

Abstract
Introduction
Apolipoproteins of demonstrated importance to brain cholesterol and ß-amyloid metabolism may serve as novel risk markers for Alzheimer's pathology.
Methods
We measured apolipoproteins (apoE, apoJ, apoA-I, and apoC-III and their uniquely defined subspecies) by enzyme-linked immunosorbent assay in plasma collected in 2000 and 2008 from 176 dementia-free participants of the Ginkgo Evaluation of Memory Study and related these to ß-amyloid on positron emission tomography scans, hippocampal volume, and white matter lesion volume in 2009.
Results
Higher apoE was associated with lower ß-amyloid deposition. Despite apoA-I being unrelated to hippocampal volume, subspecies of apoA-I containing or lacking apoJ or apoC-III showed opposite associations with hippocampal volume. Higher apoJ and apoE lacking apoJ were associated with higher hippocampal volume and higher white matter lesion volume, respectively. Associations were similar in participants without cognitive impairment or APOE ε4 noncarrier and when analyzing apolipoproteins in 2000–2002.
Discussion
Apolipoproteins may be important minimally invasive biomarkers indicative of Alzheimer's pathology.
Keywords: Apolipoproteins, High-density lipoproteins, Brain amyloid, Epidemiology, Neuroimaging
1. Background
Genetic variations in regions that encode apolipoproteins, including apoE, apoJ, apoC-III, and apoA-I, are associated with Alzheimer's disease (AD) [1], [2], [3], [4]. The relationships between plasma concentrations of apolipoproteins and AD have been difficult to disentangle [5]. Most evidence suggests that plasma apoE levels are inversely associated with AD incidence [5], and one study suggests that plasma apoC-III levels are lower in prevalent AD [6].
The relationship between apolipoproteins and AD is complicated by the fact that apolipoproteins can reside on both low-density lipoproteins and high-density lipoproteins (HDLs), two lipoprotein classes that seem to differ in their associations with AD [7]. Beyond its major structural protein component apoA-I, HDL hosts dozens of associated proteins [8]. In the brain, lipoproteins are only found in the density range similar to HDL and contain primarily apoE and some apoA-I [9]. By identifying apolipoproteins that do and do not go together, subspecies of apolipoproteins can be determined. It is possible that in the presence or absence of, for example, apoJ, plasma concentrations of apoE in HDL may show a stronger association with AD pathophysiology. In previous work, we reported that concentrations of some apolipoproteins in cerebrospinal fluid (CSF) are better reflected by measures of apolipoprotein subspecies in plasma than with the corresponding total plasma concentrations [10]. This supports the significance of studying apolipoprotein subspeciation in plasma in relation to brain structure and function.
To this end, we investigated the associations of plasma apolipoproteins and their subspecies with brain ß-amyloid deposition, hippocampal volume, and white matter lesion volumes.
2. Methods
2.1. Study population and design
Ginkgo Evaluation of Memory Study (GEMS) was a multicenter, double-blinded, placebo-controlled, randomized clinical trial (Clinicaltrials.gov identifier: NCT00010803) to evaluate the effect of Ginkgo biloba for the prevention of dementia in older adults. The design and selection criteria have been described previously [11]. Briefly, 3069 community-dwelling men and women aged 72–96 years with normal cognition or mild cognitive impairment (MCI) were recruited between 2000 and 2002 and followed up for an average of 6 years. The screening visit included the administration of a neuropsychological test battery by trained technicians, assessment of vital signs and medical history including diabetes mellitus, and blood sample collection. Participants were reevaluated at subsequent semiannual clinic visits through August 2004 and annual follow-up visits through 2008.
The present study was performed in a subsample of 197 participants from the Pittsburgh site recruited into the GEMS Imaging Study who remained dementia free throughout the GEMS trial. These participants were invited to have Pittsburgh Compound B (PiB) positron emission tomography (PET) brain scans and structural magnetic resonance imaging scans of the brain in 2009, approximately 10 ± 3 months after GEMS closeout. Three participants were excluded for technical problems with PiB PET, three were excluded who developed dementia between 2008 and imaging, and 15 participants did not have an adequate blood sample taken at the time of PET scanning; thus, 176 participants were eligible for this analysis. Written informed consent was obtained from study participants and their proxies, and the study was approved by the Human Use Subcommittee of the Radioactive Drug Research Committee and the Institutional Review Board of the University of Pittsburgh.
2.2. Biochemical measurements
Participants underwent phlebotomy at the screening visit and at study closeout in 2008. Frozen plasma samples, stored at the GEMS blood repository at the National Cell Repository for AD in Indiana, were shipped on dry ice to the lipid laboratory of the Harvard T. H. Chan School of Public Health for biochemical measurements.
We conducted two related sets of apolipoprotein measurements. First, we measured total levels of four apolipoproteins, namely, apoJ, apoE, apoA-I, and apoC-III, in whole plasma using sandwich enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN; and Academy Biomedical Company Inc, Houston, TX).
Second, to focus on apolipoproteins in the HDL fraction of plasma, we removed apoB-containing lipoproteins by dextran sulfate and magnesium chloride precipitation. Within the non-apoB fraction, concentrations of apoE, apoA-I, and apoC-III were measured using sandwich enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN; and Academy Biomedical Company Inc., Houston, TX). For measurement of apoA-I and apoE subspecies defined on coexisting apolipoproteins of interest in the non-apoB fraction, a patented modified sandwich enzyme-linked immunosorbent assay was used. For example, for measurement of apoE containing apoJ, 96-well plates were coated with anti-apoJ antibody. Diluted samples were incubated on these plates to bind lipoproteins containing apoJ. The fraction bound to this plate was released by dissociation of the lipoprotein complex with Tween-containing diluent (1 × PBS/2% BSA/0.05% Tween 20) and transferred to a second plate coated with anti-apoE antibody to quantify the concentration of apoE containing apoJ. The concentration of apoE that was not associated with apoJ was calculated as the difference of the measured total apoE concentration minus the measured concentration of apoE containing apoJ. Together, this approach provided concentrations of apoE containing apoJ and the concentration of apoE lacking apoJ (Fig. 1), as well as their sum of total apoE concentration. Both samples from baseline (2000 to 2002) and 2008 from an individual were assayed together on the same 96-well plate. HDL cholesterol was isolated for measurement by dextran sulfate/magnesium chloride precipitation of apoB-containing lipoproteins, and the resulting supernatant, total plasma cholesterol, and plasma triglycerides were measured by enzymatic assay (Thermo Fisher Scientific, Waltham, MA). Overall, the average coefficients of variation were below 20% for all apolipoproteins and apolipoprotein subspecies (Supplementary Table A.2). Samples were measured in 11 batches. Each batch contains four plasma pool samples, composed of the plasma from between 4 and 9 people depending on the pool. These pools were measured for each analyte for more than 20 times to produce an expected value. The data on each plate were adjusted using a correction factor that was the slope of the line formed by regressing the obtained values of the four control samples against their expected values.
