

Abstract
Accumulation of α-Synuclein (α-Syn) causes Parkinson's disease (PD) as well as other synucleopathies. α-Syn is the major component of Lewy bodies and Lewy neurites, the proteinaceous aggregates that are a hallmark of sporadic PD. In familial forms of PD, mutations or copy number variations in SNCA (the α-Syn gene) result in a net increase of its protein levels. Furthermore, common risk variants tied to PD are associated with small increases of wild-type α-Syn levels. These findings are further bolstered by animal studies which show that overexpression of α-Syn is sufficient to cause PD-like features. Thus, increased α-Syn levels are intrinsically tied to PD pathogenesis and underscore the importance of identifying the factors that regulate its levels. In this study, we establish a pooled RNAi screening approach and validation pipeline to probe the druggable genome for modifiers of α-Syn levels and identify 60 promising targets. Using a cross-species, tiered validation approach, we validate six strong candidates that modulate α-Syn levels and toxicity in cell lines, Drosophila, human neurons, and mouse brain of both sexes. More broadly, this genetic strategy and validation pipeline can be applied for the identification of therapeutic targets for disorders driven by dosage-sensitive proteins.
SIGNIFICANCE STATEMENT We present a research strategy for the systematic identification and validation of genes modulating the levels of α-Synuclein, a protein involved in Parkinson's disease. A cell-based screen of the druggable genome (>7,500 genes that are potential therapeutic targets) yielded many modulators of α-Synuclein that were subsequently confirmed and validated in Drosophila, human neurons, and mouse brain. This approach has broad applicability to the multitude of neurological diseases that are caused by mutations in genes whose dosage is critical for brain function.
Keywords: α-Synuclein, modifier, neurodegeneration, pooled screen, protein dosage, shRNA
Introduction
Altered protein levels are at the core of many neurodegenerative diseases. For example, altered levels of the amyloid precursor protein (APP), either due to trisomy of chromosome 21 (Down syndrome) or segmental duplications, cause early-onset Alzheimer's disease (AD) (Wisniewski et al., 1985; Rovelet-Lecrux et al., 2006; Antonarakis, 2017). Similarly, changes in the levels of proteins, such as PMP22, GRN, and Tau, contribute to a broad spectrum of neurodegenerative diseases (Gendron and Petrucelli, 2009; Li et al., 2013; Petkau and Leavitt, 2014; Ward et al., 2017). Thus, studying the molecular mechanisms that govern the levels of disease-linked proteins is essential for understanding disease pathologies and developing therapeutic approaches.
Parkinson's disease (PD) is a prototypical neurodegenerative disease that can be driven by changes in gene dosage and subsequently protein levels. It is characterized pathologically by the accumulation of wild-type α-Synuclein (α-Syn) in proteinaceous aggregates called Lewy bodies, and Lewy neurites (Spillantini et al., 1998; Wong and Krainc, 2017). Moreover, duplications and triplications of the α-Syn gene (SNCA) are sufficient to cause autosomal dominant PD (Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibáñez et al., 2004). Individuals with gene duplications present with clinical features similar to those of idiopathic PD (Chartier-Harlin et al., 2004; Ibáñez et al., 2004), whereas those with gene triplications manifest a more malignant disease phenotype characterized by early age of onset and more rapid progression, including development of dementia (Singleton et al., 2003; Devine et al., 2011). Additionally, PD-associated risk variants in regulatory elements of SNCA cause small increases in its transcript levels (Soldner et al., 2016). Thus, in humans, there is a clear connection between α-Syn levels and disease severity. This finding has been replicated in animal models, as overexpression of wild-type α-Syn is sufficient to drive pathological and behavioral abnormalities similar to those seen in PD (Kirik et al., 2002; Fleming et al., 2004; Chesselet et al., 2012; Chouhan et al., 2016). To date, studies have focused on the downstream effects of α-Syn toxicity and how its accumulation might drive degeneration. However, little is known about the upstream post-transcriptional and post-translational mechanisms that regulate α-Syn levels (Cooper et al., 2006; Kuwahara et al., 2008; Chung et al., 2013; Gonçalves et al., 2016; Yedlapudi et al., 2016; Rousseaux et al., 2017). Given these connections between α-Syn and PD pathogenesis, identifying factors that regulate its levels will shed additional insight into PD pathogenesis and open new therapeutic avenues.
We previously developed an arrayed screening strategy to monitor steady-state levels of dosage-sensitive proteins, such as Ataxin-1 (Park et al., 2013), α-Syn (Rousseaux et al., 2016), and Tau (Lasagna-Reeves et al., 2016; Rousseaux et al., 2016), based on the Global Protein Stability methodology (Yen et al., 2008). Individual samples of cells stably expressing a bicistronic reporter construct were queried by genetic disturbance (siRNA) to identify modifiers of the target proteins' levels. While these screens were successful in identifying modulators, they were limited in their scope because each gene was queried individually. To bypass this limitation, we developed a pooled screening approach for protein levels that was modeled after dropout screens previously used to identify modifiers of cell viability in several cancer types (Westbrook et al., 2005, 2008; Schlabach et al., 2008; Hu et al., 2009; Luo et al., 2009; Emanuele et al., 2011; Kessler et al., 2012). In this study, we developed a strategy that can be generalized to any protein. We describe the approach to screen and validate druggable modulators across different species, including Drosophila, human neurons, and mice. Using this approach, we highlight six among 60 previously unknown regulators of α-Syn levels that provide insight into PD pathogenesis and development of disease modifying therapeutics.
Materials and Methods
Druggable genome library generation
A whole genome shRNA library was designed based on the shERWOOD algorithm (Knott et al., 2014). Ten hairpins with the best shERWOOD score targeting each gene were selected. Hairpins were grouped into different sublibraries based on the functional categorization of their target genes, and synthesized as 97mer oligos on chip by Agilent Technologies. The chip material was amplified using primers SiRNAf: 5′CTAATTGATCTTCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG, Si-RNAr-long: 5′ATGTTACTAACTGAATTCCGAGGCAGTAGGCA and Accurime PFX (Invitrogen) with 7% DMSO. PCR product was then digested with EcoRI and XhoI, gel purified and cloned into EcoRI and XhoI digested pMSCV(Murine Stem Cell Virus)-PM-Phes vector.
Lentiviral production and titering
Library preparation.
Retrovirus was produced using standard protocols. Briefly, low-passage HEK293T (ATCC CRL-3216; RRID:CVCL_0063) cells were transfected at 80%–90% confluency in a 150 mm dish with a 7:1:1 ratio of pMSCV-library, Gag/Pol, and VSV-G (total of 48 μg of DNA, transfected using 144 μl of Mirus TransIT reagent [MIR 2706]). Media was changed the next day to low volume media (10 ml). Supernatants were collected at 48 and 72 h after transfection. The 48 and 72 h supernatants were collected and centrifuged at 4000 × g for 10 min to remove cellular debris. Supernatant was then filtered through a Polyethersulfone filter (VWR, 28145-505) before being aliquoted and snap frozen. Virus was thawed on ice before each use and 4 μg/ml of polybrene (Millipore, TR-1003-G) was added to the viral supernatant to improve infection efficiency.
Titering.
Retroviral titering was performed on Daoy cells (ATCC catalog #HTB-186, RRID:CVCL1167) using serial dilutions of the virus via a variation of the Open Biosystems pGIPZ method (Thermo Fisher Scientific). Because the libraries do not contain a fluorescent reporter, puromycin resistance-based selection was used as a tool to count-positive colonies. Briefly, Daoy cells were plated on a 24-well plate at 1 × 104 cells per well. The next day, serial dilutions of the viral stock were made in a round-bottom 96-well plate. Daoy cells in the 24-well plate were infected with different viral amounts. The next day (36–48 h after plating, 24 h after infection), media was changed to puromycin (1 μg/ml)-containing DMEM (Thermo Fisher Scientific, MT10013CM). Cells were cultured in puromycin for 3 d, changing the media once to remove dead cells. Four days after infection (3 d after selection), puromycin-resistant colonies were counted for each well and viral titer was estimated accordingly. On average, viral titers of the MSCV libraries corresponded to 2–10 × 105 transduction units/ml.
Individual lentivirus preparation.
Individual lentiviral preparations were generated as above with a few alterations. Low-passage HEK293T cells were transfected in 150 mm dish and 96-well format at 80%–90% confluency with a 4:3:1 ratio of pGIPz, psPAX2, pMD2.G (total of 45 μg and 210 ng for 150 mm dish and 96-well formats). Media was collected at 48 and 72 h and frozen down before being used for downstream applications. For viruses produced in 96-well format, viral titer was estimated for a subset of the viruses to ensure adequate viral delivery for downstream applications. For virus produced in 150 mm dishes (used for human neuron experiments), the virus was titered as described in Retroviral production. The only difference in titering method was that we used the GFP marker in the pGIPZ constructs to count colonies. Viruses produced in 150 mm dishes were concentrated 100-fold using Lenti-X concentrator (Clontech, 631231), according to the manufacturer's instructions.
Pooled shRNA screen
Cell culture.
Daoy cells stably expressing a bicistronic dual reporter construct, DsRed-IRES-SNCA-GFP (Yen et al., 2008; Rousseaux et al., 2016) were cultured in DMEM supplemented with 10% FBS (Atlanta Biological, S11150) and 1% antibiotic/antimycotic (Invitrogen, 15240-062). This medium is referred to as complete DMEM. Cells were infected at a multiplicity of infection of 0.3 with a representation of 2000x in 150 mm culture dishes. This number was used as it allows for single-copy integration of viral genomes into target cells. One day after infection, media was changed to puromycin-containing media (1 μg/ml) to select for infected cells. Three days following puromycin selection, media was changed to complete DMEM and cultured for an additional 6 d, for a total of 10 d of culture after infection. Cells were cultured in sufficient numbers to maintain 1000–2000x representation. Of note, an uninfected plate was also subjected to puromycin selection to ensure its efficacy. At each instance of sorting, a plate-containing reporter cells infected with shRNA library was trypsinized and maintained in a single-cell suspension in sorting buffer (1x PBS containing 2% FBS and 1 mM EDTA). This cell suspension was then sorted using a SH800 cell sorter equipped with a 100 μm nozzle (SONY Biotechnology). Cell populations containing the lowest 5% and the highest 5% GFP/DsRed ratio (gated from the alive, single-cell population) were sorted to at least 50–100x representation and pelleted. In each sorting instance, postsort purity confirmed that there was a significant enrichment of cells in the low and high 5% populations (≥85%). For each sorted replicate, cells were spun down for 10 min at 4000 × g at 4°C and frozen for downstream analysis.
Genomic DNA extraction, PCR amplification, and next generation sequencing.
