

Abstract
Background
Several intracellular signaling pathways that are deregulated during hepatocarcinogenesis might constitute potential targets for hepatocellular carcinoma (HCC) therapy. The aim of this study was to test the potential synergic antitumor effect of salirasib and sorafenib in a diethylnitrosamine (DEN)-induced HCC model in rat. The hypothesis of tumor phenotype changes during treatment was also analyzed.
Materials and methods
DEN was administered to Wistar rats during 9 weeks to induce cirrhosis and liver cancer. After tumor development, rats were treated with intraperitoneal injections of dimethyl sulfoxide (DMSO), or salirasib, and/or with oral sorafenib 5 days/week, during 4 weeks. At sacrifice, number and size of liver tumors as well as tumor burden were recorded, and all liver tumors were processed for histological and immunohistological analyses.
Results
Mortality rate was significantly higher in rats treated with salirasib and/or sorafenib than in the control group (P=0.001). Tumor burden was smaller in the treated group compared with the DMSO control group (P=0.044), but a synergistic effect of the two chemotherapies could not be observed. In 62.5% of rats (10/16) treated with salirasib and/or sorafenib, a cytokeratin-7 and -19-positive hepatocholangiocellular carcinoma (HCC/CHC) was found vs 20% (5/25) developing such phenotype in the DMSO control group (P=0.018). Ki67 immunostaining showed significantly reduced tumor cell proliferation in treated rats (P=0.001), whereas apoptosis as assessed by caspase-3 activity in cell lysate was similar in all groups.
Conclusions
The addition of sorafenib to salirasib did not seem to provide any synergistic therapeutic effect in this study. Both chemotherapeutic agents, administered alone or in combination, induced tumoral phenotypic changes in the majority of rats, a finding not associated with an increased tumor cell proliferation or decreased apoptosis. The rat model described in this work constitutes the first experimental tool generating putatively more aggressive combined HCC/CHC tumors following chemotherapy. Further work is required to better characterize this clinically relevant phenomenon.
Keywords: liver neoplasms, chemotherapy, disease management, liver transplantation
Introduction
Hepatocellular carcinoma (HCC) is fifth most common malignant tumor worldwide, and the second leading cause of cancer-related death.1,2 Surgical resection and liver transplantation offer encouraging results in patients with early stage HCC, with patient survival ranging from 40% to 80% at 5 years.3,4 Despite many advances in HCC management, the prognosis for intermediate and advanced stages remains poor, due to a high recurrence rate following medical or surgical treatment. Moreover, phenotypic changes of the tumor leading to increased aggressivity were also observed following chemotherapy in HCC patients.5,6 The pathophysiology of HCC involves complex processes associating genetic and epigenetic changes, including several intracellular signaling pathways, not yet fully understood.7
This complexity probably explains the lack of therapeutic response when only one signaling pathway is targeted. Several combined therapies acting on more than one signaling pathway have therefore been proposed. The aim of such strategies is to prevent the development of resistance to a single agent, as well as to improve the efficacy, putatively through apoptosis enhancement and inhibition of angiogenesis and cell proliferation. Both MAPK (Ras/Raf/MEK/ERK) and PI3k/Akt/mTOR pathways seem to play a key role in liver carcinogenesis in experimental models as well as in humans.7,8 In particular, the Ras pathway is almost always activated in human HCC; moreover, relatively recent studies have shown that overexpression of the Ras/MEK/ERK cascade is correlated with poorer survival of HCC patients.9,10 Salirasib is a S-farnesyl cysteine analog that interferes with Ras membrane anchorage and inhibits the active GTP-bound Ras in various human cancer cell lines.11,12 The tumor preventive effect of salirasib has already been demonstrated in a chemically induced HCC rodent model, by activating apoptosis and blocking hepatocyte proliferation.13 The efficacy of salirasib as a strong inhibitor of human HCC cell line proliferation was also observed in vitro.14 VEGF constitutes a key stimulus for angiogenesis in highly vascularized tumors like HCC. Sorafenib is a multikinase inhibitor targeting VEGF-receptor and PDGFR kinases as well as Raf. Its clinical effectiveness has already been demonstrated by the results of the SHARP multicentric trial in patients with advanced HCC.15
The aim of the present study was to test the antitumor effect of and tolerance to salirasib and sorafenib, alone or in combination at different doses in a diethylnitrosamine (DEN)-induced HCC model in rat. The hypothesis of changes in tumor phenotype following such chemotherapies, as observed in the clinical setting, was also analyzed in this experimental model.