Fig. 1.
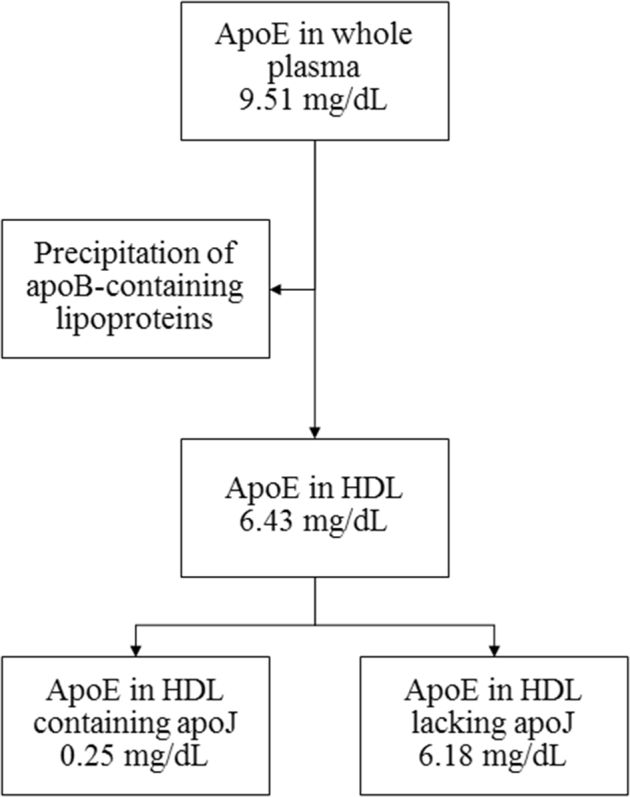
Measurement of apolipoproteins and apolipoprotein subspecies. Here, apoE in HDL containing or lacking apoJ was measured by a modified sandwich enzyme-linked immunosorbent assay approach (values are means). When measuring any apolipoprotein subspecies in the HDL fraction of plasma, apoB-containing lipoproteins are first removed. Abbreviations: HDLs, high-density lipoproteins; apo, apolipoproteins.
2.3. Neuroimaging
ß-Amyloid plaque deposition was quantified by using PiB PET scanning as published previously [12]. PET images were acquired in four 5-minute frames starting 50 minutes after injection of 11C-labeled PiB ligand (15 ± 1.5 mCi) using an ECAT HR + scanner (Siemens/CTI Modular Imaging, Knoxville, Tennessee) in 3D mode (63 planes; slide with 2.4 mm) equipped with a Neuro-Insert (CTI PET Systems, Knoxville, Tennessee). The PET data were reconstructed to 6 mm transverse and axial image resolution using Fourier rebinning and 2D back projection with Hann filter kernel full-width at half maximum = 3 mm with correction for dead-timer photon attenuation, scatter, and radioactive decay. Brain ß-amyloid deposition was quantified as the ratio of the averaged PiB retention in five bilateral brain regions (anterior cingulate gyrus, frontal cortex, lateral temporal cortex, parietal cortex, and precuneus cortex) to the PiB retention in the cerebellum.
Hippocampal volume and white matter lesion volume were quantified as the proportion of the intracranial volume within the “inner skull” *10−2. Magnetic resonance imaging scans were obtained on a 1.5-T scanner (GE Healthcare, Waukesha, WI) and standard head coil, with fluid-attenuated inversion recovery and 5 ms echo time and 25 ms repetition time. A T1-weighted volumetric spoiled gradient recalled sequence was obtained (0.937 × 0.937 mm) in sagittal (slice thickness = 1.2 mm/0 mm interslice) or coronal plane (slice thickness = 1.5 mm/0 mm interslice).Voxel-based brain morphometry was performed with the sagittal spoiled gradient recalled magnetic resonance imaging acquisition protocol. White matter volumes were quantified by using automatic segmentation software (FMRIB's Automated Segmentation Tool [13]; University of Oxford) as described previously using T1-weighted images in native anatomical space [14]. The T2-weighted fluid-attenuated inversion recovery images were used to quantify white matter lesion volume using a fuzzy connectedness algorithm. The intracranial volume was computed with an advanced option (−A) of the Brain Extraction Tool. An atlas-based automated labeling pathway segmentation technique was applied to compute hippocampal volumes using a fully deformable registration procedure to assess preselected regions of interest [15]. Anatomical regions of interest originated from the automated anatomical labeling atlas [16]. The Montreal Neurological Institute Colin 27 average reference brain was transformed to fit each individuals recorded image.
2.4. Covariates
Age, sex, years of education, and race/ethnicity was determined in interviews by trained technicians at the screening visit. Owing to limited variation, race/ethnicity was classified as white and other. At the baseline examination (2000 to 2002), a Health Habits Questionnaire was administered to collect information on smoking status (classified as never, former, and current smoker) and to collect information on alcohol intake (drinks/week). Our analyses used body mass index and systolic blood pressure measured at the study closeout in 2008. For participants with a missing body mass index or blood pressure measurement (N = 133), we replaced the missing value with the value from the closest previous visit (last visit median, 6.4 years after baseline; minimum, 5.5 years; maximum, 7.3 years after baseline examination). APOE genotype testing was performed from DNA isolated from baseline blood samples.
