

Abstract
The putative cache (Ca2+ channel and chemotaxis receptor) domain containing 1 (CACHD1) protein has predicted structural similarities to members of the α2δ voltage-gated Ca2+ channel auxiliary subunit family. CACHD1 mRNA and protein were highly expressed in the male mammalian CNS, in particular in the thalamus, hippocampus, and cerebellum, with a broadly similar tissue distribution to CaV3 subunits, in particular CaV3.1. In expression studies, CACHD1 increased cell-surface localization of CaV3.1, and these proteins were in close proximity at the cell surface, consistent with the formation of CACHD1-CaV3.1 complexes. In functional electrophysiological studies, coexpression of human CACHD1 with CaV3.1, CaV3.2, and CaV3.3 caused a significant increase in peak current density and corresponding increases in maximal conductance. By contrast, α2δ-1 had no effect on peak current density or maximal conductance in CaV3.1, CaV3.2, or CaV3.3. A comparison of CACHD1-mediated increases in CaV3.1 current density and gating currents revealed an increase in channel open probability. In hippocampal neurons from male and female embryonic day 19 rats, CACHD1 overexpression increased CaV3-mediated action potential firing frequency and neuronal excitability. These data suggest that CACHD1 is structurally an α2δ-like protein that functionally modulates CaV3 voltage-gated calcium channel activity.
SIGNIFICANCE STATEMENT This is the first study to characterize the Ca2+ channel and chemotaxis receptor domain containing 1 (CACHD1) protein. CACHD1 is widely expressed in the CNS, in particular in the thalamus, hippocampus, and cerebellum. CACHD1 distribution is similar to that of low voltage-activated (CaV3, T-type) calcium channels, in particular to CaV3.1, a protein that regulates neuronal excitability and is a potential therapeutic target in conditions such as epilepsy and pain. CACHD1 is structurally an α2δ-like protein that functionally increases CaV3 calcium current. CACHD1 increases the presence of CaV3.1 at the cell surface, forms complexes with CaV3.1 at the cell surface, and causes an increase in channel open probability. In hippocampal neurons, CACHD1 causes increases in neuronal firing. Thus, CACHD1 represents a novel protein that modulates CaV3 activity.
Keywords: CACHD1, CaV3 calcium channel, T-type calcium current, α2δ auxiliary subunit
Introduction
The putative (Ca2+ channel and chemotaxis receptor) domain containing 1 (CACHD1) gene was identified following a systematic search for proteins with structural homology to α2δ voltage-gated Ca2+ channel (VGCC) auxiliary subunits. The human CACHD1 gene on chromosome 1p31.3 encodes the putative protein CACHD1 and has many orthologues, including in speciation as early as Caenorhabditis elegans (tag-180) and Drosophila melanogaster (CG16868; Anantharaman and Aravind, 2000). Despite only a 13–16% gene homology and a <21% protein identity with the α2δ VGCC auxiliary subunits, there are several key structural similarities between CACHD1 and α2δ in terms of the arrangement of protein motifs. α2δ and CaVβ subunits are described as auxiliary or accessory VGCC subunits that modulate cell-surface expression and biophysical properties of high-voltage-activated (HVA) CaV1 (L-type Ca2+ current) and CaV2 (P/Q-, N-, and R-type Ca2+ current) VGCC major α1 subunits (Dolphin, 2012, 2013). In particular, α2δ subunits are proposed to associate with HVA channels within the secretory pathway to promote plasma membrane trafficking and, consequently, to contribute to synaptic abundance (Dolphin, 2012) and transmitter release (Hoppa et al., 2012), and to defining the extent of the active zone (Schneider et al., 2015). α2δ-1 and α2δ-2 represent molecular targets of gabapentinoid drugs (Dooley et al., 2007). However, the modulation of low-voltage-activated (LVA) CaV3 family (T-type Ca2+ current) by existing α2δ and CaVβ auxiliary subunits has not been firmly established (Dolphin et al., 1999; Lacinová et al., 1999; Dubel et al., 2004). LVA currents are activated by small depolarization to regulate excitability around the resting membrane potential and CaV3 channels have been proposed as therapeutic targets in diseases such as epilepsy and pain (Perez-Reyes, 2003; Cheong and Shin, 2013; Powell et al., 2014; Snutch and Zamponi, 2018); therefore, the knowledge of proteins that modulate CaV3 activity is paramount.
Here, we investigate the novel CACHD1 protein and test the hypothesis that CACHD1 represents an α2δ-like protein that modulates CaV3 channels. We have previously reported that, by contrast to α2δ, the CACHD1 subunit had no clear effect on CaV2.2 biophysical properties when coexpressed together with β2a in expression system studies (Soubrane et al., 2012). We characterize the expression of the CACHD1 gene in rat and human tissue at the transcriptional and translational level, and demonstrate that CACHD1, but not α2δ-1, increases CaV3 (T-type) current density and maximal conductance. CACHD1 increases CaV3.1 channel levels at the plasma membrane, and data were consistent with CACHD1 forming complexes with CaV3.1 at the cell surface to increase channel open probability. We further demonstrate that CACHD1 expression causes a functional increase in T-type current-mediated excitability in hippocampal neurons. Together, these data demonstrate that CACHD1 is structurally an α2δ-like protein that functionally modulates CaV3 activity.
Materials and Methods
RNA isolation and real-time PCR.
Tissue samples were dissected from five adult male Wistar rats (Harlan) following isoflurane overdose and cervical dislocation, according to the UK Home Office Animals (Scientific Procedures) Act 1986. Total RNA was extracted using an RNeasy kit (Qiagen) with an on-column DNase I treatment. Additional total RNA samples from AMS Biotechnology originated from human male donors that were 24–65 years of age. RNA (500 ng) was reverse transcribed, and the relative quantification of CACHD1 and α2δ-1 transcripts was performed using SYBR green and custom-made validated primers. HPRT1 (hypoxanthine phosphoribosyltransferase 1) was used as housekeeping gene. Absolute quantification of CACHD1; α2δ-1, α2δ-2, and α2δ-3; CaV2.2; and CaV3.1, CaV3.2, CaV3.3 transcripts were evaluated using “Best Coverage” TaqMan probes (Applied Biosystems) against a standard curve of plasmids containing human CACHD1 and a rat single-stranded DNA standard curve.
Sample preparation for in situ hybridization and immunohistochemistry.
Rat tissue was donated by Dr. Emilio Russo, University Magna Grecia of Catanzaro, Catanzaro, Italy. Briefly, 6-month-old male rats were killed by intraperitoneal injection of pentobarbital (200 mg/kg) according to ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (National Center for the Replacement, Refinement & Reduction of Animals in Research) and the local ethical approval committee of the University of Catanzaro, and was perfused–fixed with 4% paraformaldehyde (PFA) in RNase-free PBS, pH 7.3. Brain tissue was extracted, postfixed overnight in 4% PFA in RNase-free PBS and then cryoprotected in 30% sucrose. After being processed to wax (Tissue-Tek VIP Fixative, Sakura), 5 μm horizontal plane brain slices were cut using a microtome (Leica).
In situ hybridization.
A CACHD1 probe consisting of a cocktail of short 10–20 bp oligonucleotides spanning ∼1 kb was designed by ACD and in situ hybridization was performed on 5 μm rat brain sections using an RNAscope 2.0 Red for FFPE Kit (ACD). Positive (POLR2A, ACD) and negative (DAPB, ACD) probes were run in parallel.
Immunohistochemistry.
Chromogenic immunohistochemistry was performed using antigen retrieval in citrate buffer (Thermo Fisher Scientific) for 10 min and diaminobenzidine (DAB) staining (ImmPACT, Vector Laboratories), dehydrated and mounted with DPX. Rabbit anti-CACHD1 (1:500; catalog #AB75141, Abcam; RRID:AB_1310016) with horseradish peroxidase-coupled anti-rabbit IgG (ImmPRESS, Vector Laboratories United Kingdom) was used to detect CACHD1 protein. Qualitative expression of mRNA was evaluated with a bright-field microscope according to the color intensity of labeled mRNA.
Antibodies for biochemistry.
The following antibodies were used: mouse anti-HA.11 (clone 16B12, Lot No. B220767, Cambridge Bioscience; RRID:AB_10063630); rabbit anti-Na+/K+-ATPase (NB100-80005, Lot No. YH02206, Novus Biologicals; RRID:AB_2063297); mouse anti-c-Myc (clone 9E10, Lot No. 087M4765V, catalog #M4439, Sigma-Aldrich; RRID:AB_439694); rabbit anti-c-Myc (Lot No. 016M4762V, catalog #C3956, Sigma-Aldrich; RRID:AB_439680); mouse anti-β-actin (Lot No. 028K4826, catalog #A5441, Sigma-Aldrich; RRID:AB_476744); rabbit anti-CACHD1 (Lot No. QC22258, catalog #AV49592, Sigma-Aldrich; RRID:AB_1852421); goat anti-mouse or rabbit IgG coupled to horseradish peroxidase (Stratech Scientific); donkey anti-mouse or rabbit coupled to Alexa Fluor 488, 555, or 647 (Invitrogen). (Note: we experienced vial-to-vial variation with the rabbit anti-CACHD1 antibody for Western blotting during this study.) Although both vials were from the same lot number and specifically recognized CACHD1, the vial used for Figure 4D gave rise to more nonspecific staining on human embryonic kidney (HEK) cell lysates than vial used for Figure 4A.
Figure 4.

