

Human-associated polymicrobial communities can promote health and disease, and interbacterial interactions influence the microbial ecology of such communities. Polymicrobial infections of the cystic fibrosis respiratory tract impair lung function and lead to the death of individuals suffering from this disorder; therefore, a greater understanding of these microbial communities is necessary for improving treatment strategies. Bacteria utilize contact-dependent growth inhibition systems to kill neighboring competitors and maintain their niche within multicellular communities. Several cystic fibrosis pathogens have the potential to gain an ecological advantage during infection via contact-dependent growth inhibition systems, including Burkholderia dolosa. Our research is significant, as it has identified three functional contact-dependent growth inhibition systems in B. dolosa that may provide this pathogen a competitive advantage during polymicrobial infections.
KEYWORDS: Bcc, Burkholderia, contact-dependent inhibition, interbacterial competition, two-partner secretion
ABSTRACT
The respiratory tracts of individuals afflicted with cystic fibrosis (CF) harbor complex polymicrobial communities. By an unknown mechanism, species of the Gram-negative Burkholderia cepacia complex, such as Burkholderia dolosa, can displace other bacteria in the CF lung, causing cepacia syndrome, which has a poor prognosis. The genome of B. dolosa strain AU0158 (BdAU0158) contains three loci that are predicted to encode contact-dependent growth inhibition (CDI) systems. CDI systems function by translocating the toxic C terminus of a large exoprotein directly into target cells, resulting in growth inhibition or death unless the target cells produce a cognate immunity protein. We demonstrate here that each of the three bcpAIOB loci in BdAU0158 encodes a distinct CDI system that mediates interbacterial competition in an allele-specific manner. While only two of the three bcpAIOB loci were expressed under the in vitro conditions tested, the third conferred immunity under these conditions due to the presence of an internal promoter driving expression of the bcpI gene. One BdAU0158 bcpAIOB allele is highly similar to bcpAIOB in Burkholderia thailandensis strain E264 (BtE264), and we showed that their BcpI proteins are functionally interchangeable, but contact-dependent signaling (CDS) phenotypes were not observed in BdAU0158. Our findings suggest that the CDI systems of BdAU0158 may provide this pathogen an ecological advantage during polymicrobial infections of the CF respiratory tract.
IMPORTANCE Human-associated polymicrobial communities can promote health and disease, and interbacterial interactions influence the microbial ecology of such communities. Polymicrobial infections of the cystic fibrosis respiratory tract impair lung function and lead to the death of individuals suffering from this disorder; therefore, a greater understanding of these microbial communities is necessary for improving treatment strategies. Bacteria utilize contact-dependent growth inhibition systems to kill neighboring competitors and maintain their niche within multicellular communities. Several cystic fibrosis pathogens have the potential to gain an ecological advantage during infection via contact-dependent growth inhibition systems, including Burkholderia dolosa. Our research is significant, as it has identified three functional contact-dependent growth inhibition systems in B. dolosa that may provide this pathogen a competitive advantage during polymicrobial infections.
INTRODUCTION
Bacteria often reside in complex polymicrobial communities in which intra- and interspecies interactions influence community structure (1–4), and interbacterial competition has been proposed to have a greater impact on microbial ecology and evolution within polymicrobial environments than interbacterial cooperation (5). While complex microbial communities, such as the microbiota of the intestinal and vaginal tracts, can promote the health of their hosts (6, 7), microbial communities can also arise in diseased tissues and exacerbate morbidity, such as in the respiratory tracts of cystic fibrosis (CF) patients. Several bacterial pathogens, including Staphylococcus aureus, Pseudomonas aeruginosa, and members of the Burkholderia cepacia complex (Bcc), are notorious for dominating the CF airways, (8, 9). Infections by Bcc pathogens typically do not arise until teenage years or adulthood and can rapidly progress to “cepacia syndrome,” which is a fatal, necrotizing pneumonia accompanied by bacteremia (10, 11). Strikingly, Bcc pathogens can replace other pathogenic species as the predominant organisms during polymicrobial infections, though the mechanisms underlying this behavior remain unknown (12–14).
Contact-dependent growth inhibition (CDI) is a mechanism of interbacterial competition in which the toxic C terminus (CT) of a large exoprotein belonging to the two-partner secretion (TPS) family is delivered directly from one bacterium to another, resulting in death or growth arrest of the recipient cell (15). Autotoxicity is prevented in CDI+ cells by the production of an immunity protein that binds to the CT, blocking its activity (15). Genes encoding CDI systems are widespread among Gram-negative bacteria and are polymorphic in nature, with different alleles encoding unique CT toxins and cognate immunity proteins, and thus protection against toxicity by immunity proteins occurs in an allele-specific manner (16).
Two classes of CDI systems have been described to date: the Burkholderia type and the Escherichia coli type (15–17). E. coli-type systems are encoded by cdiBAI genes, with cdiA encoding the toxic exoprotein, cdiB encoding its TPS transporter partner protein, and cdiI encoding the immunity protein (15). Burkholderia-type systems are encoded by bcpAIOB genes (17). bcpA and bcpB encode the exoprotein and transporter TPS proteins, respectively, bcpI encodes the immunity protein, and a fourth open reading frame (ORF), bcpO, encodes a small protein of unknown function (17). In both E. coli- and Burkholderia-type CDI systems, the N-terminal ∼2,800 amino acids (aa) of CdiA and BcpA proteins, respectively, are conserved among closely related species, while the C-terminal ∼300 amino acids vary greatly, as do the CdiI and BcpI proteins, allowing for toxin-antitoxin heterogeneity across different CDI system-encoding alleles (16, 18). Burkholderia- and E. coli-type CDI systems also contain distinct motifs separating the conserved and variable domains of the toxic exoproteins; NX(E/Q)LYN in BcpA proteins and VENN in CdiA proteins (16, 17). Burkholderia-type CDI systems are further classified into two different phylogenetic groups—class I and class II—based on the amino acid sequences of the BcpB and BcpO proteins and the conserved region of BcpA (17, 19). BcpO proteins across different Burkholderia class I alleles are nearly identical, save for the N-terminal ∼20-aa-long signal sequence that covaries with BcpA-CT and BcpI, whereas the bcpO genes of class II are not similar across different alleles (17, 19).
Most information available for Burkholderia-type CDI systems is for the class I allele in B. thailandensis strain E264 (BtE264) (17, 19–21). We and others have shown that the BtE264 BcpAIOB proteins compose a functional CDI system (17) and that chimeric BtE264 strains producing the BcpA-CTs and BcpI proteins of pathogenic B. pseudomallei strains outcompete neighboring cells in a CDI-dependent manner (19, 22, 23). Our laboratory has also discovered that the BtE264 CDI system induces gene expression changes that promote cooperative behaviors between kin cells (i.e., cells that contain identical Bcp alleles) (24), a phenomenon we call “contact-dependent signaling” (CDS). We hypothesize that delivery of the BcpA-CT to a neighboring kin cell and subsequent BcpA-CT-BcpI binding forms a signaling complex that leads to expression of genes that confer community behaviors, including autoaggregation, biofilm formation, and pigment production (18, 21, 24).
Burkholderia dolosa, a member of the Bcc that caused a deadly epidemic in CF patients in Boston during the 1990s (25, 26), can transmit from human to human and lead to significant decline in lung function compared to uninfected CF patients (27, 28). The genome of B. dolosa strain AU0158 (BdAU0158) contains three bcpAIOB loci, each potentially encoding a CDI system; one class I allele and two class II alleles. The goal of this study was to characterize the bcpAIOB loci and potential CDI systems of BdAU0158 using the native pathogenic strain.
RESULTS
The Burkholderia dolosa AU0158 (BdAU0158) genome contains three bcpAIOB alleles.
By searching for homologs of the BtE264 bcpB gene, we previously detected two bcpAIOB alleles in BdAU0158 (17). Further investigation revealed a third bcpAIOB locus in BdAU0158. All three loci resemble Burkholderia-type CDI system-encoding genes, with the gene order being bcpAIOB and the presence of a fourth ORF, bcpO, between bcpI and bcpB. We will refer to these bcpAIOB loci as bcp-1 (locus tags AK34_RS22045 to AK34_RS22035, chromosome 1), bcp-2 (locus tags AK34_RS06120 to AK34_RS06110, chromosome 2), and bcp-3 (locus tags AK34_RS04315 to AK34_RS04310, chromosome 2) (Fig. 1A). The BdAU0158 bcp-1 allele belongs to the class I family of bcpAIOB alleles, whereas the BdAU0158 bcp-2 and BdAU0158 bcp-3 alleles belong to the class II family. Accordingly, the BcpA proteins encoded by the bcp-2 allele (BcpA-2) and the bcp-3 allele (BcpA-3) are more similar to one another than they are to the BcpA protein encoded by the bcp-1 allele (BcpA-1) (Fig. 1A). Strikingly, the BdAU0158 bcp-1 allele is highly similar to the bcp allele of BtE264 (Fig. 1B), which we previously determined encodes a CDI system in BtE264 (17). The BdAU0158 BcpA-1 and the BtE264 BcpA proteins share 83.0% amino acid identity overall, with 86.4% amino acid identity at the C termini (CTs). In contrast, the BdAU0158 BcpA-2 and BcpA-3 proteins share 79.7% identity overall, with only 19.9% similarity between their CTs (Fig. 1B). Phyre2 analysis (29) predicts all three BdAU0158 BcpA-CTs to be nucleases. All three BdAU0158 bcp loci contain potential bcpO genes, with the gene of the bcp-1 locus (bcpO-1) being nearly identical to BtE264 bcpO (BdAU0158 BcpO-1 and BtE264 BcpO share 94.4% identity at the amino acid level after removal of the signal sequence). ORF prediction software (30) detects two potential ORFs in the intergenic region between bcpI-2 and bcpB-2 and three potential ORFs in the intergenic region between bcpI-3 and bcpB-3 (Fig. 1A).