At each visit, history of heart disease was assessed in interviews by trained technicians. Participants were asked to bring prescribed medicine and over-the-counter drugs to each clinic visit. Participants were classified as former, current, or never user of antihypertensive and lipid-lowering medications at the last clinic visit.
The neuropsychological examination in 2009 included the administration of the Modified Mini-Mental State Examination [17] and a comprehensive neuropsychological battery of 10 tests evaluating five cognitive domains: (1) memory; (2) visual-spatial construction; (3) language; (4) attention/psychomotor speed; and (5) executive function [18]. A committee of neurologists at the University of Pittsburgh Cognitive Diagnostic Center classified participants as having MCI taking into account cognitive assessments from 2000 to 2009. MCI was defined as 1–3 tests impaired at cutoffs of 1.5 standard deviation below age- and education-adjusted means [19].
2.5. Statistical analysis
Age- and sex-adjusted partial correlation coefficients were estimated for pairwise correlations of the biological markers with each other and for the relations of the apolipoproteins to ß-amyloid and hippocampal and white matter lesion volumes. We assessed the cross-sectional associations between apolipoprotein subspecies and amyloid deposition, hippocampal volume, and white matter lesion volume, all from 2009 site visit, in robust linear regression models adjusted for age and sex, education (years), race/ethnicity (white, other), alcohol intake (drinks/week), smoking status (never, former, current), body mass index (kg/m2), MCI, history of heart disease, systolic blood pressure, antihypertensive medication use, history of diabetes mellitus, and lipid-lowering medication use. Given the novelty of the apolipoprotein subspecies, no reference values for abnormal levels exist, and we used robust linear regression analysis to prevent that a few influential data points drive associations.
In sensitivity analyses, we examined the association between apolipoprotein subspecies at baseline (2000 to 2002) with the neurobiological endpoints obtained 9 years later. We also performed analyses restricted to participants without MCI and to APOE ɛ4 noncarriers.
All analyses were performed using STATA version 12.1 (Stata Corp., College Station, TX).
3. Results
Table 1 shows the characteristics of participants by APOE ε4 carriership. None of the APOE ɛ4 allele carriers were homozygotes for the E4 allele. APOE ε4 allele carriers were slightly more likely to use lipid-lowering or antihypertensive treatment. The prevalence of MCI was similar in APOE ε4 carrier and noncarrier. ApoA-I and apoC-III were the most abundant apolipoproteins (Table 2; Supplementary Table A.2). On average, the apolipoproteins tended to be slightly higher in APOE ɛ4 allele noncarrier. Most of the apoE was found lacking apoJ, and most of the apoA-I was found lacking apoE, apoJ, or apoC-III.
Table 1.
Characteristics of 176 participants of the GEMS imaging study
| Characteristics∗ | APOE ɛ4 allele noncarrier (N = 119)† | APOE ɛ4 allele carrier (N = 29) | Total (N = 176) |
|---|---|---|---|
| Sex, male | 73 (61) | 15 (52) | 105 (60) |
| Age, y | 85 (82, 91) | 84 (82, 89) | 85 (82, 91) |
| Education, y | 14 (10, 19) | 16 (12, 18) | 14 (11, 19) |
| Body mass index, kg/m2 | 26 (20, 34) | 25 (20, 30) | 26 (20, 34) |
| Smoking status‡ | |||
| Never | 48 (41) | 13 (45) | 70 (40) |
| Former | 67 (57) | 14 (48) | 99 (57) |
| Current | 3 (3) | 2 (7) | 6 (3) |
| Alcohol intake§, drinks/week | 0.5 (0, 21) | 1 (0, 14) | 0.5 (0, 15) |
| Race, white | 115 (97) | 29 (100) | 170 (97) |
| Total cholesterol, mg/dL | 167 (107, 241) | 175 (99, 264) | 166 (104, 242) |
| HDL cholesterol, mg/dL | 60 (41, 91) | 64 (41, 87) | 60 (39, 91) |
| Triglycerides‡, mg/dL | 98 (50, 203) | 96 (39, 241) | 96 (45, 219) |
| Systolic blood pressure, mm Hg | 126 (101, 163) | 122 (105, 164) | 124 (102, 161) |
| Diastolic blood pressure, mm Hg | 68 (50, 85) | 68 (57, 86) | 67 (51, 84) |
| History of cardiovascular disease | 42 (35) | 11 (38) | 63 (36) |
| History of diabetes§ | 6 (5) | 1 (4) | 8 (5) |
| Lipid-lowering medication use | |||
| Never user | 61 (51) | 13 (45) | 87 (49) |
| Former user | 11 (9) | 3 (10) | 18 (10) |
| Current use | 47 (40) | 13 (45) | 71 (40) |
| Antihypertensive treatment | |||
| Never user | 35 (29) | 8 (28) | 52 (30) |
| Former user | 4 (3) | 1 (3) | 6 (3) |
| Current use | 80 (67) | 20 (69) | 118 (67) |
| Mild cognitive impairment | 26 (22) | 7 (24) | 39 (22) |
| 3MSE | 98 (90, 100) | 98 (91, 100) | 98 (90, 100) |
| ß-amyloid deposition, SUVR | 1.6 (1.2, 2.6) | 2.1 (1.2, 2.8) | 1.6 (1.2, 2.6) |
| Hippocampal volume¶, % of ICV | 26 (21, 31) | 25 (20, 32) | 26 (20, 31) |
| White matter lesion volume¶, % of ICV | 0.7 (0.2, 2.1) | 0.7 (0.3, 2.5) | 0.8 (0.2, 2.3) |
Abbreviations: ICV, intracranial volume; 3MSE, Modified Mini-Mental State Examination score; SUVR, standardized uptake value ratio; HDLs, high-density lipoproteins; GEMS, Ginkgo Evaluation of Memory Study; APOE, apolipoproteins E.
Values are median (Q5, Q95) or n (%).
N = 28 individuals with missing APOE genotype information were excluded.
