

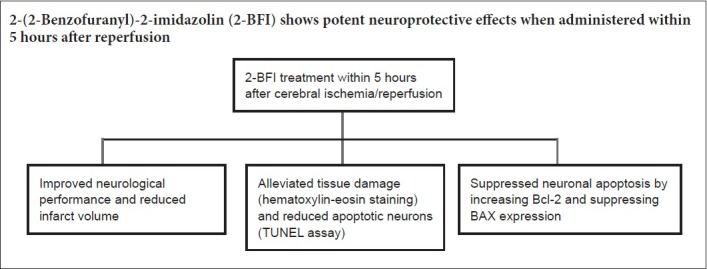
Keywords: nerve regeneration, ischemia/reperfusion, 2-(2-benzofuranyl)-2-imidazoline, neuroprotection, time window, apoptosis, Bcl-2, BAX, neural regeneration
Abstract
We previously demonstrated that administering 2-(2-benzofuranyl)-2-imidazolin (2-BFI), an imidazoline I2 receptor agonist, immediately after ischemia onset can protect the brain from ischemic insult. However, immediate administration after stroke is difficult to realize in the clinic. Thus, the therapeutic time window of 2-BFI should be determined. Sprague-Dawley rats provided by Wenzhou Medical University in China received right middle cerebral artery occlusion for 120 minutes, and were treated with 2-BFI (3 mg/kg) through the caudal vein at 0, 1, 3, 5, 7, and 9 hours after reperfusion. Neurological function was assessed using the Longa's method. Infarct volume was measured by 2,3,5-triphenyltetrazolium chloride assay. Morphological changes in the cortical penumbra were observed by hematoxylin-eosin staining under transmission electron microscopy. The apoptosis levels in the ipsilateral cortex were examined with terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay. The protein expression of Bcl-2 and BAX was detected using immunohistochemistry. We found the following: Treatment with 2-BFI within 5 hours after reperfusion obviously improved neurological function. Administering 2-BFI within 9 hours after ischemia/reperfusion decreased infarct volume and alleviated apoptosis. 2-BFI administration at different time points after reperfusion alleviated the pathological damage of the ischemic penumbra and reduced the number of apoptotic neurons, but the protective effect was more obvious when administered within 5 hours. Administration of 2-BFI within 5 hours after reperfusion remarkably increased Bcl-2 expression and decreased BAX expression. To conclude, 2-BFI shows potent neuroprotective effects when administered within 5 hours after reperfusion, seemingly by up-regulating Bcl-2 and down-regulating BAX expression. The time window provided clinical potential for ischemic stroke by 2-BFI.
Introduction
Ischemic stroke is one of the leading causes of death and long-term disability worldwide. At present, reperfusion with either thrombolysis with recombinant tissue plasminogen activator or mechanical thrombectomy is the only effective treatment for acute ischemic stroke. However, its clinical efficacy is limited by the fact that it must be applied within a narrow time window after stroke and it increases risk of bleeding (Hafeez et al., 2007; Zhang et al., 2011; Baltsavias et al., 2015). As a result, continued efforts have been made to investigate various potential neuroprotective strategies to mitigate or eliminate ischemic injury, but no alternatives have yet been validated in the clinic.
Ischemia causes rapid loss of energy stores and generalized depolarization; this induces glutamate release and a large increase of cytosolic Ca2+, which triggers apoptosis (White et al., 2000). Thus, an obvious strategy to protect against ischemic injury would be to inhibit N-methyl-D-aspartic acid receptor (NMDAR)-mediated Ca2+ influx, but generalized and lasting NMDAR inhibition results in side effects that strongly restrain its clinical application. It has been suggested that chemical compounds that transiently and reversibly block NMDARs have the greatest benefit potential to mitigate excitotoxicity-evoked brain damage (Chen et al., 2017).
Accumulating evidence suggests that imidazoline I2 receptor (I2R) ligands have various biological functions, including effective analgesia for inflammatory pain and neuroprotection (Bousquet et al., 1998; Zhu et al., 2015). We have shown in vitro and in vivo that 2-(2-benzofu-ranyl)-2-imidazoline (2-BFI) is the most effective I2R ligand for protecting neurons from ischemic damage and that it works by directly modulating NMDAR-mediated Ca2+ influx into neurons (Han et al., 2009, 2012, 2013; Thorn et al., 2016). 2-BFI triggers reversible blockade of Ca2+ influx with a relatively fast off-rate, similarly to memantine (Han et al., 2013). This suggests that 2-BFI may trigger fewer adverse effects in cognition analogously to memantine, which only works when agonist levels are pathologically high. Indeed, memantine has been clinically approved as a treatment for dementia and it is being tested in clinical trials as a treatment for vascular dementia and stroke (Orgogozo et al., 2002; Matsunaga et al., 2015). It is in great need to clarify whether 2-BFI offers the same advantages as memantine.
Prior to such work, however, it is important to determine whether 2-BFI can exert clinically useful neuroprotective effects when given up to several hours after injury. If not, the clinical usefulness of 2-BFI may be severely limited, since it is usually impossible to administer stroke medications immediately after injury. Previous studies of 2-BFI have assessed its efficacy only after immediate administration. Thus, in the present study, we examined whether 2-BFI administered with various delays after stroke onset can protect against ischemic injury in rats. Our results will help establish the potential of 2-BFI for clinical translation and begin to elucidate the drug's neuroprotective mechanism of action.