FIG 1.

bcpAIOB loci in BdAU0158. (A) Schematic of the BcpAIOB proteins encoded by the BdAU0158 bcp-1 (BdAU0158-1), bcp-2 (BdAU0158 bcp-2), and bcp-3 (BdAU0158-3) loci, as well as the BcpAIOB proteins encoded by the BtE264 bcp locus. Gray coloration corresponds to conserved regions of the proteins, whereas variable regions spanning BcpA-CT, BcpI, and BcpO are denoted in yellow (for the BtE264 and BdAU0158-1 alleles), purple (for the BdAU0158-2 allele), and blue (for the BdAU0158-3 allele). Potential BcpO proteins encoded by the BdAU0158 bcp-2 and bcp-3 loci are indicated by slashed boxes. (B) Amino acid alignments of BtE264 BcpA and BdAU0158 BcpA-1 and of BdAU0158 BcpA-2 and BdAU0158 BcpA-3. Residue similarity is denoted by grayscale, with black indicating identical residues and white indicating disparate residues. Triangles above and below alignments represent NX(E/Q)LYN motifs.
The BdAU0158 bcp-1 and bcp-2 loci encode functional CDI systems.
To determine if the bcp loci of BdAU0158 encode functional CDI systems, mutant strains containing unmarked, in-frame deletions lacking all but the first three codons of bcpA and the last three codons of bcpB in each of the bcp loci were generated (Δbcp-1, Δbcp-2, and Δbcp-3), and these mutants were competed against wild-type (WT) BdAU0158. All competitions were conducted on low-salt LB (LSLB; NaCl concentration, 5 g/liter) agar for 48 h at 37°C with 1:1 initial inhibitor/target ratios. WT BdAU0158 outcompeted the Δbcp-1 mutant by approximately 4 log (Fig. 2A) and outcompeted the Δbcp-2 mutant by approximately 2.5 log (Fig. 2B). To determine if the competitive exclusion in favor of WT BdAU0158 was CDI dependent, the Δbcp-1 and Δbcp-2 mutants were complemented at the attTn7 site with either cognate bcpI genes (bcpI-1 for the Δbcp-1 mutant, bcpI-2 for the Δbcp-2 mutant) or heterologous bcpI genes (bcpI-2 for the Δbcp-1 mutant, bcpI-1 for the Δbcp-2 mutant), with all bcpI genes present in trans under the control of the constitutive promoter of the BtE264 ribosomal S12 subunit gene (PS12). The Δbcp-1 mutant was rescued from killing by WT BdAU0158 (log10 competitive index [C.I.] ≈ 0) only when provided the cognate bcpI-1 gene in trans (Fig. 2A), and the Δbcp-2 mutant was rescued from killing by WT BdAU0158 only when provided its cognate bcpI-2 gene in trans (Fig. 2B). We did not observe growth rate differences between WT BdAU0158 and the Δbcp mutants during in vitro growth, and the lack of competition between WT inhibitor strains and target mutant strains complemented with cognate bcpI genes suggests that the ability of WT BdAU0158 to outcompete the Δbcp-1 and Δbcp-2 strains is solely due to CDI. These data indicate that the BdAU0158 bcp-1 and bcp-2 loci encode functional CDI systems that can kill or inhibit the growth of neighboring cells lacking cognate BcpI proteins.
FIG 2.

The Bcp-1 and Bcp-2 CDI systems provide BdAU0158 a competitive advantage during in vitro growth. (A) Competition assays between BdAU0158 WT inhibitor cells and Δbcp-1, Δbcp-1 attTn7::bcpI-1, and Δbcp-1 attTn7::bcpI-2 target cells. (B) Competition assays between BdAU0158 WT inhibitor cells and Δbcp-2, Δbcp-2 attTn7::bcpI-2, and Δbcp-2 attTn7::bcpI-1 target cells. (C) Competition assays between BdAU0158 WT inhibitor cells and Δbcp-3 and Δbcp-3 attTn7::bcpI-3 target cells. For each competition, results from three separate biological replicates, each with three technical replicates (except for the WT versus Δbcp-3 attTn7::bcpI-3 competitions in panel C, which show two biological replicates, each with three technical replicates). Solid horizontal lines represent mean log10 C.I. values. The dotted lines (log10 C.I. = 0) indicate no competitive advantage for inhibitor or target strain. ****, P < 0.0001, Mann-Whitney test.
Unlike the BdAU0158 Δbcp-1 and Δbcp-2 mutant strains, the Δbcp-3 mutant was not outcompeted by WT during 48 h of coculture on solid medium (Fig. 2C). Two possible explanations for this finding are that the bcp-3 locus does not encode a functional CDI system and that the bcp-3 locus is not expressed under the in vitro competition conditions used.
Unlike the BdAU0158 bcp-1 and bcp-2 loci, the bcp-3 locus is not expressed under in vitro competition conditions.
To investigate expression of the BdAU0158 bcp-1, bcp-2, and bcp-3 loci, promoter-lacZ fusions (Pbcp-1-lacZ, Pbcp-2-lacZ, and Pbcp-3-lacZ) were constructed and delivered to the attTn7 site of BdAU0158. Reporter strains were grown in monoculture under the same conditions as those used for competition experiments (LSLB agar at 37°C for 48 h), and β-galactosidase activity was measured. Consistent with results from the competition experiments, the bcp-1 and bcp-2 promoters were active under these conditions (Fig. 3A). Pbcp-1 was more active than Pbcp-2, which suggests that the bcp-1 locus is expressed to a higher degree than the bcp-2 locus, which may explain why Bcp-1-mediated CDI is more potent than Bcp-2-mediated CDI (Fig. 2A and B). β-Galactosidase activity assays showed that Pbcp-3 is not active under the conditions used for competitions (Fig. 3A), providing an explanation for why WT BdAU0158 did not outcompete the Δbcp-3 mutant.
FIG 3.

The BdAU0158 bcp-3 locus encodes a functional CDI system that is not expressed under in vitro competition conditions. (A) β-Galactosidase activity assays for promoters of the BdAU0158 bcp-1, bcp-2, and bcp-3 loci. A BdAU0158 strain harboring a constitutively expressed lacZ reporter (PS12-lacZ) served as a positive control, whereas a BdAU0158 strain harboring a lacZ reporter without a promoter (promoterless) served as a negative control. (B) Competition assays between BdAU0158 bcp-3C inhibitor cells and Δbcp-3, Δbcp-3 attTn7::bcpI-3, and WT target cells. The dotted line (log10 C.I. = 0) indicates no competitive advantage for inhibitor or target strain. The red-filled circle indicates a competition from which no target cells were recovered following 48 h of coculture. (C) β-Galactosidase activity assays for the BdAU0158 Pbcp-3 reporter strain cocultured with BdAU0158 bcp-3C (bcp-3C versus Pbcp-3-lacZ), as well as the BdAU0158 PbcpI-3 reporter strain (PbcpI-3-lacZ), with PS12-lacZ and promoterless positive and negative controls, respectively, as described for panel A. β-Galactosidase activity assays in panels A and C show results from two biological replicates, each with three technical replicates. ***, P < 0.001; ****, P < 0.0001, unpaired t test. Competition assays in in panel B show results from three biological replicates, each with three technical replicates. Solid horizontal lines represent mean log10 C.I. values. ****, P < 0.0001, Mann-Whitney test.
The BdAU0158 bcp-3 locus encodes a functional CDI system.
To determine if the BdAU0158 bcp-3 locus encodes a functional CDI system, a strain constitutively expressing the locus (bcp-3C) was generated by replacing the native bcp-3 promoter region with the PS12 constitutive promoter. BdAU0158 bcp-3C outcompeted the Δbcp-3 mutant by 5 log (Fig. 3B), with one competition resulting in no Δbcp-3 cells being recovered from the coculture (red-filled circle in Fig. 3B). The BdAU0158 Δbcp-3 mutant was protected from killing by BdAU0158 bcp-3C when bcpI-3 was supplied in trans. Surprisingly, WT BdAU0158 cells were not outcompeted by BdAU0158 bcp-3C, even though the native Pbcp-3 appears to be inactive under these conditions (Fig. 3A and B). We hypothesized three scenarios to explain this result: (i) low-level expression of the bcp-3 locus occurs and cannot be detected by promoter activity assays but leads to sufficient BcpI-3 production to resist Bcp-3-mediated CDI, (ii) CDI attack induces bcp-3 expression in target cells, or (iii) an internal promoter in the bcp-3 locus that separately drives expression of bcpI-3 exists.