Frozen cell pellets were thawed at room temperature, and genomic DNA was extracted using the Blood and Tissue DNeasy Blood and Tissue Kit (QIAGEN, 69506). The DNA was extracted according to the manufacturer's instructions with the following exceptions: (1) the tissue lysate was applied twice to the DNeasy Mini spin column (step 4 of the Quick start protocol) to increase yield; and (2) Step 8 (DNA elution) was also repeated twice for increased yield. shRNA hairpins were amplified using two consecutive PCRs with the following primers:
PCR1: amplify half-hairpin from genomic DNA.
JH353F-ext: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTAGTGAAGCCACAGATGTA and MSCV 3′ ext KK7: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTATAAACGGTTGGTCTTCCAA.
PCR2: add indices to each sample.
The following index combinations were used for indexing 12 samples at a time (Nextera Index Kit; Illumina, FC-121-1011): N503/N701, N503/N702, N503/N703, N503/N704, N503/N705, N503/N706, N504/N701, N504/N702, N504/N703, N504/N704, N504/N705, and N504/N706.
PCR2 was cleaned up using AMPure XP beads (Beckman, A63881) as per the manufacturer's protocol. Samples were then run on a Pippen prep (Sage Science; target size 339 bp), Qubit (Thermo Fisher Scientific), and Bioanalyzer (Agilent Technologies) to allow for proper quality control. Following this, samples were run on an Illumina HiSeq2500 with 150 bp single-end reads on rapid run mode using a 25% PhiX spike-in to control for sequence clustering and diversity.
Analysis of sequence data from the screen.
FASTQ files generated from the HiSeq2500 runs were analyzed through a standalone pipeline of CRISPRcloud (https://github.com/hyunhwaj/CRISPRcloud-standalone) (Jeong et al., 2017). Based on four different measures of shRNA knockdown efficiency/consistency (hit ratio, directionality score, conflict score, and enrichment score), we set the criteria of goodness of each measure as follows: hit ratio ≥ 0.35; directional score ≥ 0.75; conflict score ≤ 0.2; and enriched shRNAs in the low group ≥ 2.
From these criteria, we defined tiers of each gene with the following rules: Tier 1, genes satisfy all four criteria; Tier 2, genes fulfill three criteria; Tier 3, genes meet any of two criteria; Tier 4, genes satisfy only one criterion; and Tier 5, genes do not meet any criteria.
Results from this analysis yielded 350 Tier 1 genes. Complete analysis is presented in Table 1-1.
α-Syn ELISA
To monitor α-Syn levels, ELISA was performed on HEK293T cells previously infected with lentiviruses harboring shRNAs against the targeted gene of interest; 5 × 103 cells were plated per well in a 96-well flat-bottom dish in quadruplicates. One day later, cells were infected with 40 μl of viral supernatant (corresponding to a multiplicity of infection of ∼5). Two days after infection, media was changed to puromycin-containing media (1 μg/ml) for 3 d to allow for selection of lentivirus expression. turboGFP signal was further monitored to confirm viral expression across wells. Over a period of 11 d, cells were split into 24-well plate format and harvested using RIPA (50 mM Tris, pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate; 1% NP-40, 5 mM EDTA, pH 8.0) buffer-containing protease and phosphatase inhibitors (1×, Gendepot, P3100–100, P3200–020) as well as DNaseI (10 μg/ml, Roche Diagnostics, 10104159001). Because of the dilution step in the ELISA, cells had to be lysed in a low volume (30 μl/well for a 24-well plate) and thus adding DNaseI helped reduce viscosity. Lysis was performed on a rocking platform at 37°C for 10 min as this allowed for the best extraction. A commercial α-Syn ELISA kit (Invitrogen, KHB0061) was used to measure α-Syn levels, and a series of standards ranging from 0 to 15 ng/ml were used to quantify the absolute amount of α-Syn. Samples were then diluted 1:5 in standard dilution buffer before moving forward with the standard kit-provided protocol. Absorbance was read at 450 nm using a Synergy 2 plate reader (Biotek).
Western blot analysis
Samples (either from ELISA or prepared fresh from cells or mouse brain) were lysed in RIPA buffer, diluted 1:1 with Laemmli buffer (2x) (Sigma-Aldrich, S3401-10VL), and heated at 95°C for 10 min; 10–30 μg of sample (concentration measured using the BCA assay, Thermo Fisher Scientific, PI23225) was loaded on a 4%–12% NuPAGE gel (Invitrogen, NP0336BOX). Samples were transferred onto Immobilon-FL PVDF (0.45 μm, 26.5 cm × 3.75 m, Millipore, IPFL00010) in Tris-glycine buffer supplemented with 10% methanol using Mini Trans-Blot Electrophoretic Transfer Cells (Bio-Rad, 1703930) at 340 mA for 1 h at 4°C. Membranes were then dried for 1 h before being reactivated using 100% methanol for 15 s. Reactivated membranes were blocked in LI-COR Odyssey TBS blocking buffer (927–50003) and probed overnight with primary antibody diluted in 0.5x blocking buffer and 0.1% Tween (for antibody information and RRIDs, see Table 1-2). The next day, membranes were washed in TBS-T and incubated in fluorescent secondary antibody (1:10,000 once again diluted in 0.5× TBS blocking buffer with 0.1% Tween) before being imaged on an Odyssey CLx imager (LI-COR).
Drosophila experiments
The Drosophila lines carrying UAS-(α-Syn) optimized for expression in insect cells have been previously described (Chouhan et al., 2016). For pan-neuronal expression, we used the elav-GAL4C155 driver obtained from the Bloomington Drosophila Stock Center (BDSC). The alleles tested as candidate modifiers of the α-Syn-induced motor dysfunction were obtained from the BDSC and from the Vienna Drosophila Resource Center. To assess motor performance of fruit flies as a function of age, we used 15 age-matched virgin females per replica per genotype. Flies were collected in a 24 h period and transferred into a new vial containing 300 μl of media every day. Four replicates were used per genotype. Using an automated platform, the animals were taped to the bottom of a plastic vial and recorded for 7.5 s. Videos were analyzed using custom software to assess the speed and stumbles of each individual animal. Four trials per replicate were performed each day shown, and four replicates per genotype were evaluated. A linear mixed-effect model ANOVA was run using each four replicates to establish statistical significance across genotypes.
Control RNAi lines targeting codon-optimized α-Syn were generated as described previously (Ni et al., 2011). Briefly, shRNA sequences were selected based on the algorithm of Vert et al. (2006). Each shRNA recognizes nonoverlapping sequences of α-Syn cDNA, and no off-target gene was predicted. Hairpin constructs carrying RNAi target sequences were assembled and cloned into Valium22 vectors, then verified by Sanger sequencing. Two shRNA constructs and a Valium22 vector (served as a negative control) were injected individually into y v nanos-integrase; attP40 (y[1], v[1], P{nos-phiC31|int.NLS}X; P{CaryP}attP40) (BDSC catalog #25709, RRID:BDSC_25709) embryos, and the transgenic flies were selected. RNAi target sequences are as follows: α-Syn sh1: CGGGCTTTGTCAAGAAGGATC, and α-Syn sh2: CCAAGGAGGGAGTTCTCTACG.
For Western blot analysis, eight heads from age-matched females were used per genotype. Tissue was homogenized in NuPAGE 4x LDS sample buffer. Samples were run on 12% acrylamide gels. Membranes were stained with primary antibodies overnight (Table 1-2). Fluorescent secondary antibodies were obtained from LI-COR and blots imaged using Odyssey CLx imaging system.
Human embryonic stem cell (hESC)-derived neuron culture
We used WA09 (H9; RRID:CVCL_9773) female embryonic stem cells (ESCs) to generate human neurons as previously described (Jiang et al., 2017). Neural progenitors were differentiated into human neurons over three weeks. During this time the media was changed every 3 d. Afterwards, trypsin was used to dissociates the neurons and passage them. Three days later, cells were infected with lentiviruses containing pGIPZ shRNA clones at a multiplicity of infection of 20. We verified the tropism of the virus and infectivity using the tGFP reporter signal. At day 6 after infection, the neurons were subjected to puromycin selection (0.75–1.25 μg/ml) to select for infected cells. The selection was maintained for 6 d. The cells were cultured until 14 d after infection. Protein and RNA were extracted as described in their respective sections.
qPCR analysis
Relative RNA measurements following shRNA-mediated knockdown were performed as previously described (Rousseaux et al., 2016). Briefly, RNA was isolated from cultured cells or mouse brain (male and female) using Trizol and total RNA was purified using the miRNeasy micro kit (QIAGEN, 217004). cDNA was synthesized using the QuantiTect Reverse Transcription Kit (QIAGEN, 205313), and qPCR was performed using PowerUp SYBR Master Mix (Thermo Fisher Scientific, A25777) and gene-specific primers spanning exon junctions on a Bio-Rad CFX96 real-time PCR machine. Relative transcript abundance was measured using the ddCT method (Pfaffl, 2001) in relation to the housekeeping gene S16 (for mouse transcripts) and GAPDH (for human transcripts). qPCR primers are presented in Table 1-2.
Mouse studies
CFW (MGI catalog #5911387, RRID:MGI:5911387) and CD-1 (IMSR catalog #CRL:22, RRID:IMSR_CRL:22) female and male mice were used for neonatal (P0) viral injection due to their large litter size and good fostering capabilities. Both sexes were used, and up to 5 mice were housed per cage and kept on a 12 h light/12 h dark cycle and were given water and standard rodent chow ad libitum. All procedures performed in mice were approved by the Institutional Animal Care and Use Committee for Baylor College of Medicine and Affiliates.
Adeno-associated virus (AAV)-mediated knockdown in vivo
pAAV-YFP/miRE construct generation and virus production.
An AAV8 vector containing both YFP and a miRE cassette-containing shRNA (Fellmann et al., 2013) under the control of the chicken β actin promoter was generated using Gibson cloning as previously described (Rousseaux et al., 2018). Individual shRNA sequences were generated using the SplashRNA algorithm (Pelossof et al., 2017). Each shRNA vector was tested for efficiency in Neuro2A cells (catalog #400394/p451_Neuro-2A, RRID:CVCL_0470) before virus generation. Briefly, Neuro2A cells (1 × 105) were plated in 24-well plates and transfected with 500 ng of each vector using Lipofectamine 3000 (Thermo Fisher Scientific, L3000150). Transfection efficiency was measured by looking at YFP fluorescence (average ∼60%-75% cells infected) and knockdown efficiency was determined by qPCR.
P0 injection of AAV.
