

Abstract
We investigated whether pheophorbide A (PhA) could serve as a selective breast cancer resistance protein (BCRP) substrate (victim) to screen in vivo using fluorescent live animal imaging for transporter-mediated interactions with orally administered inhibitors (perpetrators), and whether that could be coupled with serum sampling to measure the systemic concentration of PhA with a fast-throughput in vitro fluorescent assay. PhA is a breakdown product of chlorophyll and is highly fluorescent in the near-infrared (NIR) spectrum. Whole-body NIR fluorescence was greater in the Bcrp KO compared with wild-type (WT) mice fed a regular diet containing chlorophyll and PhA, with fluorescence in WT mice confined to the intestine. PhA intestinal enterocyte fluorescence, after removing lumen contents, was greater in Bcrp knockout (KO) mice versus WT mice due to PhA enterocyte absorption and lack of PhA efflux by Bcrp. This difference was eliminated by maintaining the mice on an alfalfa (chlorophyll/PhA)-free diet. The area under the fluorescence ratio-time curve up to 6 hours (AUCFL 0–6 h) of orally administrated PhA was 3.5 times greater in the Bcrp KO mice compared with WT mice, and the PhA serum concentration was 50-fold higher in KO mice. Pretreatment with known BCRP inhibitors lapatinib, curcumin, elacridar, pantoprazole, and sorafenib, at clinically relevant doses, significantly increased PhA AUCFL 0–6 h by 2.4-, 2.3-, 2.2-, 1.5-, and 1.4-fold, respectively, whereas the area under PhA serum concentration-time curve calculated up to 6 hours (AUCSerum 0-6 h) increased by 13.8-, 7.8-, 5.2-, 2.02-, and 1.45-fold, respectively, and corresponded to their hierarchy as in vitro BCRP inhibitors. Our results demonstrate that live animal imaging using PhA can be used to identify BCRP inhibitors and to assess the potential for BCRP-mediated clinical drug-drug interactions.
Introduction
The xenobiotic efflux transporter breast cancer resistance protein (BCRP), encoded by ABCG2, is well characterized for its effect on the pharmacokinetics and pharmacodynamics of multiple drugs (Schwabedissen and Kroemer, 2011). Intestinal BCRP/ABCG2 has a significant effect limiting the oral bioavailability of its substrates (Roberts et al., 2016), which include drugs such as the nucleoside reverse-transcriptase inhibitor antiretroviral drugs, calcium channel blockers, 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, and topoisomerase I inhibitors, but also includes endogenous dietary substances such as the chlorophyll metabolite pheophorbide A (PhA) (Krishnamurthy and Schuetz, 2006; Robey et al., 2009; Schwabedissen and Kroemer, 2011). Moreover, multiple clinically used drugs, such as the tyrosine kinase inhibitor lapatinib (LPB), HIV protease inhibitor ritonavir, proton pump inhibitor pantoprazole (PPZ), as well as the dietary constituent curcumin (CCM), are known inhibitors of this transporter (Mao and Unadkat, 2015).
BCRP is recognized by the Food and Drug Administration (FDA) and International Transporter Consortium (ITC) as an important transporter that is prone to potential clinical drug-drug interactions (DDIs) (Giacomini et al., 2010; Tweedie et al., 2013; https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf; https://www.fda.gov/downloads/Drugs/Guidances/UCM581965.pdf) because of its recognition of a wide range of substrates and inhibitors. Except for biopharmaceutical classification system class I (high solubility and high permeability) compounds, BCRP substrates (victims) are prone to BCRP inhibitor (perpetrator)–mediated DDIs when they are dosed concomitantly through the oral route (Lee et al., 2015; https://www.fda.gov/downloads/Drugs/Guidances/UCM581965.pdf).
The ITC and FDA have detailed the decision tree and guidelines for determining BCRP substrates, inhibitors, and the possibility of clinical DDIs (Giacomini et al., 2010; https://www.fda.gov/downloads/Drugs/Guidances/UCM581965.pdf). In vivo assessments of BCRP substrates have used transporter knockout (KO) models or imaging techniques such as positron emission tomography and gamma-scintigraphy, which are known to have their own limitations (Giacomini et al., 2010). Although rosuvastatin and sulfasalazine have been proposed as BCRP preclinical and clinical in vivo probe substrates, they are not specific BCRP substrates (Dahan and Amidon, 2010; Ellis et al., 2013). Hence, multiple factors can complicate the interpretation of results with these probe substrates. For sulfasalazine, differences in its metabolism by colonic bacteria to sulfapyridine or genetic variation in or inhibition of N-acetyltransferase 2, which metabolizes sulfapyridine, or its transport by OATP2B1; or for rosuvastatin, the inhibition of transporters such as OATP1B1 can lead to variability in probe disposition independent of Bcrp inhibition (Lee et al., 2015). Moreover, quantitative analysis of sulfasalazine and rosuvastatin in human tissues requires expensive and sophisticated analytical instrumentation.
An ideal assay for Bcrp activity would use a specific, inexpensive, fluorescent near-infrared (NIR) substrate probe that could be monitored in real time both in vitro (high-throughput) and in vivo. PhA is a breakdown product of chlorophyll and is a specific dietary substrate of Bcrp (Jonker et al., 2002; Robey et al., 2004; Kraatz et al., 2014), with an efflux ratio of 3.15 in cell lines overexpressing Bcrp (Zhang et al., 2009). Two laboratories have developed in vitro assays for BCRP function/inhibition using PhA, also showing that PhA is not a substrate for P-glycoprotein or MRP1 in vitro (Robey et al., 2004, 2005). PhA is a low-solubility, low-permeability compound with no reported metabolic instabilities. We examined whether PhA is a BCRP probe substrate that can be used both in vitro and in vivo to investigate BCRP function according to the FDA and ITC guidelines (Giacomini et al., 2010) (https://www.fda.gov/downloads/Drugs/Guidances/UCM581965.pdf). We hypothesized that PhA can be used as an in vivo probe to assess DDIs associated with the inhibition of Bcrp at the gastrointestinal tract in mice. We used a live animal fluorescence imaging technique, using PhA, to screen in real time for orally administrated BCRP inhibitors and predict intestinal BCRP-mediated DDIs. Our results demonstrate that in vivo fluorescence imaging of PhA can be used to identify BCRP inhibitors to assess preclinically the potential for BCRP-mediated clinical DDIs.
Materials and Methods
Chemicals and Reagents.
The following chemicals and reagents were purchased: PhA (Frontier Scientific, Newark, DE); LPB and CCM (Cayman Chemicals, Ann Arbor, MI); elacridar (ECD) (Astatech Inc., Bristol, PA); PPZ (Santa Cruz Biotechnology, Dallas, TX); sorafenib (SFB) and dasatinib (DSB) (ChemieTek, Indianapolis, IN); and chlorophyll a (CLPa) and protoporphyrin IX (PPIX) (Sigma-Aldrich, St. Louis, MO).