An internal promoter in the BdAU0158 bcp-3 locus separately drives expression of bcpI-3.
To investigate the possibility that CDI induces bcp-3 expression in target cells, the BdAU0158 Pbcp-3-lacZ reporter strain was mixed at a 1:1 ratio with the BdAU0158 bcp-3C inhibitor strain (which produces all three CDI systems), and this coculture was incubated at 37°C for 48 h on LSLB agar. The Pbcp-3-lacZ reporter strain is not susceptible to killing via CDI, as it contains the bcpI genes of all three bcp loci. β-Galactosidase activity in the BdAU0158 Pbcp-3-lacZ reporter strain was no greater after coculture with the BdAU0158 bcp-3C inhibitor strain than after monoculture (Fig. 3A and C). These results indicate that CDI attack (Bcp-1-, Bcp-2-, or Bcp-3-mediated) does not induce bcp-3 expression in a target cell.
To determine if an internal promoter resides in the BdAU0158 bcp-3 locus that drives expression of bcpI-3, the last 500 bp of bcpA-3 (the sequence immediately upstream of and including the bcpI-3 start codon) was cloned into the lacZ expression cassette and the cassette was delivered to the BdAU0158 chromosome, generating the reporter strain BdAU0158 attTn7::PbcpI-3-lacZ. This strain produced ∼1,500 units of β-galactosidase activity after 48 h growth at 37°C on LSLB agar (Fig. 3C), indicating that a promoter (PbcpI-3) resides at the 3′ end of the BdAU0158 bcpA-3 gene that appears to drive expression of bcpI-3, allowing for production of the BcpI-3 antitoxin even when the remainder of the bcp-3 locus is not expressed.
CDI attack and resistance to CDI do not require the BdAU0158 BcpO proteins.
A characteristic of Burkholderia-type CDI system-encoding loci is the presence of a fourth ORF, bcpO. The function(s) of BcpO proteins has not been determined, although a BtE264 ΔbcpO mutant is partially defective at outcompeting BtE264 ΔbcpAIOB via CDI (17), indicating that the BtE264 BcpO protein is important for CDI but is not required for this activity. The BcpO protein predicted to be encoded by the BdAU0158 bcp-1 locus (BcpO-1) shares 90.4% identity at the amino acid level with BtE264 BcpO. There are two and three predicted ORFs between bcpI and bcpB in the BdAU0158 bcp-2 and bcp-3 loci, respectively (Fig. 1A).
Strains containing unmarked, in-frame deletion mutations in each of the BcpO-encoding regions of BdAU0158 were generated, resulting in ΔbcpO-1, ΔbcpO-2, and ΔbcpO-3 mutants (deletion schematics shown in Fig. 4). These mutants were competed on LSLB agar for 48 h at 37°C against their parental, bcp-intact strains or their respective locus deletion mutants to determine if the BdAU0158 BcpO proteins are required for resistance to CDI or for the ability to kill target cells via CDI, respectively. In accordance with the effect of BcpO on BtE264 CDI (17), the BdAU0158 ΔbcpO-1 mutant had approximately a 1-log defect in CDI-mediated killing of the BdAU0158 Δbcp-1 target strain compared to WT. However, BcpO-1 was not required for resistance to Bcp-1-mediated killing, as the ΔbcpO-1 mutant was not outcompeted by WT BdAU0158 (Fig. 4A). BcpO-2 was not important for CDI in BdAU0158, as the ΔbcpO-2 mutant outcompeted the Δbcp-2 mutant as well as WT outcompeted Δbcp-2, and the inability of WT to outcompete the ΔbcpO-2 mutant shows that BcpO-2 was not required for resistance to Bcp-2-mediated CDI (Fig. 4B). Competitions investigating the role of BcpO-3 had to be conducted in the bcp-3C background to ensure these genes were expressed. As shown in Fig. 4C, BdAU0158 bcp-3C and the bcp-3C strain lacking bcpO-3 (bcp-3CΔbcpO-3) were equally able to outcompete Δbcp-3 targets, indicating that BcpO-3 was not required for Bcp-3-mediated CDI. Given that target cells with the native bcp-3 locus promoter are not outcompeted by the constitutively expressing strain (Fig. 3B), a ΔbcpO-3 mutant in the WT background was used to determine if BcpO-3 is required for resistance to CDI. In agreement with results from competitions investigating BcpO-1 and BcpO-2, the BdAU0158 ΔbcpO-3 mutant was not susceptible to CDI by a BdAU0158 bcp-3C inhibitor (Fig. 4C). In fact, the ΔbcpO-3 mutant had a slight growth advantage compared to the inhibitor (similar to the Δbcp-3 attTn7::bcpI-3 and WT targets in Fig. 3B), possibly due to the energetic cost of constitutively producing the Bcp-3 proteins in the bcp-3C strain. Together, these data suggest that the BcpO proteins of BdAU0158 do not play a crucial role in CDI.
FIG 4.

BdAU0158 BcpO proteins are not required for CDI-mediated competition or resistance to CDI. (A) Competition assays between BdAU0158 WT inhibitor cells and Δbcp-1 target cells, BdAU0158 ΔbcpO-1 inhibitor cells and Δbcp-1 target cells, and BdAU0158 WT inhibitor cells and ΔbcpO-1 target cells (right of dashed vertical line). (B) Competition assays between BdAU0158 WT inhibitor cells and Δbcp-2 target cells, BdAU0158 ΔbcpO-2 inhibitor cells and Δbcp-2 target cells, and BdAU0158 WT inhibitor cells and ΔbcpO-2 target cells (right of dashed vertical line). (C) Competition assays between BdAU0158 bcp-3C inhibitor cells and Δbcp-3 target cells, BdAU0158 bcp-3CΔbcpO-3 inhibitor cells and Δbcp-3 target cells, and BdAU0158 bcp-3C inhibitor cells and ΔbcpO-3 target cells (right of dashed vertical line). bcpO gene deletion schematics are shown above each graph. For each competition, results from two separate biological replicates, each with three technical replicates. Solid horizontal lines represent mean log10 C.I. values. Dotted horizontal lines (log10 C.I. = 0) indicate no competitive advantage for inhibitor or target strain. *, P < 0.05; n.s., not significant (Mann-Whitney test).
BdAU0158 does not exhibit Bcp-dependent community behaviors.
Given that BdAU0158 produces three CDI systems, one of which is identical to the BtE264 CDI system, we hypothesized that BdAU0158 could exhibit Bcp-dependent cooperative behaviors (i.e., contact-dependent signaling [CDS]) similar to those seen in BtE264 and that these behaviors may promote infection of the CF respiratory tract. We investigated autoaggregation and pigment production by WT BdAU0158, a panel of mutants that lack one CDI system (Δbcp-1, Δbcp-2, or Δbcp-3 mutants) or all three CDI systems (labeled as AU0158 ΔΔΔ), and the strain constitutively expressing the bcp-3 locus (bcp-3C). Autoaggregation of liquid cultures that were grown rotating for 24 h at 37°C in minimal medium was assessed by measuring the optical density at 600 nm (OD600) of a culture after sitting stationary at 25°C for ∼30 min and the OD600 of the same culture following vigorous vortexing. A vortexed-to-settled OD600 ratio greater than 1 indicates that autoaggregation of cells occurred during liquid growth (as seen with WT BtE264 in Fig. 5A), whereas a vortexed/settled OD600 ratio of ∼1 indicates that cells did not autoaggregate (as seen with BtE264 ΔbcpAIOB in Fig. 5A). All BdAU0158 strains had vortexed/settled OD600 ratios of ∼1, suggesting that BdAU0158 does not autoaggregate under these conditions and that the production of CDI systems, or lack thereof, does not influence this phenotype. Pigment production was assessed by determining the growth of BdAU0158 cells on LSLB agar for 48 h at 37°C and subsequent incubation at 25°C for up to 14 days. Under these conditions, WT BtE264 produced a dark beige pigment, whereas the BtE264 ΔbcpAIOB mutant remained white (Fig. 5B). WT BdAU0158 colony biofilms appeared darker than those of the BtE264 ΔbcpAIOB mutant, though this coloration was not dependent on the Bcp proteins, as mutants lacking bcp loci and the bcp-3C strain appeared similar in color to WT BdAU0158 (Fig. 5B). These results indicate that BdAU0158 does not perform Bcp-dependent cooperative behaviors similar to those seen in BtE264; however, it is possible that the BdAU0158 Bcp proteins do mediate community-based phenotypes and that the assays used to detect BtE264 CDS and its associated phenotypes cannot detect such behaviors in BdAU0158.
FIG 5.