AAV delivery was performed in neonatal (P0) CFW or CD-1 mouse pups as previously described (Kim et al., 2013; Rousseaux et al., 2018). Briefly, neonatal pups (<8 h from birth) were separated from lactating dams and anesthetized on ice; 4 × 1010 viral genomes were injected per ventricle (total of 8 × 1010 genomes per mouse), and mice were left to recover on a heated pad before returning them to their mother. All procedures were performed in a BSL2-contained area, and mouse bedding and housing were changed 72 h after injection. Virus expression confirmation was performed using a BlueStar UV light (Electron Microscopy Sciences) at 3 d after injection (see Fig. 5B). Tissue from the caudal region of the cerebrum (cortex + hippocampus) was harvested 3 weeks after injection as this region had the maximal viral expression (YFP-positive signal) and offered optimal target-gene knockdown by qPCR. Flash frozen tissue was homogenized in a 1.5 ml Eppendorf containing 10 μl/mg of PEPI buffer (1x PBS containing 5 mM EDTA, protease inhibitor mixture, and RNase inhibitor mixture) using an electric pestle. The resulting homogenate was split in a 3:1 ratio for downstream protein (3 parts) and RNA (1 part) applications. RNA extraction was performed using the RNeasy mini kit (QIAGEN), whereas protein extraction was performed by adding equal volumes of 2× RIPA buffer (100 mM Tris, pH 8.0, 300 mM NaCl, 0.2% SDS, 1% sodium deoxycholate, 2% Nonidet P-40, 5 mm EDTA, protease and phosphatase inhibitor cocktails), vortexing and incubating samples on ice for 20 min before spinning lysates down at 16,000 × g for 20 min at 4°C.
Figure 5.

Candidate modulators regulate α-Syn levels in the mammalian brain. A, Approach to testing modulators of α-Syn levels in mouse brain. B, Confirmation of virus expression using UV light on 3-d-old pups (3 d post injection [D.P.I.]) and epifluorescence on cryosectioned tissue from 3-week-old pups (21 D.P.I.). C, Representative Western blots for α-Syn following downregulation of candidate targets Chmp4b, Acsbg1, and Lgals3bp are included above the graphical representation (n = 4–11 samples per condition). D, Each gene was targeted using two independent shRNAs (each bar represents an individual shRNA), except for Chmp4b (one caused perinatal lethality). *q < 0.1 (one-way ANOVA followed by correction for false discovery rate of 10% via the two-stage step-up method of Benjamini and Yekutieli, 2006). See also Table 1-1.
Flow cytometry following transfection of siRNAs or cDNA constructs
To measure the effect of overexpression of modulators on α-Syn levels, we used flow cytometric assisted analysis as described by Rousseaux et al. (2016). Briefly, the two bicistronic Daoy cell lines were cultured in 96-well plates (4,000 cells/well). The first is the same as was used for the pooled screen (DsRed-IRES-SNCA-EGFP); the second is a DsRed-IRES-EGFP control. On the next day, the cells were transfected with 40nMoles of siRNAs using Dharmafect (T-2001-03) or 100 ng of each cDNA construct using MIRUS TransIT-293 reagent (MIR 2706) according to the manufacturer's instructions. At 72 h after transfection, cells were trypsinized and resuspended in sorting buffer (see Cell culture). Flow cytometric measurement of EGFP and DsRed fluorescence ratio was performed using an LSRII Fortessa coupled with an HTS module (BD Biosciences).
Immunoprecipitation
HEK293T cells (500,000 cells/well in 6 well plates) were transfected with 250 ng of each construct bearing a flag tagged modifier using MIRUS TransIT-293 reagent (MIR 2706). At 48 h after transfection, cells were resuspended in 1x PBS and spun down at 5,000 rpm for 5 min at 4°C. Cell pellets were lysed in modified NETN buffer (Tris 50 mM, NaCl 170 mM, EDTA 1 mM, Triton X-100 0.5%, pH 7.4) plus 1x protease and phosphatase inhibitor cocktails (Genedepot, P3100-100, P3200-020) by careful resuspension, incubated on ice for 20 min and spun down at 15,000 rpm for 20 min at 4°C. During this time, 10 μl of Dynabeads Protein A (Invitrogen, 100–02D) per reaction were resuspended and washed with 1x modified NETN buffer 3 times using a magnetic rack (Invitrogen, 123–21D) before being coated with 1 μg of either anti-α-Syn polyclonal (C-20, Santa Cruz Biotechnology) or normal rabbit IgG (Millipore) for 1 h at 4°C (for antibodies and RRIDs, see Table 1-2). Beads were then washed 3 more times, and each sample was divided in half and incubated with 10 μl of suspended beads per reaction for 30 min at 4°C. After the incubation, all samples were washed 5 times with NETN buffer before boiling at 85°C with 1x Laemmli sample buffer (Sigma-Aldrich, S3401-10VL) and used for SDS-PAGE. SDS-PAGE and Western blot were then performed as described previously (Rousseaux et al., 2016).
Quantification and statistical analysis
FACS-based screen.
Quantification and statistical analysis in the primary screen were performed as previously described (Jeong et al., 2017). Briefly, shRNA sequence counts were compared between Low (lowest 5%) and Bulk (presort population), L:B; or high (highest 5%) and Bulk, H:B. For the each of the population comparisons, fold changes were calculated and statistical analysis was performed by using the inverted beta-binomial test (Pham and Jimenez, 2012). Inverted beta-binomial allowed for rigorous statistical interrogation while controlling for intragroup variation (because replicates were sorted on different days). shRNAs were clustered into their respective gene group, and multimetric scores were assigned to take into account number of shRNAs affecting α-Syn levels (hit ratio), directionality (directional score), and concordance within a given shRNA (conflict score). These parameters were used to assign each gene to a Tier (1–5). Of note, these tests were applied to each sublibrary, and the amalgamation of all sublibraries made the hit list for downstream validation (secondary screen).
Cell-based screen ELISA.
Quantification in the secondary screen was performed both on absolute α-Syn levels via ELISA as well as normalized α-Syn levels via Western blot analysis. In the case of α-Syn, standards of human recombinant α-Syn were run on the plate to ensure appropriate dynamic range within the lysate. Moreover, BCA analyses were run in parallel to control for effects of each shRNA on total protein levels cell viability. Results from biological quadruplicates were analyzed as a function of control (empty vector and nontargeting) shRNAs. Genes whose knockdown decreased or increased α-Syn levels by >15% in ≥2/3 shRNAs were considered hits. Moreover, each hit had to have >50% viability (measured by protein levels via BCA analysis) and be concordant with the primary screen results. As such, due to the non-normal distribution of the results (given that hits were biased toward modulators of α-Syn levels), we resorted to percentage cutoffs coupled to these other metrics to determine which gene passes as a hit to move on to Western blot analysis.
Western blot validation in human cells and mouse brain.
Quantification of α-Syn and loading control (Vinculin or βIII-Tubulin) Western blot signals obtained from a LI-COR Odyssey CLx imager was done using ImageStudio Lite (version 5.2.5, LI-COR). Relative α-Syn levels were normalized to control shRNAs and plotted as the percentage change of the control. For hit calling, we used the same percentage cutoffs as above but also performed additional statistical analysis to ensure rigor in our findings. Specifically, we used one-way ANOVA followed by correction for false discovery rate of 10% via the two-stage step-up method of Benjamini and Yekutieli (2006). We chose this statistical method because of the large number of groups being compared and the small effect size being tested. All statistical analyses for this section were performed using Prism 7 (GraphPad) software.
Mechanistic studies: qPCR, clearance assays, and flow cytometric analysis.
qPCR analysis for SNCA transcript was performed as described in qPCR analysis. For statistical analysis, we compared SNCA transcript in each condition with shLuciferase controls by one-way ANOVA followed by Dunnett's multiple-comparisons tests. In the flow cytometric measurements, we measured the percentage change α-Syn-EGFP/DsRed ratio by calculating each percentage value relative to the ratio in the empty vector control and subtracting those values to the average of the control (100%). We used one-way ANOVA followed by Dunnett's multiple-comparisons test to measure statistical significance.
Results
Pooled screens identify modulators of α-Syn levels
To gain insight into potential druggable regulators of α-Syn, we performed pooled screens targeting 7,787 genes whose protein products are potential pharmaceutical targets (Fig. 1A). To generate this list, we combined five resources: (1) Ingenuity pathway analysis (protein targets of drugs that made it to clinical trials), (2) Drugbank (protein targets of FDA-approved drugs), (3) ENSEMBL/Invitrogen (lists of protein targets curated by industry), (4) Interpro (blast of Drugbank target sequences), and (5) BioLT (a literature-based resource that measures the co-occurrence of drug and the term inhibit). Subsequently, olfactory G-protein-coupled receptors and pseudogenes were removed because it is unlikely that these categories of genes would yield druggable modifiers for protein dosage-sensitive disorders of the brain. Finally, a whole genome list of chromatin and ubiquitin modifiers was added. Using this list, we generated pooled libraries of shRNAs targeting eight different subgroups of targets (kinases and phosphatases, nonolfactory GPCRs, ion channels, transporters, ubiquitin-related, chromatin-related, enzymes, and other druggable targets; Fig. 1-1; Table 1-1) for a total of 7,787 genes. For each of these libraries, ∼10 shRNAs were generated for each gene to increase confidence of hit identification. α-Syn reporter medulloblastoma (Daoy) cells (DsRed-IRES-SNCA-EGFP) (Rousseaux et al., 2016) were infected with each pooled library tested and sorted into two bins: those with the lowest 5% GFP/DsRed ratio (lowest α-Syn stability, low group) and those with the highest 5% GFP/DsRed ratio (highest α-Syn stability, high group). Following genomic DNA extraction and half-hairpin amplification, next-generation sequencing was used to identify the relative abundance of each shRNA in relation to the unsorted population (collected on the sort day). Enrichment of a given shRNA (enriched or depleted per group) was first determined by using a low-pass p-value cutoff via the inverted beta-binomial test (Pham and Jimenez, 2012). We used four screening parameters to determine the functional significance and hierarchical tier of each gene (Fig. 1B): (1) the hit ratio, the proportion of significantly enriched shRNAs compared with the unsorted population; (2) the directional score, the inter-shRNA consistency in enrichment vs depletion; (3) the conflict score, the presence of enrichment or a depletion in both the high and/or low populations; and (4) the group enrichment score, the enrichment of more than two shRNAs in one of the populations. These collective scores were computed to allow for target identification (Jeong et al., 2017) (for details, see Materials and Methods). Tier 1 modifiers consisting of 350 genes (4.5% of the druggable genome, meeting all criteria) were selected for further downstream validation.
Figure 1.

Screening the druggable genome to identify modulators of α-Syn levels. A, Strategy used to identify modulators of α-Syn via pooled RNAi libraries in transgenic reporter cells expressing equimolar amounts of α-Syn:EGFP and DsRed (see Table 1-2). Approach was used for eight distinct libraries in quadruplicate. B, Computational prioritization of top gene candidates via tier classification. Hit ratio, directionality score, and conflict score are presented as violin plots. See also Figure 1-1; and Table 1-1.