Characterization of CACHD1 and its effects on CaV3.1 expression. HEK cells were transfected with empty vector (VC), CACHD1, Myc-CACHD1, and GFP-CaV3.1-HA alone or in combination, as shown in each panel. A, HEK cell lysates were analyzed by Western blotting (WB). An antibody to CACHD1 recognized a single protein similar to the predicted size for CACHD1, but also recognized a nonspecific protein in all lysates. B, Cell-surface proteins were biotinylated, and pull-downs were analyzed for CACHD1 and Na+/K+-ATPase (loading control). In control cells, no immunoreactive CACHD1 was detected, confirming antibody specificity. In CACHD1-expressing cells, immunoreactive CACHD1 was detected. In both cell types, immunoreactive Na+/K+-ATPase was detected. C, Cell-surface proteins were biotinylated, and pull-downs were analyzed for GFP-CaV3.1-HA (HA) and Na+/K+-ATPase (loading control). In control cells and cells only expressing CACHD1, no HA signals were detected, confirming antibody specificity. In cells expressing GFP-CaV3.1-HA, HA signals were readily detected. Quantification of the HA signals (normalized to Na+/K+-ATPase) revealed the expression of CACHD1-increased signals for GFP-CaV3.1-HA at the cell surface, *p < 0.05. Na+/K+-ATPase signals were detected in all cell types. D, Inputs of the biotin pull-down assays were analyzed by WB. Signals for HA were detected only in cells expressing GFP-CaV3.1-HA, signals for CACHD1 were detected only in cells expressing Myc-CACHD1, and signals for β-actin were detected in all cell types. All blots are representative of n ≥ 3 experiments.
Vectors and vector construction.
The human CACHD1 construct was purchased from Origene, and the truncated clone was completed by PCR. The subsequent open reading frame was then subcloned into pcDNA5/FRT. An N-terminal Myc tag was inserted after the natural signal sequence between Ala35 and Glu36 using standard PCR techniques. All constructs were sequenced to confirm identity. Construction of the vector pcDNA5/FRT-HA-CLR-Myc-RAMP1 has been described previously (Cottrell et al., 2007).
Cell maintenance and propagation.
HEK293 tsA201 (HEK) cells were cultured in DMEM (Invitrogen) containing 10% fetal bovine serum (Biosera) and maintained in 95% air and 5% CO2 at 37°C.
Cell-surface biotinylation.
HEK cells were transiently transfected in six-well plates using 3 μg DNA (ratio 2:1, GFP-CaV3.1-HA:CACHD1) using Lipofectamine 2000 (3:8 ratio, DNA/Lipofectamine 2000). HEK cells transfected with empty vector [vector control (VC)], VC + Myc-CACHD1, GFP-CaV3.1-HA + VC, or GFP-CaV3.1-HA + Myc-CACHD1 were washed (3× PBS), incubated with 0.3 mg/ml Pierce EZ-Link-Sulfo-NHS-Biotin (Thermo Fisher Scientific) in PBS (1 h, 4°C), washed (3× PBS) and cells lysed in RIPA buffer (50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 5 mm MgCl2, 1 mm EGTA, 10 mm NaF, 10 mm Na4P2O7, 0.1 mm Na3VO4, 0.5% Nonidet P-40, peptidase inhibitor cocktail; Roche) and centrifuged. Biotinylated proteins were recovered by incubation with NeutrAvidin-agarose (30 μl, overnight, 4°C), pelleted, washed with RIPA buffer (3× 1 ml), boiled in Laemmli buffer and analyzed by SDS-PAGE and Western blotting.
SDS-PAGE and Western blotting.
Immunoprecipitations and whole-cell lysates were separated by SDS-PAGE (6–9% acrylamide), and proteins were transferred to PVDF membranes [Immobilon-P, Millipore (UK)] and blocked for 1 h at room temperature [RT; 1× PBS, 0.1% Tween 20, 5% nonfat milk powder (blocking buffer)]. Membranes were incubated with antibodies to HA (1:5000), β-actin (1:20,000), CACHD1 (1:1000), rabbit or mouse Myc (1:5000), or Na+-K+-ATPase (1:20,000; overnight, 4°C; blocking buffer). Membranes were washed for 30 min (1× PBS, 0.1% Tween 20) and incubated with appropriate secondary antibodies coupled to horseradish peroxidase (1:10,000; 1 h, room temperature; blocking buffer). Immunoreactive proteins were detected using enhanced chemiluminescence (Bio-Rad). Densitometric analysis was performed using an ImageQuant-RT ECL Imaging System (GE Healthcare) and analyzed using ImageQuant TL Software.
Immunofluorescent detection of cell-surface proteins.
HEK cells were transiently transfected in 12-well plates using 1 μg of DNA (2:1 ratio, GFP-CaV3.1-HA/Myc-CACHD1) using polyethylenimine (PEI; 1:2 ratio, DNA/PEI). HEK cells transfected with empty vector (VC), VC + Myc-CACHD1, GFP-CaV3.1-HA + VC, GFP-CaV3.1-HA + Myc-CACHD1, or calcitonin receptor-like receptor (CLR)-receptor activity-modifying protein 1 (RAMP1) were seeded onto coverslips and used for experimentation after 48 h. Cells were washed twice with PBS supplemented with 0.5mm CaCl2 and 2 mm MgCl2 (PBSCM), incubated in DMEM containing 0.1% BSA and mouse anti-HA (1:100) and rabbit anti-c-Myc (1:500) antibodies (1 h, 4°C), washed twice again with PBSCM, and then fixed in 100 mm PBS containing 4% PFA (w/v), pH 7.4 (20 min, 4°C). Coverslips were incubated in blocking buffer (1× PBS, 2% normal horse serum, 0.1% saponin; 30 min, RT) and then incubated with appropriate secondary antibodies (1:2000, 2 h, RT). Coverslips were washed (blocking buffer, 30 min, RT) and mounted using VECTASHIELD (Vector Laboratories) containing DAPI.
Proximity ligation assays.
HEK cells were transiently transfected in 12-well plates using 1 μg of DNA (2:1 ratio, GFP-CaV3.1-HA/Myc-CACHD1) using PEI (1:2 ratio, DNA/PEI). HEK cells transfected with empty vector (VC), VC + Myc-CACHD1, GFP-CaV3.1-HA + VC, GFP-CaV3.1-HA + Myc-CACHD1, or CLR-RAMP1 were seeded onto coverslips and used for experimentation after 48 h. Cells were washed twice with PBSCM, incubated in DMEM containing 0.1% BSA and mouse anti-HA (1:100) and rabbit anti-c-Myc (1:500) antibodies (1 h, 4°C), washed twice again with PBSCM, and then fixed in 100 mm PBS containing 4% PFA (w/v), pH 7.4 (20 min, 4°C). After washing with PBSCM, the proximity ligation assay was conducted according to the manufacturer instructions (Duolink In Situ Red Starter Kit Mouse/Rabbit, catalog #DUO92101, Sigma-Aldrich). Briefly, cells were blocked (1 h, 37°C), washed twice (5 min, room temperature), and then incubated with appropriate secondary antibodies (1 h, 37°C). After washing (2 × 5 min, room temperature), the ligation was conducted (30 min, 37°C), and the cells were washed twice more. Coverslips were then incubated with the amplification reaction mixture (100 min, 37°C), washed, and mounted in medium containing DAPI.
Confocal microscopy.
Cells were observed with a Nikon Eclipse Ti laser-scanning confocal microscope using a 100×/1.45 oil differential interference contrast N2 objective. Images were collected at a zoom of 1–2, and at least five optical sections were captured at intervals of 0.5 μm. Single sections are shown. Images were processed using Adobe Photoshop and the NIS-Elements AR Software (Nikon).
Transformed human embryonic kidney cell culture and transfection for electrophysiology.
For electrophysiology experiments, HEK cells were transfected using 4 μl of FuGENE 6 (Promega UK) with total 2 μg of pcDNA3 at 50:1:25 for CaV3.1/pmaxGFP (Lonza), CaV3.2/pmaxGFP, or CaV3.3/pmaxGFP with or without α2δ-1 or CACHD1. Empty vector was used to compensate when α2δ or CACHD1 was omitted. Cells were maintained at 95% air and 5% CO2 at 37°C, and used for experimentation 24–48 h post-transfection.
Hippocampal neuron culture and transfection.
Low-density hippocampal cultures were prepared from male and female embryonic day 19 rat embryos, as described previously (Zhang et al., 2003). All experiments were performed in compliance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH), were approved by the University of Virginia Animal Care and Use Committee, and adhered to ARRIVE guidelines. Neurons were plated onto poly-l-lysine-coated glass coverslips at a density of ∼70 cells/mm2 and were transfected using Lipofectamine 2000 at a ratio of 2 μl of Lipofectamine 2000 to 1 μg of DNA. Neurons were transfected with either CACHD1 or pcDNA3.1 at a ratio of 10:1 excess to mVenus and moved 24 h after transfection to a new glia-feeder layer.
Electrophysiology.
Recordings from HEK cells were made as described previously (Vogl et al., 2015). Current–voltage (I–V) relationships from individual cells were fitted with a modified Boltzmann equation, as follows: I = Gmax x(V − Vrev)/(1 + exp (−(V − V1/2)/k)) where, Gmax is the peak ionic current conductance (in nanosiemens per picofarad), V1/2 is the midpoint of activation (i.e., the voltage at which 50% of the channels are open), Vrev is the null potential, and k is the slope factor. Tail currents (measured at −120 mV) were normalized to the maximal and minimal conductance, and the resultant curves were fitted with the following Boltzmann function: I = I0 + ((Imax − I0)/(1 + exp(V1/2 − V)/k))). Throughout, all comparative electrophysiological experiments were performed in transfection-matched cultures.
Recordings from hippocampal neurons were performed as described previously (Jones et al., 2007). Throughout, data are expressed as the mean ± SEM. Methods to estimate the probability of channel opening (Po) have been previously described (Shcheglovitov et al., 2008), which assumes no change in single-channel current, reducing the relationship between whole-cell current (I) to I ∼ NPo, where N is the number of channels in a cell. N is estimated by measuring the channel-gating current at the reversal potential for ionic current. The peak current represents the maximal gating charge (Qmax) and is proportional to N. Gmax was determined by fitting the I–V curve, obtained from the same cell, with a Boltzmann-function as described earlier. Gmax is used as a proxy for I since it is not affected by changes in driving force. Therefore, the Gmax/Qmax ratio can be used to estimate Po.
Experimental design and statistical analysis.
Throughout, all animal studies comply with appropriate ARRIVE and NIH guidelines and comply with country and institute guidelines (as specified in the Materials and Methods section for each animal study). Details of animal strain, sex, and method of killing and the use of anesthetics are also stated in the Materials and Methods section for each animal study.