N = 175.
N = 174.
N = 157.
Table 2.
Apolipoprotein and apolipoprotein subspecies distributions by APOE ɛ4 allele carrier status in 148 participants of the GEMS imaging study in 2008
| Apolipoprotein concentrations,∗ mg/dL | APOE ɛ4 allele noncarrier (N = 119) | APOE ɛ4 allele carrier (N = 29) |
|---|---|---|
| In whole plasma | − | |
| ApoE | 9.0 (4.9, 16.6) | 8.4 (3.6, 13.0) |
| ApoJ | 5.5 (4.4, 6.7) | 5.5 (4.3, 6.9) |
| ApoA-I | 152 (93, 235) | 160 (102, 246) |
| ApoC-III | 12.5 (6.0, 22.7) | 14.1 (7.5, 23.8) |
| In HDL fraction of plasma† | ||
| ApoE | 6.1 (3.2, 11.3) | 5.4 (2.3, 10.2) |
| ApoE containing apoJ | 0.2 (0.1, 0.4) | 0.2 (0.1, 0.4) |
| ApoE lacking apoJ | 5.9 (3.1, 11.0) | 5.3 (2.2, 9.9) |
| Proportion of apoE containing apoJ, % | 3.9 (2.2, 8.6) | 3.9 (2.2, 7.2) |
| ApoE containing apoC-III‡ | 3.0 (1.3, 6.1) | 2.5 (1.0, 6.3) |
| ApoE lacking apoC-III‡ | 2.9 (0.5, 7.3) | 2.5 (1.0, 6.4) |
| Proportion of apoE containing apoC-III,‡ % | 51 (26, 90) | 46 (18, 76) |
| ApoA-I containing apoE | 12.1 (4.5, 42.5) | 10.1 (4.0, 20.3) |
| ApoA-I lacking apoE | 138 (82, 213) | 151 (99, 237) |
| Proportion of apoA-I containing apoE, % | 7.8 (3.4, 19.7) | 6.2 (2.4, 10.8) |
| ApoA-I containing apoJ | 2.1 (1.0, 3.1) | 2.3 (1.4, 3.5) |
| ApoA-I lacking apoJ | 150 (92, 231) | 158 (101, 243) |
| Proportion of apoA-I containing apoJ, % | 1.3 (0.9, 2.1) | 1.3 (1.1, 2.0) |
| ApoA-I containing apoC-III | 9.3 (3.6, 19.2) | 10.2 (3.9, 22.3) |
| ApoA-I lacking apoC-III | 141 (83, 222) | 150 (98, 238) |
| Proportion of apoA-I containing apoC-III, % | 6.0 (3.0, 11.8) | 6.5 (3.3, 12.6) |
| ApoC-III | 8.7 (3.6, 14.7) | 10.2 (4.1, 15.5) |
Abbreviations: HDL, high-density lipoproteins; GEMS, Ginkgo Evaluation of Memory Study; APOE, apolipoproteins E; apo, apolipoproteins.
Values are median (Q5, Q95).
Plasma precipitated of apoB-containing lipoproteins.
N = 133.
The age- and sex-adjusted correlation of ß-amyloid and hippocampal volume was, as expected, inverse (r = −0.17, P = .03). White matter lesion volume was not correlated to ß-amyloid or hippocampal volume (r = −0.02, P = .80, and r = 0.04, P = .64, respectively).
In age- and sex-adjusted models, higher apoE in whole plasma and higher apoE in HDL fraction of plasma were statistically significantly associated with lower ß-amyloid deposition (Supplementary Table A.3). Furthermore, the subspecies of apoE lacking apoC-III and the subspecies of apoA-I containing apoE were related inversely to ß-amyloid deposition. Associations persisted after multivariable adjustment and were generally strengthened (Table 3). Multivariable adjustment revealed apoE subspecies lacking apoJ and apoE containing or lacking apoC-III statistically significantly inversely related to ß-amyloid deposition.
Table 3.
Mean difference (95% CI)∗ in ß-amyloid, hippocampal volume, and white matter lesion volume per 1 SD higher apolipoprotein concentration in 171 participants of the GEMS imaging study in 2009
| Exposure | Multivariable adjusted mean differences (95% CI) |
||
|---|---|---|---|
| ß-amyloid deposition SUVR (N = 171) | Hippocampal volume, % of ICV (N = 152) | White matter lesion volume, % of ICV (N = 152) | |
| Age, years | 0.01 (−0.02, 0.03) | −0.20 (−0.40, 0.01) | 0.07 (0.04, 0.10) |
| APOE ɛ4 allele carrier versus noncarrier | 0.32 (0.13, 0.52)† | −0.23 (−1.59, 1.14)‡ | 0.05 (−0.23, 0.32)‡ |
| Whole plasma | |||
| ApoE | −0.12 (−0.19, −0.06) | 0.48 (−0.12, 1.08) | 0.04 (−0.03, 0.11) |
| ApoJ | −0.01 (−0.09, 0.06) | 0.59 (0.08, 1.11) | 0.05 (−0.02, 0.12) |
| ApoA-I | −0.01 (−0.08, 0.06) | 0.36 (−0.12, 0.84) | 0.07 (−0.03, 0.16) |
| ApoC-III | −0.02 (−0.10, 0.06) | 0.03 (−0.65, 0.71) | <−0.01 (−0.10, 0.10) |
| HDL fraction of plasma§ | |||
| ApoE | −0.12 (−0.18, −0.06) | 0.32 (−0.31, 0.94) | 0.08 (−0.01, 0.16) |
| ApoE containing apoJ | −0.07 (−0.15, 0.01) | −0.33 (−1.05, 0.38) | −0.06 (−0.17, 0.05) |
| ApoE lacking apoJ | −0.09 (−0.16, −0.02) | 0.42 (−0.26, 1.11) | 0.10 (0.01, 0.18) |
| ApoE containing apoC-III | −0.10 (−0.17, −0.02)¶ | −0.23 (−0.81, 0.35)‖ | 0.06 (−0.03, 0.16)‖ |
| ApoE lacking apoC-III | −0.09 (−0.15, −0.03)¶ | 0.63 (−0.06, 1.32)‖ | 0.02 (−0.07, 0.12)‖ |
| ApoA-I containing apoE | −0.12 (−0.19, −0.05) | −0.17 (−0.65, 0.31) | 0.03 (−0.06, 0.11) |
| ApoA-I lacking apoE | 0.05 (−0.02, 0.12) | 0.45 (−0.06, 0.96) | 0.05 (−0.05, 0.16) |
| ApoA-I containing apoJ | 0.03 (−0.06, 0.12) | −0.72 (−1.31, −0.13) | 0.03 (−0.08, 0.14) |
| ApoA-I lacking apoJ | −0.02 (−0.11, 0.06) | 0.84 (0.23, 1.45) | 0.05 (−0.08, 0.18) |
| ApoA-I containing apoC-III | 0.01 (−0.06, 0.08) | −0.39 (−0.72, −0.05) | −0.04 (−0.13, 0.05) |
| ApoA-I lacking apoC-III | −0.01 (−0.09, 0.06) | 0.50 (0.01, 0.98) | 0.08 (−0.02, 0.18) |
| ApoC-III | −0.02 (−0.09, 0.06) | 0.23 (−0.41, 0.88) | 0.02 (−0.08, 0.13) |
Abbreviations: ICV, intracranial volume; SUVR, standardized uptake value ratio; HDLs, high-density lipoproteins; GEMS, Ginkgo Evaluation of Memory Study.