Animals.
Wild-type (WT) and Bcrp KO Friend Virus B (FVB) male mice (n = 4 each group, 10 weeks old) were purchased from Taconic Biosciences, Inc. (Hudson, NY) for the KO mice study. For all other Bcrp inhibitor studies, control mice of FVB background (n = 4 for control and each inhibitor treatment group) were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were fed either a regular alfalfa-containing diet (cat. no.5013; Purina, Largo, FL) or an alfalfa-free (AFF) diet (cat. no.AIN-93G; Purina) for at least 3 days, the time necessary to reduce background PhA autofluorescence (Inoue et al., 2008), and they were provided water ad libitum. The Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital, in accordance with the US National Institutes of Health guidelines, approved all experimental procedures.
Cell Lines.
Madine-Darby Canine Kidney II (MDCKII) cells overexpressing human full-length WT BCRP and the parenteral cell lines were provided by Dr. Alfred Schinkel (The Netherlands Cancer Institute, Amsterdam, Netherlands) and were cultured as described previously (Pavek et al., 2005).
Determination of PhA Concentration in Rodent Food Pellets.
Six food pellets each from regular diet and AFF diet were weighed and soaked with half parts of water. After 30 minutes, 1 g of food pellet was mixed with 5 ml of methanol and stirred overnight for PhA extraction. The following day, samples were centrifuged at 15,000g for 15 minutes and the supernatant was collected. The supernatant was further centrifuged, filtered, and used for the measurement of PhA using a modified high-performance liquid chromatography (HPLC)-fluorescence detector–based method published previously (Almela et al., 2000). Forty microliters of the final supernatant was injected into the HPLC system (Prominence; Shimadzu, Kyoto, Japan) consisting of an LC-20AB quaternary high-pressure gradient pump, SIL-20ACHT Autosampler, and RF-10 AXL Fluorescence Detector. Chromatographic separation of PhA was achieved using a Hypersil ODS C18 Column (150 × 4 mm; 5 µm particle size) fitted with a Hypersil ODS (C18) Javelin Guard Column (10 × 4 mm; 5 µm particle size) (Thermo Fisher Scientific, Waltham, MA) and a gradient elution method with a run time of 10 minutes. The mobile phase for elution was 80% methanol in 0.5 M ammonium acetate (phase A) and 100% methanol (phase B) with the following gradient: 0–2 minutes 60%–95% methanol, 2–8 minutes 95% methanol, 8–9 minutes 95%–60% methanol, and the run stopped at 10 minutes. The retention time for PhA was 3.5 minutes. The PhA peak was identified, and fluorescence was measured at 400 nm excitation (Ex) and 670 nm emission (Em) (Supplemental Fig. 1). The analyte area was used for the calibration and measurement of unknown concentrations against a known PhA concentration prepared in mobile phase A. The calibration range was 0.02–10 µM.
Treatments.
Mice were kept on an AFF diet for at least 3 days to eliminate PhA background autofluorescence. WT and Bcrp KO mice (10 weeks, n = 4/group) were administrated PhA (10 mg/kg) by oral gavage at a dose volume of 10 ml/kg b.wt., and imaging or blood collection was performed at appropriate time points up to 6 hours after dosing. For the inhibitor study, oral doses of the inhibitors were selected based on clinical relevance or previously demonstrated effects (Table 1) and were formulated for oral dosing by suspending an appropriate amount of the compound in 0.5% hydroxypropyl-methyl-cellulose and 1% Tween 80 (Sigma-Aldrich) in water by the trituration method. Bcrp inhibitors ECD (100 mg/kg), LPB (90 mg/kg), DSB (10 mg/kg), SFB (40 mg/kg), CCM (300 mg/kg), and PPZ (40 mg/kg) were administrated orally, 1 hour prior to oral dosing of PhA. Imaging and blood collection were performed at appropriate times up to 6 hours after PhA dosing. All animals were fasted up to 6 hours after PhA dosing.
TABLE 1.
Literature reported properties of selected inhibitors and final outcome of the experiment
| DSB | SFB | PPZ | ECD | CCM | LPB | ||
|---|---|---|---|---|---|---|---|
| Known parameters from literature and database | Molecular weight (g/mol) | 488.01 | 464.83 | 383.37 | 563.65 | 368.38 | 581.06 |
| Predicted solubility (µM) (DrugBank database) | 26.23 | 3.68 | 1291.18 | 4.99 | 15.61 | 38.38 | |
| Predicted logP (DrugBank database) | 2.77 | 4.12 | 2.11 | 6.81 | 3.62 | 5.18 | |
| Reported IC50 (µM) | 3.1a | 5.5b | 0.31c | 1.6d | 0.025e | ||
| Clinical information | Dose | 100 mg once dailyf | 400 mg twice a day | 40 mg Once dailyg | 400 mg twice a dayg | 2 g Once dailyg | 1250 mg dailyg |
| Cmax (µM) | 0.46f | 6.45h | 6.5g | 0.327g | 4.2g | ||
| Igut/IC50 | 1110 | 67g | 9200g | 14,000g | 21,0000g | ||
| Cmax/IC50 | 2.08 | 1.2g | 1.1g | 170g | |||
| Possible DDI | No | Yes | Yes | Yes | Yes | Yes | |
| PK information from mice | Dose (mg/kg) | 10 | 40 | 40 | 100 | 300 | 90 |
| Cmax (µM) | ∼0.7i | 21a | 0.78j | ∼0.06k | ∼10l | ||
| Tmax (h) | 0.25–2 | 1m | 8 | 1.5 | 0.5–1 | ||
| t1/2 (h) | 20 | 12 | 3 | ||||
| Prediction of possible outcome in mouse based on selected dose and observed IC50 | Observed IC50 (µM) | 22.76 | 2.18 | 11.19 | 0.21 | 2.52 | 0.04 |
| Cmax/IC50 | 0.03 | 9.63 | 3.71 | 0.02 | 250 | ||
| Oral dose for mice study (mg/kg) | 10 | 40 | 40 | 100 | 300 | 90 | |
| Cgut (µM) | 639 | 2689 | 3260 | 5544 | 25,449 | 4840 | |
| Cgut/IC50 | 28 | 1233 | 291 | 26,400 | 10,098 | 121,000 | |
| Cgut_soluble/IC50 | 1.2 | 1.7 | 115.4 | 23.7 | 6.2 | 959.5 | |
| Expected oral DDI | Yes | Yes | Yes | Yes | Yes | Yes | |
| In vivo serum concentration result | AUCSerum 0–6 h (µM/h) | 0.11 ± 0.002 | 0.13 ± 0.02 | 0.18 ± 0.01 | 0.46 ± 0.08 | 0.70 ± 0.1 | 1.24 ± 0.09 |
| Fold change over control | 1.24 | 1.45 | 2.02 | 5.17 | 7.79 | 13.78 | |
| Observed DDI? | No | No | Yes | Yes | Yes | Yes | |
| Live animal fluorescence result | AUCFL 0–6 h | 5.82 ± 1.27 | 6.98 ± 1.15 | 7.30 ± 0.74 | 10.55 ± 2.12 | 11.2 ± 1.75 | 11.66 ± 1.54 |
| Fold change over control | 1.2 | 1.44 | 1.50 | 2.17 | 2.31 | 2.4 | |
| Observed DDI? | No | No | Yes | Yes | Yes | Yes |
t1/2, half-life; Tmax, time for maximal plasma concentration; Cgut_soluble is soluble fraction of the drug in the gut, assumed to be equal to predicted solubility.