Throughout, all comparative biochemical and electrophysiological experiments were performed against transfection-matched culture controls. For electrophysiological experiments in recombinant cells, a minimum of five separate transfections were performed, and the numbers of individual replications are specified in the appropriate table. In all cases, sample size is stated in text, figure legend, or the appropriate table. Data subjected to statistical comparisons were assessed for assumptions of normality using a D'Agostino–Pearson omnibus test and were expressed as mean ± SEM throughout. Groups were compared by two-tailed paired or unpaired Student's t test, Mann–Whitney test, one- or two-way ANOVAs followed by Bonferroni post hoc tests, Kruskal–Wallis test followed by Dunn's multiple-comparison test or least-squares fits compared using extra sum of squares F test, as appropriate, using GraphPad Prism. In all cases, the statistical test used is stated in text, figure legend, or the appropriate table. Throughout, p < 0.05 was taken as being statistically significant, and, where appropriate, values of p < 0.01 and p < 0.001 are specified.
Results
The novel CACHD1 protein is an α2δ paralog
We first investigated the predicted protein domain structure of CACHD1. Figure 1A illustrates that, like α2δ-1, CACHD1 has a predicted exofacial N terminus according to its signal sequence, a von Willebrand factor A (VWA) domain, two bacterial chemosensory-like cache domains, and a short hydrophobic transmembrane domain followed by an intracellular C terminus. Although CACHD1 and α2δ-1 share limited amino acid sequence homology (<21%), the similarities in modular domain content and arrangement between the proteins suggested the possibility that CACHD1 represents an α2δ-like protein. However, there are also a number of differences between CACHD1 and α2δ-1, including the following: (1) α2δ proteins are a single gene product that is post-translationally cleaved by proteases into α2 and δ components and then associate via disulfide bonding (Calderón-Rivera et al., 2012; Segura et al., 2017), and an important 6 aa motif for proteolytic cleavage has been identified (Andrade et al., 2007) that is absent in CACHD1; (2) CACHD1 has a single predicted post-translational N-glycosylation site, while α2δ-1 is heavily glycosylated at multiple potential sites (Douglas et al., 2006); (3) CACHD1 has a variant RSR amino acid sequence at the α2δ-1 binding site for gabapentinoids; (4) despite expressing a VWA domain, the functionally important MIDAS motif in CACHD1 (DxGxS) is different from that of α2δ-1 (DxSxS); and (5) α2δs have a predicted glycophosphatidylinositol (GPI) anchoring site (Davies et al., 2010), this is absent in CACHD1 which instead has a predicted transmembrane domain and a larger intracellular C-terminus domain.
Figure 1.

A, Predicted protein sequence homology and relative expression profile of CACHD1 and α2δ-1. CACHD1 and α2δ-1 subunits both contain a N-terminal signal peptide, a VWA domain, two cache domains, and transmembrane and intracellular domains. GBP, Gabapentin binding domain (RRR); GBP*, gabapentin binding domain variant (RSR); MIDAS, metal-ion-dependent adhesion site (DxSxS); MIDAS*, Metal-ion-dependent adhesion site variant (DxGxS); Cache, Ca2+ channel and chemotaxis receptor; TM, transmembrane domain; Cys, cysteine; His, histidine (locations of domains are approximate and from data from www.Uniprot.org, figure drawn using DOG: Domain Graphics). B, Relative expression profile of CACHD1 and α2δ-1 mRNA in rat tissue determined using SYBR green real-time quantitative PCR and HPRT1 as the housekeeping gene. DRG, Dorsal root ganglion; SCG, superior cervical ganglion. (Data normalized to lowest tissue expression; n = 3 experiments using 3 animals each.) Figure 1 is supported by in situ hydridization data in different rat brain regions (Figure 1-1) and a qualitative expression profile of CACHD1 mRNA and protein in the adult rat brain (Figure 1-2).
CACHD1 mRNA expression in adult rat brain. In situ hybridization of adult rat brain. CACHD1 mRNA was labelled pink with blue counterstain (Gill’s I Haematoxylin). CA1-3: cornus ammonis 1-3; DG: dentate gyrus; g: granule cell layer; m: molecular layer; p: Purkinje cell; wm: white matter. Download Figure 1-1, DOCX file (19.2KB, docx)
Qualitative expression profile of CACHD1 mRNA and protein in the adult rat brain. + labelling similar to background; ++ weak labelling; +++ moderate labelling, ++++ strong labelling; +++++ very strong labelling. Download Figure 1-2, DOCX file (15.5KB, docx)
CACHD1 is highly expressed in brain hippocampal and thalamic regions
To obtain comparative and quantitative data on CACHD1 mRNA expression, real-time PCR was performed on rat and human mRNA from different regions of the brain and peripheral tissue. Relative expression profiles of CACHD1 and α2δ-1 transcripts in rat tissue showed high CACHD1 expression in thalamus, hippocampus, and cerebellum, while α2δ-1 transcript expression was prominent in cortex, hippocampus, and also superior cervical ganglia (Fig. 1B). We further investigated the anatomical distribution of CACHD1 at the transcriptional and protein levels using in situ hybridization and immunohistochemistry in adult mammalian brain. Rat brain regions displaying high mRNA include the hippocampus, anterodorsal thalamic nucleus, reticular thalamic nucleus, cerebellum, subiculum, medial entorhinal cortex, and zona incerta (Fig. 1-1; Fig. 1-2). Hippocampal CACHD1 mRNA staining was strong in the dentate gyrus, as well as the CA1 (cornu ammonis 1) pyramidal cell region; mRNA staining was less strong in CA3 (cornu ammonis 3). There was strong correlation between the levels of expression of CACHD1 mRNA and protein in rat brain (Fig. 1-2). In the thalamus, CACHD1 protein showed differential expression between major thalamic nuclei, in particular with prominent staining in the anterodorsal and reticular nuclei (Fig. 2). In human tissue, CACHD1 transcripts were similarly high in hippocampus, thalamus, and cerebellum (Fig. 2-1). CACHD1 transcript distribution was broadly similar to certain CaV3 subtypes, in particular to CaV3.1 (Fig. 2-1; Talley et al., 1999). CACHD1 transcript expression showed a differential distribution to α2δ-1 and α2δ-2 subtypes and was most similar to α2δ-3 (Fig. 2-1; Cole et al., 2005). In human tissue, CACHD1 protein levels were most abundant in dentate gyrus granule cells and pyramidal cells of the hippocampus cornu ammonis, cortical regions, and thalamus, in both large-diameter and small-diameter cells (Fig. 3).
Figure 2.

CACHD1 protein expression in adult rat brain. Immunoreactive protein was detected using rabbit anti-CACHD1 with peroxidase anti-rabbit secondary antibody and DAB staining (brown). AD, Anterodorsal thalamic nucleus; AVDM, anteroventral thalamic nucleus (dorsomedial); AVVL, anteroventral thalamic nucleus (ventrolateral); fi, fimbria; MD, mediodorsal thalamic nucleus; Po, posterior thalamic nucleus; sm, strai medullaris; Rt, reticular thalamus nucleus; RtSt, reticular VL, ventrolateral thalamic nucleus; VPL, ventroposterior lateral thalamus; g, granule cell layer; m, molecular layer; p, Purkinje cell; wm, white matter. Figure 2 is supported by expression profiling of CACHD1 and different voltage-gated calcium channel subunit mRNA in human tissue (Figure 2-1).
Figure 3.

CACHD1 protein expression in human brain. Immunohistochemistry of adult human brain using rabbit anti-CACHD1 with peroxidase anti-rabbit secondary antibody with (brown) DAB stain. CA1–4, Cornu ammonis 1–4; DG, dentate gyrus.
Expression profile of CACHD1 and voltage-gated calcium channel subunit mRNA in human tissue. Absolute quantification of CACHD1, α2δ-1, -2, -3, CaV2.2 and CaV1, -2, -3 transcripts was assessed in triplicate by TaqMan® qPCR using ‘Best Coverage’ Taqman probes (Applied Biosystems, UK) against a 5-point standard curve of plasmids consisting of 10-fold dilution of a known copy number of plasmid containing cDNA of the gene of interest. Total RNA was extracted using an RNeasy kit (Qiagen, UK) with an on-column DNase I treatment. Additional total RNA samples from AMS Biotechnology (Abingdon, UK) originated from human male donors aged 24-65. Download Figure 2-1, TIF file (10.9MB, tif)
CACHD1 promotes cell-surface expression of CaV3.1
Our expression data indicated high levels of CACHD1 expression in the thalamus, hippocampus, and cerebellum. As expression levels of CaV3 subunits are also high in the thalamus and hippocampus, we hypothesized that CACHD1 may modulate CaV3 subunits in a recombinant HEK cell system. As a first step, we expressed CACHD1 in HEK cells and confirmed the specificity of the CACHD1 antibody (Fig. 4A). Immunoreactive CACHD1 was detected at ∼170 kDa. We also confirmed that CACHD1 is present at the cell surface of HEK cells (Fig. 4B). Next, we determined whether the expression of CACHD1 affected the subcellular localization of CaV3.1 using a cell-surface biotinylation assay. Cell-surface proteins from HEK cells expressing empty vector, empty vector + CACHD1, GFP-CaV3.1-HA + empty vector, and GFP-CaV3.1-HA + CACHD1 were extracted, and levels of GFP-CaV3.1-HA were analyzed by Western blotting. Our data show that the coexpression of CACHD1 increased cell-surface localization of GFP-CaV3.1-HA (2.65 ± 0.40-fold over control; p < 0.05, two-tailed paired Student's t test; Fig. 4C). We also quantified the whole-cell expression of GFP-CaV3.1-HA in the same HEK cells, normalizing to levels of β-actin (Fig. 4D). Importantly, our data show that CACHD1 increases levels of GFP-CaV3.1-HA at the cell surface without affecting the total cellular level.