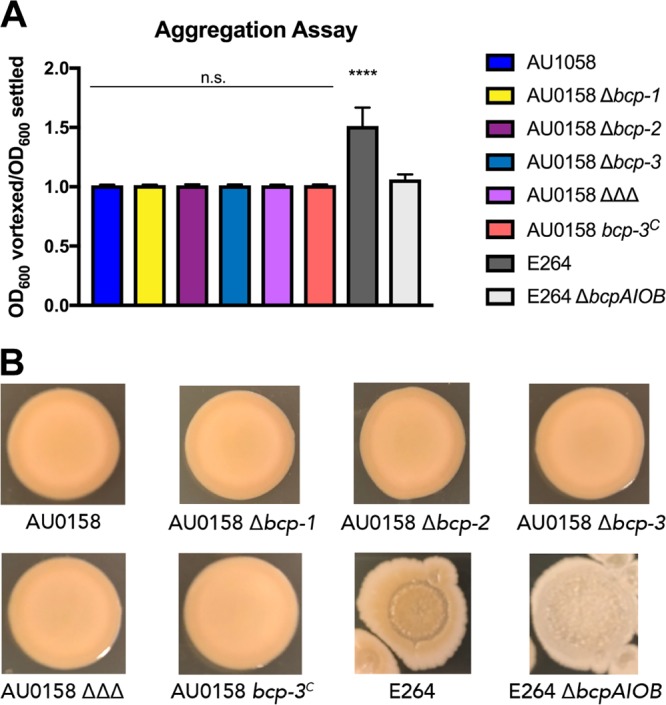
The Bcp-dependent community behaviors of autoaggregation and pigment production are not evident in BdAU0158. (A) Autoaggregation assays of BdAU0158 and BtE264 cultures grown in minimal medium, measured by determining the ratio of OD600 values of vortexed and settled cultures. Only BtE264 WT cells exhibit autoaggregation. Results from three separate biological replicates, with mean ratios plotted. Mean ratios compared to nonautoaggregating BtE264 ΔbcpAIOB to determine if cells autoaggregated. ****, P < 0.0001, Student's t test. n.s., not significant. (B) Pigment production assays of BdAU0158 and BtE264 colony biofilms grown on LSLB agar. Only BtE264 exhibits Bcp-dependent pigment production. Images are representative of at least three biological replicates.
Toxicity of BdAU0158 CDI toxins in E. coli.
To determine if the BdAU0158 BcpA-1-CT, BcpA-2-CT, and BcpA-3-CT are sufficient for toxicity, inducible expression plasmids were generated as derivatives of pET-28(a). Each plasmid contained nucleotide sequences encoding one BcpA-CT [the NX(E/Q)LYN motif through the BcpA stop codon], a CT plus its cognate BcpI [the NX(E/Q)LYN motif through the BcpI stop codon], or one BcpI alone. Escherichia coli BL21(DE3) cells harboring these plasmids were grown in LSLB broth, and expression was either induced with isopropyl-β-d-1-thiogalactopyranoside (IPTG) or repressed by d-glucose. Over the time course, OD600 was measured and aliquots were plated on solid medium to assess cell viability. To our surprise, production of the BdAU0158 BcpA-1-CT was not toxic in E. coli, but production of BcpI-1 was slightly toxic (Fig. 6B). We detected production of both the BcpA-1-CT and BcpI-1 by E. coli using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (see Fig. S1 in the supplemental material). Although induction of bcp-1 did not cause a decrease in OD600 (Fig. 6A), indicating cells were not lysing, the viability of BcpI-1-producing cells over time decreased compared to cells producing either BcpA-1-CT or BcpA-1-CT and BcpI-1 concurrently (Fig. 6B). Cells producing BcpA-1-CT and BcpI-1 concurrently were protected from the toxic effects of BcpI-1, suggesting that binding of the CT to BcpI-1 inhibits toxicity of the immunity protein. We hypothesize that the portion of BdAU0158 BcpA-1-CT being produced in E. coli is either larger or smaller than the true toxic protein and that improper folding or processing of the CT inhibits its toxic effects. The fact that BcpI-1 is toxic in E. coli is more perplexing, as it is not toxic when produced in BdAU0158 (Fig. 2A). Phyre2 analysis (29) predicts that BcpI-1 contains a DNA-binding domain. One possibility is that when BcpI-1 is produced in excess without its cognate BcpA-1-CT, it has deleterious effects in E. coli via interactions with genomic DNA.
FIG 6.

E. coli autotoxicity due to inducible production of the BdAU0158 Bcp-2 and Bcp-3 CT toxins but not the Bcp-1 CT toxin. (A, C, E) OD600 values of cultures of E. coli BL21(DE3) strains producing BdAU0158 Bcp CT toxins (red dotted lines), coproducing CT toxins and BcpI proteins (green dotted lines), and producing BcpI proteins (blue dotted lines) of the Bcp-1 (A), Bcp-2 (C), and Bcp-3 (E) CDI systems. (B, D, F) Cell viability, as measured in CFU per milliliter, of the Bcp-1 protein-producing (B), Bcp-2 protein-producing (D), and Bcp-3 protein-producing (F) E. coli BL21(DE3) cultures. Solid lines represent cultures in which BdAU0158 Bcp protein production was repressed by the addition of 0.2% glucose. For each intracellular toxicity assay, means from three biological replicates are plotted.
Unlike BdAU0158 BcpA-1-CT, BcpA-2-CT and BcpA-3-CT were toxic when produced in E. coli (Fig. 6D and F). Production of BcpA-2-CT caused the OD600 to plateau (Fig. 6C) and resulted in a 4-log decrease in cell viability (Fig. 6D). BcpA-2-CT-producing cells that concurrently produced BcpI-2 were partially rescued from the toxic effects of the BcpA-2-CT, and production of BcpI-2 alone had no deleterious effects on E. coli other than the presumed energetic cost of protein production (Fig. 6D). Similar results were found for bcp-3; induction did not cause a decrease in OD600 (Fig. 6E), but BcpA-3-CT production caused a drastic decrease in cell viability that was partially rescued by concurrent production of BcpI-3 (Fig. 6F). BcpI-3 appears to protect against the toxic effects of the BcpA-3-CT less well than BcpI-2 protects against the effects of BcpA-2-CT (Fig. 6D versus Fig. 6F), though at least partial protection was detected in each case. In these coproducing strains, the BcpA-CTs and BcpI proteins are presumably produced at 1:1 ratios, which may be the reason complete protection against CT-induced toxicity was not detected. It is possible that stoichiometry needs to favor the immunity proteins in order to fully inhibit autotoxicity—a situation that may occur under normal conditions as BcpA proteins are exported out of the cell or may occur if bcpI-specific promoters are widespread throughout bcpAIOB loci.
The BtE264 Bcp and BdAU0158 Bcp-1 alleles are functionally redundant.
The similarity of the predicted amino acid sequences of BtE264 BcpA and BdAU0158 BcpA-1 (Fig. 1B) suggests they are functionally redundant. Previous work in our laboratory identified residues required for the catalytic activity of BtE264 BcpA-CT (E3064 and K3066), and substituting alanines for these residues abrogated CDI activity (20). Glutamate and lysine residues are found at the same positions in BdAU0158 BcpA-1-CT (Fig. 7A). In addition to the similarities of BtE264 BcpA-CT and BdAU0158 BcpA-1-CT, BtE264 BcpI and BdAU0158 BcpI-1 are nearly identical (Fig. 7B). To investigate functional interchangeability, BtE264 ΔbcpAIOB and BdAU0158 Δbcp-1 mutants were provided with the heterologous bcpI gene delivered to the attTn7 site, generating BdAU0158 Δbcp-1 attTn7::bcpIE264 and BtE264 ΔbcpAIOB attTn7::bcpIAU0158-1. During 48 h of coculture on LSLB agar at 37°C, the BdAU0158 Δbcp-1 mutant was rescued from Bcp-1-mediated CDI by the BdAU0158 WT inhibitor when it constitutively expressed the BtE264 bcpI gene (log10 C.I. ≈ 0) (Fig. 7C). Similarly, during 24 h of coculture on LSLB agar at 25°C, the BtE264 ΔbcpAIOB mutant was rescued from Bcp-mediated CDI by the BtE264 WT inhibitor when it constitutively expressed the BdAU0158 bcpI-1 gene (log10 C.I. ≈ 0) (Fig. 7D). The ability of each BcpI protein to protect against CDI in the heterologous species provides experimental evidence that BdAU0158 and BtE264 share the same CDI system-encoding allele.
FIG 7.

The BdAU0158 Bcp-1 allele contains the same CT toxin-immunity pair as the BtE264 Bcp allele. (A) Amino acid sequence alignment of the BdAU0158 BcpA-1-CT and BtE264 BcpA-CT. The glutamate and lysine residues required for toxicity are boxed in orange. (B) Amino acid sequence alignment of the BdAU0158 BcpI-1 and BtE264 BcpI proteins. For panels A and B, identical residues are highlighted in black, similar residues are highlighted in gray, and disparate residues are not highlighted. (C) Competition assays between BdAU0158 WT inhibitor cells and Δbcp-1 and Δbcp-1 attTn7::bcpIE264 target cells. (D) Competition assays between BtE264 WT inhibitor cells and ΔbcpAIOB and ΔbcpAIOB attTn7::bcpIAU0158-1 target cells. For each competition, results are from three separate biological replicates, each with three technical replicates. Solid horizontal lines represent mean log10 C.I. values. Dotted lines (log10 C.I. = 0) indicate no competitive advantage for inhibitor or target strain. ****, P < 0.0001, Mann-Whitney test.