In Vivo Fluorescence Imaging.
Mice were anesthetized by isoflurane anesthesia (2%, inhalation) and placed ventrally in the chamber of a Xenogen IVIS-200 (PerkinElmer, Waltham, MA) in vitro imaging system (IVIS). Fluorescence images were obtained under anesthesia with a fixed exposure time of 1 second and a NIR filter setting of 675 nm Ex and 840 nm Em. Mice were repetitively imaged at 0 hour (baseline), and 1, 2, 3, 4, and 6 hours after PhA administration. Images were scaled to a maximum intensity of 1 × 108 photons/s per cm2 per steradian for visualization. All images were analyzed with Living Image version 4.5 (PerkinElmer) to obtain the fluorescence intensity in the selected region of interest (ROI) of fluorescence (flux/s), which was selected as a fixed rectangular area around each mouse. For the control and inhibitor treatment groups, ROI (in flux per second) for each mouse was normalized by its flux measured at 0 hour. For the WT and Bcrp KO mice, the ROI of the WT mice at 0 hour was used for normalizing all fluorescence (ROI) data. Normalized ROI data were used for the calculation of area under the fluorescence ratio time curve up to 6 hours (AUCFL 0–6 h) in GraphPad Prism version 5.02 (GraphPad Software, San Diego, CA).
Pharmacokinetic Study of PhA.
Immediately after the imaging, 50 µl of blood was collected by saphenous vein into a microvette capillary blood collection tube [Sarstedt, Nümbrecht, Germany (obtained from Thermo Fisher Scientific)], and serum was separated by centrifuging the sample at 5000 rpm for 10 minutes. Blood samples were collected at 1, 2, 4, and 6 hours for measurement of PhA concentration in the serum.
Measurement of PhA Concentration in Whole Serum.
A fluorescence-based method was used to measure PhA concentration in whole serum. A calibration of known PhA concentrations was prepared by spiking known PhA concentrations prepared in dimethylsulfoxide into 24 µl of blank serum, collected from mice on the AFF diet. Twenty-five microliters of standard serum concentrations and serum samples collected for the study were diluted to 100 µl with distilled water in a 96-well clear bottom black polystyrene plate (cat. no. 3603; Corning, Corning, NY). PhA fluorescence was measured at 400 nm (Ex) and 670 nm (Em) on a Synergy H4 Hybrid Reader (BioTek, Winooski, VT). All fluorescence values were blank corrected, and unknown sample PhA concentrations were interpolated from a freshly prepared PhA standard curve (0.0210 µM), and the PhA concentration for each mouse was used to measure area under serum concentration-time curve calculated up to 6 hours (AUCSerum 0–6 h) in GraphPad Prism version 5.02. A fluorescence emission spectral scan from 500 to 700 nm was performed for all samples with fixed Ex at 400 nm and was compared with the corresponding standard PhA concentration prepared in blank serum, with and without PhA, to confirm the identity of PhA in study samples at the emission wavelength of 670 nm (Supplemental Fig. 2).
Ex Vivo Fluorescence Imaging of Mouse Intestine.
After in vivo imaging, mice were immediately euthanized and blood was collected by cardiac puncture. The small intestine was dissected out. and fluorescence (675 nm Ex and 840 nm Em) images were obtained before and after flushing out the intestinal contents with phosphate-buffered saline (PBS) (5 ml, three times). Fluorescence images were obtained using a 1-second exposure with a filter setting for NIR. Images were scaled to a maximum intensity of 1 × 108 photons/s per cm2 per steradian.
Fluorescence Microscopy of Intestinal Sections.
Intestinal segments of WT and Bcrp KO mice on a regular diet were washed with cold PBS, filled with warm 3% low melting agarose in 0.9% (w/v) NaCl, immediately soaked in ice-cold Dulbecco’s modified Eagle’s medium to solidify the agarose, paraffin-embedded, and precision transverse slices cut onto slides. Fluoroshield mounting medium with 4′,6-diamidino-2-phenylindole dihydrochloride was applied to the tissue and spread evenly, and coverslips applied for fluorescence microscope imaging (ECLIPSE Ti; Nikon, Tokyo, Japan).
In Vitro Assay for Measuring IC50 of Bcrp Inhibitors.
A 96-well plate–based assay using control and BCRP overexpressing MDCKII cells (Weiss et al., 2007) was used for measuring the IC50 of selected BCRP inhibitor drugs using PhA as a probe BCRP substrate (Robey et al., 2004). Briefly, 1 × 104 cells/well were plated in a 96-well plate and incubated at 37°C until the cells were at least 80%–90% confluent. On the day of the experiment, media were removed, cells were washed with PBS and incubated for 30 minutes with or without BCRP inhibitors prepared in 100 µl of modified Krebs-Ringer buffer (115 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 2.5 mM CaCl2, 2.5 mM NaHCO3, and 10 mM glucose) (Mahringer et al., 2009). After preincubation with inhibitors, media were removed and cells were incubated with 1 µM PhA with or without the inhibitor prepared in 100 µl Krebs-Ringer buffer for 2 hours. Fumitremorgin C at a concentration of 5 µM was used as a standard BCRP inhibitor. After 2 hours, cells were washed with chilled Dulbecco’s PBS (DPBS) twice, 100 µl of DPBS was added, and fluorescence was measured on a Synergy H4 Hybrid Reader at 400 nm (Ex) and 670 nm (Em). Immediately after fluorescence measurement, 100 µl of Promega CellTiter-Glo Reagent (Thermo Fisher Scientific) was added to each well and incubated at 37°C for 5 minutes, and luminescence was measured. To calculate the number of cells per well, 120,000 MDCKII control and BCRP-overexpressing cells were plated per well, diluted with cell culture media 1:1 up to 937 cells/well (n = 6 for each dilution of each cell type), and incubated at 37°C for 6 hours for the cells to attach. After 6 hours, cells are washed with chilled DPBS, after which 100 µl of DPBS and CellTiter-Glo Reagent was added to the cells at room temperature and incubated for 5 minutes at 37°C, and luminescence was measured. First, the results were background corrected and normalized with cell numbers in each well to calculate PhA fluorescence/103 cells/well. The fold change of PhA accumulation in MDCKII cells in the presence of BCRP was calculated by dividing MDCKII-BCRP fluorescence/cell with that of MDCKII cells. The percentage of inhibition by BCRP inhibitors was calculated by the following equation:
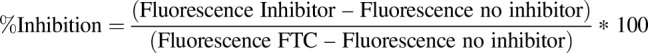 |
IC50 values of the inhibitors were calculated using GraphPad prism version 5.02. All assays were run at least three times with n = 6 wells for each concentration of inhibitor, or standard inhibitor, in each assay. The results of all experiments were combined (n = 18) to calculate final parameters.