Composition of the druggable genome and primary screen pipeline. A) Distribution of the eight sub-libraries according to their functional groups. The number in parenthesis represents the number of genes targeted (10 shRNA/gene on average) and % represents the percentage representation of the druggable genome library. B) Pipeline for the primary screen is presented as a flow diagram; 8x denotes steps that are repeated eight times (each library is done separately). Download Figure 1-1, JPG file (312KB, jpg)
{kind=link}
To test whether targets identified in the pooled screen altered endogenous α-Syn levels in an independent cell line, we tested the effect of three new shRNAs per Tier 1 modifier on α-Syn levels using ELISA in HEK293T cells (total of 1,038 shRNAs targeting the bulk of these genes). We used 293T cells given their ease of manipulation and high level of endogenous α-Syn compared with other cell lines tested (Fig. 2-1A). We used three independent shRNAs to control for false-positive hits arising from off-target effects of the RNAi. We performed high throughput virus production and cell infection in quadruplicates and monitored for infectivity using turbo-GFP reporter on the viral construct (Fig. 2-1B). Eleven days following infection, cells were lysed and processed for α-Syn as well as total protein levels (to control for effects on cell viability) (Fig. 2A). Of note, we confirmed that the targeted reduction of α-Syn was specific and did not decrease β- and γ-Syn (Fig. 2-1C). In addition, we filtered out shRNAs that altered total protein levels by >50% as this may represent altered cell viability (Table 1-1).
Figure 2.

Validation in human cells reveals modulators of endogenous α-Syn levels. A, Validation approach in HEK293T cells (see Table 1-2). Individual lentiviral infection is performed in quadruplicate using three shRNAs per gene target of the Tier 1 candidates (350 genes). Individual samples are collected and tested for α-Syn levels via ELISA (as well as total protein levels). Hits that decrease α-Syn levels are further confirmed using Western blot analysis. B, Summary chart of all shRNAs tested for Tier 1 candidates. Blue bars represent hits that decrease α-Syn levels. Red bars represent those that increase α-Syn levels (n = 4 samples per condition). C, The top 33 hits that decrease α-Syn by >15% using ≥2 shRNAs are retested by Western blot. A representative Western blot for α-Syn following downregulation of candidate targets CDK14, CHMP4B, and DUSP11 is included above the graphical representation (n = 4 samples per condition). *q < 0.1 (one-way ANOVA followed by correction for false discovery rate of 10% via the two-stage step-up method of Benjamini and Yekutieli, 2006). See also Figure 2-1; and Table 1-1.
Use of HEK293T cells as a first-pass validation step for the identification of modulators of α-Syn levels. A) ELISA for α-Syn levels in three cell lines reveals a substantial amount of α-Syn levels in HEK293T cells compared to undifferentiated neuroblastoma cell lines (SH-SY5Y and SK-N-BE(2)-C). B) Photomicrograph of HEK293T cells infected with GIPz lentivirus containing a tGFP reporter demonstrates a high level of infectivity. C) ELISA signal is specific to α-Syn and shRNA-mediated knockdown of SNCA does not decrease SNCB or SNCG expression. D) Individual data points between ELISA and western blot assays show significant level of correlation. Download Figure 2-1, JPG file (321.4KB, jpg)
{kind=link}
Of the 350 Tier 1 candidates tested, we identified 60 genes that modified endogenous α-Syn by at least 15% in two or more independent shRNAs in the same direction as the screen (Fig. 2B). Of these, 33 genes decreased α-Syn levels and 27 increased its levels. Given the therapeutic goal of decreasing α-Syn levels, we further validated the 33 genes whose knockdown resulted in a decrease in α-Syn by Western blot. Our results confirmed the ELISA findings (Fig. 2C) and show clear concordance between all data points tested (r2 = 0.32, p < 0.0001; Fig. 2-1D). Together, pooled screens targeting the druggable genome in transgenic reporter cells yields targets that can be validated in a different cell line under endogenous levels.
Assessing α-Syn-induced neuronal dysfunction to validate candidate genes in a Drosophila model of synucleinopathy
To test whether the candidates that decrease endogenous α-Syn in 293T cells can also mitigate α-Syn-mediated neuronal dysfunction, we turned to a Drosophila model of synucleinopathy. We used a recently developed fruit fly model (Chouhan et al., 2016) to drive expression of two copies of codon-optimized wild-type human α-Syn to all neurons. These animals exhibit progressive motor deficits (caused by neuronal dysfunction) over the course of 18 d. We quantified these deficits in a motor performance assay using metrics, such as average speed and stumbling, which are sensitive to α-Syn levels (Fig. 3; Fig. 3-1; Movie 1). Using this assay, we found that knocking down the Drosophila homologs of 9 of the 33 (27%) validated genes resulted in amelioration of α-Syn-induced motor dysfunction (Fig. 3; Table 1). Thus, our biochemical readout is sufficient to identify robust, phenotypically actionable effects in vivo.
Figure 3.

Cross-species validation of novel α-Syn modifiers in Drosophila. A, Approach for identifying modifiers of α-Syn toxicity in Drosophila. B, Motor performance as a function of age in control fruit flies (blue line in every panel), flies specifically expressing α-Syn in neurons (black line in every panel), and animals expressing α-Syn and carrying genetic variants reducing the function of the indicated candidate gene in neurons (red line in every panel). Two read-out metrics are represented: speed in mm/s (top panel for each gene) and stumbling events (bottom panel for each gene). Expression of α-Syn in the Drosophila nervous system (using elav-Gal4) leads to progressive motor performance deficits, resulting in decreased average speed and increased stumbling events as the fruit flies age. Reducing levels of each of the indicated candidate genes result in an amelioration of the α-Syn-induced motor impairment, seen as improved average speed and decreased number of stumbles as the animals age, compared with Drosophila expressing α-Syn alone. Error bars indicate SEM. *Statistical significance between α-Syn alone (black line) or together with a candidate gene knockdown (red line) based on a linear mixed-effect model ANOVA (α = 0.05). LOF, Loss of function; hp, hairpin RNAi. The specific genotypes are indicated in Table 1. For Additional characterization of the Drosophila model and results with additional alleles see Figure 3-1.
Table 1.
Summary of the Drosophila based candidate modifier screen
| Human gene | Drosophila homolog | Blast e-value (human to fly) | Effect on motor performance in α-Syn transgenic flies | Modifier allele |
|---|---|---|---|---|
| ACSBG1 | bgm | 2E-172 | Improves | bgm[MI14249] |
| CACNA1S | Ca-alpha1D | 0 | Improves | Ca-alpha1D[MI08906] |
| CACNA1S | cac | 0 | Improves | GD15341(hp RNAi) |
| CDK14 | Eip63E-LOF1 | 2E-159 | Improves | Eip63E[MI00413-GFSTF.1] |
| CDK14 | Eip63E-LOF2 | 2E-159 | Improves | Eip63E[81] |
| CDK14 | Eip63E | 2E-159 | Improves | Eip63E[MI04853]a |
| DDC | Ddc | 0 | Improves | Ddc[ts2] |
| DNER | Notch | 2E-65 | Improves | N[fa-swb] |
| ERBB2 | Egfr | 7E-126 | Improves | slbo(FRT.lacZ)Egfr.RNAi |
| HDAC3 | HDAC1 | 0 | Improves | HDAC1[12–37]a |
| HDAC3 | HDAC3 | 0 | Improves | GD9732(hp RNAi) |
| LGALS3BP | lox2 | 4E-17 | Improves | lox2[MI06311] |
| LOXL1 | lox2 | 8E-55 | Improves | lox2[MI06311] |
aData not shown.
Direct manipulation of α-Syn levels in the Drosophila model. A) Decreasing α-Syn levels in the Drosophila model of parkinsonism by means of inducible shRNAs targeting the α-Syn gene results in a suppression of the behavioral deficits induced by α-Syn. B) Effect of inducible shRNAs targeting the α-Syn gene in Drosophila on α-Syn levels by western blot. Download Figure 3-1, JPG file (178.7KB, jpg)
{kind=link}
Representative video of motor performance assay on α-Syn transgenic flies compared with controls.