Materials and Methods
Animals
A total of 90 adult clean male Sprague-Dawley rats aged 3–4 months and weighing 250–270 g were obtained from Wenzhou Medical College, Wenzhou, China (SYXK (Zhe) 2007-0005). For 1 week prior to surgery, rats were housed under a 12-hour light/dark cycle and allowed free access to food and water. All experimental procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication 80-23) and the Animal Use Guidelines of Wenzhou Medical University (wydw2016-0200). Every effort was made to minimize the number of animals used and their suffering. The rats were randomly assigned to eight groups: sham, 0-hour, 1-hour, 3-hour, 5-hour, 7-hour, 9-hour, and middle cerebral artery occlusion (MCAO) without treatment.
MCAO-induced ischemia and 2-BFI intervention
Rats were temporarily anesthetized using 10% chloral hydrate (Sheng Yang Ze Run Company, Beijing, China). The right middle cerebral artery was then occluded for 120 minutes using an intraluminal filament (Longa et al., 1989), with minor modifications. During and after surgery, animal body temperature was monitored using a rectal probe and maintained at 37°C using a heating pad and lamp. Sham rats (n = 6) underwent surgery but not occlusion. The MCAO group (n = 12) underwent occlusion and received the same amount of saline. Treated rats received a bolus injection of 2-BFI (3 mg/kg; Tocris Bioscience, Bristol, UK) through the tail vein at 0, 1, 3, 5, 7, or 9 hours after reperfusion (n = 12 each). Our previous study has shown that this dose of 2-BFI can efficiently protect against transient ischemia when administered immediately after ischemia (Zhao et al., 2008).
Assessment of neurological deficit
Thirty minutes before MCAO and 22 hours after reperfusion, beam balance performance was measured, as described previously (Schäbitz et al., 2004). At least three measurements were taken for each animal at each time point, and the mean and standard error were calculated. At 22 hours after occlusion, neurological deficit was measured using an expanded six-point scale (Longa et al., 1989). The balance and Longa scores were added to obtain one overall score for each rat. All assessments were performed by investigators blinded to the control or treatment. Average scores were used for statistical analysis.
Measurement of infarct volume
At 22 hours after the reperfusion, rats were sacrificed using 10% chloral hydrate. Coronal brain sections (2 mm thick) were prepared, and stained at 37°C for 20 minutes with a 4% solution of 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, USA). The sections were examined using computer-assisted Image-Pro Plus analysis (Media Cybernetics Company, Rockville, MD, USA) and standard techniques to determine infarct surface area. To minimize error introduced by edema, infarct volume in the ischemic region was normalized to the volume of the non-ischemic region and expressed as a percentage of contralateral brain volume.
Brain histopathology assay
At 22 hours after occlusion, rats were anesthetized with 10% chloral hydrate and perfused transcardially with saline, followed by 4% formaldehyde in phosphate-buffered saline (PBS). Brains were removed, post-fixed in 4% formaldehyde (pH 7.4) for 18 hours and cryoprotected in 30% sucrose for 36 hours at 4°C. Coronal sections (10 μm thick) were cut using a cryostat (Leica Microsystems, Wetzlar, Germany), mounted on superfrost slides (Fisher Scientific, Toronto, Canada), and stored at −80°C. Slides were stained with hematoxylin and eosin and examined under a light microscope (Leica Microsystems).
Electron microscopy
Brain tissue was fixed for 2 hours in 3% glutaraldehyde buffered in sodium cacodylate (pH 7.4) and post-fixed in 1% osmium tetroxide in cacodylate buffer, dehydrated in graded ethanol solutions, and embedded in Araldite. Thin sections were cut on a Reichert-Jung Ultracut ultramicrotome, mounted on 300-mesh copper grids, and stained in saturated uranyl acetate in alcohol for 10 minutes and lead citrate for 8 minutes. Sections were examined using a transmission electron microscope (H-600A-2; Hitachi, Tokyo, Japan).
TdT-mediated dUTP nick end-labeling (TUNEL) assay
Fixed frozen brain sections were permeabilized in 0.1% Triton X-100 for 15 minutes. After washing in PBS, sections were incubated at 37°C for 1 hour in a 50-μL solution containing 1× TdT buffer (catalog No. 16314015; Life Technologies, Rockville, MD, USA), 11.1 μM biotin-dUTP (catalog No. B32766; Life Technologies), and 20 U of TdT (Life Technologies). The sections were blocked with 2% bovine serum albumin in PBS, incubated in streptavidin-horseradish peroxidase (1:4000; Haoran Biotech Co., Shanghai, China) for 30 minutes, and stained with 3,3′-diaminobenzidine (1:9; Beijing Zhongshan Jinqiao Co., Beijing, China) for 15 minutes. The coverslips were then mounted using mounting medium and examined under an optical microscope (Leica, Oskar-Barnack-Straße, Germany).
Immunohistochemistry
Apoptosis-related proteins, Bcl-2 and BAX, were mainly expressed in the cytoplasm of neurons and were detected using immunohistochemistry (Yenari and Hemmen, 2010). Immunohistochemistry was performed as previously described (Han et al., 2012). Sections were brought to room temperature, washed with PBS, and incubated in 3% H2O2 in PBS to quench endogenous peroxidase. The sections were blocked for 1 hour with 5% bovine serum albumin in PBS, and incubated overnight at 4°C with rabbit anti-rat Bcl-2 monoclonal antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, TX, USA) or rabbit anti-rat BAX monoclonal antibody (1:100; Santa Cruz Biotechnology). Finally, sections were incubated with secondary antibody (goat anti-rabbit IgG-B 1:500; Santa Cruz Biotechnology) for 20 minutes at 37°C, and visualized with diaminobenzidine (Santa Cruz Biotechnology). The samples were observed under an optical microscope (Leica Microsystems).
Statistical analysis
Results are expressed as the mean ± SE. Statistical analyses were performed using SPSS 17.0 (IBM SPSS, Shanghai, China). Differences between treatment and MCAO groups were assessed for significance using paired t-tests. A value of P < 0.05 was considered as statistically significant.