Statistical Analysis.
All serum PhA concentration-time data were plotted as the mean PhA concentration ± S.E.M., and, similarly, the whole-body fluorescence data over 0 hour of each individual mouse (for the inhibitor study), or over WT mice 0-hour fluorescence (for Bcrp KO study), were plotted as the mean ratio ± S.E.M. GraphPad Prism version 5.02 was used to calculate the area under the concentration-time curve [time or fluorescence (in flux per second) ratio] for individual mice up to 6 hours (AUCSerum 0–6 h or AUCFL 0–6 h). The mean AUC ± S.E.M. values are plotted and compared with the control or WT mice using the unpaired t test; significance is calculated at P < 0.05. All calculations were performed with GraphPad Prism version 5.02.
Results
Bcrp KO Mice on a Regular Alfalfa-Containing Diet Have Higher Whole-Body Fluorescence, Arising from PhA and Chlorophyll, than WT Mice.
WT mice, fed a regular alfalfa-containing diet and imaged with an IVIS across the NIR fluorescence spectrum, had detectable autofluorescence emission signals with a 675 nm Ex and 840 nm Em setting, that was localized to the intestinal region (Fig. 1A). Bcrp KO mice fed the same alfalfa-containing diet had a higher whole-body autofluorescence (Fig. 1A). Alfalfa is known to be a source of chlorophyll, and its catabolite PhA, a known specific BCRP substrate, both of which emit in the NIR. Feeding both WT and Bcrp-KO mice an AFF diet for 3 days eliminated the autofluorescence (Fig. 1A), as reported by others (Leblond et al., 2010). To further confirm that the diet was the source of the observed fluorescence, we IVIS imaged the alfalfa with or without food pellets, using the same imaging parameters, and observed fluorescence only from the regular alfalfa food pellets (Fig. 1B). We compared the fluorescence signal intensity of CLPa, PhA, and PPIX, a fluorescent Bcrp endogenous substrate, over a concentration range of 0.31–10 μM, using the same imaging parameters (Fig. 1C). Only PhA was detectable at the lowest concentration tested (0.31 µM), almost 10-fold lower than the lowest detectable concentration of CLPa (2.5 μM), and PhA had a higher fluorescent intensity compared with chlorophyll at each concentration (Fig. 1C). PPIX fluorescence was undetectable under these imaging parameters. Hence, both chlorophyll and PhA could be the source of autofluorescence in the alfalfa-containing diet.
Fig. 1.

Autofluorescence derived from dietary PhA is higher in the Bcrp KO compared with WT mice. (A) Comparison of fluorescence (Ex, 675 nm; Em, 840 nm) in WT and Bcrp KO mice fed a regular diet or an AFF diet at least for 3 days. (B) Comparison of fluorescence (Ex, 675 nm; Em, 840 nm) in a regular and AFF diet. (C) Fluorescence comparison of the known concentrations of CLPa, PhA, and a prototypical Bcrp substrate PPIX fluorescence with filter setting (Ex, 675 nm; Em, 840 nm).
PhA is Present in Regular Mouse Diet, But Not in AFF Diet.
To further confirm the presence of PhA in the alfalfa-containing regular diet, we analyzed the amount of PhA in mouse food pellets, after methanol extraction, using an HPLC-fluorescence–based method. PhA, and another abundant fluorescent compound, was present in the regular alfalfa-containing food pellet (Supplemental Fig. 1B), but not in the AFF food pellet extract (Supplemental Fig. 1C). We determined the PhA concentration in the alfalfa-containing regular diet to be 1.71 µg/g food pellet. Assuming that the daily food intake for a 30-g mouse is 6 g, it can be estimated that a mouse eating a regular alfalfa-containing diet consumes ∼343 µg PhA/kg b.wt. daily.
Bcrp Gene Deletion Leads to Higher Absorption/Retention of PhA in Mouse Enterocytes.
During in vivo live animal imaging, we observed the maximum fluorescence in the intestinal region of Bcrp WT and KO mice (Fig. 1A), which was expected since PhA is a dietary constituent. However, unexpectedly, the fluorescence intensity was greater in the Bcrp KO mouse intestine. This result was unanticipated since we would have assumed greater absorbance of PhA from the food in the KO mouse, and hence, a lower PhA intestinal signal intensity compared with WT mice. To determine whether the higher PhA intestinal fluorescence was due to PhA trapping in the intestinal enterocytes of KO mice, we compared the localization and intensity of fluorescence in WT and Bcrp KO mice fed a regular alfalfa-containing diet, by ex vivo imaging of isolated intestines. High fluorescence intensity was observed in the small intestines of both WT and Bcrp KO mice before flushing the intestinal contents (data not shown), but after flushing, the small intestine of Bcrp KO mice, but not WT mice, retained a high level of fluorescence (Fig. 2). The intestinal segments were then filled with warm agarose and paraffin embedded, and transverse sections were cut and imaged by fluorescence microscopy. The enterocytes of BCRP KO mice showed a much higher level of fluorescence compared with those of the WT mice (Fig. 2) suggesting that the absence of Bcrp at the enterocytes was leading to higher enterocyte PhA absorption and trapping in the Bcrp KO mice that were fed a regular alfalfa-containing diet.
Fig. 2.

Absence of Bcrp leads to higher absorption of PhA in enterocytes. Ex vivo image comparison of intestinal segments of WT and Bcrp KO mice on a regular diet, after flushing out the gut contents with saline. Microscopic examination of cross-sectional images of upper intestine from WT and Bcrp KO mice on regular diet: blue is 4′,6-diamidino-2-phenylindole dihydrochloride stain for nuclear localization); red is PhA using the Cyanine5 Ex/Em wavelengths; A signifies apical and B signifies basolateral side of the intestinal section.
Oral Absorption of PhA is Greater in Bcrp KO versus WT Mice.
