

Abstract
Stac protein (named for its SH3- and cysteine-rich domains) was first identified in brain 20 years ago and is currently known to have three isoforms. Stac2, Stac1, and Stac3 transcripts are found at high, modest, and very low levels, respectively, in the cerebellum and forebrain, but their neuronal functions have been little investigated. Here, we tested the effects of Stac proteins on neuronal, high-voltage-activated Ca2+ channels. Overexpression of the three Stac isoforms eliminated Ca2+-dependent inactivation (CDI) of l-type current in rat neonatal hippocampal neurons (sex unknown), but not CDI of non-l-type current. Using heterologous expression in tsA201 cells (together with β and α2-δ1 auxiliary subunits), we found that CDI for CaV1.2 and CaV1.3 (the predominant, neuronal l-type Ca2+ channels) was suppressed by all three Stac isoforms, whereas CDI for the P/Q channel, CaV2.1, was not. For CaV1.2, the inhibition of CDI by the Stac proteins appeared to involve their direct interaction with the channel's C terminus. Within the Stac proteins, a weakly conserved segment containing ∼100 residues and linking the structurally conserved PKC C1 and SH3_1 domains was sufficient to fully suppress CDI. The presence of CDI for l-type current in control neonatal neurons raised the possibility that endogenous Stac levels are low in these neurons and Western blotting indicated that the expression of Stac2 was substantially increased in adult forebrain and cerebellum compared with neonate. Together, our results indicate that one likely function of neuronal Stac proteins is to tune Ca2+ entry via neuronal l-type channels.
SIGNIFICANCE STATEMENT Stac protein, first identified 20 years ago in brain, has recently been found to be essential for proper trafficking and function of the skeletal muscle l-type Ca2+ channel and is the site of mutations causing a severe, inherited human myopathy. In neurons, however, functions for Stac protein have remained unexplored. Here, we report that one likely function of neuronal Stac proteins is tuning Ca2+ entry via l-type, but not that via non-l-type, Ca2+ channels. Moreover, there is a large postnatal increase in protein levels of the major neuronal isoform (Stac2) in forebrain and cerebellum, which could provide developmental regulation of l-type channel Ca2+ signaling in these brain regions.
Keywords: calcium-dependent inactivation, hippocampus, l-type calcium channels, Stac protein
Introduction
Stac, so named because it contains src homology 3 (SH3)- and cysteine-rich domains, was first identified as a developmentally regulated brain transcript encoding an ∼40 kDa protein of unknown function (Suzuki et al., 1996). Subsequently, a database search identified two related proteins (Stac2 and Stac3) encoded by distinct genes and it was shown that Stac1 (i.e., “Stac”) and Stac2 are differentially expressed in subpopulations of DRG neurons (Legha et al., 2010). In adult mice, Stac3 transcript levels are high in skeletal muscle and low in cerebellum, forebrain, and eye, whereas Stac2 transcripts are found in these same three neuronally rich regions at levels comparable to those of Stac3 in skeletal muscle; Stac1 transcripts are present in cerebellum, fore/midbrain, and eye (at lower levels than Stac2) and also in the bladder and adrenal gland (Nelson et al., 2013). Therefore, it seems likely that Stac1, Stac2, and perhaps Stac3 are important for neuronal function.
The first real insight as to the function of any of the Stac proteins came from observations that the absence of Stac3 prevented differentiation of skeletal muscle (Bower et al., 2012; Reinholt et al., 2013). Subsequently, it was shown that the absence of Stac3 resulted in the failure of excitation–contraction (EC) coupling (Horstick et al., 2013; Nelson et al., 2013) and that a point mutation in Stac3 resulted in a severe, recessively inherited human myopathy (Horstick et al., 2013). Functionally, Stac3 is required for the ability of CaV1.1 to elicit the intracellular Ca2+ release underlying EC coupling (Polster et al., 2016; Linsley et al., 2017) and it modifies the gating of CaV1.1 as an l-type Ca2+ channel (Polster et al., 2015, 2016).
The interactions found between Stac3 and CaV1.1 (the principle subunit of the skeletal muscle l-type Ca2+ channel) motivated us to investigate whether functional interactions also occur between Stac proteins and neuronal voltage-gated calcium channels. These channels not only contribute to neuronal electrical activity, but also produce Ca2+ signals important for neurotransmitter release, synaptic plasticity (Moosmang et al., 2005), and activity-dependent gene regulation (Murphy et al., 1991). Given these diverse functions, it is not surprising that neuronal calcium channels exist in molecular complexes that serve to couple Ca2+ influx to downstream effectors and/or to regulate the magnitude of this influx (Rettig et al., 1996; Murphy et al., 2014). A particularly well characterized example of the latter is Ca2+-dependent inactivation (CDI), in which Ca2+ entry promotes inactivation of the channel (Brehm and Eckert, 1978; Kass and Sanguinetti, 1984). This process has been extensively documented for three types of neuronal calcium channels: the l-type channels CaV1.2 (Peterson et al., 1999; Qin et al., 1999; Zühlke et al., 1999) and CaV1.3 (Yang et al., 2006), the P/Q-type channel CaV2.1 (Lee et al., 1999), and the N-type channel CaV2.2 (Liang et al., 2003). For all three types, inactivation is greatly slowed when external Ca2+ is replaced by Ba2+.
Here, we used neonatal Sprague Dawley rat hippocampal neurons and tsA201 cells to examine the ability of Stac1, Stac2, and Stac3 to affect CDI of neuronal, voltage-gated Ca2+ channels. In control hippocampal neurons, CDI occurred both for l-type currents and for non-l-type currents, which were isolated from one another pharmacologically. Expression of all three Stac isoforms eliminated CDI of the hippocampal l-type Ca2+ currents, but not of the non-l-type currents. In tsA201 cells, the Stac proteins suppressed CDI of the l-type channels CaV1.2 and CaV1.3, but not of the P/Q channel CaV2.1. Auxiliary subunits were not required for the ability of the Stac proteins to inhibit CDI via CaV1.2, which appeared to involve an interaction between the C terminus of the channel and a segment of the Stac proteins having low homology between the isoforms. Western blotting indicated that there is a significant postnatal increase in the levels of Stac2 protein in forebrain and cerebellum, which may function as a developmental regulator of neuronal calcium signaling within these regions.
Materials and Methods
Molecular biology.
The construction of the expression plasmids for YFP-labeled CaV1.2, unlabeled β2a, unlabeled β1a, YFP-β1a unlabeled α2-δ1, Stac1-YFP, Stac2-YFP, Stac3-YFP, Stac1-tagRFP, Stac2-tagRFP, Stac3-tagRFP, unlabeled Stac1, Stac2, and Stac3, was described previously (Papadopoulos et al., 2004; Leuranguer et al., 2006; Polster et al., 2015, 2018). The plasmid YFP-CaV2.1, labeled at its N terminus with enhanced yellow fluorescent protein (YFP), was derived from the original GFP-CaV2.1 plasmid (Grabner et al., 1998) by excising the CaV2.1 encoding sequence and inserting it into the multiple-cloning site of pEYFP-C1 (Clontech) using the restriction enzymes SalI and HpaI. Unlabeled CaV1.3 (CaV1.3e[8a,11,31b,Δ32,42a] mut) was a gift from Diane Lipscombe (Addgene plasmid #26576, Xu and Lipscombe, 2001; RRID:SCR_002037). The following is a list of cytoplasmic CaV1.2 domain (fragment) constructs used in this study, their forward (fw) and reverse (rev) primers with the respective enzymes used for restriction, followed by the range of encoded rabbit CaV1.2 (GenBank number X15539.1; RRID:SCR_002760) amino acid residues.
GFP-N-term(CaV1.2), fw 5′-GCAGTCGACCTTCGAGCCCTTGTTCAGCCAG-3′, SalI, rev 5′-GGCGGATCCCTATTTCCACTCGACGATGCTTATG-3′, BamHI, residues 1–154.
GFP-I–II(CaV1.2), fw 5′-GCAGTCGACGGAGAGTTTTCCAAAGAGAGG-3′, SalI, rev 5′-GGCGGATCCCTAGTTCGACTTGACCGCTGCG-3′, BamHI, residues 436–554.
GFP-II–III(CaV1.2), fw 5′-GCAGTCGACAACCTGGCTGATGCTGAGAGC-3′, SalI, rev 5′-GGCGGATCCCTACGTGTCGTTGACGATACGG-3′, BamHI residues 784–930.
GFP-III–IV(CaV1.2), fw 5′-GCAGTCGACGTCACCTTCCAGGAGCAGG-3′, SalI, rev 5′-GGCGGATCCCTAGGTGGAGTTGACCACGTAC-3′, BamHI residues 1197–1249.
GFP-C-term(CaV1.2), fw 5′-GGAATTCCGACAACTTTGACTACCTGAC-3′, EcoRI, rev 5′-GGCGGATCCCTACAGGCTGCTGACGCC-3′, BamHI, residues 1507–2171.
GFP-1507–1839(CaV1.2), fw 5′-GGAATTCCGACAACTTTGACTACCTGAC-3′, SalI, rev 5′-GGCGGATCCCTACCATGCTGCCTCCTGTGCC-3′, BamHI, residues 1507–1839.
GFP-1840–2171(CaV1.2), fw 5′- GCAGTCGACCCCAATGAGAGTGAGGATAAG-3′, SalI, rev 5′-GGCGGATCCCTACAGGCTGCTGACGCCGGCC-3′, BamHI, residues 1840–2171.
GFP-1677–2004(CaV1.2), fw 5′-GCAGTCGACGTAGGGAAGCCGGCCCTGGAG-3′, SalI, rev 5′-GGCGGATCCCTAGGAGAGTGGCCGAGGGCGG-3′, BamHI, residues 1677–2004.
GFP-1580–1636(CaV1.2), fw 5′-GCAGTCGACCTGAACAGTGACGGGACGGTC-3′, SalI, rev 5′-GGCGGATCCCTAGGGCACCACTTGGTCCAGC-3′, BamHI, residues 1580–1636.
GFP-1637–1673(CaV1.2), fw 5′-GCAGTCGACCCTGCAGGCGATGATGAGGTC-3′, SalI, rev 5′-GGCGGATCCCTAGGGCTTGCCCACAAGCCCTTG-3′, BamHI, residues 1637–1673.
To obtain association with the cell surface, the CaV1.2 I–II-loop sequence was added as a targeting sequence to these GFP-labeled intracellular CaV1.2 domains and fragments as described previously (Polster et al., 2018). CaV1.2 aa 1580–1673 (CT1) were transferred from CyPet-CT1 (Ohrtman et al., 2008) into the vehicle vector (GFP-I–II-loop[CaV1.2]) using the restriction enzymes EcoRI and BamHI.
The expression vectors for Stac1-RFP, Stac2-RFP, and Stac3-RFP (Polster et al., 2018) were used as templates to amplify and to introduce EcoRI and KpnI sites and a stop codon flanking the coding sequence of defined domains of Stac protein via standard PCR. Subsequent digestion of the PCR products at restriction sites introduced at both ends during amplification allowed for later ligation into the expression plasmid for unlabeled Stac1 (Polster et al., 2018) that had been digested with the same enzymes. The following is a list of the Stac protein fragment constructs used in this study, the range of amino acid residues refer to encoded mouse Stac1, Stac2, and Stac3 (Gene IDs: 20840, 217154, and 237611, respectively), followed by their forward (fw) and reverse (rev) primers.