Summary of screen data. Curated data from screens in human cells, human neurons and mouse brain are presented. Each tab represents a different level of screening. Of note, qPCR data for knockdown of each candidate gene for human neurons (Figure 4) and mouse brain (Figure 5) experiments are presented on separate tabs. Figure legends are presented at the top of each table. Download Table 1-1, XLSX file (1.2MB, xlsx)
List of antibodies, cell lines and oligonucleotides used in this study. Download Table 1-2, XLSX file (12.4KB, xlsx)
Knockdown of candidate modulators in cultured human neurons confirms their effects on α-Syn protein levels
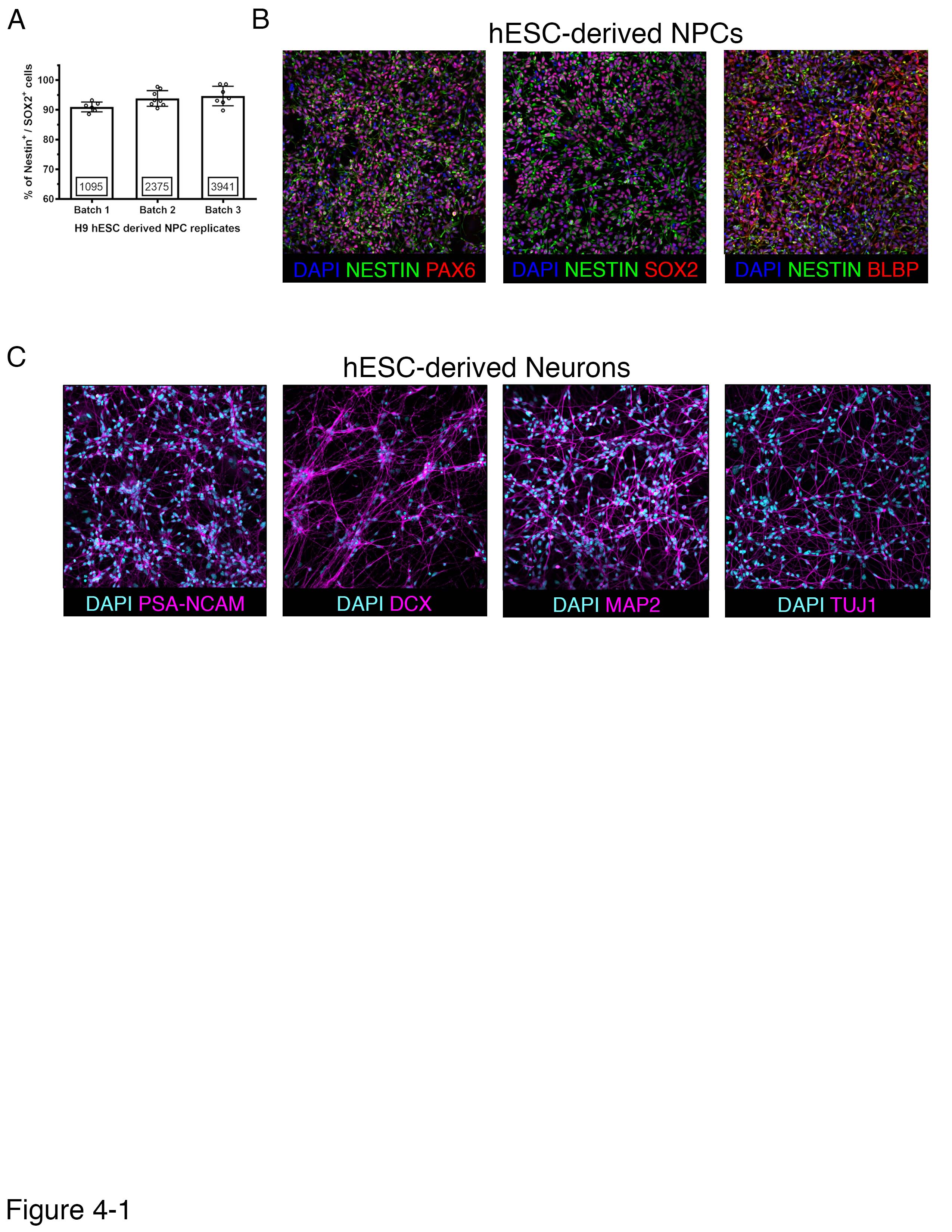
To test whether our targets regulate α-Syn levels in neurons, we turned to human neuron culture. For the scope of this study, we chose 10 of the 60 validated targets whose brain expression, biological function, or relation to PD made them attractive α-Syn modulator candidates for PD therapeutics (Zhang et al., 2014). Moreover, we chose targets whose knockdown resulted in a decrease of α-Syn levels as they would likely be easier therapeutic targets. Among the targets whose reduction decrease α-Syn levels in 293T cells are two kinases (DCLK1, and CDK14/PFTK1), enzymes involved in long-chain fatty acid synthesis (ACSBG1), Lysyl residue oxidation (LOXL1), β-galactoside binding (LGALS3BP), and deneddylation (SENP8), as well as a member of the ESCRT-III complex (CHMP4B), and an ER receptor (KDELR2). We also tested HLA-DRB1, a transmembrane receptor that is a potential PD risk gene (Nalls et al., 2014). We derived human neurons from ESCs (Fig. 4-1) and infected them with shRNA-carrying lentiviruses (Fig. 4A,B). qPCR experiments revealed differential target knockdown efficiency between each of the 3 shRNAs targeting each gene, with 50% of genes having a >40% reduction in target gene expression (Table 1-1). Notably, we used only two shRNAs to test HLA-DRB1, as these were the only two that were commercially available to us. When we knocked down each candidate in eight biological replicates, we found that all targets (with the exception of HLA-DRB1, a candidate whose neuronal expression is almost undetectable) (Uhlén et al., 2015) had multiple shRNAs that caused at least a 15% decrease in α-Syn levels (Fig. 4C,D). Thus, the biochemical effect of these modifiers on α-Syn levels are conserved in human neurons for 9 of the 10 selected (90%) modifier genes.
Figure 4.

Testing candidate α-Syn modulators in human neurons. A, Approach for identifying modulators of α-Syn levels in hESC-derived neurons (see Table 1-2). Cells are infected for 2 weeks with three independent shRNAs. Expression of virus is confirmed by presence of turboGFP (tGFP) signal (B), and candidate knockdown is confirmed using qPCR (see Table 1-1). C, Representative Western blot from human neurons infected with lentiviruses harboring the indicated shRNAs targeting candidate genes LOXL1, SENP8, and CDK14. D, Quantification of each candidate tested (n = 8 samples per condition). Each bar represents an individual shRNA. Three shRNAs were tested per gene, except for HLA-DRB1. Percentage change in α-Syn levels was quantified in relation to a scrambled shRNA. *q < 0.1 (one-way ANOVA followed by correction for false discovery rate of 10% via the two-stage step-up method of Benjamini and Yekutieli, 2006). See also Figure 4-1; and Table 1-1.
Establishment of a human neuron model to test α-Syn modulators. A) Quantification of the relative number of double positive Nestin and SOX2 cells in H9 hESC derived NPC cultures. B) Immunofluorescence staining for NESTIN and PAX6 (left panel), NESTIN and SOX2 (middle panel) and NESTIN and FABP (right panel). Nuclei are stained using DAPI. C) Representative photomicrographs of differentiated neuronal cultures, derived from H9 hESCs and stained for PSA-NCAM, doublecortin (DCX), MAP2 and TUJ1 are presented. Download Figure 4-1, JPG file (595.7KB, jpg)
{kind=link}
Candidate modulators impact α-Syn levels in the mammalian brain
To test whether target modifiers regulate α-Syn levels in a mammalian system in vivo, we tested the effect of decreasing the identified modifier genes on α-Syn levels in mouse brain (Fig. 5A). To do so, we generated shRNAs targeting the mouse homologs for 9 of 10 modifiers that were tested in human neurons (HLA-DRB1 is not conserved in the mouse). We used the recently developed SplashRNA algorithm to design effective, on-target shRNAs and cloned them into AAV-competent miR-E vectors (Fellmann et al., 2013; Pelossof et al., 2017). We next screened three shRNAs per gene for their capacity to knock down their target in a mouse neuroblastoma cell line (for details, see Materials and Methods), before taking the two most efficient ones and packing them into AAVs. We then performed intraventricular injections of AAVs carrying these shRNA constructs on neonatal mouse pups. We first confirmed effective vector delivery by imaging the YFP signal from live mice and from brain sections (Fig. 5B) and found that the virus was most strongly expressed in the caudal cortex and hippocampus (Kim et al., 2013). Three weeks following injection, we collected the caudal cortex and hippocampal brain tissue from the shRNA-expressing mice and extracted protein and RNA from both sides of the brain. Notably, of the 19 viruses tested, only the one targeting Chmp4b resulted in toxicity (animals injected with Chmp4b targeting shRNA had to be sacrificed at day 18 because they failed to thrive), likely due to its critical role in nuclear envelope formation (Olmos et al., 2015). We subsequently performed qPCR to measure target knockdown and Western blot to measure α-Syn protein levels (Fig. 5A). We found that each shRNA could knock down most of the target genes by >50% (Table 1-1). Moreover, Western blot analysis revealed that 6 of 9 genes (67%) decreased α-Syn protein levels when knocked down (Fig. 5C,D). Collectively, these experiments show that the effect of our candidate modifiers, first identified in a pooled screen on a reporter cell line, can be recapitulated in vivo and that most of these modifiers can be safely reduced in the mouse brain.
Candidates regulate α-Syn levels directly and indirectly through diverse cellular pathways
To gain insight into how these candidates regulate α-Syn levels, we chose to further study two strong modulators, DUSP11 and LGALS3BP. We first tested whether the regulation of these modifiers was bidirectional by knocking them down and overexpressing them in the α-Syn reporter cell line (DsRed-IRES-SNCA-EGFP) described previously (Fig. 6A). We found that knockdown of each modifier decreased the relative protein levels of α-Syn (measured by the EGFP/DsRed ratio) (Rousseaux et al., 2016), and that overexpression of each modifier had the opposite effect (Fig. 6B). This indicates that the regulation is indeed bidirectional. Notably, siDUSP11 and the overexpression constructs decreased GFP levels in the control cell line. In the first case, this may be due to the role of DUSP11 in regulation of RNA (Burke et al., 2016). Regardless, the effect on SNCA-GFP was significantly larger (p < 0.01) than the effect on GFP, which likely reflects a decrease in α-Syn protein. In the second case, this effect is due to the fact that both the transgene and the overexpression constructs used CMV promoters, which results in competition for transcription. This resulted in less expression of the transgene under transfected conditions but did not interfere with the increase in α-Syn protein that we observed. Next we tested if loss of these modulators altered the endogenous expression of Snca transcript. Given that the initial screen was designed to identify post-translational regulators of α-Syn levels, we surmised that knockdown of the targets would not alter Snca RNA levels. We found that depletion of Dusp11 and Lgals3bp in the mouse brain did not cause a significant decrease in Snca transcript (Fig. 6C). It is worth noting however, that one of the shRNAs against Dusp11 caused a trend toward a decrease in Snca transcript. To gain additional insight into the mechanism of regulation, we asked whether these modulators interact with α-Syn. We tested this by immunoprecipitation, and found that endogenous α-Syn interacted with DUSP11, but not with LGALS3BP or CDK14 (a candidate that did not meet our validation criteria in vivo) (Fig. 6D). Thus, our screen uncovered both direct and indirect modifiers of α-Syn protein levels.
Figure 6.

Mechanistic insight into the regulation α-Syn levels for two candidate modulators: DUSP11 and LGALS3BP. A, Experimental design for testing individual modifiers of protein stability in a bidirectional manner. B, Flow cytometry-based quantification of α-Syn stability following DUSP11 and LGALS3BP siRNA knockdown and overexpression in experimental (DsRed-IRES-SNCA-EGFP; blue) and control (DsRed-IRES-EGFP; white) cell lines (see Table 1-2). Data are presented as the percentage change compared with either siScrambled or a control plasmid (empty Flag-tag vector). C, qPCR for Snca expression following neonatal depletion of Dusp11 and Lgals3bp in mouse brain. D, Immunoprecipitation of endogenous α-Syn from HEK293T cells transfected with indicated flag-tagged constructs. α-Syn interacts with DUSP but not the other proteins; the signal in CDK14 IP is not specific as it is also seen with the IgG control. Western blot is representative of three independent experiments. ***p < 0.001, ****p < 0.0001. B, C, ANOVA followed by Dunnett's multiple-comparisons test.