DISCUSSION
CDI systems are present in a broad range of Gram-negative bacteria, including many that are pathogenic for animals or plants. Most studies of CDI function have used Escherichia coli strain EC93 as a model for E. coli-type systems or Burkholderia thailandensis strain E264 (BtE264) as a model for Burkholderia-type systems (15–17, 19, 22, 23, 31–39). Some experiments have used E. coli or B. thailandensis strains producing chimeric CdiA or BcpA proteins with toxin domains (and cognate immunity proteins) from pathogens (16, 19, 22, 23, 40), with a smaller number of studies, limited to E. coli-type systems, investigating CDI in the pathogen itself (35, 41, 42). In this work, we used the epidemic Bcc isolate B. dolosa strain AU0158 (BdAU0158) and showed that its three distinct CDI systems, including two class II Burkholderia-type systems, are capable of killing and/or arresting the growth of neighboring bacteria. A whole-genome sequence exists for two additional B. dolosa strains, PC543 and LO6 (also referred to as B. cepacia strain LO6). The genomes of these strains contain three bcpAIOB loci, and the potential proteins encoded by these loci are 100% identical at the amino acid level to the Bcp-1, Bcp-2, and Bcp-3 proteins of BdAU0158, suggesting that CDI by B. dolosa is not limited to strain BdAU0158.
While the BdAU0158 Bcp-1 and Bcp-2 CDI systems mediated interbacterial competition under laboratory conditions, Bcp-3-mediated CDI was detected only when the region 5′ to the start of bcpA-3 was replaced with the constitutively active S12 promoter. These results are consistent with our lacZ reporter fusion analyses. The 500-bp and 300-bp DNA fragments corresponding to the regions immediately 5′ to bcpA-1 and bcpA-2, respectively, resulted in substantial β-galactosidase activity when present upstream of lacZ, while the corresponding fragment from bcp-3 resulted in no detectable β-galactosidase activity under the same laboratory conditions. Together with the fact that the gene 5′ to bcpA-3 is oriented in the opposite direction, the most likely explanation for the results that we obtained for bcp-3 is that the 500-bp fragment does contain the promoter for bcp-3 but that this promoter is regulated such that it is not activated under standard laboratory conditions. Little is known about the regulation of any CDI system-encoding genes. In E. coli-type systems, the cdiBAI genes are expressed under laboratory conditions only in strain EC93 (15, 16, 41), and in B. thailandensis E264, the bcpAIOB genes appear to be tightly regulated such that only about 1 in 1,000 bacteria express the genes at a high level under laboratory conditions (17). Future experiments will be aimed at identifying transcription start sites and investigating how and why the bcp loci are differentially regulated in BdAU0158.
Because the bcp-3 locus appears to be transcriptionally silent under standard laboratory growth conditions, we were surprised to find that WT BdAU0158 was not outcompeted by the strain expressing bcp-3 constitutively (BdAU0158 bcp-3C). This result led us to search for a promoter for bcpI-3 within the 3′ end of bcpA-3. Although the transcription start site has yet to be determined, the region immediately 5′ to bcpI-3 was sufficient to drive lacZ expression, suggesting that transcription initiation in this region in the native locus results in sufficient BcpI protein production to confer protection from BcpA-3-mediated toxicity. While it would seem to be advantageous for bacteria to produce all immunity proteins at a low level constitutively, our study is the first demonstration, to our knowledge, of a promoter for bcpI (or cdiI) within a bcpAIOB (or cdiBAI) operon that is independent of that driving transcription of the rest of the operon. For E. coli-type CDI systems, “orphan” cdiA-CT/cdiI modules that encode functional CdiA-CT toxins and CdiI immunity proteins but are located outside cdiBAI loci have been identified (31). These orphan modules, which exist in the genomes of several pathogens containing CDI system-encoding genes (31, 43), share similarities with recombination hot spot (rhs) loci and can contribute to diversification of the CDI systems in these species via recombination with the cdiBAI genes. There is no evidence that promoters exist for these orphan modules or the corresponding orphan cdiI genes (31), and orphan bcpA-CT/bcpI modules have not been detected in Burkholderia spp.
The role of the additional ORF between bcpI and bcpB in Burkholderia-type CDI system-encoding loci (which we named bcpO) remains enigmatic. In class I Burkholderia-type loci, the bcpO genes are highly homologous. They are predicted to encode small lipoproteins that lack localization of lipoproteins (Lol) avoidance signals, suggesting that they localize to the inner leaflet of the outer membrane. The N-terminal halves of the 53-aa mature polypeptides are rich in prolines, and the C-terminal halves are rich in tryptophans. As with BtE264 (17), deletion of bcpO-1 in BdAU0158 resulted in a modest decrease in CDI activity, but the mechanism underlying this phenotype is unknown. Our investigations into whether BcpO contributes to CDS or cooperative behaviors have yielded inconclusive results so far. The “bcpO” genes in class II Burkholderia-type loci bear little similarity to each other or to bcpO genes in class I alleles and hence should probably be renamed. Deletion of these ORFs in BdAU0158 bcp-2 and bcp-3 had no effect on CDI activity under the conditions tested.
Although we did not detect CDS in BdAU0158, the assays used for these experiments were developed to describe CDS in BtE264 (24) and thus may not be specific for Bcp-mediated community behaviors in other strains. Future investigation into CDS by BdAU0158 and other Burkholderia spp. containing bcpAIOB loci is warranted. Given the growing appreciation for the impact of bacterial cooperation on pathogenesis (44–48), the virulence of diverse Gram-negative bacterial pathogens may be influenced by CDS.
Production of BdAU0158 BcpA-2-CT and BcpA-3-CT by E. coli resulted in autotoxicity that was partially ablated by concurrent production of BcpI-2 and BcpI-3, respectively. Conversely, E. coli producing BcpA-1-CT did not exhibit reduced viability, despite the likelihood that this polypeptide contains the toxic domain of BcpA-1. For some E. coli-type CDI systems, cytoplasmic “permissive factors” are required for activity of a delivered toxin within a target cell (32, 36, 37). A requirement for a toxicity-promoting factor specific to Burkholderia, or to BdAU0158, may explain the lack of toxicity of BdAU0158 BcpA-1-CT in E. coli. Alternatively, it is possible that the BcpA-1-CT polypeptide that we selected to produce in E. coli BL21(DE3) is not the toxic molecule delivered by the BdAU0158 Bcp-1 CDI system. Indeed, the precise BcpA or CdiA polypeptide that is delivered to the cytoplasm of target cells, and whether it is modified in any way, is not known for any CDI system. We are currently conducting experiments to identify the BcpA polypeptides that are delivered during CDI and CDS in BtE264 and other Burkholderia strains.
Also unexpected was the finding that production of BdAU0158 BcpI-1 was toxic in E. coli. BcpI-1 is not toxic when produced in BdAU0158, and the nearly identical BtE264 BcpI protein is not toxic when produced in BtE264 (17). Phyre2 analysis predicts a DNA-binding domain in BcpI-1. A possible explanation for its toxicity when massively overproduced in E. coli is that it interacts with genomic DNA in a way that blocks an essential function, such as replication or transcription of an essential gene(s). Whether BcpI from either BdAU0158 or BtE264 actually binds DNA is unknown but would be consistent with a role for BcpI, in complex with BcpA-CT, in controlling changes in gene expression during CDS (18, 24).
The high degree of similarity between bcp-1 of BdAU0158 and bcpAIOB of BtE264 and the functional redundancy of the BcpA (24) and BcpI proteins (this work) suggest that these genes represent the same allele. Genomic analyses suggest that both Burkholderia- and E. coli-type CDI system-encoding genes reside on genomic islands that were mobile at some time in the past and perhaps still are (49–53). We identified 11 Burkholderia strains potentially harboring the same CDI system as BdAU0158 and BtE264 by searching for orthologs of BdAU0158 BcpA-1-CT and BcpI-1 (Fig. 8). The Mauve software package (54) detected evidence of synteny surrounding the bcpAIOB loci between the three B. dolosa strains (BdAU0158, BdPC543, and BdLO6) only. Close examination of these orthologous BcpA-CT and BcpI proteins revealed that although they are highly similar across the different strains, variation in the amino acid sequence exists in the C-terminal portion of BcpA-CT (extreme CT, BcpA-ECT) and the N-terminal portion of BcpI (BcpI-NT) (Fig. 8). We separated these strains into three groups—the BdAU0158/BtE264 group, the BgluBGR1 group, and the BtE444 group—based on the degree of variation of the BcpA-ECTs and BcpI-NTs compared to the BdAU0158 BcpA-ECT and BcpI-NT, respectively. Variation in BcpA-ECT and BcpI-NT sequence, along with the lack of synteny between the regions surrounding bcpAIOB in these strains, suggests that if these genes were acquired horizontally, there has been substantial evolution since that time. Though we demonstrated that the BcpA and BcpI proteins of BdAU0158 and BtE264 function in the heterologous species (24; this work), it is unknown whether that holds true for all strains on this list. We hypothesize that the C terminus of BcpI is required for protection against toxicity of the BcpA-CT, given that the BcpI proteins of BdAU0158 and BtE264 block BcpA-CT toxicity in the heterologous species despite exhibiting BcpI-NT sequence variation. Though purely speculative, perhaps these variable regions of BcpA-CT and BcpI are important for CDS and delivery of an “identical” BcpA-CT, from a CDI standpoint, will not elicit CDS in a target cell producing a variable BcpI. Future comparative analyses of these variable alleles will be informative to the mechanisms of both CDI and CDS.