CACHD1 and CaV3.1 are in close proximity at the cell surface
To determine whether CaV3.1 and CACHD1 are present in a complex at the cell surface, an epitope-tagged CACHD1 (Myc-CACHD1) was used to aid cell-surface precipitation and detection. First, we tested the expression of the tagged protein and examined the ability of an anti-Myc antibody to bind to CACHD1 at the cell surface. Myc-CACHD1 was expressed in HEK cells with a similar molecular mass (∼170 kDa) to untagged CACHD1 (Fig. 5-1). Furthermore, we could detect Myc-CACHD1 at the cell surface using immunofluorescence and confocal microscopy (Fig. 5-1). Proximity ligation assays are commonly used to predict the likelihood that two proteins are sufficiently close enough to be present in the same complex. First, we determined whether we could simultaneously detect Myc-CACHD1 and GFP-CaV3.1-HA at the cell surface by confocal microscopy. Live HEK cells expressing empty vector, empty vector + CACHD1, GFP-CaV3.1-HA + empty vector, and GFP-CaV3.1-HA + CACHD1 were incubated with antibodies to the Myc and HA epitope tags of CACHD1 and CaV3.1, respectively, and immunoreactive proteins were visualized by immunofluorescence (Fig. 5A). No immunoreactive signals were detected in cells expressing empty vector, indicating antibody specificity. We were able to detect immunoreactive Myc signals only in cells expressing Myc-CACHD1. Similarly, we were able to detect signals for the HA antibody only in cells expressing GFP, indicating expression of GFP-CaV3.1-HA. We were also able to simultaneously detect CLR and RAMP1 at the cell surface of transfected cells (Fig. 5A). Next, we labeled cells from the same transfections, performed a proximity ligation assay, and visualized the cells using confocal microscopy. No proximity ligand assay (PLA) signals were detected in cells transfected with empty vector, empty vector + CACHD1, and GFP-CaV3.1-HA + empty vector (Fig. 5B). By contrast, we could readily detect PLA signals in our positive control (CLR-RAMP1) and in cells transfected with GFP-CaV3.1-HA + CACHD1. Importantly, we could detect PLA signals only in cells expressing GFP (Fig. 5B). Thus, CACHD1 and CaV3.1 are in close proximity (<40 nm) at the cell surface of HEK cells, indicating that they are likely in the same protein complex. As discussed more fully below, together, these data are consistent with CACHD1 increasing the cell-surface localization of CaV3.1 and with the formation of CACHD1–CaV3.1 complexes at the cell surface.
Figure 5.

CaV3.1 and CACHD1 are present at the cell surface and are in close proximity. Live HEK cells expressing empty vector (VC), VC + Myc-CACHD1, GFP-CaV3.1-HA + VC, Myc-CACHD1 + GFP-CaV3.1-HA, or CLR-RAMP1 (positive control) were incubated with antibodies to HA and Myc, washed, and fixed. A, Cells were then incubated with appropriate secondary antibodies and immunoreactive proteins localized by immunofluorescence and confocal microscopy. In HEK-VC cells, no signals for GFP, HA, or Myc were detected indicating specificity of detection. HA signals (white arrowheads) were detected only in cells expressing GFP-CaV3.1-HA (as determined by the GFP signal) and CLR-RAMP1. Similarly, Myc signals (yellow arrowheads) were detected only in cells expressing Myc-CACHD1 and CLR-RAMP1. Scale bar, 10 μm. B, After the proximity ligation assay, no signals were detected in cells expressing empty vector or in cells expressing only Myc-CACHD1 or GFP-CaV3.1-HA. In contrast, PLA signals were detected in cells expressing Myc-CACHD1 + GFP-CaV3.1-HA (arrows) and CLR-RAMP1 (arrows). Single optical sections are shown except for the PLA panel (CLR-RAMP1 excluded), where five optical sections are merged, two above and two below (0.5 μm increments) from the optical sections shown in the GFP/DAPI panel. Scale bar, 20 μm. All images are representative of n = 3 experiments. The figure is supported by analysis of cell-surface CACHD1 construct expression studies (Figure 5-1).
Analysis of cell-surface CACHD1 construct expression. (A, B) HEK cells were transfected with empty vector (vector control, VC) or Myc-CACHD1 and cell lysates analysed by (A) Western blotting (WB) and (B) immunofluorescence and confocal microscopy. (A) Immunoreactive signals for Myc (mouse Myc, mMyc) were detected at a similar molecular mass to that predicted for CACHD1 only in cells expressing CACHD1. (B, upper panel) Cells were incubated with antibody to Myc (rabbit Myc, rMyc), washed, fixed and then incubated with appropriate secondary antibodies. Myc signals (arrowheads) were only detected in cells expressing Myc-CACHD1. (B, lower panel) Cells were fixed, incubated with antibody to Myc (rMyc), washed and then incubated with appropriate secondary antibodies. Myc signals were detected at the cell-surface (arrowheads) and in intracellular vesicles only in cells expressing Myc-CACHD1. Scale bar, 10 µm. Download Figure 5-1, TIF file (10.9MB, tif)
CACHD1 modulates recombinant CaV3 family VGCCs
We next tested the hypothesis that CACHD1 modulates T-type Ca2+ current. Coexpression of CACHD1 with CaV3.1 caused an increase in current density around peak values (Fig. 6A,B) and a corresponding increase in maximal conductance (Fig. 6B, inset; Table 1). By contrast, in our hands, CaV3.1 peak current and conductance was not modulated by α2δ-1 in transfection-matched experiments (Fig. 6A,C; Table 1). CACHD1 effects were not accompanied by any overall change in the midpoint of activation or slope factor k (Table 1), and CACHD1 had no effect on CaV3.1 steady-state inactivation (data not shown). Neither CACHD1 nor α2δ-1 affected CaV3.1 recovery from inactivation, as measured by the lack of effect on the mid-time of recovery from inactivation (τrecovery; p > 0.1 for both, one-way ANOVA with Bonferroni post hoc test; data not shown).
Figure 6.
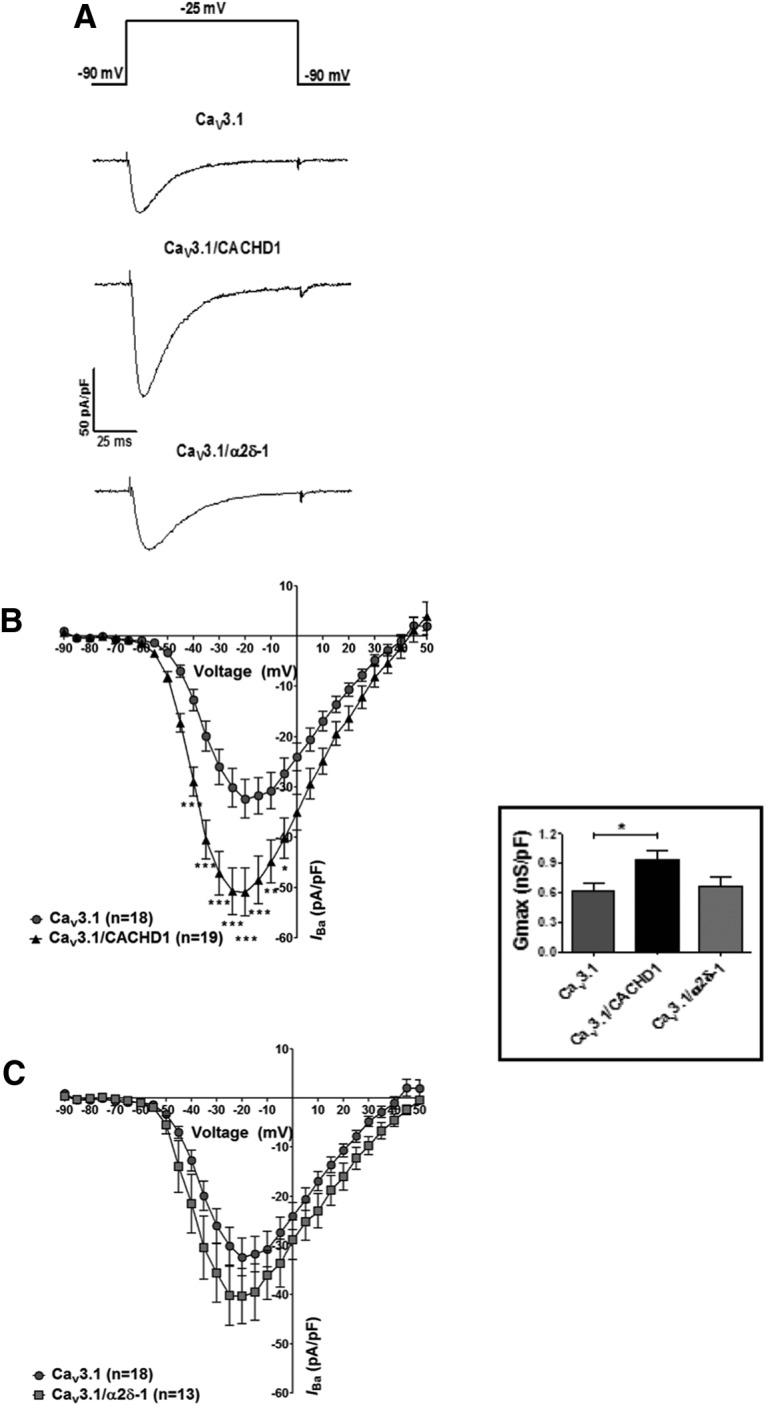
Effects of CACHD1 and α2δ-1 on CaV3.1 channels. A, B, CACHD1 significantly increased current density as shown by representative current density traces at −25 mV (A) and I–V relationships, holding potential (VH) −90 mV (B; *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA with Bonferroni post hoc test). A, C, α2δ-1 had no significant effect on current density as shown by representative current density traces at −25 mV (A) and I–V relationships, VH −90 mV (C). CACHD1, but not α2δ-1, significantly increased maximal conductance (inset, p < 0.05, one-way ANOVA with Bonferroni's post hoc test).
Table 1.