Discussion
In this study, we present a straightforward, yet effective, way to identify novel regulators of proteins levels for preclinical development. Using α-Syn as a prototype protein whose increased dosage is intrinsically tied to neurodegeneration, we provide a tiered pipeline to find brain-specific modulators of its levels (Fig. 7). Using in vivo validation on a subset of candidates, we found six strong regulators of α-Syn levels: ACSBG1, CHMP4B, DCLK1, DUSP11, KDELR2, and LGALS3BP. Several lines of evidence suggest that these modifiers are functionally related to α-Syn biology. On the one hand, KDELR2 is a member of the KDEL endoplasmic reticulum receptors, a family of proteins that function in the retrograde transport of chaperones (and other ER resident proteins) from the Golgi to the ER, thus preventing the accumulation of misfolded proteins (Giannotta et al., 2015). Interestingly, KDELRs are upregulated as part of ER stress and the unfolded protein response, and ER stress is one of the earliest events of α-Syn toxicity (Wang et al., 2011). This suggests that KDELR2-mediated increases in α-Syn levels could contribute to its toxicity. Additionally, KDELRs have been reported to regulate autophagy, a well-known mediator α-Syn clearance (Webb et al., 2003). On the other hand, LGALS3BP (lectin galactoside soluble 3 binding protein), is a membrane bound and secreted glycoprotein initially described as an interactor of LGALS3 (also known as galectin-3) (Rosenberg et al., 1991; Sasaki et al., 1998; Stampolidis et al., 2015). LGALS3 is normally present on the surface of cell and vesicular membranes. Intriguingly, exogenous α-Syn has been previously shown to rupture lysosomes coated with LGALS3 (Freeman et al., 2013). Moreover, an extensive connection exists between α-Syn accumulation and lysosomal dysfunction in PD (Sidransky and Lopez, 2012; Woodard et al., 2014; Taguchi et al., 2017). Given the clear links between α-Syn and lysosomal pathophysiology, it is plausible that α-Syn functionally interacts with LGALS3BP via LGALS3 to disrupt lysosomal function, which leads to increased levels of the toxic protein (Mazzulli et al., 2016). Together, these candidates hold promise and should be studied in further detail given their potency toward α-Syn regulation and their potential druggability. Despite these interesting clues, future studies will be required to understand the exact mechanism by which these candidates regulate α-Syn levels. One limitation to the screening approach used here is the difficulty of parsing out direct versus indirect effects of modifiers on α-Syn levels. In the future, multitiered screening approaches wherein PD-centric genomic-, transcriptomic-, proteomic-, and toxicity-based screening platforms (Tardiff et al., 2013; Dhungel et al., 2015; Chang et al., 2017; Chung et al., 2017; Khurana et al., 2017; Robak et al., 2017; Hook et al., 2018) can be integrated with protein dosage-based screens to provide mechanistic insight.
Figure 7.

Pipeline for identifying potentially druggable modulators of α-Syn levels. Validation pipeline for uncovering modulators of α-Syn levels. Primary cell-based screen is performed using pooled libraries of shRNAs targeting >7500 genes (Fig. 1). Secondary screens in HEK293T cells (Fig. 2) and Drosophila (Fig. 3) help narrow down the list and highlight strong modulators of α-Syn levels and toxicity. Validation in human neurons (Fig. 4) and mouse brain (Fig. 5) ensures robustness of hits and validates targets for preclinical nomination.
Given that even a 15%–30% decrease on neurotoxic disease protein levels often translates to a functional recovery in their respective disease models (Jafar-Nejad et al., 2011; Park et al., 2013; Lasagna-Reeves et al., 2015; Rousseaux et al., 2016), the candidates described herein may hold promise when moved into preclinical studies. Nevertheless, thorough evaluation of the modes of action and efficacy of these targets will be critical for downstream therapeutic development. Our human neuron-based validation together with our mouse-based validation offer a quick solution to identify species-relevant pathways and preliminary safety margins for candidate modifiers, respectively. By acutely knocking down a candidate gene once the brain is mostly developed (P0), this approach offers an opportunity to rapidly mimic the acute nature of postnatal pharmacological inhibition of targets, thus assessing safety and tolerability. Further studies using extensive behavioral profiling on adult knock-outs of such targets (Ruzankina et al., 2007; Rousseaux et al., 2018) will help identify safety margins for drug development.
Because this screen identified regulators that also increase α-Syn, this may allow for putative disease-driving gene identification. Several lines of evidence suggest that factors that increase levels of neurodegeneration driving proteins can be novel disease-driving genes. First, we have recently found that Pumilio-1 (Pum1) regulates Ataxin-1 levels and that loss of Pum1 leads to increased levels of Ataxin-1 and Purkinje cell degeneration in mice similar to what is seen in spinocerebellar ataxia 1 (Gennarino et al., 2015). This led us to propose that haploinsufficiency of PUM1 might cause ataxia in humans; and indeed, we found that heterozygous mutations in PUM1 cause ataxia in humans (Gennarino et al., 2018). Second, this concept was recently elegantly illustrated by the Scherzer group for α-Syn, where drugs that increase SNCA transcript correlate with increased lifetime PD risk (Mittal et al., 2017). In this study, we identified factors, including PAK2 and SIAH2, which may contribute to an increase of α-Syn levels upon their disruption. Interestingly, PAK2 was recently found to be enriched in α-Syn inclusions formed in neurons after exposure to fibrillar α-Syn (Henderson et al., 2017), whereas SIAH2 is an E3 ligase that has been previously reported to mono-ubiquitinate α-Syn and poly-ubiquitinate its binding partner Synphillin-1, leading to the degradation of the latter (Liani et al., 2004). Given that duplications and triplications of SNCA cause PD (Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibáñez et al., 2004), it is reasonable to posit that modifiers that increase α-Syn levels by 50% would be potential PD genes. This is compounded by the recently suggested finding that a lifelong increase of 10% of SNCA may predispose individuals to PD (Soldner et al., 2016). Thus, identifying factors that increase the dosage of α-Syn to 110%–150% of its normal level may provide a host of new factors contributing to PD pathogenesis.
It is important to note that only a subset of the targets identified in this screen were tested in this study. We identified 60 modulators of α-Syn (33 whose inhibition decreased and 27 whose inhibition increased its levels). Among the 60 genes, we chose 10 that met the following criteria: decreased α-Syn levels when inhibited, were expressed in the brain, and were optimal drug targets for further experimentation. Of these 10 targets, we validated six in human neurons and mice. There are other promising targets in this dataset that deserve further validation and study as they are likely to provide insight into the regulatory mechanisms for α-Syn. Nevertheless, as with most screening approaches, the attrition rates obtained at each validation step indicate that there is room for improvement. As with any shRNA screen, it is likely that these attrition rates are caused by the use of several different cellular and organismal systems together with the inherent shortcomings of the technology.
The cellular mechanisms that govern protein levels have been difficult to grasp. This is likely due to the reliance upon candidate-based approaches and assumptions as to the function of the disease-driving protein. The use of pooled genetic screens removes the need for this bias and allows for the discovery of novel modifiers of protein levels that are putative drug targets. Moreover, the basis of screens rooted in strong genetic evidence makes no assumptions as to the role of a particular protein in the cell but rather seeks to correct the root cause of disease: altered protein homeostasis. One promising therapeutic consideration is the use of antisense oligonucleotide approaches that directly lower disease-driving protein via RNase H-mediated silencing of DNA-RNA duplexes (Sztainberg et al., 2015; Finkel et al., 2017). However, because neurodegenerative diseases are chronic and age-dependent disorders, treatment will ideally start in the presymptomatic stage and must continue for decades to be effective. Thus, it is preferable to develop pharmacological-based therapy as it is easy to administer and less invasive. By targeting modifiers, and slightly decreasing (by 10%–30%) the levels of a toxic protein, a disease-modifying treatment could be achieved. This concept was recently illustrated by the finding that β2 adrenergic agonists reduce α-Syn levels, and their use is associated with lower risk of PD in a Norwegian population (Mittal et al., 2017).
A growing body of evidence suggests that the brain is exquisitely sensitive to protein dosage. Copy number variations in dosage-sensitive genes, such as APP, GRN, and SNCA, result in neurodegenerative disorders (Wisniewski et al., 1985; Baker et al., 2006; Singleton and Hardy, 2016; Ward et al., 2017), whereas altered dosage of proteins, such as RAI1, MeCP2, and SHANK3, can lead to a host of neurodevelopmental disorders (Potocki et al., 1993; Amir et al., 1999; Ramocki et al., 2010; Han et al., 2013). Thus, growing academic- and industry-led efforts have focused on finding ways to alter the levels of dosage-sensitive proteins as a therapeutic strategy. The screening pipeline outlined in this study offers one approach to do so by identifying and prioritizing candidate modifiers through steps of increasing stringency. Last, it is worth noting that the increasing accessibility to versatile gene-disruption strategies, such as CRISPR and CRISPRi (Gilbert et al., 2014; Shalem et al., 2015; Du and Qi, 2016), coupled with decreasing sequencing costs (van Dijk et al., 2014) and analysis time (Jeong et al., 2017), will help facilitate the broad use of such screening approaches to the scientific community to target virtually any protein whose dosage is critical for cellular function.
Footnotes
This was supported in part by the Hamill foundation to H.Y.Z.; UCB Pharma, and the Robert A. and Reneé Belfer Family Foundation to H.Y.Z. and J.B.; and the Howard Hughes Medical Institute to H.Y.Z. and S.J.E. S.J.E. was supported by National Institutes of Health Grant AG11083. M.W.C.R. was supported by Canadian Institutes of Health Research Fellowship 201210MFE-290072-173743 and the Parkinson's Disease Foundation Stanley Fahn Junior Faculty Award (Grant PF-JFA-1762). I.A.-R. was supported by National Institutes of Health Grant R21NS096395 and the Darrell K. Royal Research Fund for Alzheimer's Disease. J.M.S. was supported by National Institutes of Health Grant R21NS089854 and Burroughs Welcome Fund Career Award. The project was supported in part by Eunice Kennedy Shriver National Institute of Child Health and Human Development (Baylor College of Medicine IDDRC Grant U54HD083092). The IDDRC Microscopy Core was used for this project. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health and Human Development or the National Institutes of Health. We thank members of the H.Y.Z. laboratory for important discussions and critical feedback on the manuscript; Nan Lu (Baylor College of Medicine) for helping with the initial screen optimization and library construction; Christof Fellmann (University of California, Berkeley) for providing the shRNA scores (SplashRNA scores) and important discussions; the Viral Vector Core at the University of Texas M.D. Anderson; and the Gene Vector and the Genome and RNA Profiling cores at Baylor College of Medicine. Neural progenitor cells used in this study were derived from the WA09 human ESC line (H9 ESC) derived by Dr. James Thomson and distributed by WiCell Research Institute under SLA agreement.