Since both dietary chlorophyll and PhA from alfalfa-containing foods would fluoresce in the NIR, and because we could not control the amount of these constituents in the diet, the amount of chlorophyll that might be converted to PhA, or the amount that each mouse consumed, we maintained the mice on an AFF diet for at least 3 days and administered PhA by oral gavage to determine whether PhA could be a probe substrate for monitoring Bcrp function in vivo. After PhA (10 mg/kg) oral gavage, and then fasting, PhA disposition was IVIS monitored longitudinally over the next 6 hours. Representative images at 2 hours post-PhA dosing displayed significantly greater whole-body fluorescence in the Bcrp KO mice compared with WT mice (Fig. 3A). The whole-body fluorescence at different time-points, normalized to the fluorescence in WT mice at baseline, when plotted against time after dosing, exhibited significantly higher fluorescence in Bcrp KO mice across all observed time points (1, 2, 4, and 6 hours) (Fig. 3B). Quantification of the AUCFL 0–6 h showed a significant increase (3.5-fold) of fluorescence in the Bcrp KO compared with WT mice (Fig. 3C). We also measured the serum concentration of PhA in the same experiment at 1, 2, 4, and 6 hours. The serum concentration of PhA in the Bcrp KO mice was also found to be significantly higher across all observed time points compared with WT mice (Fig. 3D) with a 50-fold increase in AUCSerum 0–6 h (Fig. 3E). These results, showing a significantly higher fold increase of both whole-body and serum PhA fluorescence over baseline, in Bcrp KO mice compared with WT mice, suggest that orally administered PhA can be used to capture a range of inhibition potency by Bcrp inhibitors and thereby identify Bcrp-mediated DDIs in mice.
Fig. 3.

PhA oral absorption is significantly higher in the Bcrp KO mice. (A) Representative image comparing PhA fluorescence (Ex 675 nm; Em, 840 nm) in WT and Bcrp KO mice, 2 hours after oral administration of 10 mg/kg PhA. (B) Fluorescence intensity over 0-hour WT fluorescence-time profile up to 6 hours after 10 mg/kg PhA oral administration in Bcrp WT and KO mice (n = 3). Comparison of the AUCFL 0–6 h in (B) calculated using GraphPad Prism version 5.02 (C). Comparison of PhA serum concentration at 1, 2, 4, and 6 hours from the WT and Bcrp KO mice dosed with 10 mg/kg PhA (D) and the associated AUCSerum 0–6 h value (E). Results are presented as the mean ± S.E.M.; and significance is calculated at P < 0.05 by comparing the means with an unpaired t test. ***P < 0.001.
Selected Bcrp Inhibitors Exhibited Comparable In Vitro IC50 Values with PhA as Reported Previously.
To further test our hypothesis that PhA can be used as an in vivo probe (victim) for the identification of intestinal Bcrp-mediated DDIs, we selected known Bcrp inhibitors (perpetrators) as per FDA guidance based on their [I2]/IC50 = Igut/IC50 (clinical dose/250 ml)/IC50), where Igut is the maximum human dose in the apparent human gut fluid volume of 250 ml (Table1). Although IC50 values of the selected inhibitors (except DSB) were reported in the literature, a wide variety of Bcrp probe substrates as well as cell lines were used for these IC50 measurements. Using PhA as a BCRP probe substrate, we used MDCKII-BCRP cell–based assays to determine the IC50 values of the selected BCRP inhibitors (Fig. 4). LPB was found to be the strongest BCRP inhibitor with a measured IC50 value of 40 nM, followed by ECD (0.21 µM), SFB (2.18 µM), CCM (2.58 µM), PPZ (11.19 µM), and DSB (22.76 µM). The IC50 values of these inhibitors for Bcrp-mediated PhA transport were found to be similar to those reported in the literature using different BCRP probe substrates (Table 1), except for PPZ, for which the observed IC50 value for PhA was 2-fold higher than the reported IC50 value (5.5 µM). Assuming a 0.8 ml mouse stomach volume, the Cgut/IC50 ratio was ≥10 for all of the inhibitors (Table 1), indicating a possible DDI potential with orally dosed BCRP substrates in mice, where, Cgut is the concentration in mouse gut fluid volume (selected inhibitor dose/0.8 ml).
Fig. 4.

Determination of the IC50 for selected Bcrp inhibitors using PhA as a probe substrate. The mean percentage increase in PhA accumulation ± S.E.M. in the MDCKII cells overexpressing BCRP in the presence of selected Bcrp inhibitors was plotted against the log transformed inhibitor concentration (Conc). IC50 was determined using GraphPad Prism version 5.02.
Bcrp Inhibitors Increase Whole-Body and Serum Fluorescence of PhA.
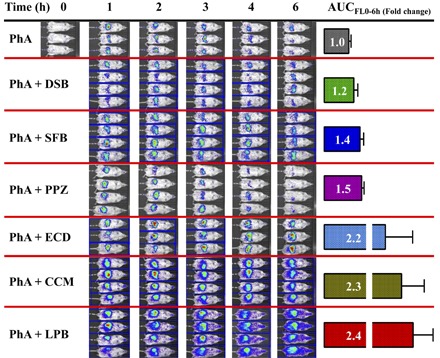
Based on the reported literature, the selected drugs were expected to cause DDIs with Bcrp substrates when administered concomitantly, via an oral route, and to increase systemic exposure (blood/plasma/serum AUC) of the substrate. Therefore, we dosed the inhibitors orally, using concentrations in mice equivalent to those recommended to screen for human BCRP DDIs in vivo (Lee et al., 2015) along with the Bcrp probe substrate PhA, and measured whole-body fluorescence and serum PhA concentrations (Fig. 5A). Whole-body fluorescence imaging of mice treated with the Bcrp inhibitors LPB, ECD, and CCM displayed higher fluorescence compared with the control-treated mice. After quantification of fluorescence flux per second and normalization with baseline fluorescence from the same mice, these values were plotted against time after dosing (Fig. 5, B and C), and AUCFL 0–6 h was calculated for all the inhibitor-treated and control mice. LPB exhibited the maximum fold increase (2.4-fold) of AUCFL 0–6 h over control mice, followed by CCM (2.3-fold), ECD (2.2-fold), PPZ (1.5-fold), and SFB (1.44-fold) (Fig. 5D). DSB did not significantly increase the AUCFL 0–6 h over control mice.
Fig. 5.

Inhibition of BCRP increased whole-body fluorescence as well as the serum concentration of PhA in control FVB mice. Six-hour representative image comparing fluorescence (Ex, 675 nm; Em, 840 nm) in control mice orally administered 10 mg/kg PhA with or without Bcrp inhibitors 1 hour prior to PhA administration, as indicated in the Materials and Methods (A). Whole-body fluorescence (flux/s) was normalized to the 0-hour fluorescence of the same mice, and the data were plotted as the mean fluorescence ratio ± S.E.M. vs. time (hours) for the tyrosine kinase inhibitors (B) and other inhibitors (C). (D) AUCFL 0–6 h calculated for each mouse and plotted as the mean ± S.E.M. PhA serum concentration-time plot for the control and tyrosine kinase inhibitors (E) and other inhibitors (F) plotted as the mean ± S.E.M. and the associated AUCSerum 0–6 h calculated using GraphPad Prism version 5.02 (G). Significance is calculated by comparing the mean AUC after inhibitor treatment with that of control AUC by unpaired t test. Significance is calculated at P < 0.05. *P < 0.05; **P < 0.01; ***P < 0.001.