NOTE. Statistically significant differences are highlighted in bold.
Each lipoprotein was modeled separately. Subspecies of a lipoprotein containing or not containing a given apolipoprotein sum to the total for that lipoprotein, and thus, all models simultaneously include the two subspecies. Models were adjusted for age, sex, education (years), race/ethnicity (white, other), alcohol intake (drinks/week), smoking status (never, former, current), body mass index (kg/m2), mild cognitive impairment, history of heart disease, systolic blood pressure, antihypertensive medication use, history of diabetes mellitus, and lipid-lowering medication use.
N = 144.
N = 127.
Plasma precipitated of apoB-containing lipoproteins.
N = 151.
N = 136.
In multivariable adjusted models, participants with higher concentrations of apoJ had higher hippocampal volume (Table 3). Although apoA-I in whole plasma was unrelated to hippocampal volume, there were differential associations of hippocampal volume with the subspecies apoA-I containing or not containing apoJ (mean difference per 1 standard deviation (SD) higher apoA-I containing apoJ: −0.72% [95% confidence interval (CI): −1.31 to −0.13] and mean difference per 1 SD higher apoA-I not containing apoJ: 0.84% [95% CI: 0.23–1.45]); the subspecies of apoA-I coexisting or not containing apoC-III (mean difference per 1 SD higher apoA-I containing apoC-III: −0.39% [95% CI: −0.72 to −0.05] and mean difference per 1 SD higher apoA-I not containing apoC-III: 0.50% [95% CI: 0.01–0.98]). For comparison, a difference of 0.60% lower hippocampal volume was approximately equivalent to the difference in hippocampal volume associated with a 3-year older age (mean difference per 1-year higher age: −0.20% [95% CI: −0.40 to 0.01]).
ApoE lacking apoJ was related to higher white matter lesion volume in multivariable adjusted regression models (Table 3) (mean difference per 1 SD higher apoE lacking apoJ: 0.10% [95% CI: 0.01–0.18]). All other apolipoproteins and apolipoprotein subspecies were not statistically significantly associated with white matter lesion volume.
In a subset of participants, we were able to differentiate apoE containing or lacking apoA-I (Supplementary Table A.2). Most of the apoE was found containing apoA-I. ApoE containing or lacking apoA-I was not statistically significantly related to ß-amyloid deposition, hippocampal volume, or white matter lesion volume in multivariable adjusted regression models.
We determined the long-term association of apolipoproteins and the imaging markers of Alzheimer's pathophysiology using apolipoprotein measures obtained at the baseline visit from 2000 to 2002 (Supplementary Table A.4). The same correlation patterns with ß-amyloid deposition were observed for the apolipoproteins assessed at baseline as analyzed at the closeout in 2008. However, apolipoproteins at the baseline visit did not correlate with hippocampal volume or white matter lesion volume.
When we restricted the study population to participants without MCI and APOE ε4 noncarriers, associations between the apolipoproteins and the imaging outcomes were very similar in direction and magnitude compared to the overall analysis (Supplementary Table A.5).
4. Discussion
In this selected sample of elderly participants without dementia, overall plasma apoE, apoE in HDL, and subspecies of apoE and apoA-I were associated with structural and functional neuroimaging parameters. Higher hippocampal volumes were found in participants with higher apoJ concentrations. The relation was generally similar after excluding participants with MCI or APOE ε4 allele carrier. These results highlight the potential importance of apolipoprotein subspecies in understanding the pathogenesis of AD.
Given the very strong associations of allelic differences in genes encoding apolipoproteins with risk of AD [2], [20], it is perhaps surprising that few studies have assessed the associations of apolipoproteins in the circulation with ß-amyloid deposition and neurodegeneration. Those that have done so have reported largely inconsistent findings. ApoE has been proposed to play key roles in the regulation of neurotoxic ß-amyloid peptide levels in the brain by limiting its deposition and increasing its clearance [21], [22], [23]. ApoE also protects the microtubule-associated protein tau from hyperphosphorylation [21], [23], [24]. Our finding of an inverse association of circulating apoE with brain ß-amyloid deposition agrees with recent cross-sectional, age- and sex-adjusted data from the Australian Imaging, Biomarkers and Lifestyle Study of Ageing [25]. Although the Baltimore Longitudinal Study of Aging (BLSA) suggested that circulating apoE levels are positively associated with ß-amyloid accumulation in the medial temporal cortex of nondemented elderly participants, the sample size of 42 participants was substantially lower than that in our study, and study participants were younger [26]. Given the nonoverlapping age range in the BLSA and our study, we cannot exclude that the association of apoE with Alzheimer's pathology differs at different age periods or at different stages of the Alzheimer's pathology continuum. Associations of apoE and AD pathology might also differ by apoE isoforms. Martínez-Morillo observed no association between total plasma apoE and apoE isoforms with biomarkers of AD including total tau, phosphorylated tau, or amyloid beta 1–42 [27]. In contrast, the Australian Imaging, Biomarkers and Lifestyle study found similar associations for total plasma apoE and apoE ɛ4 isoform concentrations with ß-amyloid deposition [28]. Future studies on apoE subspecies considering apoE isoforms are important future research avenues.