The authors declare no competing financial interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188. 10.1038/13810 [DOI] [PubMed] [Google Scholar]
- Antonarakis SE. (2017) Down syndrome and the complexity of genome dosage imbalance. Nat Rev Genet 18:147–163. 10.1038/nrg.2016.154 [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, et al. (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- Benjamini YK, Yekutieli D (2006) Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93:491–507. 10.1093/biomet/93.3.491 [DOI] [Google Scholar]
- Burke JM, Kincaid RP, Nottingham RM, Lambowitz AM, Sullivan CS (2016) DUSP11 activity on triphosphorylated transcripts promotes argonaute association with noncanonical viral microRNAs and regulates steady-state levels of cellular noncoding RNAs. Genes Dev 30:2076–2092. 10.1101/gad.282616.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet 49:1511–1516. 10.1038/ng.3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364:1167–1169. 10.1016/S0140-6736(04)17103-1 [DOI] [PubMed] [Google Scholar]
- Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR (2012) A progressive mouse model of Parkinson's disease: the Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 9:297–314. 10.1007/s13311-012-0104-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouhan AK, Guo C, Hsieh YC, Ye H, Senturk M, Zuo Z, Li Y, Chatterjee S, Botas J, Jackson GR, Bellen HJ, Shulman JM (2016) Uncoupling neuronal death and dysfunction in Drosophila models of neurodegenerative disease. Acta Neuropathol Commun 4:62. 10.1186/s40478-016-0333-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, Mungenast AE, Muffat J, Mitalipova M, Pluth MD, Jui NT, Schüle B, Lippard SJ, Tsai LH, Krainc D, Buchwald SL, et al. (2013) Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 342:983–987. 10.1126/science.1245296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Khurana V, Yi S, Sahni N, Loh KH, Auluck PK, Baru V, Udeshi ND, Freyzon Y, Carr SA, Hill DE, Vidal M, Ting AY, Lindquist S (2017) In situ peroxidase labeling and mass-spectrometry connects alpha-synuclein directly to endocytic trafficking and mRNA metabolism in neurons. Cell Syst 4:242–250.e4. 10.1016/j.cels.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S (2006) Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313:324–328. 10.1126/science.1129462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Gwinn K, Singleton A, Hardy J (2011) Parkinson's disease and alpha-synuclein expression. Mov Disord 26:2160–2168. 10.1002/mds.23948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhungel N, Eleuteri S, Li LB, Kramer NJ, Chartron JW, Spencer B, Kosberg K, Fields JA, Stafa K, Adame A, Lashuel H, Frydman J, Shen K, Masliah E, Gitler AD (2015) Parkinson's disease genes VPS35 and EIF4G1 interact genetically and converge on alpha-synuclein. Neuron 85:76–87. 10.1016/j.neuron.2014.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du D, Qi LS (2016) CRISPR technology for genome activation and repression in mammalian cells. Cold Spring Harb Protoc 2016:pdb prot090175. 10.1101/pdb.prot090175 [DOI] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AE, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen YN, Rush J, Hsu PW, Yen HC, Elledge SJ (2011) Global identification of modular cullin-RING ligase substrates. Cell 147:459–474. 10.1016/j.cell.2011.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, Sinha N, Zuber J (2013) An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 5:1704–1713. 10.1016/j.celrep.2013.11.020 [DOI] [PubMed] [Google Scholar]
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, et al. (2017) Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 377:1723–1732. 10.1056/NEJMoa1702752 [DOI] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF (2004) Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci 24:9434–9440. 10.1523/JNEUROSCI.3080-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O'Connor C, Wiethoff CM, Campbell EM (2013) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8:e62143. 10.1371/journal.pone.0062143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L (2009) The role of tau in neurodegeneration. Mol Neurodegener 4:13. 10.1186/1750-1326-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino VA, Palmer EE, McDonell LM, Wang L, Adamski CJ, Koire A, See L, Chen CA, Schaaf CP, Rosenfeld JA, Panzer JA, Moog U, Hao S, Bye A, Kirk EP, Stankiewicz P, Breman AM, McBride A, Kandula T, Dubbs HA, et al. (2018) A mild PUM1 mutation is associated with adult-onset ataxia, whereas haploinsufficiency causes developmental delay and seizures. Cell 172:924–936.e11. 10.1016/j.cell.2018.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino VA, Singh RK, White JJ, De Maio A, Han K, Kim JY, Jafar-Nejad P, di Ronza A, Kang H, Sayegh LS, Cooper TA, Orr HT, Sillitoe RV, Zoghbi HY (2015) Pumilio1 haploinsufficiency leads to SCA1-like neurodegeneration by increasing wild-type Ataxin1 levels. Cell 160:1087–1098. 10.1016/j.cell.2015.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannotta M, Fragassi G, Tamburro A, Vanessa C, Luini A, Sallese M (2015) Prohibitin: a novel molecular player in KDEL receptor signalling. Biomed Res Int 2015:319454. 10.1155/2015/319454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159:647–661. 10.1016/j.cell.2014.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves SA, Macedo D, Raquel H, Simões PD, Giorgini F, Ramalho JS, Barral DC, Ferreira Moita L, Outeiro TF (2016) shRNA-based screen identifies endocytic recycling pathway components that act as genetic modifiers of alpha-synuclein aggregation, secretion and toxicity. PLoS Genet 12:e1005995. 10.1371/journal.pgen.1005995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Holder JL Jr, Schaaf CP, Lu H, Chen H, Kang H, Tang J, Wu Z, Hao S, Cheung SW, Yu P, Sun H, Breman AM, Patel A, Lu HC, Zoghbi HY (2013) SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503:72–77. 10.1038/nature12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson MX, Chung CH, Riddle DM, Zhang B, Gathagan RJ, Seeholzer SH, Trojanowski JQ, Lee VMY (2017) Unbiased proteomics of early Lewy body formation model implicates active microtubule affinity-regulating kinases (MARKs) in synucleinopathies. J Neurosci 37:5870–5884. 10.1523/JNEUROSCI.2705-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook PW, McClymont SA, Cannon GH, Law WD, Morton AJ, Goff LA, McCallion AS (2018) Single-cell RNA-seq of mouse dopaminergic neurons informs candidate gene selection for sporadic Parkinson disease. Am J Hum Genet 102:427–446. 10.1016/j.ajhg.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ (2009) A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev 23:837–848. 10.1101/gad.1769609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet 364:1169–1171. 10.1016/S0140-6736(04)17104-3 [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad P, Ward CS, Richman R, Orr HT, Zoghbi HY (2011) Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14–3-3epsilon haploinsufficiency in mice underscores complex pathogenicity in neurodegeneration. Proc Natl Acad Sci U S A 108:2142–2147. 10.1073/pnas.1018748108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong HH, Kim SY, Rousseaux MW, Zoghbi HY, Liu Z (2017) CRISPRcloud: a secure cloud-based pipeline for CRISPR pooled screen deconvolution. Bioinformatics 33:2963–2965. 10.1093/bioinformatics/btx335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Chen J, Bajic A, Zhang C, Song X, Carroll SL, Cai ZL, Tang M, Xue M, Cheng N, Schaaf CP, Li F, MacKenzie KR, Ferreon AC, Xia F, Wang MC, Maletic-Savatic M, Wang J (2017) Quantitative real-time imaging of glutathione. Nat Commun 8:16087. 10.1038/ncomms16087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, Rao M, Yu P, Dominguez-Vidana R, Liang AC, Solimini NL, Bernardi RJ, Yu B, Hsu T, Golding I, Luo J, et al. (2012) A SUMOylation-dependent transcriptional subprogram is required for myc-driven tumorigenesis. Science 335:348–353. 10.1126/science.1212728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V, Peng J, Chung CY, Auluck PK, Fanning S, Tardiff DF, Bartels T, Koeva M, Eichhorn SW, Benyamini H, Lou Y, Nutter-Upham A, Baru V, Freyzon Y, Tuncbag N, Costanzo M, San Luis BJ, Schöndorf DC, Barrasa MI, Ehsani S, et al. (2017) Genome-scale networks link neurodegenerative disease genes to alpha-synuclein through specific molecular pathways. Cell Syst 4:157–170.e14. 10.1016/j.cels.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Ash RT, Ceballos-Diaz C, Levites Y, Golde TE, Smirnakis SM, Jankowsky JL (2013) Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualising and manipulating neuronal circuits in vivo. Eur J Neurosci 37:1203–1220. 10.1111/ejn.12126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A (2002) Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci 22:2780–2791. 10.1523/JNEUROSCI.22-07-02780.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott SR, Maceli A, Erard N, Chang K, Marran K, Zhou X, Gordon A, Demerdash OE, Wagenblast E, Kim S, Fellmann C, Hannon GJ (2014) A computational algorithm to predict shRNA potency. Mol Cell 56:796–807. 10.1016/j.molcel.2014.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T, Koyama A, Koyama S, Yoshina S, Ren CH, Kato T, Mitani S, Iwatsubo T (2008) A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum Mol Genet 17:2997–3009. 10.1093/hmg/ddn198 [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Rousseaux MW, Guerrero-Munoz MJ, Vilanova-Velez L, Park J, See L, Jafar-Nejad P, Richman R, Orr HT, Kayed R, Zoghbi HY (2015) Ataxin-1 oligomers induce local spread of pathology and decreasing them by passive immunization slows spinocerebellar ataxia type 1 phenotypes. Elife 4:e10891. 10.7554/eLife.10891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, de Haro M, Hao S, Park J, Rousseaux MW, Al-Ramahi I, Jafar-Nejad P, Vilanova-Velez L, See L, De Maio A, Nitschke L, Wu Z, Troncoso JC, Westbrook TF, Tang J, Botas J, Zoghbi HY (2016) Reduction of Nuak1 decreases tau and reverses phenotypes in a tauopathy mouse model. Neuron 92:407–418. 10.1016/j.neuron.2016.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Parker B, Martyn C, Natarajan C, Guo J (2013) The PMP22 gene and its related diseases. Mol Neurobiol 47:673–698. 10.1007/s12035-012-8370-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D, Bornemann A, Riess O, Ross CA, Rott R, Engelender S (2004) Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson's disease. Proc Natl Acad Sci U S A 101:5500–5505. 10.1073/pnas.0401081101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the ras oncogene. Cell 137:835–848. 10.1016/j.cell.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D (2016) α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 113:1931–1936. 10.1073/pnas.1520335113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal S, Bjørnevik K, Im DS, Flierl A, Dong X, Locascio JJ, Abo KM, Long E, Jin M, Xu B, Xiang YK, Rochet JC, Engeland A, Rizzu P, Heutink P, Bartels T, Selkoe DJ, Caldarone BJ, Glicksman MA, Khurana V, et al. (2017) β2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson's disease. Science 357:891–898. 10.1126/science.aaf3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 46:989–993. 10.1038/ng.3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JQ, Zhou R, Czech B, Liu LP, Holderbaum L, Yang-Zhou D, Shim HS, Tao R, Handler D, Karpowicz P, Binari R, Booker M, Brennecke J, Perkins LA, Hannon GJ, Perrimon N (2011) A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods 8:405–407. 10.1038/nmeth.1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos Y, Hodgson L, Mantell J, Verkade P, Carlton JG (2015) ESCRT-III controls nuclear envelope reformation. Nature 522:236–239. 10.1038/nature14503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Al-Ramahi I, Tan Q, Mollema N, Diaz-Garcia JR, Gallego-Flores T, Lu HC, Lagalwar S, Duvick L, Kang H, Lee Y, Jafar-Nejad P, Sayegh LS, Richman R, Liu X, Gao Y, Shaw CA, Arthur JSC, Orr HT, Westbrook TF, Botas J, et al. (2013) RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature 498:325–331. 10.1038/nature12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelossof R, Fairchild L, Huang CH, Widmer C, Sreedharan VT, Sinha N, Lai DY, Guan Y, Premsrirut PK, Tschaharganeh DF, Hoffmann T, Thapar V, Xiang Q, Garippa RJ, Rätsch G, Zuber J, Lowe SW, Leslie CS, Fellmann C (2017) Prediction of potent shRNAs with a sequential classification algorithm. Nat Biotechnol 35:350–353. 10.1038/nbt.3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkau TL, Leavitt BR (2014) Progranulin in neurodegenerative disease. Trends Neurosci 37:388–398. 10.1016/j.tins.2014.04.003 [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TV, Jimenez CR (2012) An accurate paired sample test for count data. Bioinformatics 28:i596–i602. 10.1093/bioinformatics/bts394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocki L, Neira-Fresneda J, Yuan B (1993) Potocki-Lupski syndrome. In: GeneReviews (Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, eds). Seattle: NCBI Bookshelf. [PubMed] [Google Scholar]