Results
2-BFI alleviates the neurological impairment in cerebral ischemia/reperfusion rats
After MCAO, rats showed signs of neurological impairment, such as retracting the left forepaw, circling to the left, and/or falling when walking, while the sham group showed no sign of neurological deficits. Treatment with 2-BFI improved neurological outcomes, and the extent of improvement depended on when the drug was administered; the earlier 2-BFI was given after reperfusion, the better the behavioral outcome (Figure 1). At 22 hours after MCAO, neurological deficit scores were significantly lower in rats treated with 2-BFI within 5 hours after reperfusion than those in MCAO rats (all P < 0.05; Figure 1A).
Figure 1.
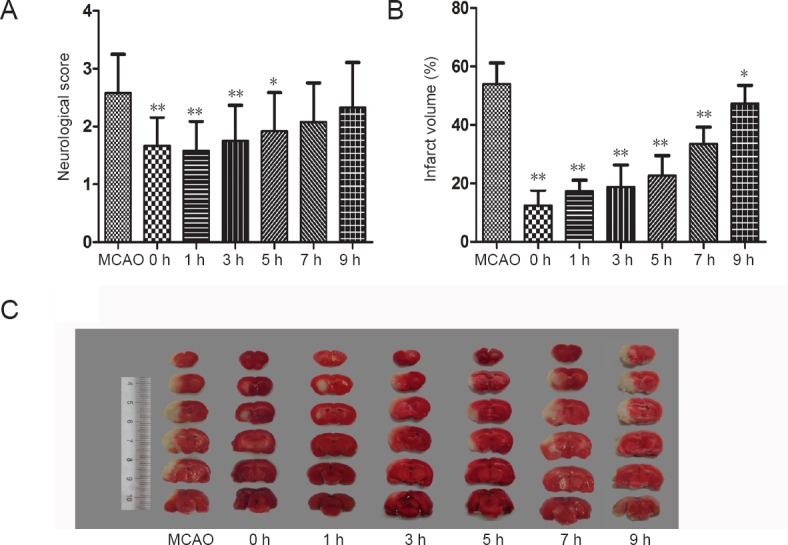
Time window for 2-BFI therapeutic efficacy against cerebral ischemia/reperfusion in rat models.
Rats were subjected to 2-hour MCAO followed by reperfusion. At 0, 1, 3, 5, 7, or 9 hours after reperfusion, 2-BFI (3 mg/kg) was injected intravenously and rats were killed at 22 hours of reperfusion. (A) Neurological deficits were less apparent in rats treated with 2-BFI (n = 12) than rats in the MCAO group. Neurological deficit was measured using an expanded 6-point scale. (B) Percent reduction in infarct volume was significantly greater in rats treated with 2-BFI (n = 12). (C) 2,3,5-Triphenyl-2H-tetrazolium chloride staining of coronal brain slices indicated the reduction in brain infarction (whitish area on the left hemisphere). Data are expressed as the mean ± SE (paired t-test). *P < 0.05, **P < 0.01, vs. MCAO group. 2-BFI: 2-(2-Benzofu-ranyl)-2-imidazoline; MCAO: middle cerebral artery occlusion; h: hour(s).
However, according to behavioral measures, 2-BFI administered at 7 and 9 hours after reperfusion had no protective effect (Figure 1A). Brain infarct volume compared with the contralateral side was significantly smaller in rats treated with 2-BFI than that of in the MCAO group (Figure 2). The protective effect of 2-BFI was greater when it was administered earlier after injury (Figure 1B, C). The infarct volumes were reduced by 2-BFI after reperfusion by 77.1%, 67.8%, 65.4%, 59.2%, 38.2%, and 12.4% at 0, 1, 3, 5, 7, and 9 hours, respectively, compared with the MCAO group (all P < 0.05; Figure 1B).
Figure 2.

Histology and ultrastructural analysis of mitigation of neuronal injury in cerebral ischemia/reperfusion rats by 2-BFI.
Rats were subjected to 2-hour MCAO followed by reperfusion. At 0, 1, 3, 5, 7, or 9 hours after reperfusion, 2-BFI (3 mg/kg) was injected intravenously and rats were killed at 22 hours of reperfusion. (A) Brains were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. MCAO led to pyknosis, homogeneous cytoplasm, and increased numbers of vacuoles. 2-BFI treatment mitigated these negative effects. Arrowheads indicate vacuoles. (B) Neurons from the peri-infarct cortex of the ipsilateral side were examined using electron microscopy. The double nuclear membrane is clearly visible in the boxed area in sham rats, and there is no condensed chromatin next to the membrane. Injury to the neurons causes thickened membrane and condensed chromatin along membrane. Neurons from the brains exposed to MCAO show a thickened nuclear membrane and condensed chromatin (white arrowheads). In rats given 2-BFI within 9 hours after injury, the membrane was less thick and there were fewer chromatin deposits near the nuclear membrane. 2-BFI: 2-(2-Benzofu-ranyl)-2-imidazoline; MCAO: middle cerebral artery occlusion; h: hour(s). Scale bars: 10 μm.
2-BFI treatment reduces brain cell death in cerebral ischemia/reperfusion rats
Hematoxylin-eosin staining was used to determine the severity of neuronal injury in the ischemic penumbra after ischemia/reperfusion (Han et al., 2012). In sham-operated rats, cortical neurons had an orderly arrangement, distinct outlines, and a compact structure (Figure 2A). In contrast, the ischemic penumbra of MCAO rats showed numerous, irregularly arranged eosinophilic neurons with homogeneous cytoplasm that lacked subcellular structures and/or that showed a triangulated karyopyknosis. 2-BFI treatment at all time points appeared to alleviate these effects, with the greatest protection observed when the drug was administered within 5 hours after reperfusion (Figure 2A).