Given that findings on apoE in the circulation and neurodegeneration have been inconsistent [29], [30], [31], [32], our findings that apoE subspecies lacking apoJ might be more closely related to white matter lesion volume than total apoE concentration support further investigation of apoE subspecies in relation to AD risk. Because circulatory and brain apoE pools are distinct [33] and plasma and CSF concentrations tend to correlate poorly [27], plasma apoE has been hypothesized to impact ß-amyloid deposition and neurodegeneration indirectly, by protecting blood brain barrier integrity and cerebrovascular function [34].
In our study, plasma apoJ and apoC-III concentrations were unrelated to neuroimaging parameters with the exception of an association of apoJ with hippocampal volume. In 60 dementia-free participants from the BLSA, plasma apoJ concentration 10 years before PET correlated positively with ß-amyloid deposition taking age and sex into account [35]. However, the baseline circulating apoJ concentrations were not related to changes in hippocampal volume over 6 years of follow-up in the BLSA [36]. In the Australian Imaging, Biomarkers and Lifestyle study, plasma apoJ also correlated directly with ß-amyloid deposition assessed by positron emission tomography and inversely with hippocampal volume in the combined study sample which included participants with normal cognition, MCI, or AD [37]. At this juncture, particularly given the low concentrations of apoJ found in plasma, we view overall results as still inconclusive. Despite some evidence that apoC-III can bind circulatory ß-amyloid, our understanding of the relationship of apoC-III and AD pathology is limited [38]. ApoC-III levels were unrelated to hippocampal volume in age- and sex-adjusted analyses of the Sydney Memory and Aging Study [30]. Further studies of apoJ and apoC-III and neuropathology with repeated imaging are needed to clarify their roles in AD pathology.
We found no evidence that plasma apoA-I was associated with brain amyloid deposition, hippocampal volume, or white matter lesion volume. In vitro experimental studies have shown that human apoA-I can bind amyloid β, and prevents amyloid aggregation as well as amyloid β–induced neuronal damage to hippocampal cultures [39]. Mice that overexpress human apoA-I have similar amounts of Aβ in the brain compared with control mice but have less cerebral amyloid angiopathy, suggesting selective effects of apoA-I on vascular amyloid deposition [40]. In our study, the resolution of the PET images did not allow distinction between parenchymal and vascular amyloid burden.
When we separated apoA-I in subspecies containing or lacking apoJ or apoC-III, the concentration of apoA-I containing apoJ and apoC-III was inversely related to hippocampal volume. These subspecies could be functionally more relevant than overall apoA-I concentration, or they could be markers for specific brain apolipoproteins. Recently, we reported that that the CSF concentration of apoC-III might be better approximated by plasma apoA-I containing apoC-III than total plasma apoC-III. Similarly, CSF apoJ correlated most strongly with apoA-I lacking apoJ [10]. Together, these results suggest that the relation of apoA-I and hippocampal volume is likely more complex and warrants further study [41]. A previous cross-sectional study in 1282 middle-aged and elderly study participants found an inverse association between apoA-I concentration and white matter lesion volume, which was not replicated in our study possibly because of smaller sample size and the special nature of this study population free of clinical dementia around the age of 85 years [42].
There were some specific limitations of this study. The study sample was a highly selective sample of oldest old, dementia-free survivors—limiting the generalizability to younger populations. None of the study participants were homozygotes for the APOE ɛ4 allele, and we had limited statistical power to investigate associations by APOE genotype. Thus, additional studies are needed to further confirm these associations and to test if associations differ by APOE isoforms. No CSF samples were available, and plasma and CSF lipoproteins differ in composition and metabolism. We precipitated for apoB-containing lipoproteins to narrow our investigations to lipoproteins in the range of densities found in the central nervous system. HDL-like particles found in the brain contain apoE along with apoA-I, but no apoB [43]. Nevertheless, CSF biomarkers are of limited use in large-scale studies and in routine clinical practice, and enormous interest in the identification of blood markers related to AD exists. It is a limitation of our study that we do not formally test whether the different apolipoproteins are present on separate lipoprotein particles or if they are bound to each other and carry phospholipid for the constellation of a lipoprotein. Strength of this study is the uniquely detailed assessment of neurological status and the assessment of an unprecedented variety of apolipoproteins and their subspecies as well as the comprehensive control for potential confounders.
5. Conclusion
In this selective sample of elderly participants lacking dementia, overall apoE concentrations were associated with AD pathology. Subspecies of apoE and apoA-I were differentially associated with neuropathology. The relations were generally similar after excluding participants with MCI or APOE ε4 carrier. Given the observed differential associations of apolipoproteins and their subspecies, further study of these in relation to AD pathology in younger populations and in relation to AD and cognitive decline in larger prospective samples is needed.
Research in Context.
-
1.
Systematic review: PubMed search identified several genome-wide association studies that linked genetic variation in regions that encode apolipoproteins, including apoE, apoJ, apoC-III, and apoA-I, to Alzheimer's disease and few studies examining plasma concentrations of apolipoproteins and Alzheimer's disease.
-
2.
Interpretation: Apolipoproteins may be important minimally invasive biomarkers indicative of Alzheimer's pathology. Subspecies of apoA-I containing either apoJ or apoC-III might be more closely related to hippocampal volume than overall apoA-I.
-
3.
Future directions: Given the observed differential associations of apolipoproteins and their subspecies, further study of these in relation to Alzheimer's disease pathology in younger populations and in relation to Alzheimer's disease and cognitive decline in larger prospective samples is needed.