- Ramocki MB, Tavyev YJ, Peters SU (2010) The MECP2 duplication syndrome. Am J Med Genet A 152A:1079–1088. 10.1002/ajmg.a.33184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, Heutink P, Shulman JM (2017) Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain 140:3191–3203. 10.1093/brain/awx285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg I, Cherayil BJ, Isselbacher KJ, Pillai S (1991) Mac-2-binding glycoproteins. putative ligands for a cytosolic beta-galactoside lectin. J Biol Chem 266:18731–18736. [PubMed] [Google Scholar]
- Rousseaux MW, Shulman JM, Jankovic J (2017) Progress toward an integrated understanding of Parkinson's disease. F1000Res 6:1121. 10.12688/f1000research.11820.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseaux MW, de Haro M, Lasagna-Reeves CA, De Maio A, Park J, Jafar-Nejad P, Al-Ramahi I, Sharma A, See L, Lu N, Vilanova-Velez L, Klisch TJ, Westbrook TF, Troncoso JC, Botas J, Zoghbi HY (2016) TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. Elife 5:e19809. 10.7554/eLife.19809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseaux MW, Revelli JP, Vázquez-Vélez GE, Kim JY, Craigen E, Gonzales K, Beckinghausen J, Zoghbi HY (2018) Depleting Trim28 in adult mice is well tolerated and reduces levels of alpha-synuclein and tau. Elife 7:e36768. 10.7554/eLife.36768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38:24–26. 10.1038/ng1718 [DOI] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ (2007) Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1:113–126. 10.1016/j.stem.2007.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Brakebusch C, Engel J, Timpl R (1998) Mac-2 binding protein is a cell-adhesive protein of the extracellular matrix which self-assembles into ring-like structures and binds beta1 integrins, collagens and fibronectin. EMBO J 17:1606–1613. 10.1093/emboj/17.6.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, Westbrook TF, Liang AC, Chang K, Hackett JA, Harper JW, Hannon GJ, Elledge SJ (2008) Cancer proliferation gene discovery through functional genomics. Science 319:620–624. 10.1126/science.1149200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Zhang F (2015) High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 16:299–311. 10.1038/nrg3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Lopez G (2012) The link between the GBA gene and parkinsonism. Lancet Neurol 11:986–998. 10.1016/S1474-4422(12)70190-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton A, Hardy J (2016) The evolution of genetics: Alzheimer's and Parkinson's diseases. Neuron 90:1154–1163. 10.1016/j.neuron.2016.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. (2003) α-Synuclein locus triplication causes Parkinson's disease. Science 302:841. 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R (2016) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533:95–99. 10.1038/nature17939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473. 10.1073/pnas.95.11.6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampolidis P, Ullrich A, Iacobelli S (2015) LGALS3BP, lectin galactoside-binding soluble 3 binding protein, promotes oncogenic cellular events impeded by antibody intervention. Oncogene 34:39–52. 10.1038/onc.2013.548 [DOI] [PubMed] [Google Scholar]
- Sztainberg Y, Chen HM, Swann JW, Hao S, Tang B, Wu Z, Tang J, Wan YW, Liu Z, Rigo F, Zoghbi HY (2015) Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 528:123–126. 10.1038/nature16159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, Keutzer J, Mistry PK, Chandra SS (2017) Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson's disease. J Neurosci 37:9617–9631. 10.1523/JNEUROSCI.1525-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff DF, Jui NT, Khurana V, Tambe MA, Thompson ML, Chung CY, Kamadurai HB, Kim HT, Lancaster AK, Caldwell KA, Caldwell GA, Rochet JC, Buchwald SL, Lindquist S (2013) Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science 342:979–983. 10.1126/science.1245321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, et al. (2015) Proteomics : tissue-based map of the human proteome. Science 347:1260419. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C (2014) Ten years of next-generation sequencing technology. Trends Genet 30:418–426. 10.1016/j.tig.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Vert JP, Foveau N, Lajaunie C, Vandenbrouck Y (2006) An accurate and interpretable model for siRNA efficacy prediction. BMC Bioinformatics 7:520. 10.1186/1471-2105-7-520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Li B, Zhou L, Fei E, Wang G (2011) The KDEL receptor induces autophagy to promote the clearance of neurodegenerative disease-related proteins. Neuroscience 190:43–55. 10.1016/j.neuroscience.2011.06.008 [DOI] [PubMed] [Google Scholar]
- Ward ME, Chen R, Huang HY, Ludwig C, Telpoukhovskaia M, Taubes A, Boudin H, Minami SS, Reichert M, Albrecht P, Gelfand JM, Cruz-Herranz A, Cordano C, Alavi MV, Leslie S, Seeley WW, Miller BL, Bigio E, Mesulam MM, Bogyo MS, et al. (2017) Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med 9:eaah5642. 10.1126/scitranslmed.aah5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278:25009–25013. 10.1074/jbc.M300227200 [DOI] [PubMed] [Google Scholar]
- Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, Zhao JJ, Roberts TM, Mandel G, Hannon GJ, Depinho RA, Chin L, Elledge SJ (2005) A genetic screen for candidate tumor suppressors identifies REST. Cell 121:837–848. 10.1016/j.cell.2005.03.033 [DOI] [PubMed] [Google Scholar]
- Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, Leng Y, Maehr R, Shi Y, Harper JW, Elledge SJ (2008) SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature 452:370–374. 10.1038/nature06780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski KE, Wisniewski HM, Wen GY (1985) Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol 17:278–282. 10.1002/ana.410170310 [DOI] [PubMed] [Google Scholar]
- Wong YC, Krainc D (2017) α-Synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23:1–13. 10.1038/nm.4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard CM, Campos BA, Kuo SH, Nirenberg MJ, Nestor MW, Zimmer M, Mosharov EV, Sulzer D, Zhou H, Paull D, Clark L, Schadt EE, Sardi SP, Rubin L, Eggan K, Brock M, Lipnick S, Rao M, Chang S, Li A, et al. (2014) iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson's disease. Cell Rep 9:1173–1182. 10.1016/j.celrep.2014.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedlapudi D, Joshi GS, Luo D, Todi SV, Dutta AK (2016) Inhibition of alpha-synuclein aggregation by multifunctional dopamine agonists assessed by a novel in vitro assay and an in vivo Drosophila synucleinopathy model. Sci Rep 6:38510. 10.1038/srep38510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ (2008) Global protein stability profiling in mammalian cells. Science 322:918–923. 10.1126/science.1160489 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Composition of the druggable genome and primary screen pipeline. A) Distribution of the eight sub-libraries according to their functional groups. The number in parenthesis represents the number of genes targeted (10 shRNA/gene on average) and % represents the percentage representation of the druggable genome library. B) Pipeline for the primary screen is presented as a flow diagram; 8x denotes steps that are repeated eight times (each library is done separately). Download Figure 1-1, JPG file (312KB, jpg)
Use of HEK293T cells as a first-pass validation step for the identification of modulators of α-Syn levels. A) ELISA for α-Syn levels in three cell lines reveals a substantial amount of α-Syn levels in HEK293T cells compared to undifferentiated neuroblastoma cell lines (SH-SY5Y and SK-N-BE(2)-C). B) Photomicrograph of HEK293T cells infected with GIPz lentivirus containing a tGFP reporter demonstrates a high level of infectivity. C) ELISA signal is specific to α-Syn and shRNA-mediated knockdown of SNCA does not decrease SNCB or SNCG expression. D) Individual data points between ELISA and western blot assays show significant level of correlation. Download Figure 2-1, JPG file (321.4KB, jpg)
Direct manipulation of α-Syn levels in the Drosophila model. A) Decreasing α-Syn levels in the Drosophila model of parkinsonism by means of inducible shRNAs targeting the α-Syn gene results in a suppression of the behavioral deficits induced by α-Syn. B) Effect of inducible shRNAs targeting the α-Syn gene in Drosophila on α-Syn levels by western blot. Download Figure 3-1, JPG file (178.7KB, jpg)
Representative video of motor performance assay on α-Syn transgenic flies compared with controls.
Summary of screen data. Curated data from screens in human cells, human neurons and mouse brain are presented. Each tab represents a different level of screening. Of note, qPCR data for knockdown of each candidate gene for human neurons (Figure 4) and mouse brain (Figure 5) experiments are presented on separate tabs. Figure legends are presented at the top of each table. Download Table 1-1, XLSX file (1.2MB, xlsx)
List of antibodies, cell lines and oligonucleotides used in this study. Download Table 1-2, XLSX file (12.4KB, xlsx)
Establishment of a human neuron model to test α-Syn modulators. A) Quantification of the relative number of double positive Nestin and SOX2 cells in H9 hESC derived NPC cultures. B) Immunofluorescence staining for NESTIN and PAX6 (left panel), NESTIN and SOX2 (middle panel) and NESTIN and FABP (right panel). Nuclei are stained using DAPI. C) Representative photomicrographs of differentiated neuronal cultures, derived from H9 hESCs and stained for PSA-NCAM, doublecortin (DCX), MAP2 and TUJ1 are presented. Download Figure 4-1, JPG file (595.7KB, jpg)