Effects of CACHD1 and α2δ-1 on biophysical properties of CaV3 subtypes
| Gmax (pS/pF) | V1/2 (mV) | k (mV) | τ Activation (ms)a | τ Inactivation (ms)b | |
|---|---|---|---|---|---|
| CaV3.1 (18) | 628 ± 70 | −34.5 ± 0.8 (30) | 5.4 ± 0.1 (30) | 2.0 ± 0.1 | 25.8 ± 2.0 |
| CaV3.1/CACHD1 (19) | 944 ± 90* | −36.3 ± 0.9 (29) | 5.6 ± 0.2 (29) | 2.0 ± 0.2 | 22.2 ± 4.8 |
| CaV3.1/α2δ-1 (13) | 672 ± 90 | −35.7 ± 1.4 | 5.6 ± 0.3 | 3.3 ± 0.2** | 18.9 ± 0.86*** |
| CaV3.2 (13) | 596 ± 120 | −34.4 ± 2.4 | 5.7 ± 0.2 | 7.1 ± 0.40 | 33.3 ± 0.97 |
| CaV3.2/CACHD1 (15) | 1060 ± 140**** | −33.4 ± 0.8 | 5.9 ± 0.2 | 5.9 ± 0.38 | 32.0 ± 1.6 |
| CaV3.3 (12) | 573 ± 88 | −36.1 ± 1.2 | 4.3 ± 0.2 | 24.4 ± 1.9 | 134 ± 12 |
| CaV3.3/CACHD1 (10) | 849 ± 78***** | −38.9 ± 1.6 | 4.0 ± 0.3 | 28.5 ± 3.4 | 126 ± 8.3 |
In all cases, comparisons were performed in culture-matched experiments. Numbers in parenthesis represents the number of cells each from a minimum of five separate transfections.
aτ activation was measured at −25 mV in all cases.
bτ inactivation was measured at −20 mV for CaV3.1 and CaV3.2, and at −30 mV for CaV3.3.
*p < 0.05 vs CaV3.1 (one-way ANOVA with Bonferroni post hoc test);
**p < 0.05,
***p < 0.05, vs CaV3.1 (two-way ANOVA with Bonferroni post hoc test);
****p < 0.05 vs CaV3.2 (two-tailed unpaired Student's t test);
*****p < 0.05 vs CaV3.3 (two-tailed unpaired Student's t test).
We next investigated the potential modulation of CaV3.2 and CaV3.3 by CACHD1. The peak current densities of CaV3.2 (Fig. 7A,C) and CaV3.3 (Fig. 7B,D) were increased by CACHD1 with corresponding increases in maximal conductance (Table 1). CACHD1 had no significant effect on the midpoint of activation or slope factor k for either CaV3.2 or CaV3.3 (Table 1) or for steady-state inactivation (p > 0.1, Kruskal–Wallis test with Dunn's multiple-comparison test; data not shown). CACHD1 was without effect on CaV3 activation or inactivation kinetics (Fig. 7-1; Table 1). In our hands, α2δ-1 was without effect on current density in CaV3.2 (Fig. 7E) or CaV3.3 (Fig. 7F). α2δ-1 was without effect on CaV3.2 activation kinetics or on CaV3.2 and CaV3.3 inactivation kinetics (Fig. 7-1; Table 1). α2δ-1 had subtle effects on CaV3.1 activation and inactivation kinetics (Table 1) and CaV3.3 activation kinetics (Fig. 7-1; Table 1). Overall, these data suggest that CACHD1, but not α2δ-1, has a major effect on recombinant CaV3 VGCCs in terms of increased Ca2+ current density and maximal conductance.
Figure 7.

Effects of CACHD1 and α2δ-1 on CaV3.2 and CaV3.3 channels. A–D, CACHD1 significantly increased current density, as shown by representative current density traces at −20 mV for CaV3.2 (A) and CaV3.3 (C), and I–V relationships for CaV3.2 (B) and CaV3.3 (D), VH −90 mV (*p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA with Bonferroni post hoc test). α2δ-1 had no effect on CaV3.2 (E) and CaV3.3 (F) I–V relationships, holding potential (VH) −90 mV. The figure is supported by analysis of effects of CACHD1 and α2δ-1 on CaV3 channel kinetic properties (Figure 7-1).
Effects of CACHD1 and α2δ-1 on CaV3 channel kinetic properties. CACHD1 co-expression had no significant effect on tactivation in (Aa) CaV3.1, (Ba) CaV3.2 and (Ca) CaV3.3. α2δ-1 significantly increased CaV3.1 tactivation at all voltages tested (Aa) (*p<0.05, **p<0.01, ***p<0.001, two-way ANOVA with Bonferroni post-hoc test); α2δ-1 had no effect on CaV3.2 tactivation (Ba); α2δ-1 significantly decreased CaV3.3 tactivation at -35 and -30 mV (Ca) (*p<0.05, ***p<0.001, two-way ANOVA with Bonferroni post-hoc test). CACHD1 co-expression had no significant effect on tinactivation in (Ab) CaV3.1, (Bb) CaV3.2 and (Cb) CaV3.3. α2δ-1 co-expression with CaV3.1 (Ab) resulted in significantly faster inactivation kinetics (*p<0.05, one-way ANOVA with Bonferroni post-hoc test), but had no effect on tinactivation in (Bb) CaV3.2 and (Cb) CaV3.3. Inactivation traces at -20 mV or -30 mV were fitted with a single exponential function. Download Figure 7-1, TIF file (10.9MB, tif)
To determine the mechanism by which CACHD1 increased T-type channel currents, we estimated channel opening probability by measuring CaV3.1 gating currents at the reversal potential for the ionic current (Fig. 8Aab). In these experiments, the CACHD1-mediated increase in current density was recapitulated; thus, CaV3.1 maximal conductance of 280 ± 30 pS/pF was significantly increased to 860 ± 15 pA/pF (n = 12 for each condition from three separate transfections; p < 0.001, Mann–Whitney test; data not shown). Measurement of the area under the gating current provides a measure of the Qmax value. A plot of conductance versus gating current amplitude of the ionic current of the same cell (Fig. 8B) provides a measure of Po (Agler et al., 2005). Under these conditions, there was an ∼1.4-fold increase in CaV3.1 Po in CACHD1-expressing cells (p < 0.001; Fig. 8C). These findings are consistent with the CACHD1 interaction with CaV3.1 at the cell surface, causing a functional increase in Po as a major contribution to CACHD1-mediated increases in Ca2+ current density.
Figure 8.

CACHD1 expression increases CaV3.1 gating currents and Po. Aa, Ab, Representative gating currents recorded from CaV3.1 (Aa) and CaV3.1 + CACHD1 (Ab) at the observed reversal potential. Expanded time scale illustrates the increase in the area under the gating current for CACHD1-expressed cells. B, Conductance vs gating current plot for multiple cells. Line represents linear regression to data points. The slopes (Gmax/Qmax) were significantly different (p = 0.0004, least-squares fits compared using extra sum of squares F test; CaV3.1: 0.09 ± 0.003, n = 10; CaV3.1 + CACHD1: 0.14 ± 0.090, n = 11). C, Plot showing the slopes (i.e., relative Po) and SEM for fits shown in B (***p < 0.001).
CACHD1 increase CaV3-mediated excitability in hippocampal neurons
CaV3 channels are predicted to affect neuronal excitability around the resting membrane potential (Perez-Reyes, 2003; Cheong and Shin, 2013). To investigate the role of CACHD1 in controlling neuronal excitability, we expressed CACHD1 (vs empty vector controls) in hippocampal neurons. Transfected neurons were identified by coexpression of the biomarker mVenus (Fig. 9A). At a depolarizing current injection step of 220 pA, CACHD1-expressing neurons fired at a higher frequency than control neurons (Fig. 9B,C,D; Table 2). To further determine the role of T-type currents in establishing the increase in neuronal firing frequencies, we used the selective CaV3 channel blocker TTA-P2 (Dreyfus et al., 2010). TTA-P2 (1 μm) reversed the firing frequency in CACHD1-expressing neurons back to control levels, but was without effect on control neurons (Fig. 9D; Table 2). To increase the contribution of T-type current to neuronal excitability, a hyperpolarizing prepulse was used to recover LVA Ca2+ channels from inactivation, followed by a short depolarizing pulse to evoke an action potential (AP) (Eckle et al., 2014). Under these conditions, CACHD1 expression caused a more profound increase in rebound firing frequency in CACHD1-transfected neurons, but not in control neurons (Fig. 9E,F,G; Table 2). TTA-P2 (1 μm) reversed the increase in rebound AP firing in CACHD1-expressing neurons back to control levels, but was without effect on control neurons (Fig. 9G; Table 2). Throughout these experiments, CACHD1 had no significant effects on AP waveform properties (Fig. 9-1). These data support a CACHD1-mediated selective increase in T-type Ca2+ current, which leads to an increase in AP firing frequency and excitability in native neurons.
Figure 9.

Effects of CACHD1 in hippocampal neurons. A, Colabeling of hippocampal neurons with CACHD1 and mVenus. B, CACHD1 increased the firing frequency of hippocampal neurons. C, Example traces representing depolarizing current injections steps of −20, 70, and 140 pA. D, Summary data from separate experiments confirming CACHD1-mediated increased firing frequency and also showing that TTA-P2 (1 μm) reduced firing rates in CACHD1-expressing neurons, but not in controls. E, Rebound APs were evoked using a −50 pA hyperpolarizing prepulse followed by a depolarizing step from 0 to 200 pA in steps of 10 pA for 200 ms, CACHD1-expressing neurons displayed a significantly greater number of rebound APs compared with controls. F, Example traces representing depolarizing current injection steps of 40, 90 and 140 pA. G, Summary data from separate experiments confirming CACHD1-mediated increased in rebound APs and also showing that TTA-P2 (1 μm) reduced firing rates in CACHD1-expressing neurons, but not in controls. *p < 0.05 throughout, two-tailed paired Student's t test or one-way ANOVA with Bonferroni post hoc test. Figure 9 is supported by analysis of the effects of CACHD1 and TTA-P2 on biophysical properties of hippocampal neurons (Figure 9-1).
Table 2.
Effects of CACHD1 and TTA-P2 on hippocampal neuronal firing
| Firing frequency (Hz) | Rebound firing frequency (Hz) | |
|---|---|---|
| Control | 6.0 ± 1.2 (41/6) | 7.2 ± 1.2 (32/5) |
| CACHD1 | 9.8 ± 1.1* (29/5) | 12.1 ± 0.9* (28/5) |
| Control | 8.5 ± 1.4 (6/3) | 10.0 ± 1.8 (6/3) |
| Control + TTA-P2 | 6.5 ± 1.2 (6/3) | 9.2 ± 1.5 (6/3) |
| CACHD1 | 14.1 ± 1.7 (7/3) | 16.7 ± 0.8 (10/3) |
| CACHD1 + TTA-P2 | 6.9 ± 1.4* (7/3) | 10.0 ± 1.2* (10/3) |
Values represent the mean ± SEM; the numbers in parentheses represent the number of neurons/number of separate transfections.
