values had returned to baseline on Day 8. There were no other notable changes in mean laboratory values over time or appreciable trends overall or within subjects observed during this study.
4. DISCUSSION
Race and/or ethnicity have long been identified and known as important determinants of drug metabolism and response and therefore contribute to inter‐individual variability in drug pharmacokinetics and pharmacodynamics.23, 24 Ethnic differences in pharmacokinetics can be divided into the traditional pharmacokinetic categories of bioavailability, distribution, metabolism, and elimination. First, there are many examples of racial differences in absorption with drugs undergoing active absorption.25, 26 Second, racial differences in drug distribution have been demonstrated in the published literature.27, 28 Plasma protein binding is an important determinant of drug distribution, the determinants of which are protein concentration, the number of binding sites per unit of protein, and binding affinity.24 It has been found that the plasma free fractions of propranolol, disopyramide, and diphenhydramine were approximately 17%, 26% and 44% higher (all P < 0.05), respectively, in Chinese than in Caucasians and plasma α1‐acid glycoprotein concentrations were significantly lower in Chinese than in Caucasians resulting in different drug volume of distributions.27, 28 Last and more important, racial differences in hepatic drug metabolism are fairly common and account for the majority of literature on racial differences in PK.24 Zhou et al. studied oral propranolol pharmacokinetics at steady state in Chinese and Caucasians and found the oral clearance of propranolol to be more than 100% higher in Chinese than Caucasians.29 A study showed that the area under the plasma concentration time curve (AUC) following a single 20 mg oral dose of nifedipine was 148% higher in South Asians than in Caucasians.30
Genetic variations may provide a molecular basis for racial and ethnic differences in drug metabolizing enzymes (CYP 2C9, 2C19, 2D6, and 3A4), drug transporter (P‐glycoprotein), and drug receptors (adrenoceptors).23 These drug metabolizing CYP enzymes contribute more than 50% of total hepatic P‐450 content and the genes encoding these enzymes are moderately/highly polymorphic in different ethnic groups. In vitro studies showed that metabolism of Enasidenib is mediated by multiple CYP enzymes (eg, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4), therefore, the influence of race on Enasidenib PK disposition due to different CYPs polymorphic frequencies or different phenotype distribution of CYPs in different ethnic groups cannot be ignored.
An understanding of how an investigational agent behaves in a variety of racial and ethnic groups is a key component of a global development program.31 The concern of the effects of race or ethnicity on PK and pharmacodynamics is especially important as the U.S. population becomes increasingly non‐Caucasian and as marketing of drugs by American and European companies extends worldwide.24 Specifically in response to such concern about the underrepresentation of ethnic minorities in clinical trials and to support the inclusion of Japanese sites in the global phase 3 studies, the PMDA specifically requires that at least one single‐dose phase 1 study to be conducted to confirm the safety and PK of an investigational product in Japanese healthy volunteers (HVs); results should be compared to those obtained in similar studies with non‐Japanese individuals; and the possible ethnic differences should be assessed prior to Japanese people joining a global clinical trial.21, 22 Therefore, conducting a phase 1 single dose study to evaluate the PK and safety of Enasidenib in healthy adult male Japanese subjects relative to healthy adult male Caucasian subjects was necessary to support the inclusion of Japanese sites in the Enasidenib global phase 3 clinical trial.
In the present study, we found that Enasidenib demonstrated linear PK over the dose range tested in Japanese healthy subjects, with a mean t1/2 ranging from 19.5 to 28.3 hours, and a systemic exposure that was dose proportional. These findings are in line with results from Caucasian subjects from the present study, and from previous Caucasian PK studies. The AGI‐16903 to Enasidenib plasma concentration ratio (M/P ratio) vs time profiles (Figure 4) clearly demonstrated a similar time course in vivo Enasidenib metabolism pattern between these two populations.
Regarding safety, Enasidenib was well tolerated when administered to healthy male subjects at the doses of 50 mg, 100 mg, and 300 mg. The incidence of TEAEs was similar in Japanese and Caucasian subjects. There was an increased incidence of TEAEs in both Japanese and Caucasian subjects at the 300 mg Enasidenib dose level compared to the 50 and 100 mg dose levels predominantly represented by mild and moderate AEs of headache, and in lesser extent by mild AEs of nausea and vomiting. There were no clinically significant changes or findings in clinical laboratory evaluations, vital sign measurements, or ECGs for this study.
5. CONCLUSION
Taken together, PK exposures of Enasidenib and its metabolite AGI‐16903 for Caucasians and Japanese subjects were similar and Enasidenib was safe and well tolerated with no apparent differences between Japanese and Caucasian subjects when administered as single oral doses of 50 mg, 100 mg, and 300 mg.
DISCLOSURES
Yan Li, Liangang Liu, Diana Gomez, Jian Chen, Zeen Tong, Maria Palmisano, and Simon Zhou are employees of and hold equity ownership in Celgene Corporation.