Electron microscopy revealed that the cortical neurons of sham-operated rats had an orderly arrangement, clear outlines, and a compact structure. The electronic density of the nucleus and cytoplasm was even, nuclear membranes were clear, and chromatins were well distributed. In the MCAO and 2-BFI treatment groups, the ultrastructure showed condensed chromatin arranged along the nuclear membranes, thickened nuclear membranes, and uneven electronic density of the nucleus and cytoplasm (Figure 2B). 2-BFI treatment reduced the numbers of condensed chromatins and alleviated thickened nuclear membrane at all time points. Good protection was observed in groups that received 2-BFI within 7 hours after reperfusion (Figure 2B).
To determine the DNA injury of the neurons in penumbra area after focal brain ischemia/reperfusion, TUNEL staining was performed in paraffin sections (Han et al., 2010). A few apoptotic neurons were observed in sham-operated rats, while markedly increased apoptotic neurons were found in the penumbra area of both MCAO rats and all 2-BFI treated rats (Figure 3). However, 2-BFI used within 5 hours after reperfusion remarkably reduced the number of TUNEL-positive cells compared with the MCAO group (all P < 0.01; Figure 3B). The protective effects could still be detected when 2-BFI was administered at 7 and 9 hours after reperfusion (all P < 0.05; Figure 3B).
Figure 3.

Effects of 2-BFI on apoptosis in cerebral ischemia/reperfusion rats.
Rats were subjected to 2-hour MCAO followed by reperfusion. At 0, 1, 3, 5, 7, or 9 hours after reperfusion, 2-BFI (3 mg/kg) was injected intravenously and rats were killed at 22 hours of reperfusion. (A) Frozen rat brains were sectioned and fixed for TUNEL as described in the Methods. Arrowheads indicate TUNEL-positive cells on the ischemic side of the brain. These cells were counted in randomly selected fields under a 40× objective. The number of TUNEL-positive cells (brown) was obviously higher than in sham-treated rats, and 2-BFI reversed this increase in a time-dependent manner. Scale bar: 10 μm. (B) Quantitative analysis of the number of TUNEL-positive cells/40-fold field. The number was markedly smaller in 2-BFI-treated rats than in control rats, particularly in rats that received 2-BFI within 5 hours after injury. Data are expressed as the mean ± SE (n = 6 per group; paired t-test). *P < 0.05, **P < 0.01, vs. MCAO group. 2-BFI: 2-(2-Benzofu-ranyl)-2-imidazoline; MCAO: middle cerebral artery occlusion; TUNEL: terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling; h: hour(s).
2-BFI treatment up-regulates Bcl-2 expression and down-regulates BAX expression in the ischemic brain of cerebral ischemia/reperfusion rats
In the sham-operated rats, few Bcl-2- and BAX-positive cells were detected. More Bcl-2- and BAX-positive cells were observed in the MCAO and 2-BFI treatment groups (Figure 4A and B). Moderated Bcl-2-positive cells were found in the peri-ischemic brain tissue in the MCAO group and rats treated with 2-BFI after 5 hours. However, the number of Bcl-2-positive cells in rats treated with 2-BFI was significantly higher than in the MCAO group when the compound was given within 5 hours after reperfusion, but similar to the MCAO group when the compound was given at 7 hours or later (all P > 0.05; Figure 4C). Similar numbers of Bcl-2-positive cells were observed when 2-BFI was administered at 0 or 1 hour after reperfusion.
Figure 4.
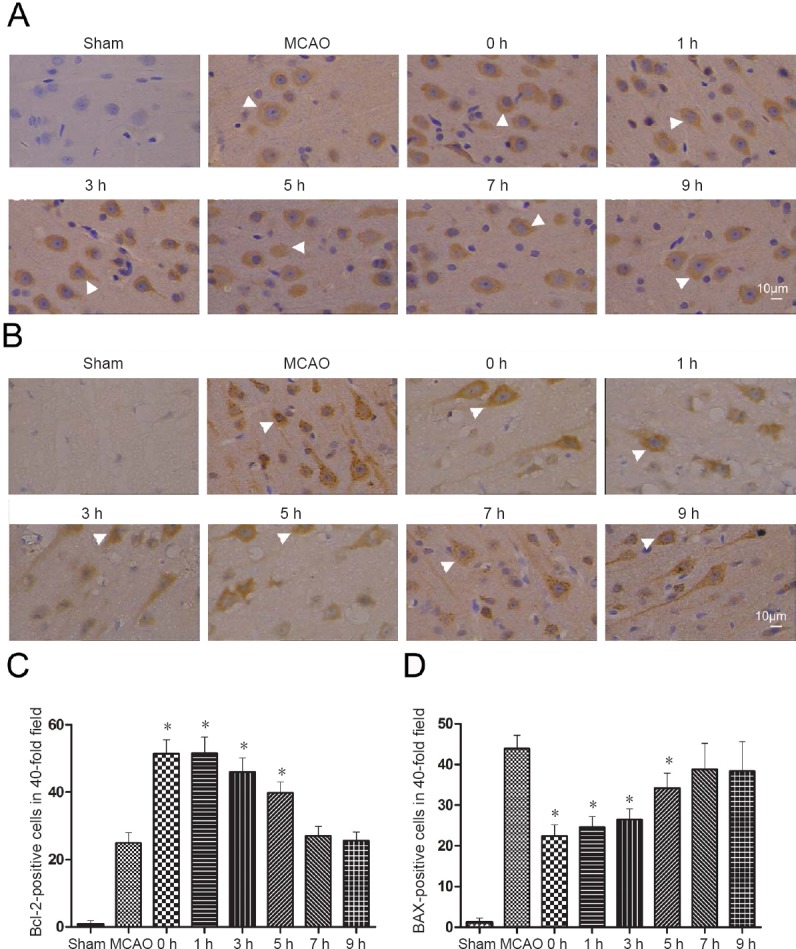
Modulation of apoptosis-related proteins by 2-BFI in cerebral ischemia/reperfusion rats.