*p < 0.05 vs control two-tailed paired Student's t test.
Effects of CACHD1 and TTA-P2 on biophysical properties of hippocampal neurons. Download Figure 9-1, TIF file (10.9MB, tif)
Discussion
This study characterizes the protein CACHD1, encoded by the cache domain containing 1 gene, and presents evidence that it represents a novel protein that modulates CaV3 VGCC activity. These data also provide further evidence that the major α2δ-1 auxiliary calcium channel subunit does not fulfil a similar role for CaV3 channels. Detailed examination of CaV3.1 channels suggests an underlying mechanism whereby CACHD1 promotes increased CaV3.1 levels at the plasma membrane. In addition, data were consistent with CACHD1 forming a complex with the channel at the cell surface to increase open probability and potentiate T-type current.
CACHD1 protein modulates CaV3 VGCCs
At a cellular level, CACHD1 transcripts were localized to granule and pyramidal cells of the hippocampus, and specific thalamic nuclei, notably the anterodorsal thalamic nucleus and reticular nucleus. Compared with the gene expression of the major α2δ-1 and α2δ-2 subunits, CACHD1 protein displayed a unique expression signature with, in particular, high expression in the thalamus and hippocampus and also in some regions of the cerebellum and cortex. CACHD1 was largely coincident with the expression pattern of the CaV3.1 channel in the CNS (Talley et al., 1999). CACHD1 cotransfection with CaV3.1 in recombinant cells increased cell surface expression and Ca2+ current levels and maximal conductance. CACHD1 similarly modulated CaV3.2 and CaV3.3 current levels. Under equivalent conditions, α2δ-1 was without significant effect on current levels in any CaV3 subtype. Proximity ligation assays were consistent with CACHD1 being able to form complexes with CaV3.1 at the cell surface. Mechanistically, CACHD1 effects on CaV3.1 were associated with increases in channel Po. A similar role has been reported for α2δ auxiliary subunit interactions with CaV1 channels; thus, α2δ-1 increased channel Po and channel number, and allosterically regulated drug binding (Shistik et al., 1995; Wei et al., 1995). Other studies have reported either an α2δ-mediated reduction in Po (Wakamori et al., 1999) or a lack of effect on Po (Brodbeck et al., 2002). The latter study suggested that α2δ predominantly performs a VGCC trafficking function to increase the number of active channels at the membrane (for review, see Dolphin, 2012). The demonstrated CACHD1-mediated increase in CaV3.1 cell-surface expression is proposed to contribute to an increase in cell Ca2+ current levels and maximal conductance. Here, the ∼1.4-fold increase in Po is insufficient to fully account for the approximately threefold increase in current density seen in this set of experiments; channel number is predicted to increase [according to I = iNPo, where I is the whole-cell current, i is the single channel current (predicted to be constant), and N is the number of functional channels]. Thus, increase in channel number may be attributable to either CACHD1-mediated increases in forward trafficking or reduced endocytosis of CaV3.1. With respect to α2δ auxiliary subunits, HVA CaVα1–α2δ interactions are reported to occur during early maturation at an intracellular site to drive forward trafficking to the plasma membrane (Cantí et al., 2005). While CaV2.2 proteomic data have reported only a low appreciable amount of copurified α2δ, with detection dependent on the solubilizing agent used (Müller et al., 2010), recent work using exofacial tags and antigen-stripping techniques has supported α2δ also remaining associated with CaV2.2 at the plasma membrane (Cassidy et al., 2014). In the present study, a clear indication of CACHD1 and Cav3.1 complex formation at the cell surface was obtained using proximity ligand assays. Moreover, α2δ has the propensity to sequester into lipid raft compartments, as reported by us (Ronzitti et al., 2014) and others; this may also limit efficient detection of α2δ–CaVα1 complexes, and it will be of interest to determine whether CACHD1 similarly localizes to lipid rafts. Overall, we propose that CACHD1 acts to increase CaV3 expression at the plasma membrane; at the cell surface, CACHD1 can form a complex with the channel to increase Po and, consequently, to increase T-type current.
Potential functional impact of CACHD1 on CaV3 VGCCs
T-type Ca2+ currents are active around the resting membrane potential, where noninactivating channels generate low-threshold Ca2+ spikes and the consequential triggering of Na+-dependent APs (Llinás 1988; Cheong and Shin, 2013). Of further interest here is that multiple mechanisms and proteins involved in folding and trafficking are reported to be involved in CaV3 expression at the cell surface. For example, proteins such the actin binding protein kelch-like 1 (Aromolaran et al., 2010), stac1 (Rzhepetskyy et al., 2016), and calnexin (Proft et al., 2017) have a proposed role in CaV3 expression. Moreover, the glycosylated form of CaV3 represents the mature, correctly folded protein that is associated with higher Po (Weiss et al., 2013; Ondacova et al., 2016). T-type current has also been implicated in regulating presynaptic transmitter release in hippocampal and nociceptive circuitry (Huang et al., 2011; Jacus et al., 2012). Increases in CaV3 current are predicted to have profound effects on neuronal firing (McCormick and Huguenard, 1992). Correspondingly, the overexpression of CACHD1 caused a pronounced increase in T-type current-mediated spike firing in hippocampal neurons. This activity was enhanced using a protocol to trigger the recovery of CaV3 channels from their inactivated states, thereby increasing the contribution of T-type current to neuronal excitability. CaV3 subtypes have been suggested as targets for antiepileptic drugs (Powell et al., 2014). In models of temporal lobe epilepsy (TLE), selective upregulation of T-type current in hippocampal neurons causes intrinsic bursting activity (Sanabria et al., 2001; Su et al., 2002). CaV3.2 transcripts were upregulated in TLE models, and intrinsic burst firing was reduced in CaV3.2 knock-out mice (Becker et al., 2008). Moreover, the deubiquitinating enzyme USP5 (García-Caballero et al., 2014) and the prevention of CaV3.2 deubiquitination were suggested to be beneficial in neuropathic and inflammatory pain. Our data suggest CACHD1 as a potential future target in hyperexcitability disorders associated with CaV3 dysfunction, such as epilepsy and pain. Moreover, CACHD1 gene expression has been shown to be modulated in patients with type 1 diabetes (Rassi et al., 2008) and Parkinson's disease (Aguiar and Severino, 2010).
CACHD1 protein structure dictates α2δ-like function
There are clear similarities in protein structural motifs between CACHD1 and α2δ, namely, the presence of an N-terminal signal sequence, VWA, and two downstream cache domains, and these similarities suggest a conserved evolution (Anantharaman and Aravind, 2000). However, a number of important differences are also present. CACHD1 has an RSR variant at the gabapentin binding motif; α2δ-1 and α2δ-2 were found to bind to gabapentinoids via their RRR binding motif and α2δ-3 and α2δ-4 have variant RNR sites that do not bind gabapentin (Wang et al., 1999; Marais et al., 2001). Earlier studies also identified porcine α2δ-1 residues 516–537 within the first cache domain and residues 583–603 as also contributing to gabapentin binding (Wang et al., 1999). It will be of interest to determine whether CACHD1 binds gabapentinoids. Despite sharing a common VWA domain, CACHD1 has a variant MIDAS motif. The α2δ-1 MIDAS motif is functionally important in Ca2+ channel trafficking and synaptic function (Cantí et al., 2005; Hoppa et al., 2012). However, it has been suggested that MIDAS is unlikely to represent a key CaV2.2/α2δ-1 interaction site, and, rather, other regions are more likely involved (Cassidy et al., 2014); such regions may include cache domains, for example, rat α2δ-1 residues 751–755, which are within a modeled cache region, were implicated in CaV2.2/α2δ-1 interaction (Cassidy et al., 2014). By contrast, comparative data investigating α2δ effects on CaV1.2 point to aspartate and the first serine residue within the DxSxS MIDAS site as molecular determinants for interaction and correct modulation of CaV1.2 (Briot et al., 2018). Of interest here is that CACHD1 contains a variant MIDAS with a glycine residue at the equivalent position of the critical serine residue identified by Briot et al. (2018). It has also been proposed that the α2δ N terminal (amino acids 26–230, termed the R-domain) contains all the machinery required to support α2δ-1-mediated current enhancement in CaV2.2 channels (Song et al., 2015). This study identified a tryptophan residue (W205), which is conserved across all four α2δ isoforms, as an important molecular determinant for these R-domain effects; it is of note that CACHD1 also contains a conserved tryptophan residue at the equivalent position.
In bacteria, the cache domain is proposed to arise from bacterial small-molecule binding domains PAS and GAF (Anantharaman et al., 2001) and to play a key role in chemotaxis by acting as an extracellular receptor (Anantharaman and Aravind, 2000). Computational work has suggested that cache domains represent the dominant extracellular sensor in prokaryotes; by contrast, cache domains are largely limited to only α2δ subunits in metazoa (Upadhyay et al., 2016). The present study adds CACHD1 to this classification. While the functional relevance of mammalian cache domains remains to be fully established, deletions within the cache domain of α2δ-4 have been associated with familial bipolar disorder (Van Den Bossche et al., 2012). Roles for 'free' α2δ (not associated with VGCCs) have also been extended to functions including synaptogenesis and neurodegeneration via interaction with alternative ligands such as thrombospondins and prion proteins, respectively (Eroglu et al., 2009, Lana et al., 2016; Senatore et al., 2012); it will also be of interest to see whether CACHD1 possesses similar functionality.
Overall, our data are consistent with CACHD1 structurally representing an α2δ-like protein that acts to increase CaV3 cell surface expression and current. Identification of the CACHD1 protein as a modulator of Cav3 activity expands the range of VGCC-associated proteins and may provide an additional target itself, or via its modulation of T-type current, in different disease states.
Footnotes
This work was supported by a Biotechnology and Biological Sciences Research Council Departmental Training Grant with a CASE Award from Pfizer UK to G.J.S. and E.B.S. to support C.H.S. This work was also supported by National Institutes of Health (NIH)/NINDS Grant NS-075157 to M.K.P. and NIH Grant NS-069524 to E.P.R.
The authors declare no competing financial interests.