Li Y, Liu L, Gomez D, et al. Pharmacokinetics and safety of Enasidenib following single oral doses in Japanese and Caucasian subjects. Pharmacol Res Perspect. 2018;e00436 10.1002/prp2.436
References
- 1. Amaya ML, Pollyea DA. Targeting the IDH2 pathway in acute myeloid leukemia. Clin Cancer Res. 2018. 10.1158/1078-0432.CCR-18-0536 [DOI] [PubMed] [Google Scholar]
- 2. DiNardo C. Enasidenib for patients with relapsed acute myeloid leukemia and the IDH2 mutation. Clin Adv Hematol Oncol. 2018;16:247‐249. [PubMed] [Google Scholar]
- 3. Dugan J, Pollyea D. Enasidenib for the treatment of acute myeloid leukemia. Expert Rev Clin Pharmacol. 2018;11:755‐760. 10.1080/17512433.2018.1477585. [DOI] [PubMed] [Google Scholar]
- 4. Sharma H. Development of novel therapeutics targeting isocitrate dehydrogenase mutations in cancer. Curr Top Med Chem. 2018;18:505‐524. 10.2174/1568026618666180518091144. [DOI] [PubMed] [Google Scholar]
- 5. Stein EM. Enasidenib, a targeted inhibitor of mutant IDH2 proteins for treatment of relapsed or refractory acute myeloid leukemia. Future Oncol. 2018;14:23‐40. [DOI] [PubMed] [Google Scholar]
- 6. Nassereddine S, Lap CJ, Haroun F, Tabbara I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann Hematol. 2017;96:1983‐1991. [DOI] [PubMed] [Google Scholar]
- 7. Gross S, Cairns RA, Minden MD, et al. Cancer‐associated metabolite 2‐hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clark O, Yen K, Mellinghoff IK. Molecular Pathways: isocitrate Dehydrogenase Mutations in Cancer. Clin Cancer Res. 2016;22:1837‐1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dang L, White DW, Gross S, et al. Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature. 2010;465:966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27:599‐608. [DOI] [PubMed] [Google Scholar]
- 11. Figueroa ME, Abdel‐Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Losman JA, Looper RE, Koivunen P, et al. (R)‐2‐hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621‐1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia‐associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha‐ketoglutarate to 2‐hydroxyglutarate. Cancer Cell. 2010;17:225‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. ClinicalTrials.gov. 2018. https://clinicaltrials.gov/ct2/results?cond=&term=enasidenib&cntry=&state=&city=&dist=. Accessed July 1, 2018.
- 16. ClinicalTrials.gov . 2018. https://clinicaltrials.gov/ct2/show/NCT02273739. Accessed July 1, 2018.
- 17. IDHIFA® (enasidenib) . 2017. PI [package insert] Celgene Corporation, Sumit, NJ, USA. Accessed July 1, 2018.
- 18. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol. 2018;4:1106‐1110. 10.1001/jamaoncol.2017.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patel SA. Enasidenib‐induced differentiation syndrome in IDH2‐12 mutant acute myeloid leukemia. JAMA Oncol. 2018;4:1110‐1111. 10.1001/jamaoncol.2017.4724. [DOI] [PubMed] [Google Scholar]
- 20. Tong Z, Atsriku C, Yerramilli U, et al. Absorption, distribution, metabolism and excretion of an isocitrate dehydrogenase‐2 inhibitor enasidenib in rats and humans. Xenobiotica. 2018. 10.1080/00498254.2018.1425511 [DOI] [PubMed] [Google Scholar]
- 21. Agency PaMD . 2007. Basic principles on global clinical trials. https://www.pmda.go.jp/english/rs-sb-std/standards-development/cross-sectional-project/0010.html. Accessed July 1, 2018.
- 22. Baverel P, She D, Piper E, et al. A randomized, placebo‐controlled, single ascending‐dose study to assess the safety, tolerability, pharmacokinetics, and immunogenicity of subcutaneous tralokinumab in Japanese healthy volunteers. Drug Metab Pharmacokinet. 2017;33:150‐158. [DOI] [PubMed] [Google Scholar]
- 23. Xie H‐G, Kim RB, Kim RB, Wood AJ, Stein ACM. Molecular basis of ethnic differences in drug disposition and response. Annu Rev Pharmacol Toxicol. 2001;41:815‐850. [DOI] [PubMed] [Google Scholar]
- 24. Johnson JA. Influence of race or ethnicity on pharmacokinetics of drugs. J Pharm Sci. 1997;86(12):1328‐1333. [DOI] [PubMed] [Google Scholar]
- 25. Abrams SA, O'Brien KO, Liang LK, Stuff JE. Differences in calcium absorption and kinetics between black and white girls aged 5‐16 years. J Bone Miner Res. 1995;10:829‐833. [DOI] [PubMed] [Google Scholar]
- 26. Lindholm A, Welsh M, Alton C, Kahan BD. Demographic factors influencing cyclosporine pharmacokinetic parameters in patients with uremia: racial differences in bioavailability. Clin Pharmacol Ther. 1992;52:359‐371. [DOI] [PubMed] [Google Scholar]
- 27. Johnson JA, Livingston TN. Differences between blacks and whites in plasma protein binding of drugs. Eur J Clin Pharmacol. 1997;51:485‐488. [DOI] [PubMed] [Google Scholar]
- 28. Zhou HH, Adedoyin A, Wilkinson GR. Differences in plasma binding of drugs between Caucasians and Chinese subjects. Clin Pharmacol Ther. 1990;48:10‐17. [DOI] [PubMed] [Google Scholar]
- 29. Zhou HH, Koshakji RP, Silberstein DJ, Wilkinson GR, Wood AJ. Racial differences in drug response. Altered sensitivity to and clearance of propranolol in men of Chinese descent as compared with American whites. N Engl J Med. 1989;320:565‐570. [DOI] [PubMed] [Google Scholar]
- 30. Ahsan CH, Renwick AG, Waller DG, Challenor VF, George CF, Amanullah M. The influences of dose and ethnic origins on the pharmacokinetics of nifedipine. Clin Pharmacol Ther. 1993;54:329‐338. [DOI] [PubMed] [Google Scholar]
- 31. Krishna R, Gheyas F, Liu Y, et al. Pharmacokinetics and pharmacodynamics of anacetrapib following single doses in healthy, young Japanese and white male subjects. J Clin Pharmacol. 2018;58:254‐262. [DOI] [PubMed] [Google Scholar]