Rats were subjected to 2 hours MCAO followed by reperfusion. At 0, 1, 3, 5, 7, or 9 hours after reperfusion, 2-BFI (3 mg/kg) was injected intravenously and rats were killed at 22 hours after reperfusion. (A) Representative micrographs of tissue immunostained against Bcl-2 (white arrowheads indicate Bcl-2-positive neurons in the ischemic penumbra). (B) Representative micrographs of tissue immunostained against BAX (white arrowheads indicate BAX-positive neurons in the ischemic penumbra). Scale bars: 10 μm. (C) Numbers of Bcl-2-positive neurons in randomly selected fields of brain slices under a 40× objective. The number of positive neurons was higher in the MCAO rats than in sham-operated rats (n = 5 for each group). (D) Numbers of BAX-positive cells in randomly selected fields of brain slices under a 40× objective. The number of positive neurons was higher in the MCAO rats than in sham-operated rats. Data are expressed as the mean ± SE (n = 6 per group; paired t-test). *P < 0.05, vs. MCAO group. 2-BFI: 2-(2-Benzofu-ranyl)-2-imidazoline; MCAO: middle cerebral artery occlusion; h: hour(s).
The opposite results were observed in BAX-positive cells. The MCAO group showed a remarkable increase of BAX-positive cells in the peri-ischemic brain tissue. Of the rats subjected to occlusion, rats treated with 2-BFI within 5 hours showed significantly fewer BAX-positive cells than the MCAO group (all P < 0.05). The rats treated with 2-BFI at 7 and 9 hours also showed a lower number of BAX-positive cells, but this difference was not significant (Figure 4D).
Discussion
Research on neuroprotective compounds that target NMDAR has been a high research priority for decades. The greatest challenge is to selectively block NMDAR-mediated neuronal toxicity while reserving the normal physiological functions of the glutamate receptor. Memantine, a noncompetitive NMDAR antagonist, is widely used in the clinic (Vogels et al., 1997; Lipton, 2006). The I2R ligand 2-BFI reversibly blocks NMDAR in a way similar to memantine (Jiang et al., 2010; Han et al., 2013), thereby protecting the brain from ischemic insult (Gustafson et al., 1990; Maiese et al., 1992; Han et al., 2009). Previous studies have focused only on the neuroprotective effects of 2-BFI administered immediately after ischemia onset, which is rarely feasible in clinical settings (Gustafson et al., 1990; Maiese et al., 1992; Han et al., 2009). To successfully translate 2-BFI from the laboratory to the clinic, it is necessary to determine whether the drug is effective when given with a certain delay after stroke onset. The findings of our study support our hypothesis that 2-BFI can effectively protect neurons from ischemic injury when administered within 5 hours after reperfusion. Under these conditions, the drug was associated with better neurological motor function, greater reduction in infarct volume, and less apoptosis at 22 hours after reperfusion compared with the MCAO group. However, initiating 2-BFI treatment after 5 hours conferred very limited protection. Further studies showed that the observed anti-apoptotic effect of 2-BFI is strongly related to the down-regulation of pro-apoptotic BAX and up-regulation of anti-apoptotic Bcl-2 expression.
In the intraluminal rat model of MCAO, ischemia triggers hypoxic injury and necrosis, which leads to varying degrees of neurological deficit in the contralateral limb. Brain injury is then exacerbated by reperfusion which involves complex pathophysiological processes, including apoptosis, necrosis, and ischemia surrounding the ischemic core area, as well as complex changes in gene expression (Friedlander, 2003). The present study shows that 2-BFI can remarkably improve neurological performance of rats and reduce infarct volume. Our findings are in accordance with previous findings that 2-BFI and another I2R ligand, idazoxan, can reduce infarct volume, improve neurological deficits, and decrease the volume of cell death in the penumbra after transient focal cerebral ischemia (Maiese et al., 1992; Han et al., 2009, 2012). To our knowledge, this is the first study to elucidate the effective therapeutic time window of 2-BFI for ischemic injury. Our findings suggest that drug administration with a certain delay can still provide neuroprotection against ischemic stroke, which increases its potential for clinical use, where delays are common. Although preclinical studies have examined at least 1026 neuroprotective strategies, none have resulted in approved stroke treatments (Sutherland et al., 2012). One important reason for this failure is the lack of a clear therapeutic time window for neuroprotectants. In our study, 2-BFI used within 5 hours after reperfusion reduced neurological deficit and infarct volume in a time-dependent manner, whereby the earlier the administration of 2-BFI, the greater the protection. Nevertheless, we were still able to detect some protective effects even at 9 hours after reperfusion. This demonstrates the importance of starting 2-BFI treatment during the latent phase of recovery, before secondary deterioration with seizures and cytotoxic edema.
Our TUNEL staining results revealed that 2-BFI mitigates apoptosis after ischemia. Not only did 2-BFI remarkably decreased the numbers of apoptotic cells in the penumbra area, but levels of apoptosis also increased rapidly with longer delays. This finding is in accordance with the fact that neuroprotection therapies exert better effect when administrated earlier after stroke by earlier interrupting the cascade of biochemical events after ischemia. Although a single neuroprotective agent may not be enough to provide remarkable protection alone, the time window of 2-BFI efficacy means that it could potentially be combined with other protective treatments, such as thrombolysis, to protect the brain from ischemic insult.