Materials and methods
Animals
Four-week-old male Wistar rats (mean body weight: 152 g, range: 101–170 g) were obtained from the Janvier Labs, Le Genest-St-Isle, France. Animals were housed in barrier facilities on a 12-hour light/dark cycle and kept in a temperature and humidity-controlled environment; standard food (Usine d’Alimentation Rationnelle, Villemoisson-sur-Orges, France) and water were supplied ad libitum. Animal care was provided in accordance with the guidelines for laboratory animals established by the Université Catholique de Louvain in accordance with European regulations and in conformity with ARRIVE guidelines. The study protocol was approved by the University Ethics Committee (2012UCLMD026).
Cirrhosis and liver tumor induction
Animals (n=76) were acclimated for 2 weeks, then a solution of DEN (0.01%; Sigma Chemical Co., Saint Louis, MO, USA) was continuously administered via drinking water for 9 weeks followed by a 3-week washout period. DEN concentration was adapted weekly, according to the weight of the animals as previously described by us and others.16
A laparotomy was planned after a washout period of 3 weeks, in order to confirm cirrhotic evolution and liver tumor development. Thereafter, surviving animals were randomly assigned to one of six groups of treatment, as detailed in Table 1. Sorafenib (Nexavar, Bayer Schering Pharma AG, Berlin, Germany) and salirasib (S-trans-trans-farnesylthiosalicyclic acid, Concordia Pharmaceuticals, Ft. Lauderdale, FL, USA) were diluted in 0.1% dimethyl sulfoxide (DMSO) to a final volume of 500 µL and daily administered, by gavage or intraperitoneal injections, respectively, 5 days per week during 4 weeks, under short sedation with isoflurane, using doses determined by the in vitro study described hereafter. DMSO (500 µL) was administered intraperitoneally with the same schedule in the control group (n=30).
Table 1.
Details of treatment for each group of rats alive after development of chemically induced cirrhosis and liver cancer (n=67)
| Group | Number of rats | Dose | Administration |
|---|---|---|---|
|
| |||
| DMSO (control group) | 30 | 500 µL | i.p. |
| Salirasib | 9 | 10 mg/kg | i.p. |
| Sorafenib | 6 | 7.5 mg/kg | Gavage |
| Sorafenib | 6 | 15 mg/kg | Gavage |
| Salirasib+sorafenib | 8 | 10 mg/kg+7.5 mg/kg | i.p.+gavage |
| Salirasib+sorafenib | 8 | 10 mg/kg+15 mg/kg | i.p.+gavage |
Abbreviations: DMSO, dimethyl sulfoxide; i.p., intraperitoneally.
In vitro study
FTO-2B cells (HCC rat line; Thermo Fisher Scientific, Waltham, MA, USA) were incubated in DMEM, supplemented with DMSO or different concentrations of salirasib (50 and 150 µM), alone or in combination with sorafenib (1, 2.5, and 5 µM), in order to define the effectiveness and toxicity of these molecules.17 Briefly, cell viability was evaluated 48 hours later using a colorimetric WST-1 assay (Roche, Vilvoorde, Belgium), this method allows evaluation of the proportion of metabolically active cells, labeled by a higher optical density (absorbance) in spectroscopic analysis. IC50 values for each drug combination and dosage (value at which cell growth is inhibited by 50% compared with the control group) were calculated by nonlinear regression analysis using GraphPad Prism software (San Diego, CA, USA) (Figure 1).
Figure 1.

Cellular viability as assessed by colorimetric WST-1 assay (Y-axis: relative absorbance, determined at 450 nm) on cultured rat’s FTO-2B cells in vitro, submitted to DMSO (control group) and different concentrations of salirasib (50 and 150 µM) and sorafenib (1, 2.5, and 5 µM), alone or in combination.
Note: These data allowed extrapolation of IC50 value for the different doses of drugs (data not shown).
Abbreviation: DMSO, dimethyl sulfoxide.