FIG 8.
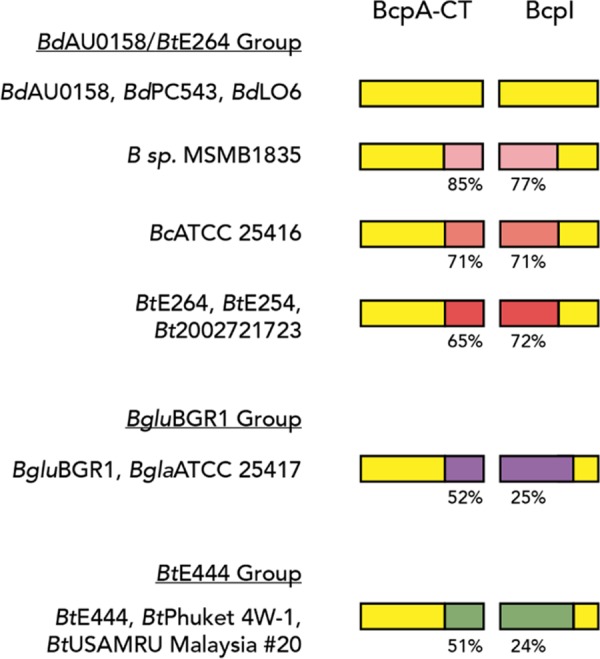
Orthologs of the BdAU0158 BcpA-CT and BcpI across several Burkholderia strains. Yellow indicates regions of amino acid sequence identity. Regions of amino acid sequence variation are indicated by shades of pink (BdAU0158/BtE264 group), purple (BgluBGR1 group), and green (BtE444 group), with percent identity to the corresponding BdAU0158 sequence shown below. Members of the BdAU0158/BtE264 group have variable sequences that are more similar to each other than they are to members of the BgluBGR1 and BtE444 groups. BdPC543, B. dolosa strain PC543; BdLO6, B. dolosa strain LO6 (also called B. cepacia LO6); BcATCC 25416, B. cepacia strain ATCC 25416; BtE254, B. thailandensis strain E254; Bt2002721723, B. thailandensis strain 2002721723; BgluBGR1, B. glumea strain BGR1; BglaATCC 25417, B. gladioli strain ATCC 25417; BtE444, B. thailandensis strain E444; BtPhuket 4W-1, B. thailandensis strain Phuket 4W-1; BtUSAMRU Malaysia #20, B. thailandensis strain USAMRU Malaysia #20; B. sp. MSMB1835, species unknown.
Aside from the investigation of P. aeruginosa CDI by Melvin et al. (42), our study is the only demonstration of multiple functional CDI systems in a single pathogenic species. Given the polymicrobial nature of the CF respiratory tract, CDI may provide BdAU0158, as well as P. aeruginosa, a competitive advantage during host infection. The genomes of strains of several Bcc species, including B. cenocepacia, B. multivorans, and B. vietnamiensis, contain possible CDI system-encoding genes, and thus these pathogens may benefit from CDI activity during infection. Evolutionary genomic analysis of B. dolosa isolates from CF patients over a 16-year period (all originating from the Boston epidemic) revealed that identical nonsynonymous mutations arose in bcpA-2 across multiple patients, suggesting there is strong selective pressure promoting parallel adaptive evolution of this CDI-encoding gene within the human host (26). Additionally, Bcc species are found ubiquitously in the environment, especially in soil (55), and thus BdAU0158 could employ its CDI systems to outcompete potential competitors in diverse settings. It is hypothesized that competitive behaviors are the strongest force shaping microbial ecology (5); therefore, BdAU0158 and other Bcp-producing Bcc pathogens may utilize CDI to gain a foothold in the complex polymicrobial environments of the CF respiratory tract and establish long-term, devastating infections in these patients.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains used in this study are listed in Table S1 in the supplemental material. B. dolosa strains were maintained in either LB (NaCl concentration, 10 g/liter) or low-salt LB (LSLB; NaCl concentration, 5 g/liter), while B. thailandensis strains were exclusively maintained in LSLB. Antibiotics were added to select for growth of various Burkholderia strains at the following concentrations: 250 μg/ml kanamycin, 50 μg/ml tetracycline, and/or 20 μg/ml chloramphenicol. E. coli strains were maintained in LB, and antibiotics were added at the following concentrations when appropriate: 100 μg/ml ampicillin, 50 μg/ml kanamycin, 10 μg/ml tetracycline, and/or 35 μg/ml chloramphenicol. Diaminopimelic acid (DAP) was added at a concentration of 200 μg/ml to cultures maintaining E. coli strain RHO3. All strains were grown overnight with aeration at 37°C (unless indicated otherwise).
Construction of plasmids and mutant strains.
All plasmids used in this study are listed in Table S2 in the supplemental material and were delivered to BdAU0158 or BtE264 cells via conjugation with E. coli RHO3 strains harboring these plasmids. BdAU0158 Δbcp-1, BdAU0158 Δbcp-2, BtE264 ΔbcpAIOB, BtE264 attTn7::Cm, and BtE264 ΔbcpAIOB attTn7::Km were constructed previously (17, 24). Allelic exchange plasmids for generating BdAU0158 in-frame deletion mutants were constructed on the pEXKm5 backbone (56). Briefly, ∼500 bp upstream from and including the first three codons of the ORF(s) to be deleted were fused via overlap extension PCR to the last three codons of the ORF(s) and ∼500 bp downstream sequence, and constructs were cloned into pEXKm5.
Plasmids to deliver cassettes to the attTn7 sites of the BdAU0158 and BtE264 chromosomes were constructed using the pUC18Tmini-Tn7T backbone (57). Cassettes for generating antibiotic-resistant BdAU0158 strains were delivered to the BdAU0158 chromosome via triparental mating with E. coli RHO3 strains harboring either pUC18T-miniTn7-Km (for kanamycin resistance) or pUC18T-miniTn7-Tet (for tetracycline resistance), as well as the RHO3 strain harboring the transposase-encoding helper plasmid pTNS3 (17). To complement BdAU0158 and BtE264 bcp locus deletion mutants with various bcpI genes, the genes of interest were cloned into pUCS12Km (pUC18T-miniTn7-Km plasmid with the constitutive BtE264 ribosomal S12 subunit gene promoter cloned immediately 5′ to the multiple cloning site), and these PS12-driven constructs were delivered to a neutral site in the chromosome via triparental mating with E. coli RHO3 harboring pTNS3. Plasmids containing lacZ reporter cassettes were generated on the pUClacZ backbone (pUC18T-miniTn7-Km with promoterless lacZ cloned into the multiple cloning site), with promoters of interest inserted immediately 5′ to the lacZ gene. lacZ reporter cassettes were delivered to the BdAU0158 chromosome via triparental mating with E. coli RHO3 harboring pTNS3.
Interbacterial competition experiments.
All competitions between BdAU0158 strains followed our previously developed protocol (17), with minor adjustments. Cells from overnight cultures were washed in phosphate-buffered saline (PBS) and diluted to an OD600 value of 0.2. Equal volumes of inhibitor and target cells were mixed, and 20-μl spots of cell suspensions were plated on LSLB agar and allowed to dry. Once spots had dried, competitions were incubated at 37°C for 48 h. To determine the starting ratios for competitions, the cell suspensions containing inhibitor and target bacteria were serially diluted and 20-μl spots were plated onto selective medium (LSLB/Km250 or LSLB/Tet50) and incubated at 37°C. Following 48 h of coculture, cells were sampled from the edge of the colony biofilms, resuspended in 1 ml PBS, and serially diluted, and 20-μl spots of serial dilutions were plated onto selective medium (LSLB/Km250 or LSLB/Tet50) and incubated at 37°C. Competitions involving BtE264 were conducted similarly, except that cocultures were incubated at room temperature for 24 h. Colony counts for inhibitor and target bacteria at the starting and 24-h or 48-h time points were used to determine the competitive index (C.I.) for each competition experiment, according to the equation C.I. = (inhibitortx/targettx)/(inhibitort0/targett0), with tx representing either the 24-h or 48-h time point and t0 representing starting time point. A positive log10 C.I. indicates that the inhibitor strain outcompeted the target strain, a negative log10 C.I. indicates that the target strain outcompeted the inhibitor strain, and a log10 C.I. of ∼0 indicates no competition in favor of either strain.
β-Galactosidase activity assays.