References
- Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT (2005) G protein-gated inhibitory module of N-type (CaV2.2) Ca2+ channels. Neuron 46:891–904. 10.1016/j.neuron.2005.05.011 [DOI] [PubMed] [Google Scholar]
- Aguiar PM, Severino P (2010) Biomarkers in Parkinson disease: global gene expression analysis in peripheral blood from patients with and without mutations in PARK2 and PARK8. Einstein (Sao Paulo) 8:291–297. 10.1590/s1679-45082010ao1674 [DOI] [PubMed] [Google Scholar]
- Anantharaman V, Aravind L (2000) Cache - a signaling domain common to animal Ca2+-channel subunits and a class of prokaryotic chemotaxis receptors. Trends Biochem Sci 25:535–537. 10.1016/S0968-0004(00)01672-8 [DOI] [PubMed] [Google Scholar]
- Anantharaman V, Koonin EV, Aravind L (2001) Regulatory potential, phyletic distribution and evolution of ancient, intracellular small-molecule-binding domains. J Mol Biol 307:1271–1292. 10.1006/jmbi.2001.4508 [DOI] [PubMed] [Google Scholar]
- Andrade A, Sandoval A, Oviedo N, De Waard M, Elias D, Felix R (2007) Proteolytic cleavage of the voltage-gated Ca2+ channel α2δ subunit: structural and functional features. Eur J Neurosci 25:1705–1710. 10.1111/j.1460-9568.2007.05454.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aromolaran KA, Benzow KA, Cribbs LL, Koob MD, Piedras-Rentería ES (2010) T-type current modulation by the actin-binding protein kelch-like 1. Am J Physiol Cell Physiol 298:C1353–C1362. 10.1152/ajpcell.00235.2009 [DOI] [PubMed] [Google Scholar]
- Becker AJ, Pitsch J, Sochivko D, Opitz T, Staniek M, Chen CC, Campbell KP, Schoch S, Yaari Y, Beck H (2008) Transcriptional upregulation of CaV3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. J Neurosci 28:13341–13353. 10.1523/JNEUROSCI.1421-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briot J, Mailhot O, Bourdin B, Tétreault MP, Najmanovich R, Parent L (2018) A three-way inter-molecular network accounts for the CaVα2δ1-induced functional modulation of the pore-forming CaV1.2 subunit. J Biol Chem 293:7176–7188. 10.1074/jbc.RA118.001902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbeck J, Davies A, Courtney JM, Meir A, Balaguero N, Canti C, Moss FJ, Page KM, Pratt WS, Hunt SP, Barclay J, Rees M, Dolphin AC (2002) The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated α2δ2 protein with abnormal function. J Biol Chem 277:7684–7693. 10.1074/jbc.M109404200 [DOI] [PubMed] [Google Scholar]
- Calderón-Rivera A, Andrade A, Hernández-Hernández O, González-Ramírez R, Sandoval A, Rivera M, Gomora JC, Felix R (2012) Identification of a disulfide bridge essential for structure and function of the voltage-gated Ca2+ channel α2δ-1 auxiliary subunit. Cell Calcium 51:22–30. 10.1016/j.ceca.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantí C, Nieto-Rostro M, Foucault I, Heblich F, Wratten J, Richards MW, Hendrich J, Douglas L, Page KM, Davies A, Dolphin AC (2005) The metal-ion-dependent adhesion site in the von willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A 102:11230–11235. 10.1073/pnas.0504183102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy JS, Ferron L, Kadurin I, Pratt WS, Dolphin AC (2014) Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary α2δ-1 subunits. Proc Natl Acad Sci U S A 111:8979–8984. 10.1073/pnas.1403731111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong E, Shin HS (2013) T-type Ca2+ channels in normal and abnormal brain functions. Physiol Rev 93:961–992. 10.1152/physrev.00010.2012 [DOI] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA, Gu G (2005) Differential distribution of voltage-gated calcium channel alpha-2 delta (α2δ) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491:246–269. 10.1002/cne.20693 [DOI] [PubMed] [Google Scholar]
- Cottrell GS, Padilla B, Pikios S, Roosterman D, Steinhoff M, Grady EF, Bunnett NW (2007) Post-endocytic sorting of calcitonin receptor-like receptor and receptor activity-modifying protein 1. J Biol Chem 282:12260–12271. 10.1074/jbc.M606338200 [DOI] [PubMed] [Google Scholar]
- Davies A, Kadurin I, Alvarez-Laviada A, Douglas L, Nieto-Rostro M, Bauer CS, Pratt WS, Dolphin AC (2010) The α2δ subunits of voltage-gated calcium channels form GPI anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci U S A 107:1654–1659. 10.1073/pnas.0908735107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. (2012) Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci 13:542–555. 10.1038/nrn3311 [DOI] [PubMed] [Google Scholar]
- Dolphin AC. (2013) The α2δ subunits of voltage-gated calcium channels. Biochim Biophys Acta 1828:1541–1549. 10.1016/j.bbamem.2012.11.019 [DOI] [PubMed] [Google Scholar]
- Dolphin AC, Wyatt CN, Richards J, Beattie RE, Craig P, Lee JH, Cribbs LL, Volsen SG, Perez-Reyes E (1999) The effect of α2δ and other accessory subunits on expression and properties of the calcium channel α1G. J Physiol 519:35–45. 10.1111/j.1469-7793.1999.0035o.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley DJ, Taylor CP, Donevan S, Feltner D (2007) Ca2+ channel α2δ ligands: novel modulators of neurotransmission. Trends Pharmacol Sci 28:75–82. 10.1016/j.tips.2006.12.006 [DOI] [PubMed] [Google Scholar]
- Douglas L, Davies A, Wratten J, Dolphin AC (2006) Do voltage-gated calcium channel α2δ subunits require proteolytic processing into α2 and δ to be functional? Biochem Soc Trans 34:894–898. 10.1042/BST0340894 [DOI] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N (2010) Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of ITwindow. J Neurosci 30:99–109. 10.1523/JNEUROSCI.4305-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubel SJ, Altier C, Chaumont S, Lory P, Bourinet E, Nargeot J (2004) Plasma membrane expression of T-type calcium channel α1 subunits is modulated by high voltage-activated auxiliary subunits. J Biol Chem 279:29263–29269. 10.1074/jbc.M313450200 [DOI] [PubMed] [Google Scholar]
- Eckle VS, Shcheglovitov A, Vitko I, Dey D, Yap CC, Winckler B, Perez-Reyes E (2014) Mechanisms by which a CACNA1H mutation found in epilepsy patients increases seizure susceptibility. J Physiol 592:795–809. 10.1113/jphysiol.2013.264176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA (2009) Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139:380–392. 10.1016/j.cell.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, Bladen C, Chen L, Hamid J, Pizzoccaro A, Deage M, François A, Bourinet E, Zamponi GW (2014) The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 83:1144–1158. 10.1016/j.neuron.2014.07.036 [DOI] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA (2012) α2δ couples calcium channels to neurotransmitter release sites to control release probability. Nature 486:122–125. 10.1038/nature11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, Dolphin AC, Shah MM (2011) Presynaptic HCN1 channels regulate CaV3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci 14:478–486. 10.1038/nn.2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacus MO, Uebele VN, Renger JJ, Todorovic SM (2012) Presynaptic CaV3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J Neurosci 32:9374–9382. 10.1523/JNEUROSCI.0068-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PJ, Wang Y, Smith MD, Hargus NJ, Eidam HS, White HS, Kapur J, Brown ML, Patel MK (2007) Hydroxyamide analogs of propofol exhibit state-dependent block of sodium channels in hippocampal neurons: implications for anticonvulsant activity. J Pharmacol Exp Ther 320:828–836. 10.1124/jpet.106.111542 [DOI] [PubMed] [Google Scholar]
- Lacinová L, Klugbauer N, Hofmann F (1999) Absence of modulation of the expressed calcium channel α1G subunit by α2δ subunits. J Physiol 516:639–645. 10.1111/j.1469-7793.1999.0639u.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana B, Page KM, Kadurin I, Ho S, Nieto-Rostro M, Dolphin AC (2016) Thrombospondin-4 reduces binding affinity of [3H]-gabapentin to calcium-channel α2δ-1 subunit but does not interact with α2δ-1 on the cell-surface when co-expressed. Sci Rep 6:24531. 10.1038/srep24531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás RR. (1988) The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science 242:1654–1664. 10.1126/science.3059497 [DOI] [PubMed] [Google Scholar]
- Marais E, Klugbauer N, Hofmann F (2001) Calcium channel α2δ subunits-structure and gabapentin binding. Mol Pharmacol 59:1243–1248. 10.1124/mol.59.5.1243 [DOI] [PubMed] [Google Scholar]
- McCormick DA, Huguenard JR (1992) A model of the electrophysiological properties of thalamocortical relay neurons. J Neurophysiol 68:1384–1400. 10.1152/jn.1992.68.4.1384 [DOI] [PubMed] [Google Scholar]
- Müller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B, Schulte U (2010) Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A 107:14950–14957. 10.1073/pnas.1005940107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondacova K, Karmazinova M, Lazniewska J, Weiss N, Lacinova L (2016) Modulation of Cav3.2 T-type calcium channel permeability by asparagine-linked glycosylation. Channels (Austin) 10:175–184. 10.1080/19336950.2016.1138189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. (2003) Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev 83:117–161. 10.1152/physrev.00018.2002 [DOI] [PubMed] [Google Scholar]