Acknowledgments
We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible.
Author disclosures: All authors report no disclosures.
Funding: The study was supported by U01 AT000162 from the National Center for Complementary and Alternative Medicine (NCCAM) and the Office of Dietary Supplements, and the study received support from the National Institute on Aging, National Heart, Lung, and Blood Institute; the University of Pittsburgh Alzheimer's Disease Research Center (P50AG05133); the Roena Kulynych Center for Memory and Cognition Research; and National Institute of Neurological Disorders and Stroke. Samples from the National Cell Repository for AD (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. This work was supported by the NIH/NINDS (1R01NS089638-01A1). Manja Koch is a recipient of a Postdoctoral Research Fellowship from the German Research Foundation (Deutsche Forschungsgemeinschaft [DFG], KO 5187/1-1). The funding sources had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.dadm.2018.07.001.
Supplementary data
References
- 1.Sadigh-Eteghad S., Talebi M., Farhoudi M. Association of apolipoprotein E epsilon 4 allele with sporadic late onset Alzheimer`s disease. A meta-analysis. Neurosciences (Riyadh) 2012;17:321–326. [PubMed] [Google Scholar]
- 2.Lambert J.C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 3.Vollbach H., Heun R., Morris C.M., Edwardson J.A., McKeith I.G., Jessen F. APOA1 polymorphism influences risk for early-onset nonfamiliar AD. Ann Neurol. 2005;58:436–441. doi: 10.1002/ana.20593. [DOI] [PubMed] [Google Scholar]
- 4.Sun Y., Shi J., Zhang S., Tang M., Han H., Guo Y. The APOC3 SstI polymorphism is weakly associated with sporadic Alzheimer's disease in a Chinese population. Neurosci Lett. 2005;380:219–222. doi: 10.1016/j.neulet.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 5.Koch M., Jensen M.K. HDL-cholesterol and apolipoproteins in relation to dementia. Curr Opin Lipidol. 2016;27:76–87. doi: 10.1097/MOL.0000000000000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llano D.A., Devanarayan V., Simon A.J. Evaluation of plasma proteomic data for Alzheimer disease state classification and for the prediction of progression from mild cognitive impairment to Alzheimer disease. Alzheimer Dis Assoc Disord. 2013;27:233–243. doi: 10.1097/WAD.0b013e31826d597a. [DOI] [PubMed] [Google Scholar]
- 7.Schilling S., Tzourio C., Soumare A., Kaffashian S., Dartigues J.F., Ancelin M.L. Differential associations of plasma lipids with incident dementia and dementia subtypes in the 3C Study: A longitudinal, population-based prospective cohort study. PLoS Med. 2017;14:e1002265. doi: 10.1371/journal.pmed.1002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaisar T., Pennathur S., Green P.S., Gharib S.A., Hoofnagle A.N., Cheung M.C. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–756. doi: 10.1172/JCI26206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roheim P.S., Carey M., Forte T., Vega G.L. Apolipoproteins in human cerebrospinal fluid. Proc Natl Acad Sci U S A. 1979;76:4646–4649. doi: 10.1073/pnas.76.9.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koch M., Furtado J.D., Falk K., Leypoldt F., Mukamal K.J., Jensen M.K. Apolipoproteins and their subspecies in human cerebrospinal fluid and plasma. Alzheimers Dement (Amst) 2017;6:182–187. doi: 10.1016/j.dadm.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeKosky S.T., Fitzpatrick A., Ives D.G., Saxton J., Williamson J., Lopez O.L. The Ginkgo Evaluation of Memory (GEM) study: design and baseline data of a randomized trial of Ginkgo biloba extract in prevention of dementia. Contemp Clin Trials. 2006;27:238–253. doi: 10.1016/j.cct.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Mathis C.A., Kuller L.H., Klunk W.E., Snitz B.E., Price J.C., Weissfeld L.A. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Ann Neurol. 2013;73:751–761. doi: 10.1002/ana.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y., Brady M., Smith S. Segmentation of brain MR images through a hidden Markov random field model and the expectation-maximization algorithm. IEEE Trans Med Imaging. 2001;20:45–57. doi: 10.1109/42.906424. [DOI] [PubMed] [Google Scholar]
- 14.Lopez O.L., Klunk W.E., Mathis C., Coleman R.L., Price J., Becker J.T. Amyloid, neurodegeneration, and small vessel disease as predictors of dementia in the oldest-old. Neurology. 2014;83:1804–1811. doi: 10.1212/WNL.0000000000000977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y., Storey P., Cohen B.A., Epstein L.G., Edelman R.R., Ragin A.B. Diffusion alterations in corpus callosum of patients with HIV. AJNR Am J Neuroradiol. 2006;27:656–660. [PMC free article] [PubMed] [Google Scholar]
- 16.Carmichael O.T., Aizenstein H.A., Davis S.W., Becker J.T., Thompson P.M., Meltzer C.C. Atlas-based hippocampus segmentation in Alzheimer's disease and mild cognitive impairment. Neuroimage. 2005;27:979–990. doi: 10.1016/j.neuroimage.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teng E.L., Chui H.C. The Modified Mini-Mental State (3MS) examination. J Clin Psychiatry. 1987;48:314–318. [PubMed] [Google Scholar]
- 18.Snitz B.E., O'Meara E.S., Carlson M.C., Arnold A.M., Ives D.G., Rapp S.R. Ginkgo biloba for preventing cognitive decline in older adults: A randomized trial. JAMA. 2009;302:2663–2670. doi: 10.1001/jama.2009.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snitz B.E., Weissfeld L.A., Lopez O.L., Kuller L.H., Saxton J., Singhabahu D.M. Cognitive trajectories associated with beta-amyloid deposition in the oldest-old without dementia. Neurology. 2013;80:1378–1384. doi: 10.1212/WNL.0b013e31828c2fc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertram L., Tanzi R.E. Genome-wide association studies in Alzheimer's disease. Hum Mol Genet. 2009;18:R137–R145. doi: 10.1093/hmg/ddp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holtzman D.M. Role of apoe/Abeta interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J Mol Neurosci. 2001;17:147–155. doi: 10.1385/JMN:17:2:147. [DOI] [PubMed] [Google Scholar]