Cells from overnight cultures were washed in PBS and diluted to an OD600 value of 0.2. Twenty-microliter spots of cell suspensions were plated on LSLB agar, and after spots had dried, cells were incubated at 37°C for 48 h. For the coculture with BdAU0158 bcp-3C and BdAU0158 attTn7::Pbcp-3-lacZ, strains were mixed at a 1:1 ratio, and 20-μl spots were plated onto LSLB and incubated at 37°C for 48 h. Following incubation, entire colony biofilms were resuspended in 1 ml PBS and diluted 1:10 in Z-buffer plus 0.27% β-mercaptoethanol (Fisher Scientific), and 250 μl was removed to measure OD600 values. To permeabilize cells, 50 μl chloroform and 10 μl 0.1% sodium dodecyl sulfate were added to the remaining 750-μl cell suspensions and samples were vortexed and allowed to settle. Fifty microliters of permeabilized cells and 50 μl 4-mg/ml ortho-nitrophenyl-β-galactoside were added to 150 μl Z-buffer, and OD420 values were measured every minute over a 20-min time course. OD420 values for two time points within the linear range and the corresponding change in time were used to calculate β-galactoside activity with the following formula: β-galactoside activity = [ΔOD420/(Δt × 0.05 ml cells × OD600)] × 1,000.
Z-buffer was prepared (per liter) as follows: 50 mM Na2HPO4 (anhydrous), 40 mM NaH2PO4, 10 mM KCl, and 1 mM MgSO4 (anhydrous).
Intracellular toxicity assays.
E. coli BL21(DE3) cells harboring IPTG-inducible plasmids were grown overnight in LB plus kanamycin. Cells were washed in PBS, subcultured in 50 ml LSLB plus kanamycin at a starting OD600 of 0.02 (with d-glucose added at a final concentration of 0.2% for noninduced cultures), and grown at 37°C with aeration for 7 h. Samples were taken hourly to determine OD600 values of the cultures, as well as to plate serial dilutions on LB agar plus kanamycin supplemented with 0.2% d-glucose to determine cell viability. At the 2-h time point, IPTG was added at a final concentration of 0.5 mM to induce expression of bcp constructs. Production of BcpA-1-CT and BcpI-1 was assessed via separation in a 12% SDS-PAGE gel and Coomassie blue staining.
Aggregation assays.
Cells from overnight cultures were washed in PBS and inoculated into 2 ml M63 minimal medium (3 g/liter KH2PO4, 7 g/liter K2HPO4, 2 g/liter (NH4)2SO4, 0.5 g/liter FeSO4, 0.2% glucose, 1 mM MgSO4, 0.4% glycerol, 0.01% Casamino Acids) at a starting OD600 of 0.2. Aggregation cultures were grown on a rotator drum at 37°C for 24 h. After 24 h, culture tubes were taken off the rotator drum and incubated statically at 25°C for ∼30 min to allow cells to settle, and the OD600 values of settled cultures were measured. The tubes were then vigorously vortexed to homogenize the cultures, and the OD600 values of vortexed cultures were measured.
Pigment production assays.
Cells from overnight cultures were washed twice in PBS and diluted to an OD600 value of 0.2, and 20-μl spots were plated onto LSLB agar. After spots had dried, plates were wrapped in paraffin film and incubated at 25°C for 14 days.
Bioinformatic analyses.
BtE264 BcpB homologs were identified using the National Center for Biotechnology Information BLASTP suite. Burkholderia strains harboring the same BcpA-CT as the allele shared by BtE264 and BdAU0158 were identified using the Burkholderia Genome Database (58). ORF predictions were conducted with the Geneious 8 software package (30), and genome comparisons using the Mauve plug-in (54) were conducted on Geneious 8. Protein alignments were conducted using the Clustal Omega online server (59). Protein structure predictions were conducted using the Phyre2 online server (29). Signal sequence identification was performed using the SignalP 4.1 online server (60).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Cotter Laboratory for support, thoughtful discussion, and critical reading of the manuscript. We also thank Erin Garcia for insight and technical assistance.
This work was supported by NIH awards NIHR01GM121110 and NIHR21AI112764 to P.A.C. and Cystic Fibrosis Foundation award COTTER1810 to P.A.C.
Funders had no role in the design, execution, or analysis of this study or in the preparation and submission of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00428-18.
REFERENCES
- 1.Hansen SK, Rainey PB, Haagensen JAJ, Molin S. 2007. Evolution of species interactions in a biofilm community. Nature 445:533–536. doi: 10.1038/nature05514. [DOI] [PubMed] [Google Scholar]
- 2.Ren D, Madsen JS, Sørensen SJ, Burmølle M. 2015. High prevalence of biofilm synergy among bacterial soil isolates in cocultures indicates bacterial interspecific cooperation. ISME J 9:81–89. doi: 10.1038/ismej.2014.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rendueles O, Ghigo J-M. 2015. Mechanisms of competition in biofilm communities. Microbiol Spectr 3:319–342. doi: 10.1128/microbiolspec.MB-0009-2014. [DOI] [PubMed] [Google Scholar]
- 4.Flemming H-C, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. 2016. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol 14:563–575. doi: 10.1038/nrmicro.2016.94. [DOI] [PubMed] [Google Scholar]
- 5.Foster KR, Bell T. 2012. Competition, not cooperation, dominates interactions among culturable microbial species. Curr Biol 22:1845–1850. doi: 10.1016/j.cub.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Sekirov I, Russell SL, Antunes LCM, Finlay BB. 2010. Gut microbiota in health and disease. Physiol Rev 90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 7.Ma B, Forney LJ, Ravel J. 2012. Vaginal microbiome: rethinking health and disease. Annu Rev Microbiol 66:371–389. doi: 10.1146/annurev-micro-092611-150157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filkins LM, O'Toole GA. 2015. Cystic fibrosis lung infections: polymicrobial, complex, and hard to treat. PLoS Pathog 11:e1005258. doi: 10.1371/journal.ppat.1005258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, Young VB, Lipuma JJ. 2012. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci U S A 109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lipuma JJ. 2010. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev 23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isles A, Maclusky I, Corey M, Gold R, Prober C, Fleming P, Levison H. 1984. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr 104:206–210. doi: 10.1016/S0022-3476(84)80993-2. [DOI] [PubMed] [Google Scholar]
- 12.Schwab U, Abdullah LH, Perlmutt OS, Albert D, Davis CW, Arnold RR, Yankaskas JR, Gilligan P, Neubauer H, Randell SH, Boucher RC. 2014. Localization of Burkholderia cepacia complex bacteria in cystic fibrosis lungs and interactions with Pseudomonas aeruginosa in hypoxic mucus. Infect Immun 82:4729–4745. doi: 10.1128/IAI.01876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carmody LA, Zhao J, Kalikin LM, LeBar W, Simon RH, Venkataraman A, Schmidt TM, Abdo Z, Schloss PD, Lipuma JJ. 2015. The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome 3:12. doi: 10.1186/s40168-015-0074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernier SP, Workentine ML, Li X, Magarvey NA, O'Toole GA, Surette MG. 2016. Cyanide toxicity to Burkholderia cenocepacia is modulated by polymicrobial communities and environmental factors. Front Microbiol 7:725. doi: 10.3389/fmicb.2016.00725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. 2005. Contact-dependent inhibition of growth in Escherichia coli. Science 309:1245–1248. doi: 10.1126/science.1115109. [DOI] [PubMed] [Google Scholar]
- 16.Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, Jones AM, Webb JS, Hayes CS, Cotter PA, Low DA. 2010. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468:439–442. doi: 10.1038/nature09490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson MS, Garcia EC, Cotter PA. 2012. The Burkholderia bcpAIOB genes define unique classes of two-partner secretion and contact dependent growth inhibition systems. PLoS Genet 8:e1002877. doi: 10.1371/journal.pgen.1002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danka ES, Garcia EC, Cotter PA. 2017. Are CDI systems multicolored, facultative, helping greenbeards? Trends Microbiol 25:391–401. doi: 10.1016/j.tim.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson MS, Garcia EC, Cotter PA. 2014. Kind discrimination and competitive exclusion mediated by contact-dependent growth inhibition systems shape biofilm community structure. PLoS Pathog 10:e1004076. doi: 10.1371/journal.ppat.1004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia EC, Anderson MS, Hagar JA, Cotter PA. 2013. Burkholderia BcpA mediates biofilm formation independently of interbacterial contact-dependent growth inhibition. Mol Microbiol 89:1213–1225. doi: 10.1111/mmi.12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia EC. 2018. Contact-dependent interbacterial toxins deliver a message. Curr Opin Microbiol 42:40–46. doi: 10.1016/j.mib.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikolakakis K, Amber S, Wilbur JS, Diner EJ, Aoki SK, Poole SJ, Tuanyok A, Keim PS, Peacock S, Hayes CS, Low DA. 2012. The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol Microbiol 84:516–529. doi: 10.1111/j.1365-2958.2012.08039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koskiniemi S, Garza-Sánchez F, Edman N, Chaudhuri S, Poole SJ, Manoil C, Hayes CS, Low DA. 2015. Genetic analysis of the CDI pathway from Burkholderia pseudomallei 1026b. PLoS One 10:e0120265. doi: 10.1371/journal.pone.0120265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia EC, Perault AI, Marlatt SA, Cotter PA. 2016. Interbacterial signaling via Burkholderia contact-dependent growth inhibition system proteins. Proc Natl Acad Sci U S A 113:8296–8301. doi: 10.1073/pnas.1606323113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vermis K, Coenye T, Lipuma JJ, Mahenthiralingam E, Nelis HJ, Vandamme P. 2004. Proposal to accommodate Burkholderia cepacia genomovar VI as Burkholderia dolosa sp. nov. Int J Syst Evol Microbiol 54:689–691. doi: 10.1099/ijs.0.02888-0. [DOI] [PubMed] [Google Scholar]