- Powell KL, Cain SM, Snutch TP, O'Brien TJ (2014) Low threshold T-type calcium channels as targets for novel epilepsy treatments. Br J Clin Pharmacol 77:729–739. 10.1111/bcp.12205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proft J, Rzhepetskyy Y, Lazniewska J, Zhang FX, Cain SM, Snutch TP, Zamponi GW, Weiss N (2017) The Cacna1h mutation in the GAERS model of absence epilepsy enhances T-type Ca2+ currents by altering calnexin-dependent trafficking of CaV3.2 channels. Sci Rep 7:11513. 10.1038/s41598-017-11591-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassi DM, Junta CM, Fachin AL, Sandrin-Garcia P, Mello S, Silva GL, Evangelista AF, Magalhães DA, Wastowski IJ, Crispim JO, Martelli-Palomino G, Fernandes AP, Deghaide NN, Foss-Freitas MC, Foss MC, Soares CP, Sakamoto-Hojo ET, Passos GA, Donadi EA (2008) Gene expression profiles stratified according to type 1 diabetes mellitus susceptibility regions. Ann N Y Acad Sci 1150:282–289. 10.1196/annals.1447.064 [DOI] [PubMed] [Google Scholar]
- Ronzitti G, Bucci G, Emanuele M, Leo D, Sotnikova TD, Mus LV, Soubrane CH, Dallas ML, Thalhammer A, Cingolani LA, Mochida S, Gainetdinov RR, Stephens GJ, Chieregatti E (2014) Exogenous α-synuclein decreases raft partitioning of CaV2.2 channels inducing dopamine release. J Neurosci 34:10603–10615. 10.1523/JNEUROSCI.0608-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzhepetskyy Y, Lazniewska J, Proft J, Campiglio M, Flucher BE, Weiss N (2016) A CaV3.2/Stac1 molecular complex controls T-type channel expression at the plasma membrane. Channels (Austin) 10:346–354. 10.1080/19336950.2016.1186318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanabria ER, Su H, Yaari Y (2001) Initiation of network bursts by Ca2+-dependent intrinsic bursting in the rat pilocarpine model of temporal lobe epilepsy. J Physiol 532:205–216. 10.1111/j.1469-7793.2001.0205g.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Hosy E, Kohl J, Klueva J, Choquet D, Thomas U, Voigt A, Heine M (2015) Mobility of calcium channels in the presynaptic membrane. Neuron 86:672–679. 10.1016/j.neuron.2015.03.050 [DOI] [PubMed] [Google Scholar]
- Segura E, Bourdin B, Tétreault MP, Briot J, Allen BG, Mayer G, Parent L (2017) Proteolytic cleavage of the hydrophobic domain in the CaVα2δ1 subunit improves assembly and activity of cardiac CaV1.2 channels. J Biol Chem 292:11109–11124. 10.1074/jbc.M117.784355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatore A, Colleoni S, Verderio C, Restelli E, Morini R, Condliffe SB, Bertani I, Mantovani S, Canovi M, Micotti E, Forloni G, Dolphin AC, Matteoli M, Gobbi M, Chiesa R (2012) Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC α2δ-1 subunit. Neuron 74:300–313. 10.1016/j.neuron.2012.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheglovitov A, Vitko I, Bidaud I, Baumgart JP, Navarro-Gonzalez MF, Grayson TH, Lory P, Hill CE, Perez-Reyes E (2008) Alternative splicing within the I-II loop controls surface expression of T-type CaV3.1 calcium channels. FEBS Lett 582:3765–3770. 10.1016/j.febslet.2008.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shistik E, Ivanina T, Puri T, Hosey M, Dascal N (1995) Ca2+ current enhancement by α2/δ and β subunits in xenopus oocytes: contribution of changes in channel gating and α1 protein level. J Physiol 489:55–62. 10.1113/jphysiol.1995.sp021029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snutch TP, Zamponi GW (2018) Recent advances in the development of T-type calcium channel blockers for pain intervention. Br J Pharmacol 175:2375–2383. 10.1111/bph.13906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Espinoza-Fuenzalida IA, Etheridge S, Jones OT, Fitzgerald EM (2015) The R-domain: identification of an N-terminal region of the α2δ-1 subunit which is necessary and sufficient for its effects on CaV2.2 calcium currents. Curr Mol Pharmacol 8:169–179. 10.2174/1874467208666150507104555 [DOI] [PubMed] [Google Scholar]
- Soubrane CH, Stevens EB, Stephens GJ (2012) Expression and functional studies of the novel CNS protein CACHD1. Paper presented at Physiology 2012, Edinburgh, UK, July. [Google Scholar]
- Su H, Sochivko D, Becker A, Chen J, Jiang Y, Yaari Y, Beck H (2002) Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci 22:3645–3655. 10.1523/JNEUROSCI.22-09-03645.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA (1999) Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 19:1895–1911. 10.1523/JNEUROSCI.19-06-01895.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay AA, Fleetwood AD, Adebali O, Finn RD, Zhulin IB (2016) Cache domains that are homologous to, but different from PAS domains comprise the largest superfamily of extracellular sensors in prokaryotes. PLoS Comput Biol 12:e1004862. 10.1371/journal.pcbi.1004862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bossche MJ, Strazisar M, De Bruyne S, Bervoets C, Lenaerts AS, De Zutter S, Nordin A, Norrback KF, Goossens D, De Rijk P, Green EK, Grozeva D, Mendlewicz J, Craddock N, Sabbe BG, Adolfsson R, Souery D, Del-Favero J (2012) Identification of a CACNA2D4 deletion in late onset bipolar disorder patients and implications for the involvement of voltage-dependent calcium channels in psychiatric disorders. Am J Med Genet B Neuropsychiatr Genet 159B:465–475. 10.1002/ajmg.b.32053 [DOI] [PubMed] [Google Scholar]
- Vogl C, Tanifuji S, Danis B, Daniels V, Foerch P, Wolff C, Whalley BJ, Mochida S, Stephens GJ (2015) Synaptic vesicle glycoprotein 2A modulates vesicular release and calcium channel function at peripheral sympathetic synapses. Eur J Neurosci 41:398–409. 10.1111/ejn.12799 [DOI] [PubMed] [Google Scholar]
- Wakamori M, Mikala G, Mori Y (1999) Auxiliary subunits operate as a molecular switch in determining gating behaviour of the unitary N-type Ca2+ channel current in Xenopus oocytes. J Physiol 517:659–672. 10.1111/j.1469-7793.1999.0659s.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Offord J, Oxender DL, Su TZ (1999) Structural requirement of the calcium-channel subunit α2δ for gabapentin binding. Biochem J 342:313–320. 10.1042/bj3420313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Pan S, Lang W, Kim H, Schneider T, Perez-Reyes E, Birnbaumer L (1995) Molecular determinants of cardiac Ca2+ channel pharmacology. subunit requirement for the high affinity and allosteric regulation of dihydropyridine binding. J Biol Chem 270:27106–27111. 10.1074/jbc.270.45.27106 [DOI] [PubMed] [Google Scholar]
- Weiss N, Black SA, Bladen C, Chen L, Zamponi GW (2013) Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch 465:1159–1170. 10.1007/s00424-013-1259-3 [DOI] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Horwitz AF (2003) Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol 161:131–142. 10.1083/jcb.200211002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CACHD1 mRNA expression in adult rat brain. In situ hybridization of adult rat brain. CACHD1 mRNA was labelled pink with blue counterstain (Gill’s I Haematoxylin). CA1-3: cornus ammonis 1-3; DG: dentate gyrus; g: granule cell layer; m: molecular layer; p: Purkinje cell; wm: white matter. Download Figure 1-1, DOCX file (19.2KB, docx)
Qualitative expression profile of CACHD1 mRNA and protein in the adult rat brain. + labelling similar to background; ++ weak labelling; +++ moderate labelling, ++++ strong labelling; +++++ very strong labelling. Download Figure 1-2, DOCX file (15.5KB, docx)
Expression profile of CACHD1 and voltage-gated calcium channel subunit mRNA in human tissue. Absolute quantification of CACHD1, α2δ-1, -2, -3, CaV2.2 and CaV1, -2, -3 transcripts was assessed in triplicate by TaqMan® qPCR using ‘Best Coverage’ Taqman probes (Applied Biosystems, UK) against a 5-point standard curve of plasmids consisting of 10-fold dilution of a known copy number of plasmid containing cDNA of the gene of interest. Total RNA was extracted using an RNeasy kit (Qiagen, UK) with an on-column DNase I treatment. Additional total RNA samples from AMS Biotechnology (Abingdon, UK) originated from human male donors aged 24-65. Download Figure 2-1, TIF file (10.9MB, tif)
Analysis of cell-surface CACHD1 construct expression. (A, B) HEK cells were transfected with empty vector (vector control, VC) or Myc-CACHD1 and cell lysates analysed by (A) Western blotting (WB) and (B) immunofluorescence and confocal microscopy. (A) Immunoreactive signals for Myc (mouse Myc, mMyc) were detected at a similar molecular mass to that predicted for CACHD1 only in cells expressing CACHD1. (B, upper panel) Cells were incubated with antibody to Myc (rabbit Myc, rMyc), washed, fixed and then incubated with appropriate secondary antibodies. Myc signals (arrowheads) were only detected in cells expressing Myc-CACHD1. (B, lower panel) Cells were fixed, incubated with antibody to Myc (rMyc), washed and then incubated with appropriate secondary antibodies. Myc signals were detected at the cell-surface (arrowheads) and in intracellular vesicles only in cells expressing Myc-CACHD1. Scale bar, 10 µm. Download Figure 5-1, TIF file (10.9MB, tif)
Effects of CACHD1 and α2δ-1 on CaV3 channel kinetic properties. CACHD1 co-expression had no significant effect on tactivation in (Aa) CaV3.1, (Ba) CaV3.2 and (Ca) CaV3.3. α2δ-1 significantly increased CaV3.1 tactivation at all voltages tested (Aa) (*p<0.05, **p<0.01, ***p<0.001, two-way ANOVA with Bonferroni post-hoc test); α2δ-1 had no effect on CaV3.2 tactivation (Ba); α2δ-1 significantly decreased CaV3.3 tactivation at -35 and -30 mV (Ca) (*p<0.05, ***p<0.001, two-way ANOVA with Bonferroni post-hoc test). CACHD1 co-expression had no significant effect on tinactivation in (Ab) CaV3.1, (Bb) CaV3.2 and (Cb) CaV3.3. α2δ-1 co-expression with CaV3.1 (Ab) resulted in significantly faster inactivation kinetics (*p<0.05, one-way ANOVA with Bonferroni post-hoc test), but had no effect on tinactivation in (Bb) CaV3.2 and (Cb) CaV3.3. Inactivation traces at -20 mV or -30 mV were fitted with a single exponential function. Download Figure 7-1, TIF file (10.9MB, tif)
Effects of CACHD1 and TTA-P2 on biophysical properties of hippocampal neurons. Download Figure 9-1, TIF file (10.9MB, tif)