- 22.Zlokovic B.V., Deane R., Sallstrom J., Chow N., Miano J.M. Neurovascular pathways and Alzheimer amyloid beta-peptide. Brain Pathol. 2005;15:78–83. doi: 10.1111/j.1750-3639.2005.tb00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Q., Lee C.Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnes S.R., Ng T.S., Montagne A., Law M., Zlokovic B.V., Jacobs R.E. Optimal acquisition and modeling parameters for accurate assessment of low Ktrans blood-brain barrier permeability using dynamic contrast-enhanced MRI. Magn Reson Med. 2016;75:1967–1977. doi: 10.1002/mrm.25793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta V.B., Wilson A.C., Burnham S., Hone E., Pedrini S., Laws S.M. Follow-up plasma apolipoprotein E levels in the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing (AIBL) cohort. Alzheimers Res Ther. 2015;7:16. doi: 10.1186/s13195-015-0105-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thambisetty M., Tripaldi R., Riddoch-Contreras J., Hye A., An Y., Campbell J. Proteome-based plasma markers of brain amyloid-beta deposition in non-demented older individuals. J Alzheimers Dis. 2010;22:1099–1109. doi: 10.3233/JAD-2010-101350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Morillo E., Hansson O., Atagi Y., Bu G., Minthon L., Diamandis E.P. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer's disease patients and controls. Acta Neuropathol. 2014;127:633–643. doi: 10.1007/s00401-014-1266-2. [DOI] [PubMed] [Google Scholar]
- 28.Gupta V.B., Laws S.M., Villemagne V.L., Ames D., Bush A.I., Ellis K.A. Plasma apolipoprotein E and Alzheimer disease risk: the AIBL study of aging. Neurology. 2011;76:1091–1098. doi: 10.1212/WNL.0b013e318211c352. [DOI] [PubMed] [Google Scholar]
- 29.Teng E., Chow N., Hwang K.S., Thompson P.M., Gylys K.H., Cole G.M. Low plasma ApoE levels are associated with smaller hippocampal size in the Alzheimer's disease neuroimaging initiative cohort. Demen Geriatr Cogn Disord. 2015;39:154–166. doi: 10.1159/000368982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song F., Poljak A., Crawford J., Kochan N.A., Wen W., Cameron B. Plasma apolipoprotein levels are associated with cognitive status and decline in a community cohort of older individuals. PLoS One. 2012;7:e34078. doi: 10.1371/journal.pone.0034078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Dijk E.J., Prins N.D., Vermeer S.E., Hofman A., van Duijn C.M., Koudstaal P.J. Plasma amyloid beta, apolipoprotein E, lacunar infarcts, and white matter lesions. Ann Neurol. 2004;55:570–575. doi: 10.1002/ana.20050. [DOI] [PubMed] [Google Scholar]
- 32.Hughes T.M., Lopez O.L., Evans R.W., Kamboh M.I., Williamson J.D., Klunk W.E. Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol Aging. 2014;35:802–807. doi: 10.1016/j.neurobiolaging.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linton M.F., Gish R., Hubl S.T., Butler E., Esquivel C., Bry W.I. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88:270–281. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stukas S., Robert J., Lee M., Kulic I., Carr M., Tourigny K. Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J Am Heart Assoc. 2014;3:e001156. doi: 10.1161/JAHA.114.001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thambisetty M., Simmons A., Velayudhan L., Hye A., Campbell J., Zhang Y. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry. 2010;67:739–748. doi: 10.1001/archgenpsychiatry.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thambisetty M., An Y., Kinsey A., Koka D., Saleem M., Guntert A. Plasma clusterin concentration is associated with longitudinal brain atrophy in mild cognitive impairment. Neuroimage. 2012;59:212–217. doi: 10.1016/j.neuroimage.2011.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta V.B., Doecke J.D., Hone E., Pedrini S., Laws S.M., Thambisetty M. Plasma apolipoprotein J as a potential biomarker for Alzheimer's disease: Australian Imaging, Biomarkers and Lifestyle study of aging. Alzheimers Dement (Amst) 2016;3:18–26. doi: 10.1016/j.dadm.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shih Y.H., Tsai K.J., Lee C.W., Shiesh S.C., Chen W.T., Pai M.C. Apolipoprotein C-III is an amyloid-beta-binding protein and an early marker for Alzheimer's disease. J Alzheimers Dis. 2014;41:855–865. doi: 10.3233/JAD-140111. [DOI] [PubMed] [Google Scholar]
- 39.Paula-Lima A.C., Tricerri M.A., Brito-Moreira J., Bomfim T.R., Oliveira F.F., Magdesian M.H. Human apolipoprotein A-I binds amyloid-beta and prevents Abeta-induced neurotoxicity. Int J Biochem Cell Biol. 2009;41:1361–1370. doi: 10.1016/j.biocel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 40.Lewis T.L., Cao D., Lu H., Mans R.A., Su Y.R., Jungbauer L. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem. 2010;285:36958–36968. doi: 10.1074/jbc.M110.127829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pascoal T.A., Dadar M., Manitsirikul S., Breitner J.C.S., Collins D.L., Poirier J. Association between apolipoprotein a-i levels and white matter hyperintensities depends on CSF tau levels in a high-risk cohort of aging cognitively normal persons: The prevent-alzheimer's disease study. Alzheimers Dement. 2015;11:P103. [Google Scholar]
- 42.Yin Z.G., Li L., Cui M., Zhou S.M., Yu M.M., Zhou H.D. Inverse relationship between apolipoprotein A-I and cerebral white matter lesions: a cross-sectional study in middle-aged and elderly subjects. PLoS One. 2014;9:e97113. doi: 10.1371/journal.pone.0097113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pitas R.E., Boyles J.K., Lee S.H., Hui D., Weisgraber K.H. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E (LDL) receptors in the brain. J Biol Chem. 1987;262:14352–14360. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.