Stac1[168–403], fw 5′-GCAGAATTCCGATGTTTCGGCGTTACTACAGC-3′, rev 5′-GTGGTACCTACACGTCTACCAGTACATCC-3′,
Stac1[286–403], fw 5′-GCAGAATTCCGATGTTACAGATGAACACCTACG-3′, rev 5′-GTGGTACCTACACGTCTACCAGTACATCC-3′,
Stac1[1–285], fw 5′-GCAGAATTCCGATGATTCCTCCAAGTGGCGCCC-3′, rev 5′-GTGGTACCTATGGGTCTTTGGAAAGAGGTCCC-3′,
Stac1[1–167], fw 5′-GCAGAATTCCGATGATTCCTCCAAGTGGCGCCC-3′, rev 5′-GTGGTACCTATCCCTTTGGCAACTTGCCCATG-3′,
Stac1[168–285], fw 5′-GCAGAATTCCGATGTTTCGGCGTTACTACAGC-3′, rev 5′-GTGGTACCTATGGGTCTTTGGAAAGAGGTCCC-3′,
Stac1[168–217], fw 5′-GCAGAATTCCGATGTTTCGGCGTTACTACAGC-3′, rev 5′-GTGGTACCTAGCCCTTCTTCGTTCTCTGGGCC-3′,
Stac1[168–240], fw 5′-GCAGAATTCCGATGTTTCGGCGTTACTACAGC-3′, rev 5′-GTGGTACCTACTCTTCAGGGACGTCTACAAGC-3′,
Stac1[218–285], fw 5′-GCAGAATTCCGATGGGCTCAGGCAGTGGTTCTG-3′, rev 5′-GTGGTACCTATGGGTCTTTGGAAAGAGGTCCC-3′,
Stac2[169–408], fw 5′-GCAGAATTCCGATGTTTCGACGCAACTTCAGC-3′, rev 5′-GTGGTACCTAGATCTCTGCCAAGGAGTCG-3′,
Stac2[289–408], fw 5′-GCAGAATTCCGATGGGGCCCATGTACTCCTACG-3′, rev 5′-GTGGTACCTAGATCTCTGCCAAGGAGTCG-3′,
Stac2[1–288], fw 5′-GCAGAATTCCGATGACCGAAATGAGCGAGAAGG-3′, rev 5′-GTGGTACCTACACGTCCTTCCGCAGGGGCAGC-3′,
Stac2[1–168], fw 5′-GCAGAATTCCGATGACCGAAATGAGCGAGAAGG-3′, rev 5′-GTGGTACCTAAGATGTGGATGTCTTGCCCGGG-3′,
Stac2[169–288], fw 5′-GCAGAATTCCGATGTTTCGACGCAACTTCAGC-3′, rev 5′-GTGGTACCTACACGTCCTTCCGCAGGGGCAGC-3′,
Stac2[169–221], fw 5′-GCAGAATTCCGATGTTTCGACGCAACTTCAGC-3′, rev 5′-GTGGTACCTAGCTGGAACGGTTCATCAGTGCC-3′,
Stac2[169–242], fw 5′-GCAGAATTCCGATGTTTCGACGCAACTTCAGC-3′, rev 5′-GTGGTACCTAGTCTTCTGTTAGCTCGTCACGC-3′,
Stac2[222–288], fw 5′-GCAGAATTCCGATGTTCAGCAGCACATCTGAGTC-3′, rev 5′-GTGGTACCTACACGTCCTTCCGCAGGGGCAGC-3′,
Stac3[147–360], fw 5′-GCAGAATTCATGTTCCGTCGGGCCTATAGC-3′, rev 5′-GTGGTACCTAAATCTCCTCCAGGAAGTCG-3′,
Stac3[243–360], fw 5′-GCAGAATTCCGATGCAGCAGTCTCATTACTTTG-3′, rev 5′-GTGGTACCTAAATCTCCTCCAGGAAGTCG-3′,
Stac3[1–242], fw 5′-GCAGAATTCCGATGACAGAAAAGGAAGTGGTGG-3′, rev 5′-GTGGTACCTAGAAGCCAGGCTGCTTGTTTTTG-3′,
Stac3[1–146], fw 5′-GCAGAATTCCGATGACAGAAAAGGAAGTGGTGG-3′, rev 5′-GTGGTACCTAACCAGGTGGGATCTTGCCGAAG-3′,
Stac3[147–242], fw 5′-GCAGAATTCCGATGTTCCGTCGGGCCTATAGC-3′, rev 5′-GTGGTACCTAGAAGCCAGGCTGCTTGTTTTTG-3′,
Stac3[147–189], fw 5′-GCAGAATTCCGATGTTCCGTCGGGCCTATAGC-3′, rev 5′-GTGGTACCTACTTCTTCCGTTCCTTGTTTGCC-3′.
Stac3[147–242], fw 5′-GCAGAATTCCGATGTTCCGTCGGGCCTATAGC-3′, rev 5′-GTGGTACCTAGAAGCCAGGCTGCTTGTTTTTG-3′,
Stac3[147–213], fw 5′-GCAGAATTCCGATGTTCCGTCGGGCCTATAGC-3′, rev 5′-GTGGTACCTACTCCTCTGGCCTGGCTGACTC-3′,
Stac3[190–242], fw 5′-GCAGAATTCCGATGGGGCAGGCAGATAAGAAAAATC-3′, rev 5′-GTGGTACCTAGAAGCCAGGCTGCTTGTTTTTG-3′,
All constructs were verified by enzyme digestion and sequence analysis (Eton Bioscience).
Cell culture and expression of cDNA for electrophysiology.
Primary cultures of neonatal hippocampal neurons were prepared as described previously (Gomez et al., 2002; Smith et al., 2006). Specifically, hippocampi were dissected from neonatal postnatal day 0 (P0) to P2 Sprague Dawley rats (RRID:RGD_734476; sex unknown) and then dissociated. For transfection, the dissociated neurons were pelleted and resuspended (3.5–4 × 106 cells per transfection) in AMAXA buffer (Lonza, RRID:SCR_000377), together with 6–8 μg of cDNA for Stac1-YFP, Stac2-YFP, or Stac3-YFP and electroporated with an AMAXA Nucleofector (Lonza). Neurons (either transfected or not) were plated at 6–8 × 105 cells per dish (25 mm with a poly-d-lysine-coated glass coverslip bottom) in glutamine-supplemented MEM (Thermo Fisher Scientific; RRID:SCR_008452) plus 10% (v/v) fetal bovine serum, which was replaced by fresh medium of the same composition 4 h later. One day later, the medium was changed to Neurobasal A medium supplemented with B27 and GlutaMax (all from Thermo Fisher Scientific). Primary cultures of dysgenic (CaV1.1-null) myotubes were prepared and injected in single nuclei with plasmid cDNA for YFP-CaV1.2 (150 ng/μl) as described previously (Beam and Franzini-Armstrong, 1997).
tsA201 cells (ATCC catalog #CRL-3216, RRID:CVCL_0063) were propagated in high-glucose DMEM (Corning Mediatech) supplemented with 10% (v/v) fetal bovine serum and 2 mm glutamine in a humidified incubator with 5% CO2. Cells were plated at a density of 2 × 105 cells in 35 mm dishes and transiently transfected 24 h later. Except for experiments involving CaV1.3, this was accomplished with jetPRIME (Polyplus Transfection) and various combinations of cDNAs: YFP-CaV1.2, YFP-CaV2.1 (1 μg/dish), β1a, β2a, α2-δ1, GFP- and CaV1.2 I–II loop-labeled intracellular CaV1.2 regions, and unlabeled or RFP-labeled Stac protein isoforms or fragments (0.5 μg/dish). Alternatively, the cells were transfected with lipofectamine 2000 (Thermo Fisher Scientific) and cDNAs: CaV1.3 (0.5 μg/dish) and YFP-β1a, and α2-δ1 alone or in combination with one of the three Stac isoforms (0.25 μg/dish). Four hours following transfection, cells were removed from the dish using Trypsin EDTA (Corning Mediatech) and replated at ∼1 × 104 cells per 35 mm dish to obtain isolated cells that were used for electrophysiology either ∼45 or ∼25 h (experiments on CaV1.3) later. Following replating, 2 μm nifedipine was added to the medium of the dishes containing cells transfected with CaV1.3, YFP-β1a, α2-δ1, and one of the Stac proteins, with the nifedipine-containing medium replaced by nifedipine-free medium 30–45 min before the recording of currents. As a control, this same nifedipine pretreatment was also applied to a subset of the cells transfected only with CaV1.3, YFP-β1a, and α2-δ1. Cells transfected with any of the three Ca2+ channels (CaV1.2, CaV1.3, and CaV2.1) were selected for recording by the pattern of yellow fluorescence.
Confocal microscopy, photobleaching, and colocalization.
tsA201 cells were superfused with rodent Ringer's solution, which consisted of the following (in mm): 146 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4, with NaOH and examined using a Zeiss LSM 710 confocal microscope. Excitation and emission for the fluorescent proteins were as follows: GFP (excitation: 488 nm, emission: 493–590 nm), and tagRFP (excitation: 543, emission: 582–754). Relative to full power output, the excitation was attenuated to ∼1% (488 nm) and ∼2–4% (543 nm). Images were obtained with a 40× (1.3 numerical aperture) oil-immersion objective as a single, optical slice that was halfway between the substrate and upper cell surface. Cells were selected for subsequent analysis on the basis of isolation from surrounding cells and an initial midlevel optical scan displaying close association of the tagged cytoplasmic CaV1.2 domain with the perimeter of the cell and high expression of the tagged Stac construct as judged by a uniformly strong fluorescence throughout the entire optical section. The mobile faction of the tagged Stac was then partially photobleached by repeatedly scanning for 15–45 s, with nonattenuated excitation within a region of interest that was designated to avoid the cell surface. A second midlevel scan acquired afterward was used to assess colocalization, which was quantified by means of Pearson's coefficients as described previously (Polster et al., 2018) using ZEN Blue (ZEN Digital Imaging for Light Microscopy; RRID:SCR_013672).
Measurement of ionic currents.
All experiments were performed at room temperature (∼25°C) on cells displaying yellow fluorescence. After 4–5 d in vitro, control neurons and transfected neurons were whole-cell clamped with heat-polished borosilicate patch pipets of 3–5 MΩ resistance filled with the following (in mm): 120 Cs-MeSO3, 30 tetraethylammonium-Cl (TEA-Cl), 10 (l-type currents) or 0.5 (non-l-type currents) EGTA, 5 MgCl2, 5 Na2ATP and 10 HEPES, pH 7.2 with TEA-OH. The bath solution contained the following (in mm): 125 NaCl, 10 CaCl2 or BaCl2, 5.85 KCl, 22.5 TEA-Cl, 1.2 MgCl2, 10 Na-HEPES, 11 d-glucose, and 0.001 tetrodotoxin, pH 7.4 with HCl. Currents were recorded with an Axopatch 200A amplifier (Molecular Devices), low-pass filtered at 2 kHz, and sampled at 10 kHz using PatchMaster software (HEKA). Analog compensation was used to reduce the effective series resistance and partially cancel cell capacitance. Data were analyzed only if the calculated series resistance error was <3 mV. Test currents were recorded first with Ba2+ as the charge carrier and then with Ca2+ and corrected for residual, linear components of leak current by P/4 subtraction. Holding potential was −60 mV and currents were evoked by 200 ms (l-type) or 500 ms (non-l-type) step depolarizations once every 15 s. To isolate l-type currents, cells were incubated just before recording for 30 min in 1 μm ω-CTx-GVIA and 5 μm ω-CTx-MVIIC to block N- and P/Q-type channels and data were collected within 1 h thereafter (Oliveria et al., 2007). Non-l-type currents were recorded in the presence of 5 μm nimodipine.