The PhA serum concentration-time plots after treatment with Bcrp inhibitors displayed a trend similar to that observed with whole-body fluorescence (Fig. 5, E and F), with a similar rank order of inhibitor-mediated increase in AUCSerum 0–6 h observed as was seen for AUCFL 0–6 h (Fig. 5G). LPB treatment caused the largest increase in PhA serum fluorescence (13.8-fold), followed by treatment with CCM (7.79-fold), ECD (5.2-fold), PPZ (2.02-fold), and SFB (1.45-fold). No significant effect of DSB was found on serum PhA concentration consistent with the lack of effect on PhA whole-body fluorescence. Although the Cgut/IC50 ratio for all inhibitors was ≥10, Cgut_soluble/IC50 values (Table 1) displayed a better correlation with the observed fold change by Bcrp inhibitors, where Cgut_soluble is assumed to be equal to the predicted solubility of the inhibitors obtained from DrugBank database (https://www.drugbank.ca). Although PhA, and related porphyrins, are not substrates of P-glycoprotein or multidrug resistance protein 1 in vitro (Robey et al., 2004; Bakhsheshian et al., 2013), since many of the inhibitors tested can also affect Pgp-mediated transport, we compared the effect of ECD on PhA disposition in Bcrp WT and KO mice. A very small effect of ECD was seen on PhA serum fluorescence in the Bcrp KO mice, but there was no significant effect of ECD on PhA whole-body fluorescence in Bcrp KO mice (Supplemental Fig. 3).
Discussion
These Studies Demonstrate that the DDI Potential of Intestinal Bcrp Inhibitors Can Be Characterized Preclinically In Vivo Using the Bcrp Probe Substrate PhA.
PhA whole-body fluorescence imaging over time, coupled with serum sampling for PhA systemic quantification, allows for longitudinal, real-time kinetic analysis of the effect of Bcrp inhibitors directly on changes in tissue exposure. Murine Bcrp limited PhA intestinal oral absorption, as evidenced by the significant difference in PhA AUC between Bcrp KO and WT mice (50-fold higher PhA serum concentration in KO mice). Indeed, the enterocytes themselves, after washout of intestinal food contents, showed a significantly higher concentration of PhA that was retained in the absence of the Bcrp efflux transporter (Fig. 2). The dynamic range of PhA fluorescence, both whole body and in serum, between WT and Bcrp KO mice, allowed us to rapidly examine the effect of Bcrp inhibitors on the whole-body and serum PhA fluorescence. We specifically chose drugs that, when administered at clinically relevant doses to mice, would be predicted to cause a Bcrp-mediated DDI in vivo. For an orally administered Bcrp inhibitor drug, if the calculated dose (dose/250 ml)/IC50 for Bcrp is ≥10 (i.e., Igut/IC50 ≥10), the FDA recommends analysis for a possible oral Bcrp inhibition-mediated DDI (https://www.fda.gov/downloads/Drugs/Guidances/UCM581965.pdf). Indeed, for PPZ, ECD, CCM, and LPB the predicted clinical DDI liability, Igut/IC50, obtained from literature reports, are significantly higher than 10 (Lee et al., 2015).
Taken Together, the Serum Concentration and Imaging Data Provide Complimentary Evidence that PhA Can Be Used for Identification of Oral DDIs Associated with Bcrp.
Indeed, the rank order of efficacy for the series of Bcrp inhibitors increasing the PhA serum concentration over time [LPB (13.8-fold) > CCM (7.8-fold) > ECD (5.17-fold) > PPZ (2.02-fold) > SFB (1.45-fold)] precisely matched the rank order for the same BCRP inhibitors increasing the AUCFL 0–6 h whole-body fluorescence of PhA in the same mice. Indeed, for this series of perpetrators, the Bcrp inhibitor log transformed gut concentration/IC50 versus fold change in either the AUC of PhA body fluorescence or the AUC of PhA serum fluorescence gives a linear relationship (Supplemental Fig. 4), further validating that this assay is measuring the oral Bcrp DDI. Moreover, the rank order of intestinal perpetrator Bcrp inhibition potential (when victim drugs, such as sulfasalazine, have been tested) was similar to the experimentally determined rank order of PhA inhibitor efficacy. Hence, the hierarchy of intestinal Bcrp inhibition was similar when the perpetrators were the same, but the victim was either sulfasalazine or PhA. For example, when mice were orally administered CCM (300 mg/kg) with PhA, we observed a 7.79-fold increase in the serum fluorescence of PhA, whereas others observed an 8-fold increase in sulfasalazine plasma exposure in the presence of CCM (300–400 mg/kg) (Kusuhara et al., 2012), further supporting our proposal that PhA is useful as an in vivo probe for Bcrp function. Notably, FDA Drug Interactions and Labeling guidelines indicate that a ≥1.5-fold increase in sulfasalazine AUC (with concomitant dosing with a Bcrp inhibitor) is considered as evidence for in vivo inhibition of Bcrp. According to these criteria, our studies indicate that PPZ, ECD, CCM, and LPB produced a PhA AUC ≥1.5-fold in vivo, thus indicating the inhibition of Bcrp.
Our Experimental Findings Indicate that PhA that Could Be Useful for Pre-Clinical Evaluation of Clinical Bcrp-Mediated DDI Using WT Mice in the Presence of Bcrp Inhibitors.
Mouse Abcg2/Bcrp has 81% sequence homology with human ABCG2/BCRP (Allen et al., 1999), and there is significant functional overlap in substrate and inhibitor specificity (Bakhsheshian et al., 2013). Multiple lines of evidence demonstrate that PhA is transported by Bcrp, but not by other ABC transporters. First, PhA was identified as a unique Bcrp substrate in an initial Bcrp KO mouse report (Jonker et al., 2002), and this has been confirmed and extended to mouse Bcrp and human BCRP cell lines (Robey et al., 2004; Bakhsheshian et al., 2013). Indeed, Bakhsheshian et al. (2013) demonstrated that a series of porphyrins, including PhA, behaved similarly with both mouse Bcrp and human BCRP displaying similar substrate and inhibitor specificity. Second, ECD did not cause a significant increase in PhA whole-body NIR fluorescence or serum concentration in Bcrp KO mice. Third, among all of the ABC transporter KO mice, and numerous publications with each of them, only the absence of Bcrp resulted in an accumulation of PhA (a breakdown product of alfalfa in all standard rodent chow) and phototoxicity (Jonker et al., 2002).
The PhA Ex and Em wavelengths in mice in vivo (Ex, 670 nm; Em, 840 nm) differed from those in vitro (Ex, 400–410 nm; Em, 670 nm). This is not unexpected because live animal fluorescence at any wavelength is routinely affected by absorbance and light scattering due to the complex environment composed of plasma proteins and tissue barriers (Leblond et al., 2010). Thus, it is important to consider this a priori when selecting Ex/Em wavelength settings to use in vivo. Of note, if we had used the in vitro Ex/Em PhA wavelength parameters, we would have missed the in vivo PhA fluorescence signal.