Our previous study found that 2-BFI is a non-competitive inhibitor of NMDAR with a fast-off rate, and that, during excitotoxicity, it reduces intracellular overload of Ca2+ in neurons, which is mediated by NMDAR (Han et al., 2013). Excessive intracellular Ca2+ triggers intrinsic stimulated apoptosis through the mitochondrial signaling pathway (Liu et al., 2016). It is unclear when cell death becomes irreversible in the ischemic process. Apoptosis, a basic characteristic of all rat cells, is necessary for normal development and tissue homeostasis (Nagata, 1997; Henson and Hume, 2006). Two main pathways, the intrinsic and extrinsic pathways, contribute to post-ischemia apoptosis. The intrinsic pathway is thought to be triggered intracellularly by mitochondria dysfunction (Green and Reed, 1998), whereas the extrinsic pathway is induced through receptors on cell surfaces (Ashkenazi and Dixit, 1998). We hypothesize that 2-BFI may protect the brain by suppressing Ca2+ influx, thereby reducing the triggering of the intrinsic (mitochondrial) pathway of apoptosis. Simultaneously, 2-BFI may inhibit apoptosis by binding to I2R on the outer membrane of mitochondria (Regunathan et al., 1993), thereby inhibiting mitochondrial nitric oxide synthase and cytochrome c oxidase, which in turn alters energy production (Sastre et al., 1996). This pathway is similar to the intrinsic apoptotic pathway that leads to caspase activation and ultimately apoptosis.
While decreasing neurological deficit and infarct volume, 2-BFI also dramatically up-regulated Bcl-2 expression, which is consistent with our previous study (Han et al., 2010). In the present study, this up-regulation was seen even when 2-BFI was administered at 5 hours after reperfusion. These results suggest that 2-BFI exerts neuroprotective effects not only by transiently and reversibly binding to NMDA receptors, but also by stimulating anti-apoptotic pathways. Bcl-2, located mainly on the outer membrane of mitochondria, regulates the release of cytochrome c, a key component of apoptosis, from mitochondria (Yang et al., 1997; Shimizu et al., 1999). The expression of Bcl-2, which blocks translocation of apoptosis-inducing factor from the mitochondria to the nucleus, has been found to increase neuronal survival after focal brain infarction (Zhao et al., 2004; Hu et al., 2017). Our previous study also found that overexpression of Bcl-2 prevents hippocampal neuron death caused by NMDAR-mediated excitotoxicity (Wong et al., 2005) and that its expression can inhibit apoptosis of newborn neurons and reduce infarct volume after MCAO (Martinou et al., 1994; Zhang et al., 2006). In this study, Bcl-2 expression was up-regulated when 2-BFI was used within 5 hours after reperfusion, which was associated with better neurological outcomes, a smaller infarct volume, and reduced apoptosis level. These findings strengthen the argument that 2-BFI protects against ischemic damage by inhibiting apoptosis.
Similar to Bcl-2, the pro-apoptotic BAX plays a role in neuronal apoptosis, which is caused by various insults to neurons, including excitotoxicity, oxidative stress, and deprivation of trophic factors (Jin et al., 2016; Sun et al., 2017). BAX functions together with the Bcl-2 antagonist/killer (BAK) protein to trigger mitochondrial dysfunction, which is strongly associated with apoptosis (Wei et al., 2001). The balance between pro-apoptotic factors (e.g. BAK and BAX) and anti-apoptotic factors (e.g Bcl-2 and Bcl-XL) in the BCL-2 superfamily is finely regulated by the intrinsic apoptotic pathway. Our results revealed an inverse trend of Bcl-2 and BAX change with the delay of 2-BFI use, which is similar to the report that higher ratios of Bcl-2 to BAX promote neuronal survival after ischemia (Sulejczak et al., 2004). Our study is the first to report that 2-BFI affects BAX expression after ischemia, and that it simultaneously increases Bcl-2 and decreases BAX expression. The effects of 2-BFI on BAX expression were greatest when the compound was given within 3 hours after stroke, although effects were still measurable up to 5 hours after reperfusion.
Our study has some limitations. First, our animal model is a reperfusion model and 2-BFI may exert neuroprotection based on reperfusion. It is unclear if 2-BFI would have a similar effect on a brain without reperfusion. Our next step is to investigate the effect of 2-BFI on permanent ischemia. Second, the protection was observed at 24 hours after reperfusion and the long-term protective effect is still unclear. In clinical practice, long-term protection may be associated with the outcome of stroke. Further studies should investigate the effects of 2-BFI on the long-term recovery of stroke.
In conclusion, we proposed that 2-BFI exerts strong neuroprotective effects when administered up to 5 hours after reperfusion, and even stronger effects when given within 3 hours. Our study provided the optimal time window of 2-BFI use after infarction. Because of the time window, 2-BFI treatment could be potentially used in clinical practice. Further studies may investigate combination of 2-BFI with recanalization or other neuroprotective strategies.
Additional file: Open peer review reports 1 (12.8KB, pdf) and 2 (12.8KB, pdf) .
Acknowledgments
We thank Doctor Guo-Ling Xiao from Scientific Research Center, the Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University in China for revision of the manuscript and statistical analysis.
Footnotes
Conflicts of interest: The authors report no conflicts.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 81571114 and 81771267 (to ZH); the National Science Funds for Distinguished Youth Scholars of China, No. 81325007 (to XMJ); the Distinguished Professor of Cheung Kong Scholars Program in China, No. T2014251 (to XMJ); the Wenzhou Municipal Sci-Tec Bureau Programs in China, No. Y20120154 (to ZZ) and Y20140686 (to ZH); the Projects of International Cooperation and Exchanges National Natural Science Foundation of China, No. 81620108011 (to XMJ). The funders had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement: The study was approved by the Animal Use Guidelines of Wenzhou Medical University (wydw2016-0200).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Cheung-Ter Ong, Chia-Yi Christian Hospital, China; Ramon Na-varro, Cleveland Clinic Abu Dhabi, United Arab Emirates.