For tsA201 cells and dysgenic myotubes, pipettes (∼2.0 MΩ) were fabricated from borosilicate glass and were filled with internal solution consisting of the following (in mm): 140 Cs-aspartate, 10 Cs2-EGTA, 5 MgCl2, and 10 HEPES, pH 7.4 with CsOH except that the Cs-aspartate was increased to 155 mm and the Cs2-EGTA was reduced to 0.5 mm for experiments with CaV2.1. The bath solution contained the following (in mm): 145 TEA-Cl, 10 CaCl2 or 10 BaCl2, and 10 HEPES, pH 7.4 with TEA-OH. Electronic compensation was used to reduce the effective series resistance (time constant of ∼500 μs or less). Cell capacitance was determined by integration of a transient elicited by stepping from the holding potential (−80 mV) to −70 mV using pClamp 8.2 (Molecular Devices; RRID:SCR_011323) and was used to normalize ionic currents (pA/pF). Test currents were obtained by stepping from the holding potential either directly to 8 varying test potentials (tsA201 cells) or (for myotubes) by stepping from the holding potential to −30 mV for 1 s (to inactivate endogenous T-type Ca2+ current), to −50 mV for 25 ms, and then to more positive test potentials. Linear components of leak and capacitive current were corrected with −P/4 online subtraction protocols. Filtering was at 2–5 kHz and digitization was at 10 kHz. Peak I–V relationships were fitted according to the following:
Where I is the peak current for the test potential V, Vrev is the reversal potential, Gmax is the maximum Ca2+ channel conductance, V1/2 is the half-maximal activation potential and kG is the slope factor. All data reported from tsA201 cells were obtained from cells in which both Ca2+ and Ba2+ currents were recorded. For solution changes, ∼18 ml of solution was perfused at a rate of ∼6 ml min−1.
Western blotting.
To obtain recombinant Stac2-YFP, tsA201 cells were plated at a density of 6 × 105 per 10 cm plastic tissue culture dish (BD Falcon), grown to 80% confluency, and then transfected with Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer's instructions with 10 μg of the cDNA of interest per 10 cm dish. Sixteen hours after transfection, tsA201 cells in 10 cm dishes were washed twice with cold PBS, scraped from the dish, resuspended in 0.5 ml of ice-cold homogenization buffer (300 mm sucrose, 20 mm Tris-Maleate and 1% (v/v) P8340 protease inhibitor Sigma-Aldrich; RRID:SCR_008988), and subjected to 3× 100 strokes (at 3 min intervals) in a Teflon/glass homogenizer on ice. For brain tissue, mice (sex unknown) were killed by isoflurane overdose, after which the cerebellum and forebrain were isolated and frozen on dry ice and stored at −80°C. Frozen cerebella and forebrains from 4 neonatal mice (∼5 d) and 3 adult mice (∼6 month) were finely minced with a razor blade, disrupted with a Polytron tissue homogenizer (Ultra-Turrax T25; IKA) at 9500 rpm for 30 s, and then subjected to Teflon/glass homogenization as described for the tsA201 cells. The homogenates (both tsA201 cells and nervous tissue) were placed in an ultrasonic water bath for 15 s and then centrifuged for 10 min at 5000 × g, retaining the supernatant for subsequent analysis. Protein concentration was determined with the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific) using bovine serum albumin as the standard.
Proteins in the ts201, forebrain, and cerebellar homogenates were separated by SDS-PAGE on a continuous-gradient 4–15% gel (Mini protean TGX; Bio-Rad; RRID:SCR_008426), followed either by staining with Coomassie blue R-250 (Thermo Fisher Scientific) or transfer onto polyvinylidene difluoride membranes (Bio-Rad Laboratories). The membranes were incubated for 1 h in TBS-T (20 mm Tris, 500 mm NaCl, pH 7.4, plus 0.1% [v/v] Tween 20) plus 5% (w/v) non-fat dry milk (Bio-Rad), washed with TBS-T, incubated overnight at 4°C in a 1:1000 dilution of rabbit polyclonal anti-Stac2 (N-terminal region; Aviva Systems Biology, catalog #ARP69675_P050, RRID:AB_2737299), washed with TBS-T, and then incubated 1 h at room temperature with a 1:10,000 dilution of HRP-conjugated goat anti-rabbit antibody (Thermo Fisher Scientific). Immunoreactive proteins were detected via enhanced chemiluminescence (SuperSignal West Femto substrate; Thermo Fisher Scientific). In some cases, the anti-Stac2 antibody was preincubated 3 h at room temperature with a 30-fold molar excess of its immunogenic peptide (Aviva Systems Biology, catalog #AAP69675) as the blocking control.
Analysis.
SigmaPlot (version 11.0; SYSTAT Software; RRID:SCR_003210) was used for curve fitting and preparation of figures. Unless otherwise specified, all data are presented as mean ± SEM. Statistical significance was assessed with one-way ANOVA.
Results
Stac proteins suppress CDI of l-type Ca2+ currents but not of non-l-type currents in neurons
Our previous experiments using heterologous expression in tsA201 cells (Polster et al., 2015) showed that the expression of either Stac2 or Stac3 slowed the inactivation of Ca2+ current via CaV1.2, which is the predominant isoform (∼75%) of neuronal l-type Ca2+ channel in cerebral cortex and hippocampus (Hell et al., 1993). Here, we tested whether the overexpression of the Stac proteins would similarly affect endogenous l-type channels in hippocampal neurons isolated from neonatal rats and maintained several days in culture. l-type currents were isolated by pretreatment with ω-CTx-GVIA and ω-CTx-MVIIC (Oliveria et al., 2007) and were measured both in control cells and in cells that had been transfected with Stac1-YFP, Stac2-YFP, or Stac3-YFP. Figure 1 compares representative currents at +10 mV (Fig. 1A), and fraction of peak current remaining 150 ms after the peak (R150; Fig. 1B) of whole-cell Ca2+ and Ba2+ currents in neurons either without or with Stac1, Stac2, or Stac3 (Fig. 1, left to right, respectively). In the absence of any coexpressed Stac protein, the Ca2+ currents decayed to a greater extent than the Ba2+ currents, a difference that was greatest at test potentials of ∼+10 mV and which became progressively smaller for both weaker and stronger test depolarizations (Fig. 1, left). This “U-shaped” voltage dependence of fractional decay with Ca2+ as charge carrier and its significant suppression by Ba2+ are characteristic of CDI. In contrast to the situation for control neurons, the decay of Ca2+ and Ba2+ currents was almost identical in neurons expressing Stac1-YFP, Stac2-YFP or Stac3-YFP (Fig. 1A,B, right three panels), indicating that all three Stac proteins suppressed the Ca2+-dependent inactivation of the l-type channels present in these neonatal hippocampal neurons.
Figure 1.

Stac proteins suppress Ca2+-dependent inactivation of l-type Ca2+ currents in neonatal rat hippocampal neurons. Representative whole-cell Ca2+ (red, vertically scaled by indicated factors) and Ba2+ (black) currents at +10 mV (A) and fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential (B) are shown for either control neurons or neurons transfected with Stac1-, Stac2-, or Stac3-YFP (left to right, respectively). l-type currents were isolated by pharmacological blockade of non-l-type currents (see Materials and Methods). Numbers of cells (Ca2+, Ba2+): (9,4), (7,6), (6,3), and (6,4) for control, Stac1-, Stac2-, and Stac3-transfected cells, respectively. Here and in subsequent figures, the error bars indicate ± SEM. Scale bars in A: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal).
To test the effects of the Stac proteins on high-voltage-activated neuronal Ca2+ channels that were not l-type, we recorded currents with 5 μm nimodipine present. With this treatment and the use of a holding potential of −60 mV, the measured Ca2+ channel currents should have arisen mainly from CaV2.1 (P/Q-type) and CaV2.2 (N-type). These have both been reported to undergo Ca2+-dependent inactivation, although weaker than that for l-type channels (DeMaria et al., 2001; Liang et al., 2003). Accordingly, we reduced the EGTA concentration of the pipette solution (from 10 to 0.5 mm) and increased the test pulse duration (from 200 to 500 ms). Under these conditions, the non-l-type currents displayed prominent calcium-dependent inactivation both in control hippocampal neurons and in neurons expressing Stac1-YFP, Stac2-YFP, or Stac3-YFP (Fig. 2). Therefore, the Stac proteins did not appear to affect the Ca2+-dependent inactivation of non-l-type channels expressed endogenously in hippocampal neurons.
Figure 2.

Stac proteins do not affect Ca2+-dependent inactivation of non-l-type Ca2+ currents in neonatal rat hippocampal neurons. Representative whole-cell Ca2+ (red, vertically scaled by indicated factors) and Ba2+ (black) currents at +10 mV (A) and ratio of current 400 ms after the peak inward current to the peak inward current (R400) as a function of test potential (B) are shown for control neurons and neurons transfected with Stac1-, Stac2-, or Stac3-YFP (left to right, respectively). Non-l-type currents were isolated by the presence of 5 μm nimodipine in the bathing solution. Numbers of cells (Ca2+, Ba2+): (5,6), (6,5), (6,5) and (6,7) for control, Stac1-, Stac2-, and Stac3-transfected cells, respectively. Scale bars in A: 10 pA/pF (vertical, unscaled Ba2+ currents), 100 ms (horizontal).
Stac proteins suppress CDI of the l-type channel CaV1.2 in tsA201 cells
To obtain information about the effects of the Stac proteins on Ca2+ channels of known molecular composition, we used heterologous expression in tsA201 cells. Figure 3 compares representative peak currents (Fig. 3A), fraction of peak current remaining 150 ms after the peak (R150; Fig. 3B) and peak I–V relationships (Fig. 3C) of whole-cell Ca2+ and Ba2+ currents in tsA201 cells transfected with YFP-CaV1.2, β2a and α2-δ1, either without or with Stac1, Stac2, or Stac3 (Fig. 3, left to right, respectively). The behavior of the currents in the tsA201 cells expressing CaV1.2 was qualitatively similar to that of the l-type currents in the hippocampal neurons. Therefore, in the cells transfected with YFP-CaV1.2, β2a and α2-δ1 without any Stac protein, the Ca2+ current decayed to a greater extent than the Ba2+ current, as evident both in the peak currents (Fig. 3A) and in the R150 values across a range of test potentials (Fig. 3B). Moreover, R150 for Ca2+ reached a minimum at approximately the same potential as that eliciting the maximal inward current (Fig. 3C). In contrast, in cells transfected with YFP-CaV1.2, β2a and α2-δ1 together with any one of the three Stac isoforms, the peak Ca2+ and Ba2+ currents had a similar time course (Fig. 3A) and the values of R150 for Ca2+ did not differ greatly from those for Ba2+ (Fig. 3B). All three Stac isoforms appeared to be essentially equivalent at suppressing calcium-dependent inactivation. However, peak current densities were ∼30% smaller for Stac3 than without Stac or than with Stac1 or Stac2 (Fig. 3C).