PhA has low solubility and permeability, and, as a Bcrp substrate, is poorly absorbed, which was reflected in PhA being detected in the intestine of PhA-gavaged mice, but was undetectable in the serum of WT mice gavaged with PhA. PhA has been reported to be mainly cleared unabsorbed in feces (Kraatz et al., 2014). Interestingly, we routinely found that ex vivo fluorescence comparison of the intestinal segments of WT mice fed a normal PhA-containing diet, after flushing out the intestinal contents, revealed a lower PhA fluorescence in the duodenum compared with a higher PhA fluorescence in the jejunum and ileum. The gradient of Bcrp expression in the mouse intestine (i.e., higher in the duodenum and lowest in the ileum) (Gutmann et al., 2005) appears to be inverse to the PhA fluorescence in the enterocytes of WT mice (i.e., lowest in the duodenum and higher in the jejunum and ileum). The increased fluorescence in the intestinal region in Bcrp KO mice (Fig. 2A), despite increased systemic absorption from the lumen, was due to trapping of PhA in the enterocytes in the absence of Bcrp-mediated efflux (Fig. 2B), and this was captured in the in vivo imaging.
Serum PhA Fluorescence after Oral Coadministration of PhA and Inhibitors Might Be a More Useful Tool than Whole-Body Fluorescence for Elucidating the DDI of Weak Bcrp Inhibitors.
We found a significantly higher (3.5-fold) AUCFL 0–6 h value (Fig. 3, B and C) in BCRP KO mice compared with WT mice. However, in these same animals, the PhA AUCSerum 0–6 h value (Fig. 3, D and E) was increased over 50-fold. The discrepancy in the increase in PhA serum concentration (50-fold) versus whole-body fluorescence (3.5-fold) is likely due to a higher background fluorescence from the unabsorbed dietary PhA in the intestine. Nevertheless, there are unique advantages to using whole-body fluorescence of the Bcrp substrate PhA to screen for oral DDI including the following: 1) the ease and noninvasive nature of its detection; 2) that animals do not need to be sacrificed and can be used repeatedly; and 3) the ability to longitudinally monitor changes in Bcrp substrate tissue distribution into other organs (ex vivo) in the presence of systemic inhibition of Bcrp (the subject of another manuscript).
To our knowledge, this is the first report showing that the fluorescent Bcrp substrate PhA can be used for the identification of in vivo DDI due to the inhibition of Bcrp at the gastrointestinal tract and creates an opportunity for a high-throughput in vivo Bcrp inhibitor assay. Also, the use of serum fluorescence by simple spectrometry makes this method unique in that minimal technical resources are required for the identification of Bcrp substrates. Furthermore, this method is in conformation with the “3R” principal (replacement, reduction, and refinement), as it would reduce the requirement of invasive studies thus significantly reducing the number of laboratory animals, at the same time creating high-quality reliable data. In conclusion, our study shows that dynamic fluorescence imaging in live animals using PhA as a Bcrp probe can be used for the identification of Bcrp inhibitors and to understand the DDI liability of such inhibitors. Further studies are required to determine whether we can use PhA as a probe substrate of BCRP activity in the clinic, and whether it may be further used to understand systemic Bcrp inhibition.
Acknowledgments
We thank the St. Jude Center for In Vivo Imaging and Therapeutics Core (Dr. Walter Akers and Cheryl Winter) in this research.
Abbreviations
- AFF
alfalfa-free
- AUCFL 0–6 h
area under the fluorescence ratio-time curve up to 6 hours
- AUCSerum 0–6 h
area under PhA serum concentration-time curve calculated up to 6 hours
- BCRP
breast cancer resistance protein
- CCM
curcumin
- Cgut
mouse gut fluid drug concentration (mouse dose/0.8 ml)
- Cgut_soluble
soluble drug concentration in gut fluid assumed to be predicted solubility
- CLPa
chlorophyll a
- DDI
drug-drug interaction
- DPBS
Dulbecco’s phosphate-buffered saline
- DSB
dasatinib
- ECD
elacridar
- Em
emission
- Ex
excitation
- FDA
Food and Drug Administration
- HPLC
high-performance liquid chromatography
- Igut
maximum human dose in the apparent human gut fluid volume of 250 ml
- ITC
International Transporter Consortium
- IVIS
in vivo imaging system
- KO
knockout
- LPB
lapatinib
- MDCKII
Madine-Darby Canine Kidney II
- NIR
near-infrared
- PBS
phosphate-buffered saline
- PhA
pheophorbide A
- PPIX
protoporphyrin IX
- PPZ
pantoprazole
- ROI
region of interest
- SFB
sorafenib
- WT
wild type
Authorship Contributions
Participated in research design: Yasuda, Ganguly, Schuetz.
Conducted experiments: Yasuda, Ganguly.
Performed data analysis: Yasuda, Ganguly, Schuetz.
Wrote or contributed to the writing of the manuscript: Yasuda, Ganguly, Schuetz.
Footnotes
The work was supported by the National Cancer Institute [Cancer Center Support Grant P30-CA-21765]; and the American Lebanese Syrian Associated Charities (ALSAC).