Funding: This study was supported by the National Natural Science Foundation of China, No. 81571114 and 81771267 (to ZH); the National Science Funds for Distinguished Youth Scholars of China, No. 81325007 (to XMJ); the Distinguished Professor of Cheung Kong Scholars Program in China, No. T2014251 (to XMJ); the Wenzhou Municipal Sci-Tec Bureau Programs in China, No. Y20120154 (to ZZ) and Y20140686 (to ZH); the Projects of International Cooperation and Exchanges National Natural Science Foundation of China, No. 81620108011 (to XMJ).
P-Reviewer: Ong CT, Navarro R; C-Editor: Zhao M; S-Editor: Yu J, Li CH; L-Editor: Cason N, Yajima W, Qiu Y, Song LP; T-Editor: Liu XL
References
- 1.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 2.Baltsavias G, Yella S, Al Shameri RA, Luft A, Valavanis A. Intra-arterial administration of papaverine during mechanical thrombectomy for acute ischemic stroke. J Stroke Cerebrovasc Dis. 2015;24:41–47. doi: 10.1016/j.jstrokecerebrovasdis.2014.07.052. [DOI] [PubMed] [Google Scholar]
- 3.Bousquet P, Dontenwill M, Greney H, Feldman J. I1-imidazoline receptors: an update. J Hypertens. 1998;Suppl 16:S1–5. [PubMed] [Google Scholar]
- 4.Chen J, Li Z, Hatcher JT, Chen QH, Chen L, Wurster RD, Chan SL, Cheng Z. Deletion of TRPC6 attenuates NMDA receptor-mediated Ca2+ entry and Ca2+-induced neurotoxicity following cerebral ischemia and oxygen-glucose deprivation. Front Neurosci. 2017;11:138. doi: 10.3389/fnins.2017.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 6.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 7.Gustafson I, Westerberg E, Wieloch T. Protection against ischemia-induced neuronal damage by the alpha 2-adrenoceptor antagonist idazoxan: influence of time of administration and possible mechanisms of action. J Cereb Blood Flow Metab. 1990;10:885–894. doi: 10.1038/jcbfm.1990.145. [DOI] [PubMed] [Google Scholar]
- 8.Hafeez F, Razzaq MA, Levine RL, Ramirez MA. Reperfusion seizures: a manifestation of cerebral reperfusion injury after administration of recombinant tissue plasminogen activator for acute ischemic stroke. J Stroke Cerebrovasc Dis. 2007;16:273–277. doi: 10.1016/j.jstrokecerebrovasdis.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Han Z, Zhang HX, Tian JS, Zheng RY, Hou ST. 2-(-2-benzofuranyl)-2-imidazoline induces Bcl-2 expression and provides neuroprotection against transient cerebral ischemia in rats. Brain Res. 2010;1361:86–92. doi: 10.1016/j.brainres.2010.09.029. [DOI] [PubMed] [Google Scholar]
- 10.Han Z, Yang JL, Jiang SX, Hou ST, Zheng RY. Fast, non-competitive and reversible inhibition of NMDA-activated currents by 2-BFI confers neuroprotection. PLoS One. 2013;8:e64894. doi: 10.1371/journal.pone.0064894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han Z, Xiao MJ, Shao B, Zheng RY, Yang GY, Jin K. Attenuation of ischemia-induced rat brain injury by 2-(-2-benzofuranyl)-2-imidazoline, a high selectivity ligand for imidazoline I(2) receptors. Neurol Res. 2009;31:390–395. doi: 10.1179/174313209X444116. [DOI] [PubMed] [Google Scholar]
- 12.Han Z, Cheng ZH, Liu S, Yang JL, Xiao MJ, Zheng RY, Hou ST. Neurovascular protection conferred by 2-BFI treatment during rat cerebral ischemia. Biochem Biophys Res Commun. 2012;424:544–548. doi: 10.1016/j.bbrc.2012.06.152. [DOI] [PubMed] [Google Scholar]
- 13.Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 2006;27:244–250. doi: 10.1016/j.it.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Hu GQ, Du X, Li YJ, Gao XQ, Chen BQ, Yu L. Inhibition of cerebral ischemia/reperfusion injury-induced apoptosis: nicotiflorin and JAK2/STAT3 pathway. Neural Regen Res. 2017;12:96–102. doi: 10.4103/1673-5374.198992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang SX, Zheng RY, Zeng JQ, Li XL, Han Z, Hou ST. Reversible inhibition of intracellular calcium influx through NMDA receptors by imidazoline I(2) receptor antagonists. Eur J Pharmacol. 2010;629:12–19. doi: 10.1016/j.ejphar.2009.11.063. [DOI] [PubMed] [Google Scholar]
- 16.Jin XL, Li PF, Zhang CB, Wu JP, Feng XL, Zhang Y, Shen MH. Electroacupuncture alleviates cerebral ischemia and reperfusion injury via modulation of the ERK1/2 signaling pathway. Neural Regen Res. 2016;11:1090–1098. doi: 10.4103/1673-5374.187041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 18.Liu CL, Zhang K, Chen G. Hydrogen therapy: from mechanism to cerebral diseases. Med Gas Res. 2016;6:48–54. doi: 10.4103/2045-9912.179346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 20.Maiese K, Pek L, Berger SB, Reis DJ. Reduction in focal cerebral ischemia by agents acting at imidazole receptors. J Cereb Blood Flow Metab. 1992;12:53–63. doi: 10.1038/jcbfm.1992.7. [DOI] [PubMed] [Google Scholar]