Figure 3.

Stac proteins eliminate Ca2+-dependent inactivation of the l-type channel CaV1.2. Representative peak Ca2+ (red, vertically scaled by indicated factors) and Ba2+ (black) currents (A), fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential (B), and peak I–V relationships (C) in tsA201 cells transfected with YFP-CaV1.2, β2a, and α2-δ1 either without or with Stac1, Stac2, or Stac3 (left to right, respectively). In A, test potentials for Ca2+ were +20 mV (without Stacs) or +30 mV (with Stac1, 2 or 3); the test potentials for Ba2+ were +10 mV (without Stacs, with Stac1 or 2) or +20 mV (with Stac3). Scale bars: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal).
Stac proteins suppress calcium-dependent inactivation of CaV1.3 in tsA201 cells
Although less abundant than CaV1.2, CaV1.3 also comprises a significant fraction (∼20%) of l-type channels in cerebral cortex and hippocampus (Hell et al., 1993). Moreover, the more negative activation range of CaV1.3 (Koschak et al., 2001) might be expected to amplify its relative contribution to the l-type currents illustrated for hippocampal neurons in Figure 1. Therefore, we also tested the effects of Stac proteins on CaV1.3. Figure 4 compares representative peak currents (Fig. 4A), fraction of peak current remaining 150 ms after the peak (R150; Fig. 4B) and peak I–V relationships (Fig. 4C) of whole-cell Ca2+ and Ba2+ currents in tsA201 cells transfected with CaV1.3, YFP-β1a, and α2-δ1 either alone or with Stac1, Stac2, or Stac3 (Fig. 4, left to right, respectively). As for CaV1.2 in the presence of β2a and α2-δ1 (Fig. 3), the Ca2+ current of CaV1.3 decayed to a greater extent than the Ba2+ current, as evident both in the peak currents (Fig. 3A) and in the R150 values, which had a U-shaped dependence on test potential (Fig. 3B) that mirrored the peak I–V relationship (Fig. 3C). This behavior of currents via CaV1.3 is similar to that described in previous work (Yang et al., 2006). Unlike the rapid decay of Ca2+ current observed in cells transfected only with CaV1.3, YFP β1a, and α2-δ1, there was little inactivation of Ca2+ currents in cells transfected with this channel complex plus Stac1, Stac2, or Stac3 and the peak Ca2+ and Ba2+ currents had a similar time course and the values of R150 for Ca2+ did not differ from those for Ba2+ (Fig. 4A,B). Therefore, all three Stac proteins appeared to be similar in suppressing Ca2+-dependent inactivation of both CaV1.2 and CaV1.3.
Figure 4.

Stac proteins suppress Ca2+-dependent inactivation of the l-type channel CaV1.3. Representative peak Ca2+ (red, vertically scaled by indicated factors) and Ba2+ (black) currents (A), fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential (B), and peak I–V relationships (C) in tsA201 cells transfected with CaV1.3, YFP-β1a, and α2-δ1 either without or with Stac1, Stac2, or Stac3 (left to right, respectively). In A, test potentials for Ca2+ were +10 mV (with or without Stac proteins); the test potentials for Ba2+ were 0 mV (without Stacs or with Stac1) or −10 mV (with Stac2 or Stac3). Scale bars: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal). All cells transfected with Stac proteins were bathed for 24 h in medium containing 2 μm nifedipine. Recordings were obtained ∼30 min after washing off the nifedipine. Figure 4-1 compares R150 values obtained from control cells either with or without this pretreatment with nifedipine.
Nifedipine pre-treatment does not alter the fractional inactivation of Ca2+ current in tsA201 cells transfected with CaV1.3, YFP-β1a, and α2-δ1. R150 plotted as a function of test potential for 3 cells pre-treated with 2 µM nifedipine (see Methods) and 3 cells not pre-treated with nifedipine. Download Figure 4-1, TIF file (2.6MB, tif)
As described in the Materials and Methods section, the cells cotransfected with CaV1.3 and Stac proteins were maintained in a medium containing 2 μm nifedipine, which was then washed away before recording. Without this pretreatment, we found it very difficult to obtain cells producing measureable currents, which we hypothesize was a consequence of Ca2+-induced cell death that resulted from the loss of inactivation produced by the Stac proteins and the leftward shift of activation of CaV1.3 (compared with CaV1.2), which resulted in persistent Ca2+ entry due to the relatively depolarized resting potentials of tsA201 cells. In control experiments on cells transfected with CaV1.3, YFP-β1a and α2-δ1 without Stac, we found that the nifedipine pretreatment had no effect on inactivation (Fig. 4-1).
Stac proteins do not affect calcium-dependent inactivation of CaV2.1 in tsA201 cells
As shown in Figure 2, Stac proteins did not noticeably affect the calcium-dependent inactivation of the non-l-type, high-voltage-activated Ca2+ channels that were present in neonatal hippocampal neurons. Figure 5 illustrates an experiment to test whether similar behavior occurred for one representative of these channels, CaV2.1, after heterologous expression together with β1a and α2-δ1. Because Ca2+-dependent inactivation for CaV2.1 (DeMaria et al., 2001) is much slower than for the l-type channels, the duration of the test depolarizations was increased from 200 to 500 ms and the extent of inactivation was quantified as the fraction of peak current remaining 400 ms after the peak (R400). With the longer test pulses, it was evident that the peak Ca2+ current via CaV2.1 inactivated more rapidly than the peak Ba2+ current regardless of whether it was cotransfected with any one of the three Stac proteins (Fig. 5A). Furthermore, the relationship between R400 and test potential both for Ca2+ and for Ba2+ was little affected by whether a Stac protein was also transfected (Fig. 5B,C). Therefore, the Stac proteins did not appear to alter calcium-dependent inactivation of CaV2.1.
Figure 5.

Stac proteins do not affect Ca2+-dependent inactivation of the P/Q-type channel CaV2.1. Representative peak Ca2+ (red, vertically scaled by indicated factors) and Ba2+ (black) currents (A), fraction of peak current remaining 400 ms after the peak (R400) as a function of test potential (B), and peak I–V relationships (C) in tsA201 cells transfected with YFP-CaV2.1, β1a, and α2-δ1 either without or with Stac1, Stac2, or Stac3 (left to right, respectively). Ca2+ and Ba2+ test potentials in A: +25 mV and +10 mV, respectively. Scale bars in A: 5 pA/pF (unscaled Ba2+ currents, vertical), 100 ms (horizontal).
Interaction between the Stac proteins and cytoplasmic domains of CaV1.2 appears to be responsible for the suppression of calcium-dependent inactivation
To determine whether the auxiliary β2a and α2-δ1 subunits were required for the ability of Stac proteins to suppress calcium-dependent inactivation of CaV1.2, we removed their cDNAs from the transfection mixtures. Figure 6 compares representative peak currents (Fig. 6A), fraction of peak current remaining 150 ms after the peak (R150; Fig. 6B), and peak I–V relationships (Fig. 6C) of whole-cell Ca2+ and Ba2+ currents in tsA201 cells transfected with YFP-CaV1.2 either alone or with Stac1, Stac2, or Stac3 (Fig. 6, left to right, respectively). Consistent with previous work on tsA201 cells (Zong et al., 1994; Pérez-García et al., 1995), expression of CaV1.2 without auxiliary subunits resulted in small-amplitude currents (Fig. 6A,C). Despite this ∼10-fold reduced current density, calcium-dependent inactivation for YFP-CaV1.2 alone differed little from that of YFP-CaV1.2 plus β2a and α2-δ1 (Fig. 6A,B, left, and Fig. 3A,B, left, respectively). The additional transfection of Stac1, Stac2, or Stac3 was very effective in eliminating the calcium-dependent inactivation of CaV1.2 lacking auxiliary subunits (Fig. 6A,B, right three panels). Additionally, Stac2 and Stac3 caused an ∼50% increase in the Ba2+ current density (Fig. 6C).
Figure 6.

Stac proteins inhibit Ca2+-dependent inactivation of CaV1.2 in the absence of auxiliary subunits. Representative peak Ca2+ (red, vertically scaled by indicated factors) and Ba2+ (black) currents (A), ratio of current 150 ms after the peak inward current to the peak inward current (R150) plotted as a function of test potential (B) and peak I–V relationships (C) in tsA201 cells transfected with YFP-CaV1.2 (in the absence of auxiliary subunits) either without or with Stac1, Stac2, or Stac3 (left to right, respectively). Ca2+ test potentials in A: +40 mV. Ba2+ test potentials in A: +30 mV (without Stacs or with Stac3) or +20 mV (with Stac1 or Stac2). Scale bars in A: 2 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal).
Based on the results illustrated in Figure 6, it seemed likely that all three Stac proteins interact directly with CaV1.2 to suppress its calcium-dependent inactivation. To identify potential sites of interaction, we tested whether the Stac proteins colocalized in tsA201 cells with specific cytoplasmic domains of CaV1.2. For this, the CaV1.2 cytoplasmic domains were linked at their N termini to a surface-targeting sequence, the GFP-tagged I–II loop of CaV1.2 (Polster et al., 2018), tagRFP was linked to the C termini of the Stac proteins, and the mobile pool of the RFP labeled Stac proteins was partially photobleached by intense illumination of a region of interest in the cell interior. Figure 7 illustrates the results of such an experiment for Stac1, which associated with the surface-localized II–III loop and C terminus of CaV1.2, but not with the other cytoplasmic domains. Stac2 displayed a similar association with the II–III loop, in agreement with the observation of Wong King Yuen et al. (2017) that the isolated SH3 tandem from Stac2 interacts with a core segment of the CaV1.2 II–III loop, and with the C terminus (Fig. 7-1). Of these two domains, an interaction with the II–III loop seems less likely to be important for the ability of the Stac proteins to suppress calcium-dependent inactivation because Stac3 appeared only to associate with the CaV1.2 C terminus and not with the II–III loop (Fig. 7-2). Pearson's colocalization coefficients for each of the three Stac proteins and cytoplasmic CaV1.2 domains are provided in Figure 7-3. Our previous work (Polster et al., 2018) had also shown that Stac3 fails to associate with the CaV1.2 II–III loop either as an isolated fragment in tsA201 cells or when substituted for the II–III loop of CaV1.1 in myotubes.
Figure 7.