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Ahmed-Belkacem A, Pozza A, Muñoz-Martínez F, Bates SE, Castanys S, Gamarro F, Di Pietro A, Pérez-Victoria JM. (2005) Flavonoid structure-activity studies identify 6-prenylchrysin and tectochrysin as potent and specific inhibitors of breast cancer resistance protein ABCG2. Cancer Res 65:4852–4860. [DOI] [PubMed] [Google Scholar]
- Allen JD, Brinkhuis RF, Wijnholds J, Schinkel AH. (1999) The mouse Bcrp1/Mxr/Abcp gene: amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res 59:4237–4241. [PubMed] [Google Scholar]
- Almela L, Fernández-López JA, Roca MJ. (2000) High-performance liquid chromatographic screening of chlorophyll derivatives produced during fruit storage. J Chromatogr A 870:483–489. [DOI] [PubMed] [Google Scholar]
- Bakhsheshian J, Hall MD, Robey RW, Herrmann MA, Chen JQ, Bates SE, Gottesman MM. (2013) Overlapping substrate and inhibitor specificity of human and murine ABCG2. Drug Metab Dispos 41:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan A, Amidon GL. (2010) MRP2 mediated drug-drug interaction: indomethacin increases sulfasalazine absorption in the small intestine, potentially decreasing its colonic targeting. Int J Pharm 386:216–220. [DOI] [PubMed] [Google Scholar]
- Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U, Wolf J, Jaehde U. (2011) Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin Pharmacokinet 50:551–603. [DOI] [PubMed] [Google Scholar]
- Ellis LC, Hawksworth GM, Weaver RJ. (2013) ATP-dependent transport of statins by human and rat MRP2/Mrp2. Toxicol Appl Pharmacol 269:187–194. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann H, Hruz P, Zimmermann C, Beglinger C, Drewe J. (2005) Distribution of breast cancer resistance protein (BCRP/ABCG2) mRNA expression along the human GI tract. Biochem Pharmacol 70:695–699. [DOI] [PubMed] [Google Scholar]
- Hu S, Chen Z, Franke R, Orwick S, Zhao M, Rudek MA, Sparreboom A, Baker SD. (2009) Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin Cancer Res 15:6062–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudachek SF, Gustafson DL. (2013) Incorporation of ABCB1-mediated transport into a physiologically-based pharmacokinetic model of docetaxel in mice. J Pharmacokinet Pharmacodyn 40:437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Izawa K, Kiryu S, Tojo A, Ohtomo K. (2008) Diet and abdominal autofluorescence detected by in vivo fluorescence imaging of living mice. Mol Imaging 7:21–27. [PubMed] [Google Scholar]
- Jonker JW, Buitelaar M, Wagenaar E, Van Der Valk MA, Scheffer GL, Scheper RJ, Plosch T, Kuipers F, Elferink RP, Rosing H, et al. (2002) The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci USA 99:15649–15654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath AV, Wang J, Lee FY, Marathe PH. (2008) Preclinical pharmacokinetics and in vitro metabolism of dasatinib (BMS-354825): a potent oral multi-targeted kinase inhibitor against SRC and BCR-ABL. Cancer Chemother Pharmacol 61:365–376. [DOI] [PubMed] [Google Scholar]
- Kraatz M, Whitehead TR, Cotta MA, Berhow MA, Rasmussen MA. (2014) Effects of chlorophyll-derived efflux pump inhibitor pheophorbide a and pyropheophorbide a on growth and macrolide antibiotic resistance of indicator and anaerobic swine manure bacteria. Int J Antibiotic 2014:185068. [Google Scholar]
- Krishnamurthy P, Schuetz JD. (2006) Role of ABCG2/BCRP in biology and medicine. Annu Rev Pharmacol Toxicol 46:381–410. [DOI] [PubMed] [Google Scholar]
- Kusuhara H, Furuie H, Inano A, Sunagawa A, Yamada S, Wu C, Fukizawa S, Morimoto N, Ieiri I, Morishita M, et al. (2012) Pharmacokinetic interaction study of sulphasalazine in healthy subjects and the impact of curcumin as an in vivo inhibitor of BCRP. Br J Pharmacol 166:1793–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagas JS, van Waterschoot RA, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. (2010) Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther 9:319–326. [DOI] [PubMed] [Google Scholar]
- Leblond F, Davis SC, Valdés PA, Pogue BW. (2010) Pre-clinical whole-body fluorescence imaging: review of instruments, methods and applications. J Photochem Photobiol B 98:77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CA, O’Connor MA, Ritchie TK, Galetin A, Cook JA, Ragueneau-Majlessi I, Ellens H, Feng B, Taub ME, Paine MF, et al. (2015) Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: practical recommendations for clinical victim and perpetrator drug-drug interaction study design. Drug Metab Dispos 43:490–509. [DOI] [PubMed] [Google Scholar]
- Mahringer A, Delzer J, Fricker G. (2009) A fluorescence-based in vitro assay for drug interactions with breast cancer resistance protein (BCRP, ABCG2). Eur J Pharm Biopharm 72:605–613. [DOI] [PubMed] [Google Scholar]
- Mao Q, Unadkat JD. (2015) Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—an update. AAPS J 17:65–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavek P, Merino G, Wagenaar E, Bolscher E, Novotna M, Jonker JW, Schinkel AH. (2005) Human breast cancer resistance protein: interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine. J Pharmacol Exp Ther 312:144–152. [DOI] [PubMed] [Google Scholar]
- Polli JW, Humphreys JE, Harmon KA, Castellino S, O’Mara MJ, Olson KL, John-Williams LS, Koch KM, Serabjit-Singh CJ. (2008) The role of efflux and uptake transporters in [N-3-chloro-4-[(3-fluorobenzyl)oxy]phenyl-6-[5-([2-(methylsulfonyl)ethyl]aminomethyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos 36:695–701. [DOI] [PubMed] [Google Scholar]
- Roberts JK, Birg AV, Lin T, Daryani VM, Panetta JC, Broniscer A, Robinson GW, Gajjar AJ, Stewart CF. (2016) Population pharmacokinetics of oral topotecan in infants and very young children with brain tumors demonstrates a role of ABCG2 rs4148157 on the absorption rate constant. Drug Metab Dispos 44:1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Steadman K, Polgar O, Bates SE. (2005) ABCG2-mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther 4:187–194. [PubMed] [Google Scholar]
- Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, Bates SE. (2004) Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res 64:1242–1246. [DOI] [PubMed] [Google Scholar]
- Robey RW, To KK, Polgar O, Dohse M, Fetsch P, Dean M, Bates SE. (2009) ABCG2: a perspective. Adv Drug Deliv Rev 61:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane R, Agarwal S, Elmquist WF. (2012) Brain distribution and bioavailability of elacridar after different routes of administration in the mouse. Drug Metab Dispos 40:1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabedissen HEMz, Kroemer HK. (2011) In vitro and in vivo evidence for the importance of breast cancer resistance protein transporters (BCRP/MXR/ABCP/ABCG2), in Drug Transporters (Fromm MF, Kim RB. eds), pp 325–371, Springer, Berlin, Heidelberg. [DOI] [PubMed] [Google Scholar]
- Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, Hirte HW, Eder JP, Lenz HJ, Schwartz B. (2007) Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 12:426–437. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Doki K, Homma M, Tamaki H, Hori S, Ohtani H, Sawada Y, Kohda Y. (2009) Co-administration of proton pump inhibitors delays elimination of plasma methotrexate in high-dose methotrexate therapy. Br J Clin Pharmacol 67:44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Polli JW, Berglund EG, Huang SM, Zhang L, Poirier A, Chu X, Feng B, International Transporter Consortium (2013) Transporter studies in drug development: experience to date and follow-up on decision trees from the International Transporter Consortium. Clin Pharmacol Ther 94:113–125. [DOI] [PubMed] [Google Scholar]
- Weiss J, Rose J, Storch CH, Ketabi-Kiyanvash N, Sauer A, Haefeli WE, Efferth T. (2007) Modulation of human BCRP (ABCG2) activity by anti-HIV drugs. J Antimicrob Chemother 59:238–245. [DOI] [PubMed] [Google Scholar]
- Zhang W, Li J, Allen SM, Weiskircher EA, Huang Y, George RA, Fong RG, Owen A, Hidalgo IJ. (2009) Silencing the breast cancer resistance protein expression and function in caco-2 cells using lentiviral vector-based short hairpin RNA. Drug Metab Dispos 37:737–744. [DOI] [PubMed] [Google Scholar]
- Zhongfa L, Chiu M, Wang J, Chen W, Yen W, Fan-Havard P, Yee LD, Chan KK. (2012) Enhancement of curcumin oral absorption and pharmacokinetics of curcuminoids and curcumin metabolites in mice. Cancer Chemother Pharmacol 69:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]