- 21.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, et al. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 22.Matsunaga S, Kishi T, Iwata N. Memantine monotherapy for Alzheimer's disease: a systematic review and meta-analysis. PLoS One. 2015;10:e0123289. doi: 10.1371/journal.pone.0123289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 24.Orgogozo JM, Rigaud AS, Stoffler A, Möbius HJ, Forette F. Efficacy and safety of memantine in patients with mild to moderate vascular dementia: a randomized, placebo-controlled trial (MMM 300) Stroke. 2002;33:1834–1839. doi: 10.1161/01.str.0000020094.08790.49. [DOI] [PubMed] [Google Scholar]
- 25.Regunathan S, Feinstein DL, Reis DJ. Expression of non-adrenergic imidazoline sites in rat cerebral cortical astrocytes. J Neurosci Res. 1993;34:681–688. doi: 10.1002/jnr.490340611. [DOI] [PubMed] [Google Scholar]
- 26.Sastre M, Regunathan S, Galea E, Reis DJ. Agmatinase activity in rat brain: a metabolic pathway for the degradation of agmatine. J Neurochem. 1996;67:1761–1765. doi: 10.1046/j.1471-4159.1996.67041761.x. [DOI] [PubMed] [Google Scholar]
- 27.Schäbitz WR, Berger C, Kollmar R, Seitz M, Tanay E, Kiessling M, Schwab S, Sommer C. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke. 2004;35:992–997. doi: 10.1161/01.STR.0000119754.85848.0D. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 29.Sun XZ, Liao Y, Li W, Guo LM. Neuroprotective effects of ganoderma lucidum polysaccharides against oxidative stress-induced neuronal apoptosis. Neural Regen Res. 2017;12:953–958. doi: 10.4103/1673-5374.208590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sulejczak D, Czarkowska-Bauch J, Macias M, Skup M. Bcl-2 and Bax proteins are increased in neocortical but not in thalamic apoptosis following devascularizing lesion of the cerebral cortex in the rat: an immunohistochemical study. Brain Res. 2004;1006:133–149. doi: 10.1016/j.brainres.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 31.Sutherland BA, Minnerup J, Balami JS, Arba F, Buchan AM, Kleinschnitz C. Neuroprotection for ischaemic stroke: translation from the bench to the bedside. Int J Stroke. 2012;7:407–418. doi: 10.1111/j.1747-4949.2012.00770.x. [DOI] [PubMed] [Google Scholar]
- 32.Thorn DA, Zhang Y, Li JX. Effects of the imidazoline I2 receptor agonist 2-BFI on the development of tolerance to and behavioural/physical dependence on morphine in rats. Br J Pharmacol. 2016;173:1363–1372. doi: 10.1111/bph.13435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogels BA, Maas MA, Daalhuisen J, Quack G, Chamuleau RA. Memantine, a noncompetitive NMDA receptor antagonist improves hyperammonemia-induced encephalopathy and acute hepatic encephalopathy in rats. Hepatology. 1997;25:820–827. doi: 10.1002/hep.510250406. [DOI] [PubMed] [Google Scholar]
- 34.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1–33. doi: 10.1016/s0022-510x(00)00386-5. [DOI] [PubMed] [Google Scholar]
- 36.Wong LF, Ralph GS, Walmsley LE, Bienemann AS, Parham S, Kingsman SM, Uney JB, Mazarakis ND. Lentiviral-mediated delivery of Bcl-2 or GDNF protects against excitotoxicity in the rat hippocampus. Mol Ther. 2005;11:89–95. doi: 10.1016/j.ymthe.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 38.Yenari MA, Hemmen TM. Therapeutic hypothermia for brain ischemia: where have we come and where do we go? Stroke. 2010;41:S72–74. doi: 10.1161/STROKEAHA.110.595371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang B, Sun XJ, Ju CH. Thrombolysis with alteplase 4,5-6 hours after acute ischemic stroke. Eur Neurol. 2011;65:170–174. doi: 10.1159/000324291. [DOI] [PubMed] [Google Scholar]
- 40.Zhang R, Xue YY, Lu SD, Wang Y, Zhang LM, Huang YL, Signore AP, Chen J, Sun FY. Bcl-2 enhances neurogenesis and inhibits apoptosis of newborn neurons in adult rat brain following a transient middle cerebral artery occlusion. Neurobiol Dis. 2006;24:345–356. doi: 10.1016/j.nbd.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Zhao H, Yenari MA, Cheng D, Barreto-Chang OL, Sapolsky RM, Steinberg GK. Bcl-2 transfection via herpes simplex virus blocks apoptosis-inducing factor translocation after focal ischemia in the rat. J Cereb Blood Flow Metab. 2004;24:681–692. doi: 10.1097/01.WCB.0000127161.89708.A5. [DOI] [PubMed] [Google Scholar]
- 42.Zhao J, Zhao Y, Zheng W, Lu Y, Feng G, Yu S. Neuroprotective effect of curcumin on transient focal cerebral ischemia in rats. Brain Res. 2008;1229:224–232. doi: 10.1016/j.brainres.2008.06.117. [DOI] [PubMed] [Google Scholar]
- 43.Zhu YB, Xia NG, Zhang YT, Wang XS, Liang SS, Yin WY, Xu HQ, Hou ST, Zheng RY. Brain protection conferred by long-term administration of 2-(2-benzofuranyl)-2-imidazoline against experimental autoimmune encephalomyelitis. Neurochem Res. 2015;40:572–578. doi: 10.1007/s11064-014-1502-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.