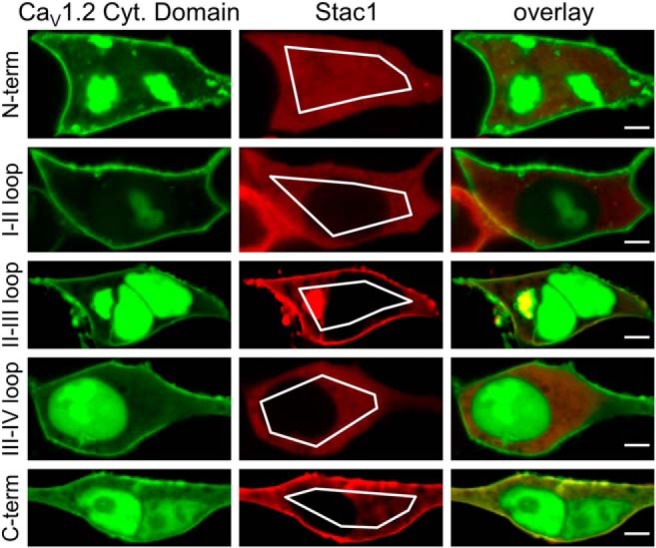
Association of Stac1 with cytoplasmic CaV1.2 domains in tsA201 cells. Representative images are shown of cells cotransfected with Stac1-tagRFP and the indicated cytoplasmic domains of CaV1.2, which were targeted to the cell surface by being linked at the N-terminal to the GFP-tagged I–II loop of CaV1.2 (Polster et al., 2018). The images were obtained after partial photobleaching of the mobile pool of Stac1-tagRFP by repeated scans within the indicated regions of interest with nonattenuated excitation at 543 nm. Bars, 5 μm. Figure 7-1 and Figure 7-2 illustrate similarly obtained images for Stac2 and Stac3. Figure 7-3 and Figure 7-4 present the Pearson colocalization coefficients for the different combinations of Stac proteins and cytoplasmic CaV1.2 domains or CaV1.2 C terminus segments.
Association of Stac2 with cytoplasmic CaV1.2 domains in tsA201 cells. Representative images are shown of cells co-transfected with Stac2-tagRFP and the indicated cytoplasmic domains of CaV1.2, which were targeted to the cell surface by being linked at the N-terminal to the GFP-tagged I-II loop of CaV1.2. The images were obtained after partial photo-bleaching of the mobile pool of Stac1-tagRFP by repeated scans within the indicated regions of interest with non-attenuated excitation at 543 nm. Bars, 5 μm. Download Figure 7-1, TIF file (1.4MB, tif)
Association of Stac3 with cytoplasmic CaV1.2 domains in tsA201 cells. Representative images are shown of cells co-transfected with Stac3-tagRFP and the indicated cytoplasmic domains of CaV1.2, which were targeted to the cell surface by being linked at the N-terminal to the GFP-tagged I-II loop of CaV1.2. The images were obtained after partial photo-bleaching of the mobile pool of Stac1-tagRFP and GFP-tagged CaV1.2 C-terminus by repeated scans within the indicated regions of interest with non-attenuated excitation at 543 nm and 515 nm, respectively. Bars, 5 μm. Download Figure 7-2, TIF file (1.4MB, tif)
Pearson’s coefficients (mean ± SEM) for colocalization of the Stac proteins and the cytoplasmic domains of CaV1.2. The number of images analyzed is indicated for each combination of constructs. ***, P < 0.001 by one-way ANOVA. Download Figure 7-3, TIF file (946KB, tif)
The interaction between Stac proteins and the CaV1.2 C-terminus. (A) Schematic representation and domain architecture (not to scale) of the full length C-terminus of rabbit CaV1.2 (top, residues 1507-2171) and different sub-segments (numbers refer to the first and last residues of CaV1.2 sequence) tested for Stac protein interaction, with several regions indicated that have been implicated as important for calcium-dependent inactivation (EF hand motif, pre-IQ region consisting of regions A and C, IQ domain; for references, see Results), (B) Pearson’s coefficients (mean ± SEM) for co-localization of indicated CaV1.2 C-terminus segments (linked to the GFP-tagged I-II loop of CaV1.2) and RFP-labelled Stac proteins after cotransfection in tsA201 cells (the number of images analyzed is indicated for each combination of constructs). Download Figure 7-4, TIF file (1.7MB, tif)
In an attempt to identify subregions of the CaV1.2 C terminus important for its interaction with the Stac proteins, we applied the approach described above to the fragments of the C terminus, which are illustrated schematically in Figure 7-4A. We began with large, overlapping fragments corresponding to the N-terminal half (rabbit CaV1.2 residues 1507–1839), center (residues 1674–2004), or C-terminal half (residues 1840–2171). For each of the Stac isoforms, the Pearson's coefficients for these three large fragments are grouped together beneath the corresponding coefficient for the full-length C terminus (Fig. 7-4B). Although displaying variability between the Stac isoforms, the Pearson's coefficients for the N-terminal half (residues 1507–1839) were closer in value to those for the full-length C-terminal than for the other large fragments. Therefore, we next tested three smaller N-terminal fragments containing one or more of the regions (“pre-IQ”, “IQ”), which other studies have shown to be important for calcium-dependent inactivation (Peterson et al., 1999; Pate et al., 2000; Pitt et al., 2001; Van Petegem et al., 2005; Kim et al., 2010). These fragments were also of interest because Campiglio et al. (2018) found that the colocalization of Stac3 with CaV2.1/CaV1.2 chimeras in dysgenic myotubes depended on CaV1.2 sequence within the IQ domain. We found that for a given Stac isoform, the Pearson's coefficients were similar to one another for the three small fragments whether or not the IQ domain was present. Additionally, we found that the Pearson's coefficients for the N-terminal fragments were smaller than the coefficient for the larger, N-terminal half construct. Therefore, our results are consistent with the idea that Stac binding involves multiple regions of the CaV1.2 C terminus, including the IQ domain.
Within the Stac proteins, the region of ∼100 residues that links the PKC C1 and SH3_1 domains is sufficient to suppress calcium-dependent inactivation of CaV1.2
Having found that an interaction with the CaV1.2 C terminus most likely accounts for the ability of all three Stac isoforms to suppress calcium-dependent inactivation, we next attempted to identify the region(s) of the Stac proteins required for this suppression. Toward this end, we measured Ca2+ and Ba2+ currents in tsA201 cells after expression of YFP-CaV1.2, β2a, and α2-δ1, together with Stac constructs lacking one or more regions of the full-length proteins: the N-terminal region, a PKC C1 domain, a linking region, and two SH3 domains. Results with this approach for Stac1 are illustrated in Figure 8. The only region that was required for inhibition of calcium-dependent inactivation was the region linking the PKC C1 and tandem SH3 domains and a construct of this region alone (118 residues in Stac1) was also sufficient for this inhibition (Fig. 8B–F). Similarly for Stac2 and Stac3, the region linking the PKC C1 and tandem SH3 domains (120 and 96 residues, respectively) was sufficient for the inhibition of calcium-dependent inactivation (Fig. 8-1, and Fig. 8-2) and, as for Stac1, neither the PKC C1 domain nor the two SH3 domains appeared to be important for such inhibition.
Figure 8.

The unstructured region of Stac1 (residues 168–285), which links the PKC C1 and SH3_1 domains, is sufficient to suppress calcium-dependent inactivation of CaV1.2. A, Schematic representation and domain architecture of full-length mouse Stac1 (residue numbers adapted from UniProt). B–F, for tsA201 cells transfected with CaV1.2, β2a, α2-δ1, and the indicated Stac1 construct. The top row illustrates representative peak currents carried by Ca2+ (red, vertically scaled by indicated factors, test potential of +30 mV except +20 mV for D) or Ba2+ (black, test potential of +10 mV), the middle row shows the fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential, and the bottom row illustrates the peak I–V relationships. Scale bars: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal). Figure 8-1 and Figure 8-2 show that the region linking the PKC C1 and SH3_1 domains of both Stac2 and Stac3, respectively, is also sufficient to completely suppress calcium-dependent inactivation. Figure 8-3 shows that smaller, N- or C-terminal segments of this region either do not, or only partially, suppress calcium-dependent inactivation.
The unstructured region of Stac2 (residues 169-288), which links the PKC C1 and SH3_1 domains, is sufficient to suppress calcium-dependent inactivation of CaV1.2. (A) Schematic representation and domain architecture of full-length mouse Stac2 (residue numbers adapted from UniProt). (B-D) for tsA201 cells transfected with CaV1.2, β2a, α2-δ1 and the indicated Stac2 construct, the top row illustrates representative peak currents carried by Ca2+ (red, vertically scaled by indicated factors, test potential of +20 mV) or Ba2+ (black, test potential of +10 mV), the middle row shows the fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential, and the bottom row illustrates the peak I-V relationships. Calibrations: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal). Download Figure 8-1, TIF file (402.7KB, tif)
The unstructured region of Stac3 (residues 147-242), which links the PKC C1 and SH3_1 domains, is sufficient to suppress calcium-dependent inactivation of CaV1.2. (A) Schematic representation and domain architecture of full-length mouse Stac3 (residue numbers adapted from UniProt). (B-D) for tsA201 cells transfected with CaV1.2, β2a, α2-δ1 and the indicated Stac3 construct, the top row illustrates representative peak currents carried by Ca2+ (red, vertically scaled by indicated factors, test potential of +20 mV for B and D, and +30 mV for C, E and F) or Ba2+ (black, test potential of +10 mV), the middle row shows the fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential, and the bottom row illustrates the peak I-V relationships. Calibrations: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal). Download Figure 8-2, TIF file (7.1MB, tif)
Effect on calcium-dependent inactivation of subdivisions of the unstructured region between the PKC C1 and SH3_1 domains of Stac1, Stac2 and Stac3. (A) Alignment is shown of the PKC C1-SH3_1 linker region of mouse Stac1 (top, residues 168-285, Gene ID 20840), and the corresponding sequences of mouse Stac2 (middle, residues 169-288, Gene ID 217154), and mouse Stac3 (bottom, residues 147-242, Gene ID 237611). Identical residues are shown in white font on a black background and conserved residues are shown in black on a gray background. The colored lines underneath the sequences indicate segments of the PKC C1-SH3_1 linker regions tested for their effects on calcium-dependent inactivation. (B-K) The top row illustrates representative peak Ba2+ currents (black) and peak Ca2+ currents (vertically scaled by indicated factors, colors corresponding to underlined segments). The middle row plots the fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential, and the bottom row illustrates and peak I-V relationships. Data from tsA201 cells transfected with YFP-CaV1.2, β2a and α2-δ1 either with Stac1[168-217] (B), Stac2[169-221] (C), Stac3[147-189] (D), Stac1[218-285] (E), Stac2[222-288] (F), Stac3[190-242] (G), Stac1[168-240] (H), Stac2[169-242] (I), or Stac3[147-213] (K). In (B-K), test potentials for Ca2+ and Ba2+ were +20 mV and +10 mV, respectively. Calibrations: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal). Download Figure 8-3, TIF file (7.3MB, tif)
Amino acid alignment of the three Stac isoforms reveals only weak conservation within the PKC C1-to-SH3_1 linker regions (Fig. 8-3A, making it difficult to infer which elements within this region are likely to be important for inhibiting the calcium-dependent inactivation of CaV1.2. Therefore, somewhat arbitrarily, we divided the linker into two halves, with the first and second half indicated, respectively, by the orange and teal lines in Figure 8-3A. Calcium-dependent inactivation was not noticeably affected by either the first half, which contained the majority of the conserved/identical/residues, or the second half, which contained substantially fewer conserved residues (Fig. 8-3B–G). Therefore, we extended the first-half segment to produce a construct (pink line in Figure 8-3A), which contained all but two of the identical/conserved residues of the linker. Although this extended construct failed to produce the essentially complete suppression of calcium-dependent inactivation produced by the full-length linker (Fig. 8-1, and Fig. 8-2), it did produce a partial inhibition (Fig. 8-3H–K). Apparently, this suppression depends upon structural features of the PKC C1-to-SH3_1 linker that are not apparent from the sequence alignment.