- 26.Lieberman TD, Michel J-B, Aingaran M, Potter-Bynoe G, Roux D, Davis MR, Skurnik D, Leiby N, Lipuma JJ, Goldberg JB, McAdam AJ, Priebe GP, Kishony R. 2011. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet 43:1275–1280. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biddick R, Spilker T, Martin A, Lipuma JJ. 2003. Evidence of transmission of Burkholderia cepacia, Burkholderia multivorans and Burkholderia dolosa among persons with cystic fibrosis. FEMS Microbiol Lett 228:57–62. doi: 10.1016/S0378-1097(03)00724-9. [DOI] [PubMed] [Google Scholar]
- 28.Kalish LA, Waltz DA, Dovey M, Potter-Bynoe G, McAdam AJ, Lipuma JJ, Gerard C, Goldmann D. 2006. Impact of Burkholderia dolosa on lung function and survival in cystic fibrosis. Am J Respir Crit Care Med 173:421–425. doi: 10.1164/rccm.200503-344OC. [DOI] [PubMed] [Google Scholar]
- 29.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poole SJ, Diner EJ, Aoki SK, Braaten BA, t' Kint de Roodenbeke C, Low DA, Hayes CS. 2011. Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet 7:e1002217. doi: 10.1371/journal.pgen.1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diner EJ, Beck CM, Webb JS, Low DA, Hayes CS. 2012. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev 26:515–525. doi: 10.1101/gad.182345.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruhe ZC, Wallace AB, Low DA, Hayes CS. 2013. Receptor polymorphism restricts contact-dependent growth inhibition to members of the same species. mBio 4:e00480-. doi: 10.1128/mBio.00480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webb JS, Nikolakakis KC, Willett JLE, Aoki SK, Hayes CS, Low DA. 2013. Delivery of CdiA nuclease toxins into target cells during contact-dependent growth inhibition. PLoS One 8:e57609. doi: 10.1371/journal.pone.0057609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beck CM, Willett JLE, Cunningham DA, Kim JJ, Low DA, Hayes CS. 2016. CdiA effectors from uropathogenic Escherichia coli use heterotrimeric osmoporins as receptors to recognize target bacteria. PLoS Pathog 12:e1005925. doi: 10.1371/journal.ppat.1005925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson PM, Beck CM, Morse RP, Garza-Sánchez F, Low DA, Hayes CS, Goulding CW. 2016. Unraveling the essential role of CysK in CDI toxin activation. Proc Natl Acad Sci U S A 113:9792–9797. doi: 10.1073/pnas.1607112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones AM, Garza-Sánchez F, So J, Hayes CS, Low DA. 2017. Activation of contact-dependent antibacterial tRNase toxins by translation elongation factors. Proc Natl Acad Sci U S A 114:E1951–E1957. doi: 10.1073/pnas.1619273114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruhe ZC, Nguyen JY, Xiong J, Koskiniemi S, Beck CM, Perkins BR, Low DA, Hayes CS. 2017. CdiA effectors use modular receptor-binding domains to recognize target bacteria. mBio 8:e00290-. doi: 10.1128/mBio.00290-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh A, Baltekin Ö, Wäneskog M, Elkhalifa D, Hammarlöf DL, Elf J, Koskiniemi S. 2018. Contact-dependent growth inhibition induces high levels of antibiotic-tolerant persister cells in clonal bacterial populations. EMBO J 37:e98026. doi: 10.15252/embj.201798026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morse RP, Willett JLE, Johnson PM, Zheng J, Credali A, Iniguez A, Nowick JS, Hayes CS, Goulding CW. 2015. Diversification of β-augmentation interactions between CDI toxin/immunity proteins. J Mol Biol 427:3766–3784. doi: 10.1016/j.jmb.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beck CM, Morse RP, Cunningham DA, Iniguez A, Low DA, Goulding CW, Hayes CS. 2014. CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure 22:707–718. doi: 10.1016/j.str.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melvin JA, Gaston JR, Phillips SN, Springer MJ, Marshall CW, Shanks RMQ, Bomberger JM. 2017. Pseudomonas aeruginosa contact-dependent growth inhibition plays dual role in host-pathogen interactions. mSphere 2:e00336-. doi: 10.1128/mSphere.00336-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arenas J, Schipper K, van Ulsen P, van der Ende A, Tommassen J. 2013. Domain exchange at the 3′ end of the gene encoding the fratricide meningococcal two-partner secretion protein A. BMC Genomics 14:622. doi: 10.1186/1471-2164-14-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griffin AS, West SA, Buckling A. 2004. Cooperation and competition in pathogenic bacteria. Nature 430:1024–1027. doi: 10.1038/nature02744. [DOI] [PubMed] [Google Scholar]
- 45.Rumbaugh KP, Diggle SP, Watters CM, Ross-Gillespie A, Griffin AS, West SA. 2009. Quorum sensing and the social evolution of bacterial virulence. Curr Biol 19:341–345. doi: 10.1016/j.cub.2009.01.050. [DOI] [PubMed] [Google Scholar]
- 46.Raymond B, West SA, Griffin AS, Bonsall MB. 2012. The dynamics of cooperative bacterial virulence in the field. Science 337:85–88. doi: 10.1126/science.1218196. [DOI] [PubMed] [Google Scholar]
- 47.Stacy A, McNally L, Darch SE, Brown SP, Whiteley M. 2016. The biogeography of polymicrobial infection. Nat Rev Microbiol 14:93–105. doi: 10.1038/nrmicro.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nadell CD, Drescher K, Foster KR. 2016. Spatial structure, cooperation and competition in biofilms. Nat Rev Microbiol 14:589–600. doi: 10.1038/nrmicro.2016.84. [DOI] [PubMed] [Google Scholar]
- 49.Holden MTG, Titball RW, Peacock SJ, Cerdeño-Tárraga AM, Atkins T, Crossman LC, Pitt T, Churcher C, Mungall K, Bentley SD, Sebaihia M, Thomson NR, Bason N, Beacham IR, Brooks K, Brown KA, Brown NF, Challis GL, Cherevach I, Chillingworth T, Cronin A, Crossett B, Davis P, DeShazer D, Feltwell T, Fraser A, Hance Z, Hauser H, Holroyd S, Jagels K, Keith KE, Maddison M, Moule S, Price C, Quail MA, Rabbinowitsch E, Rutherford K, Sanders M, Simmonds M, Songsivilai S, Stevens K, Tumapa S, Vesaratchavest M, Whitehead S, Yeats C, Barrell BG, Oyston PCF, Parkhill J. 2004. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci U S A 101:14240–14245. doi: 10.1073/pnas.0403302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu Y, Kim HS, Chua HH, Lin CH, Sim SH, Lin D, Derr A, Engels R, DeShazer D, Birren B, Nierman WC, Tan P. 2006. Genomic patterns of pathogen evolution revealed by comparison of Burkholderia pseudomallei, the causative agent of melioidosis, to avirulent Burkholderia thailandensis. BMC Microbiol 6:46. doi: 10.1186/1471-2180-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuanyok A, Auerbach RK, Brettin TS, Bruce DC, Munk AC, Detter JC, Pearson T, Hornstra H, Sermswan RW, Wuthiekanun V, Peacock SJ, Currie BJ, Keim P, Wagner DM. 2007. A horizontal gene transfer event defines two distinct groups within Burkholderia pseudomallei that have dissimilar geographic distributions. J Bacteriol 189:9044–9049. doi: 10.1128/JB.01264-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tuanyok A, Leadem BR, Auerbach RK, Beckstrom-Sternberg SM, Beckstrom-Sternberg JS, Mayo M, Wuthiekanun V, Brettin TS, Nierman WC, Peacock SJ, Currie BJ, Wagner DM, Keim P. 2008. Genomic islands from five strains of Burkholderia pseudomallei. BMC Genomics 9:566. doi: 10.1186/1471-2164-9-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruhe ZC, Nguyen JY, Chen AJ, Leung NY, Hayes CS, Low DA. 2016. CDI systems are stably maintained by a cell-contact mediated surveillance mechanism. PLoS Genet 12:e1006145. doi: 10.1371/journal.pgen.1006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darling ACE, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahenthiralingam E, Urban TA, Goldberg JB. 2005. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol 3:144–156. doi: 10.1038/nrmicro1085. [DOI] [PubMed] [Google Scholar]
- 56.López CM, Rholl DA, Trunck LA, Schweizer HP. 2009. Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl Environ Microbiol 75:6496–6503. doi: 10.1128/AEM.01669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi K-H, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 58.Winsor GL, Khaira B, Van Rossum T, Lo R, Whiteside MD, Brinkman FSL. 2008. The Burkholderia Genome Database: facilitating flexible queries and comparative analyses. Bioinformatics 24:2803–2804. doi: 10.1093/bioinformatics/btn524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539–539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nielsen H. 2017. Predicting secretory proteins with SignalP. Methods Mol Biol 1611:59–73. doi: 10.1007/978-1-4939-7015-5_6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.