Postnatal increase of Stac2 in forebrain and cerebellum.
Based on qPCR, levels of Stac2 transcript in adult mouse forebrain exceed those of Stac3 in skeletal muscle (Nelson et al., 2013), where Stac3 is required for the function of CaV1.1 in EC coupling (Polster et al., 2016; Linsley et al., 2017). Therefore, the qPCR indicates the likely importance of Stac2 in forebrain function but also raises a conundrum. In particular, the level of Stac3 in skeletal muscle appears to be sufficient to suppress the Ca2+-dependent inactivation of CaV1.2 after its expression in dysgenic (CaV1.1-null) myotubes (Fig. 9), whereas the l-type current in control, neonatal hippocampal neurons displayed prominent Ca2+-dependent inactivation (Fig. 1, left). One way to accommodate these results would be to postulate that the high levels of Stac2 present in adult forebrain are not present in neonatal forebrain. We tested this idea by Western blotting for Stac2 protein in forebrain and also cerebellum, another brain region having high levels of Stac2 transcript in the adult (Nelson et al., 2013). To allow comparison with existing data from qPCR and in situ hybridization, these two brain regions were obtained from neonatal and adult mice. Figure 10A illustrates a Western blot probing developmental changes in Stac2 protein in forebrain and cerebellar homogenates, in which the total protein loaded per gel lane was calculated from the measured protein concentration in those homogenates. Coomassie blue staining of a similarly loaded gel indicated that this procedure provided a good estimate of the protein loaded per lane (Fig. 10B). Figure 10A reveals that Stac2, relative to total protein, increased substantially in both cerebellum and forebrain between neonate and adult. Densitometric analysis of the Western blot shown in Figure 10A and of two additional Western blots indicated that Stac2 relative to total protein increased between neonate and adult by 3.84 ± 2.32-fold (mean ± SD) in cerebellum and by 4.78 ± 1.44-fold in forebrain. Control experiments for the specificity of the Stac2 antibody are illustrated in Figure 10-1.
Figure 9.
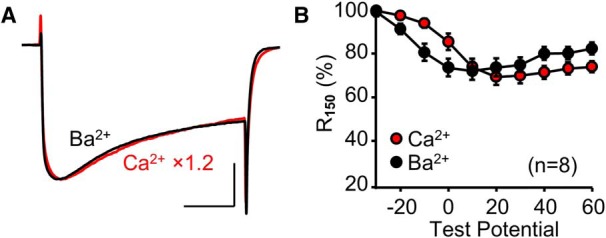
Endogenous Stac3 in myotubes eliminates Ca2+-dependent inactivation of heterologously expressed CaV1.2. Superimposed peak Ca2+ (red, vertically scaled by indicated factor) and Ba2+ (black) currents (A) and R150 vs test potential (B) are shown for dysgenic (CaV1.1-null) myotubes expressing YFP-CaV1.2. In A, test potentials were +30 mV and +10 mV for Ca2+ and Ba2+, respectively. Scale bars: 10 pA/pF (unscaled Ba2+ currents, vertical) and 50 ms (horizontal).
Figure 10.
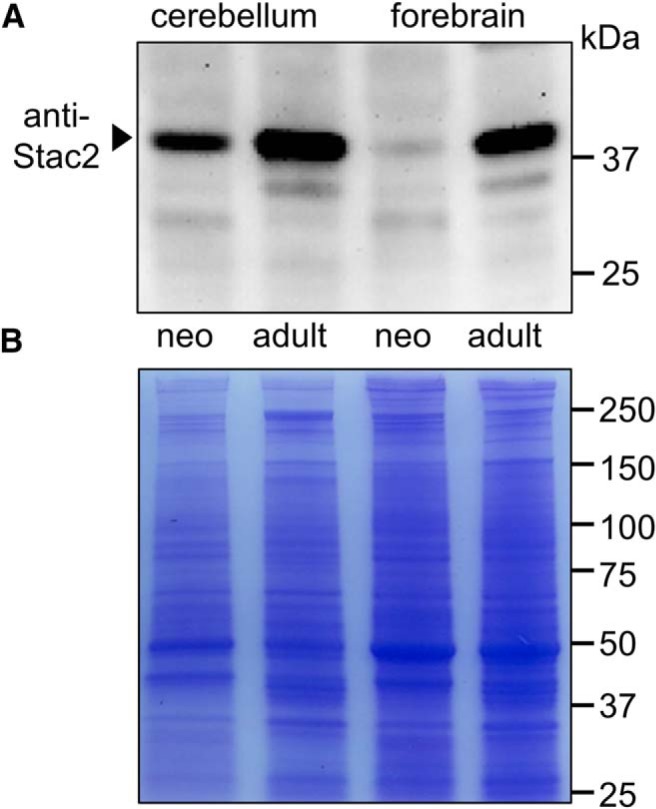
Expression of Stac2 protein in mouse cerebellum and forebrain increases greatly during postnatal development. Western blotting for Stac2 revealed an ∼42 kDa band that was present at much higher levels in homogenates of adult (∼6 mo) mouse cerebellum and forebrain than in the corresponding neonatal (∼5 d) homogenates (A). Based on a BCA assay of the homogenates, the protein loaded per lane differed little between neonate and adult (56 and 50 μg/ lane, respectively, for cerebellum and 32 and 31 μg/lane, respectively, for forebrain). The BCA assay was additionally confirmed by Coomassie blue staining of the same four samples (20 μg loaded per lane; B). Figure 10-1 illustrates the effects of preincubating the anti-Stac2 antibody with its immunogenic peptide.
Immunogenic peptide blocks anti-Stac2 binding in Western blots of both tsA201 cells heterologously expressing Stac2-YFP and native tissue. In the absence of the blocking peptide, a polyclonal antibody against Stac2 did not produce a band in non-transfected tsA201 cells (lane 1) but gave bands of the expected ∼72 and ∼42 kDa, respectively, in homogenates of tsA201 cells transfected with Stac2-YFP (lane 2) and adult mouse forebrain (lane 3). Prior incubation of the anti-Stac2 antibody with its immunogenic peptide eliminated both the Stac2-YFP band in transfected tsA201 cells (lane 4) and the presumptive Stac2 band in forebrain (lane 5). Download Figure 10-1, TIF file (458.9KB, tif)
Discussion
Here, we examined the effects of Stac proteins on high-voltage activated Ca2+ channels both in neonatal rat hippocampal neurons and after heterologous expression in tsA201 cells. In control neurons, both l-type (Fig. 1) and non-l-type (Fig. 2) Ca2+ channels displayed calcium-dependent inactivation: with Ca2+ as charge carrier, the extent of current decay showed a U-shaped dependence on test potential and the extent of decay was greatly reduced when Ba2+ was the charge carrier. Overexpression of the Stac proteins in neonatal rat hippocampal neurons suppressed calcium-dependent inactivation of the l-type current, but did not affect the calcium-dependent inactivation of the non-l-type current. This differential effect of the Stac proteins on l-type and non-l-type channels in hippocampal neurons was also observed after heterologous expression in tsA201 cells: all three Stac isoforms eliminated calcium-dependent inactivation of the l-type channels CaV1.2 (Fig. 3) and CaV1.3 (Fig. 4), but not that of the non-l-type channel CaV2.1 (Fig. 5). Because the Stac proteins suppressed the calcium-dependent inactivation of CaV1.2 expressed without auxiliary subunits (Fig. 6), it seems likely this suppression involves a direct interaction with CaV1.2 itself. In support of this idea, all three Stac proteins interacted with one or more cytoplasmic domains of CaV1.2 expressed in tsA201 cells as isolated fragments attached to a surface-targeting construct. Specifically, Stac1 (Fig. 7) and Stac2 (Fig. 7-1) colocalized with both the II–III loop and C terminus of CaV1.2, whereas Stac3 (Fig. 7-2) appeared only to associate with the C terminus. A variety of evidence, discussed below, indicates that the interaction with the C terminus is the one that is relevant for suppressing calcium-dependent inactivation. Surprisingly, the ability of the Stac proteins to suppress calcium-dependent inactivation appeared to reside in the segment linking the PKC C1 domain to the SH3 domains (Fig. 8-1, and Fig. 8-2). This segment is short (118, 120, and 96 residues in Stac1, Stac2, and Stac3, respectively), lacks obvious structure, and is only weakly conserved between the Stac isoforms.
Although CaV1.2 displayed calcium-dependent inactivation in transfected tsA201 cells, it did not do so in dysgenic myotubes (Fig. 9), which can be attributed to the presence of endogenously expressed Stac3 in these cultured muscle cells. By contrast, the endogenous levels of Stac proteins were sufficiently low in neonatal rat hippocampal neurons that l-type currents in these cells displayed prominent calcium-dependent inactivation (Fig. 1). Western blots revealed that protein levels of Stac2, the Stac isoform with the most abundant transcript in adult mouse brain (Nelson et al., 2013), were substantially higher in adult mouse forebrain and cerebellum compared with neonate (Fig. 10). In principle, this increased expression might cause a reduced calcium-dependent inactivation of l-type currents in adult brain, but this remains an open question. In particular, calcium-dependent inactivation has been documented in dissociated hippocampal pyramidal neurons of adult rats (Johnson and Byerly, 1994) and guinea pigs (Kay, 1991), but these studies did not determine whether this arose from l-type channels. Moreover, developmental changes, which were unrelated to Stac proteins, could also affect calcium-dependent inactivation, including channel clustering and expression of endogenous calcium buffers. The importance of the latter is highlighted by the finding of Campiglio et al. (2018) that the concentration of exogenous buffer affected the ability of Stac1 and Stac2 to inhibit the calcium-dependent inactivation of CaV1.2 expressed in tsA201 cells: the extent of inhibition was much lower for 0.5 mm EGTA compared with 10.0 mm EGTA.
Structural and functional interactions between Stac proteins and voltage-gated Ca2+ channels
To date, at least three different categories of functional interactions have been reported to occur between Stac proteins and CaV channels: regulation of CaV1.1 trafficking and function, inhibition of calcium-dependent inactivation of CaV1.2 and CaV1.3, and increased membrane trafficking of CaV3.2. These functional interactions appear to depend on distinct cytoplasmic regions of the CaV channels and also upon distinct domains of the Stac proteins, which consist of the following: (1) an N-terminal region, (2) a PKC C1 domain, (3) a linking region, and (4) tandem SH3 domains (SH3_1 and SH3_2). The functional interaction between Stac3 and CaV1.1 results in increased membrane trafficking of CaV1.1 (Polster et al., 2015, 2016; Linsley et al., 2017), alters the kinetics and amplitude of the l-type current that CaV1.1 produces (Polster et al., 2015, 2016), and is required for the ability of CaV1.1 to mediate skeletal-type EC coupling (Horstick et al., 2013; Nelson et al., 2013; Polster et al., 2016; Linsley et al., 2017). A primary structural determinant for these effects is an interaction between the SH3_1 domain of Stac3 and a polyproline domain in the II–III loop of CaV1.1 (Wong King Yuen et al., 2017; Polster et al., 2018). However, this II–III loop interaction does not appear to be important for the ability of the Stac proteins to suppress calcium-dependent inhibition of CaV1.2. In particular, Stac3 abolishes calcium-dependent inactivation of CaV1.2 (Fig. 3; Campiglio et al., 2018), but does not appear to bind to the CaV1.2 II–III loop (Polster et al., 2018). Moreover, calcium-dependent inactivation of CaV1.2 was effectively inhibited by a Stac3 construct that consisted of the N-terminal, PKC C1, and linking domain, but which lacked the SH3 domains important for interacting with the II–III loop (Wong King Yuen et al., 2017). In fact, we found that even smaller Stac constructs, consisting only of the linking domain, were effective at inhibiting calcium-dependent inactivation (Fig. 8-1, and Fig. 8-2).
Different groups using different methodologies have arrived at different conclusions about the site(s) of interaction between the Stac proteins and the CaV1 C terminus. Campiglio et al. (2018) found that colocalization of Stac3-GFP with CaV2.1/CaV1.2 chimeras expressed in dysgenic myotubes (which express endogenous Stac3) depended on the presence of the 22 residues constituting the IQ motif of CaV1.2. By contrast, Niu et al. (2018) found with a FRET assay of tagged constructs expressed in HEK293 cells that Stac3 interacted with the EF hand motif of CaV1.1 (the position of the EF hand relative to the IQ and pre-IQ motifs is illustrated in Fig. 7-4). In our previous and present work, using a colocalization assay in tsA201 cells, we found that the interactions varied between Stac proteins and differed between CaV1.1 (Figs. 4, S2, and S3 of Polster et al., 2018) and CaV1.2 (Fig. 7-1, Fig. 7-2, and Fig. 7-3). Despite these discrepancies, all of these studies indicate that interactions occur near or at the IQ motif. This motif is identical in CaV1.2 and CaV1.3 (Fujita et al., 1993) and has to be important for binding of calmodulin and thus essential for calcium-dependent inactivation (Peterson et al., 1999; Qin et al., 1999; Zühlke et al., 1999; Yang et al., 2006). Therefore, it is reasonable to suggest (Campiglio et al., 2018) that interaction of Stac3 with this region alters the interactions of calmodulin with the channel that are necessary for calcium-dependent inactivation. Although CaV2.1 also contains an IQ motif, its structure is sufficiently different that bound CaM has the opposite orientation from CaV1.2 (Kim et al., 2008) and Stac3 does not bind to it.
As described above, the Stac proteins appear to interact with at least two distinct regions of the l-type channels. As discussed above, we can ascribe functions to the interaction of the three Stac proteins with the C terminus of CaV1.2 (and probably CaV1.3), to that of suppressing calcium-dependent inactivation, and also to that of Stac3 with the II–III loop of CaV1.1 (increased membrane trafficking and altered channel function). However, we are not currently able to ascribe an obvious function to the interaction of Stac1 and Stac2 with the II–III loop of CaV1.2. Yet another CaV cytoplasmic domain appears to be important for an interaction between Stac1 and the low-voltage-activated channel, CaV3.2. In particular, Stac1 coimmunoprecipitates with residues 50–100 of the CaV3.2 N terminus and increases the current density of full-length CaV3.2 heterologously expressed in tsA201 cells (Rzhepetskyy et al., 2016).
Although calcium channels appear to be one important target for neuronally expressed Stac proteins, they seem unlikely to be the only target given the diversity of interaction sites that have already been identified. As a recent example, Jeong et al. (2018) demonstrated that Stac2 negatively regulates osteoclast formation by interaction with “RANK,” a protein that affects signaling cascades that include gene transcription factors. This effect of Stac2 appeared to involve interaction with the sequence IVVY (residues 535–538) in RANK (Jeong et al., 2018), a sequence not present in the three regions of CaV channels to which the Stac proteins appear to bind. The regulation of transcription factors by Stac2 is intriguing given that in neurons Stac2 interacts with l-type Ca2+ channels, which themselves regulate nuclear signaling via several transcription factors including cAMP/Ca2+-response element binding protein (CREB), nuclear factor of activated T-cells (NFAT), myocyte enhancer factor 2 (MEF2), and CREB-regulated transcription coactivator 1 (CRTC1) (Bading et al., 1993; Graef et al., 1999; Mao et al., 1999; Ch'ng et al., 2012). Therefore, an important goal for future research will be to determine whether Stac2 can affect neuronal transcription independent of its effects on l-type Ca2+ channels.
Footnotes
This work was supported by National Institutes of Health (Grant AR070298 to K.G.B.), the Muscular Dystrophy Association (Grant 479598 to A.P.), and the Departments of Physiology and Pharmacology, University of Colorado (Basic Science Pilot Grant to P.J.D.). All authors discussed the results and approved the final version of the manuscript. We thank O. Moua for expert technical assistance.
The authors declare no competing financial interests.
References
- Bading H, Ginty DD, Greenberg ME (1993) Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260:181–186. 10.1126/science.8097060 [DOI] [PubMed] [Google Scholar]
- Beam KG, Franzini-Armstrong C (1997) Functional and structural approaches to the study of excitation-contraction coupling. Methods Cell Biol 52:283–306. 10.1016/S0091-679X(08)60384-2 [DOI] [PubMed] [Google Scholar]
- Bower NI, de la Serrana DG, Cole NJ, Hollway GE, Lee HT, Assinder S, Johnston IA (2012) Stac3 is required for myotube formation and myogenic differentiation in vertebrate skeletal muscle. J Biol Chem 287:43936–43949. 10.1074/jbc.M112.361311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm P, Eckert R (1978) Calcium entry leads to inactivation of calcium channel in paramecium. Science 202:1203–1206. 10.1126/science.103199 [DOI] [PubMed] [Google Scholar]
- Campiglio M, Costé de Bagneaux P, Ortner NJ, Tuluc P, Van Petegem F, Flucher BE (2018) STAC proteins associate to the IQ domain of CaV1.2 and inhibit calcium-dependent inactivation. Proc Natl Acad Sci U S A 115:1376–1381. 10.1073/pnas.1715997115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng TH, Uzgil B, Lin P, Avliyakulov NK, O'Dell TJ, Martin KC (2012) Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell 150:207–221. 10.1016/j.cell.2012.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT (2001) Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411:484–489. 10.1038/35078091 [DOI] [PubMed] [Google Scholar]
- Fujita Y, Mynlieff M, Dirksen RT, Kim MS, Niidome T, Nakai J, Friedrich T, Iwabe N, Miyata T, Furuichi T, Furutama D, Mikoshiba K, Mori Y, Beam KG (1993) Primary structure and functional expression of the ω-conotoxin-sensitive N-type calcium channel from rabbit brain. Neuron 10:585–598. 10.1016/0896-6273(93)90162-K [DOI] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML (2002) Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci 22:7027–7044. 10.1523/JNEUROSCI.22-16-07027.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner M, Dirksen RT, Beam KG (1998) Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-l-type Ca2+ channels expressed in dysgenic myotubes. Proc Natl Acad Sci U S A 95:1903–1908. 10.1073/pnas.95.4.1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR (1999) L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401:703–708. 10.1038/44378 [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA (1993) Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. J Cell Biol 123:949–962. 10.1083/jcb.123.4.949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstick EJ, Linsley JW, Dowling JJ, Hauser MA, McDonald KK, Ashley-Koch A, Saint-Amant L, Satish A, Cui WW, Zhou W, Sprague SM, Stamm DS, Powell CM, Speer MC, Franzini-Armstrong C, Hirata H, Kuwada JY (2013) Stac3 is a component of the excitation-contraction coupling machinery and mutated in native american myopathy. Nat Commun 4:1952. 10.1038/ncomms2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong E, Choi HK, Park JH, Lee SY (2018) STAC2 negatively regulates osteoclast formation by targeting the RANK signaling complex. Cell Death Differ 25:1364–1374. 10.1038/s41418-017-0048-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BD, Byerly L (1994) Ca2+ channel Ca2+-dependent inactivation in a mammalian central neuron involves the cytoskeleton. Pflugers Arch 429:14–21. 10.1007/BF02584025 [DOI] [PubMed] [Google Scholar]
- Kass RS, Sanguinetti MC (1984) Inactivation of calcium channel current in the calf cardiac purkinje fiber. evidence for voltage- and calcium-mediated mechanisms. J Gen Physiol 84:705–726. 10.1085/jgp.84.5.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR. (1991) Inactivation kinetics of calcium current of acutely dissociated CA1 pyramidal cells of the mature guinea-pig hippocampus. J Physiol 437:27–48. 10.1113/jphysiol.1991.sp018581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Rumpf CH, Fujiwara Y, Cooley ES, Van Petegem F, Minor DL Jr (2008) Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure 16:1455–1467. 10.1016/j.str.2008.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Rumpf CH, Van Petegem F, Arant RJ, Findeisen F, Cooley ES, Isacoff EY, Minor DL Jr (2010) Multiple C-terminal tail Ca2+/CaMs regulate CaV1.2 function but do not mediate channel dimerization. EMBO J 29:3924–3938. 10.1038/emboj.2010.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J (2001) α1D (CaV1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem 276:22100–22106. 10.1074/jbc.M101469200 [DOI] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA (1999) Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 399:155–159. 10.1038/20194 [DOI] [PubMed] [Google Scholar]
- Legha W, Gaillard S, Gascon E, Malapert P, Hocine M, Alonso S, Moqrich A (2010) stac1 and stac2 genes define discrete and distinct subsets of dorsal root ganglia neurons. Gene Expr Patterns 10:368–375. 10.1016/j.gep.2010.08.003 [DOI] [PubMed] [Google Scholar]
- Leuranguer V, Papadopoulos S, Beam KG (2006) Organization of calcium channel β1a subunits in triad junctions in skeletal muscle. J Biol Chem 281:3521–3527. 10.1074/jbc.M509566200 [DOI] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT (2003) Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron 39:951–960. 10.1016/S0896-6273(03)00560-9 [DOI] [PubMed] [Google Scholar]
- Linsley JW, Hsu IU, Groom L, Yarotskyy V, Lavorato M, Horstick EJ, Linsley D, Wang W, Franzini-Armstrong C, Dirksen RT, Kuwada JY (2017) Congenital myopathy results from misregulation of a muscle Ca2+ channel by mutant Stac3. Proc Natl Acad Sci U S A 114:E228–E236. 10.1073/pnas.1619238114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME (1999) Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286:785–790. 10.1126/science.286.5440.785 [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T (2005) Role of hippocampal CaV1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci 25:9883–9892. 10.1523/JNEUROSCI.1531-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell'Acqua ML (2014) AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep 7:1577–1588. 10.1016/j.celrep.2014.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Baraban JM (1991) L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron 7:625–635. 10.1016/0896-6273(91)90375-A [DOI] [PubMed] [Google Scholar]
- Nelson BR, Wu F, Liu Y, Anderson DM, McAnally J, Lin W, Cannon SC, Bassel-Duby R, Olson EN (2013) Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc Natl Acad Sci U S A 110:11881–11886. 10.1073/pnas.1310571110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Yang W, Yue DT, Inoue T, Ben-Johny M (2018) Duplex signaling by CaM and Stac3 enhances CaV1.1 function and provides insights into congenital myopathy. J Gen Physiol 150:1145–1161. 10.1085/jgp.201812005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohrtman J, Ritter B, Polster A, Beam KG, Papadopoulos S (2008) Sequence differences in the IQ motifs of CaV1.1 and CaV1.2 strongly impact calmodulin binding and calcium-dependent inactivation. J Biol Chem 283:29301–29311. 10.1074/jbc.M805152200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA (2007) AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55:261–275. 10.1016/j.neuron.2007.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos S, Leuranguer V, Bannister RA, Beam KG (2004) Mapping sites of potential proximity between the dihydropyridine receptor and RyR1 in muscle using a cyan fluorescent protein-yellow fluorescent protein tandem as a fluorescence resonance energy transfer probe. J Biol Chem 279:44046–44056. 10.1074/jbc.M405317200 [DOI] [PubMed] [Google Scholar]
- Pate P, Mochca-Morales J, Wu Y, Zhang JZ, Rodney GG, Serysheva II, Williams BY, Anderson ME, Hamilton SL (2000) Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J Biol Chem 275:39786–39792. 10.1074/jbc.M007158200 [DOI] [PubMed] [Google Scholar]
- Pérez-García MT, Kamp TJ, Marbán E (1995) Functional properties of cardiac L-type calcium channels transiently expressed in HEK293 cells. roles of alpha 1 and beta subunits. J Gen Physiol 105:289–305. 10.1085/jgp.105.2.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP, Yue DT (1999) Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22:549–558. 10.1016/S0896-6273(00)80709-6 [DOI] [PubMed] [Google Scholar]
- Pitt GS, Zühlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW (2001) Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem 276:30794–30802. 10.1074/jbc.M104959200 [DOI] [PubMed] [Google Scholar]
- Polster A, Perni S, Bichraoui H, Beam KG (2015) Stac adaptor proteins regulate trafficking and function of muscle and neuronal L-type Ca2+ channels. Proc Natl Acad Sci U S A 112:602–606. 10.1073/pnas.1423113112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polster A, Nelson BR, Olson EN, Beam KG (2016) Stac3 has a direct role in skeletal muscle-type excitation-contraction coupling that is disrupted by a myopathy-causing mutation. Proc Natl Acad Sci U S A 113:10986–10991. 10.1073/pnas.1612441113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polster A, Nelson BR, Papadopoulos S, Olson EN, Beam KG (2018) Stac proteins associate with the critical domain for excitation-contraction coupling in the II–III loop of CaV1.1. J Gen Physiol 150:613–624. 10.1085/jgp.201711917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L (1999) Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci U S A 96:2435–2438. 10.1073/pnas.96.5.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinholt BM, Ge X, Cong X, Gerrard DE, Jiang H (2013) Stac3 is a novel regulator of skeletal muscle development in mice. PLoS One 8:e62760. 10.1371/journal.pone.0062760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA (1996) Isoform-specific interaction of the α1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci U S A 93:7363–7368. 10.1073/pnas.93.14.7363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzhepetskyy Y, Lazniewska J, Proft J, Campiglio M, Flucher BE, Weiss N (2016) A CaV3.2/Stac1 molecular complex controls T-type channel expression at the plasma membrane. Channels 10:346–354. 10.1080/19336950.2016.1186318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML (2006) cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci 26:2391–2402. 10.1523/JNEUROSCI.3092-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kawai J, Taga C, Yaoi T, Hara A, Hirose K, Hayashizaki Y, Watanabe S (1996) Stac, a novel neuron-specific protein with cysteine-rich and SH3 domains. Biochem Biophys Res Commun 229:902–909. 10.1006/bbrc.1996.1900 [DOI] [PubMed] [Google Scholar]
- Van Petegem F, Chatelain FC, Minor DL Jr (2005) Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol 12:1108–1115. 10.1038/nsmb1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong King Yuen SM, Campiglio M, Tung CC, Flucher BE, Van Petegem F (2017) Structural insights into binding of STAC proteins to voltage-gated calcium channels. Proc Natl Acad Sci U S A 114:E9520–E9528. 10.1073/pnas.1708852114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Lipscombe D (2001) Neuronal CaV1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21:5944–5951. 10.1523/JNEUROSCI.21-16-05944.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, Fuchs PA, Yue DT (2006) Switching of Ca2+-dependent inactivation of CaV1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci 26:10677–10689. 10.1523/JNEUROSCI.3236-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong S, Zhou J, Tanabe T (1994) Molecular determinants of calcium-dependent inactivation in cardiac L-type calcium channels. Biochem Biophys Res Commun 201:1117–1123. 10.1006/bbrc.1994.1821 [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H (1999) Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399:159–162. 10.1038/20200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nifedipine pre-treatment does not alter the fractional inactivation of Ca2+ current in tsA201 cells transfected with CaV1.3, YFP-β1a, and α2-δ1. R150 plotted as a function of test potential for 3 cells pre-treated with 2 µM nifedipine (see Methods) and 3 cells not pre-treated with nifedipine. Download Figure 4-1, TIF file (2.6MB, tif)
Association of Stac2 with cytoplasmic CaV1.2 domains in tsA201 cells. Representative images are shown of cells co-transfected with Stac2-tagRFP and the indicated cytoplasmic domains of CaV1.2, which were targeted to the cell surface by being linked at the N-terminal to the GFP-tagged I-II loop of CaV1.2. The images were obtained after partial photo-bleaching of the mobile pool of Stac1-tagRFP by repeated scans within the indicated regions of interest with non-attenuated excitation at 543 nm. Bars, 5 μm. Download Figure 7-1, TIF file (1.4MB, tif)
Association of Stac3 with cytoplasmic CaV1.2 domains in tsA201 cells. Representative images are shown of cells co-transfected with Stac3-tagRFP and the indicated cytoplasmic domains of CaV1.2, which were targeted to the cell surface by being linked at the N-terminal to the GFP-tagged I-II loop of CaV1.2. The images were obtained after partial photo-bleaching of the mobile pool of Stac1-tagRFP and GFP-tagged CaV1.2 C-terminus by repeated scans within the indicated regions of interest with non-attenuated excitation at 543 nm and 515 nm, respectively. Bars, 5 μm. Download Figure 7-2, TIF file (1.4MB, tif)
Pearson’s coefficients (mean ± SEM) for colocalization of the Stac proteins and the cytoplasmic domains of CaV1.2. The number of images analyzed is indicated for each combination of constructs. ***, P < 0.001 by one-way ANOVA. Download Figure 7-3, TIF file (946KB, tif)
The interaction between Stac proteins and the CaV1.2 C-terminus. (A) Schematic representation and domain architecture (not to scale) of the full length C-terminus of rabbit CaV1.2 (top, residues 1507-2171) and different sub-segments (numbers refer to the first and last residues of CaV1.2 sequence) tested for Stac protein interaction, with several regions indicated that have been implicated as important for calcium-dependent inactivation (EF hand motif, pre-IQ region consisting of regions A and C, IQ domain; for references, see Results), (B) Pearson’s coefficients (mean ± SEM) for co-localization of indicated CaV1.2 C-terminus segments (linked to the GFP-tagged I-II loop of CaV1.2) and RFP-labelled Stac proteins after cotransfection in tsA201 cells (the number of images analyzed is indicated for each combination of constructs). Download Figure 7-4, TIF file (1.7MB, tif)
The unstructured region of Stac2 (residues 169-288), which links the PKC C1 and SH3_1 domains, is sufficient to suppress calcium-dependent inactivation of CaV1.2. (A) Schematic representation and domain architecture of full-length mouse Stac2 (residue numbers adapted from UniProt). (B-D) for tsA201 cells transfected with CaV1.2, β2a, α2-δ1 and the indicated Stac2 construct, the top row illustrates representative peak currents carried by Ca2+ (red, vertically scaled by indicated factors, test potential of +20 mV) or Ba2+ (black, test potential of +10 mV), the middle row shows the fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential, and the bottom row illustrates the peak I-V relationships. Calibrations: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal). Download Figure 8-1, TIF file (402.7KB, tif)
The unstructured region of Stac3 (residues 147-242), which links the PKC C1 and SH3_1 domains, is sufficient to suppress calcium-dependent inactivation of CaV1.2. (A) Schematic representation and domain architecture of full-length mouse Stac3 (residue numbers adapted from UniProt). (B-D) for tsA201 cells transfected with CaV1.2, β2a, α2-δ1 and the indicated Stac3 construct, the top row illustrates representative peak currents carried by Ca2+ (red, vertically scaled by indicated factors, test potential of +20 mV for B and D, and +30 mV for C, E and F) or Ba2+ (black, test potential of +10 mV), the middle row shows the fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential, and the bottom row illustrates the peak I-V relationships. Calibrations: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal). Download Figure 8-2, TIF file (7.1MB, tif)
Effect on calcium-dependent inactivation of subdivisions of the unstructured region between the PKC C1 and SH3_1 domains of Stac1, Stac2 and Stac3. (A) Alignment is shown of the PKC C1-SH3_1 linker region of mouse Stac1 (top, residues 168-285, Gene ID 20840), and the corresponding sequences of mouse Stac2 (middle, residues 169-288, Gene ID 217154), and mouse Stac3 (bottom, residues 147-242, Gene ID 237611). Identical residues are shown in white font on a black background and conserved residues are shown in black on a gray background. The colored lines underneath the sequences indicate segments of the PKC C1-SH3_1 linker regions tested for their effects on calcium-dependent inactivation. (B-K) The top row illustrates representative peak Ba2+ currents (black) and peak Ca2+ currents (vertically scaled by indicated factors, colors corresponding to underlined segments). The middle row plots the fraction of peak current remaining 150 ms after the peak (R150) as a function of test potential, and the bottom row illustrates and peak I-V relationships. Data from tsA201 cells transfected with YFP-CaV1.2, β2a and α2-δ1 either with Stac1[168-217] (B), Stac2[169-221] (C), Stac3[147-189] (D), Stac1[218-285] (E), Stac2[222-288] (F), Stac3[190-242] (G), Stac1[168-240] (H), Stac2[169-242] (I), or Stac3[147-213] (K). In (B-K), test potentials for Ca2+ and Ba2+ were +20 mV and +10 mV, respectively. Calibrations: 10 pA/pF (unscaled Ba2+ currents, vertical), 50 ms (horizontal). Download Figure 8-3, TIF file (7.3MB, tif)
Immunogenic peptide blocks anti-Stac2 binding in Western blots of both tsA201 cells heterologously expressing Stac2-YFP and native tissue. In the absence of the blocking peptide, a polyclonal antibody against Stac2 did not produce a band in non-transfected tsA201 cells (lane 1) but gave bands of the expected ∼72 and ∼42 kDa, respectively, in homogenates of tsA201 cells transfected with Stac2-YFP (lane 2) and adult mouse forebrain (lane 3). Prior incubation of the anti-Stac2 antibody with its immunogenic peptide eliminated both the Stac2-YFP band in transfected tsA201 cells (lane 4) and the presumptive Stac2 band in forebrain (lane 5). Download Figure 10-1, TIF file (458.9KB, tif)