Histology and immunohistochemistry
All surviving rats were sacrificed 1 week after the end of the therapeutic phase (Figure 2). Rat and liver weight as well as number and diameter of all whitish liver nodules measuring more than 2 mm and visualized after slicing the liver every 2 mm were recorded. Tumor burden was calculated according to the WHO definition: length × width for each target lesion and sum of the value of each tumor.18 All suspicious liver lesions were resected and fixed overnight in 10% buffered formalin and embedded in paraffin; a specimen of each of all these lesions was also frozen for further analysis. Five-micrometer sections of formalin-fixed and paraffin-embedded liver tumoral specimen were stained with H&E for conventional histology. HCC was diagnosed at conventional histology in presence of thick trabecular growth pattern composed of cells showing eosinophilic cytoplasm with irregular nucleus and distinct nucleoli. The diagnosis of combined hepatocholangiocellular carcinoma (HCC/CHC) tumor was made if a glandular structure was clearly present.19 Immunohistochemical staining was performed on deparaffinized 4 µm tissue sections with the Dako Envision system (Dako Japan Co. Ltd, Kyoto, Japan). Mouse monoclonal antibodies against CK19 (dilution 1:200; Sigma), CK7 (dilution 1:50; Abcam, Cambridge, UK), alpha-fetoprotein (AFP) (dilution 1:500; Abcam), and Ki67 (dilution 1:50; Abcam) were used. The two former permitted to confirm the biliary nature of the tumoral glandular proliferation, AFP the hepatocellular nature of the tumoral proliferation, and the latter used as an index of tumoral cell proliferation. Computer image analyses of immunostained sections for Ki67 were performed using Zeiss microscope coupled to an Axiocam camera (MR3, Carl Zeiss, Munich, Germany) and the AxioVision software (Zeiss). A morphometrical quantification was therefore done, leading to a mean percentage of labeled nuclei for each specimen of liver tissue.20 Calculation of the percentage of labeled nuclei by the specific antibody on slides was performed via a computer algorithm (Ki67 index).
Figure 2.

Experimental design including tumoral induction with DEN (during 9 weeks), assessment of tumor induction (week 12th), randomization to therapeutic groups (week 13th), mortality in each group, and sacrifice (week 18th).
Abbreviations: DEN, diethylnitrosamine; DMSO, dimethyl sulfoxide.
Apoptosis assessment
Apoptosis was assessed by fluorogenic activity assays of effector caspase-3 in cell lysates, using the Caspase-Glo® 3/7 Assay (Promega Corporation, Fitchburg, WI, USA) as indicated by the manufacturer. Cell lysates were prepared according to the procedure as described previously.21
Statistical analysis
Analysis was run using SPSS (version 25.0; IBM Corp., Armonk, NY, USA). All the quantitative variables were tested for normality with the Shapiro–Wilk test. Accordingly, all normality distributed variables were tested with Student’s t-test and the other ones with the Mann–Whitney U test. One-way ANOVA test was used to compare tumor burden in different groups of rats after randomization. Animal survival was analyzed with the Kaplan–Meier method and the log-rank test was used to compare survival between subgroups. The significance of statistical tests was taken at a P-value ≤0.05.
Results
Impact of low and high doses of salirasib and sorafenib on cell viability of tumor cells in vitro
As shown in Figure 1, high doses of salirasib as single therapy did not seem to impact cellular viability, as assessed by WST-1 assay. By contrast, the combination of these two drugs clearly showed enhanced cytotoxicity when sorafenib concentration was increased, already with the low dose of salirasib (50 µM). Accordingly, a dose-dependent cytotoxic effect of sorafenib in combination with salirasib was suspected, and it was decided to include two subgroups of animals treated with low and high doses of sorafenib in the in vivo experiment.
Tumoral induction, mortality, and antitumor treatment outcome
Five of the 76 rats given DEN for 9 weeks died during the washout period of 3 weeks, leaving a study population of 71 animals at the end of the tumoral induction phase (Figure 2). As originally planned, exploratory laparotomy was performed at week 12, starting with 17 animals randomly selected. Macroscopic examination confirmed cirrhosis in 100% of these animals as well as whitish tumoral nodules on the livers (Figure 3A). Laparotomy was poorly tolerated with an early (<24 hours) postsurgical mortality of 23.5% (4/17). Given such unexpected high mortality rate and based on the development of cirrhosis and HCC tumors observed in all 17 laparotomized animals, the laparotomy was not performed in the remaining 54 rats. Moreover, postmortem histology of macroscopically identified tumors in the four rats that died after laparotomy confirmed HCC development (Figure 3B). It was therefore assumed that cirrhosis and HCC had likely developed in all rats before randomization. At week 13, surviving rats (n=67) were randomized into six groups, which received salirasib (10 mg/kg), sorafenib (7.5 or 15 mg/kg), salirasib (10 mg/kg) combined with sorafenib (7.5 or 15 mg/kg), or DMSO in the control group, for a duration of 4 weeks, 5 days per week (Table 1).
Figure 3.

(A) Macroscopic appearance of dyschromic, whitish tumor (black arrows) developed on cirrhotic liver in rats given DEN for 9 weeks and which died after exploratory laparotomy at week 12. (B) Representative H&E section of HCC in rats that died after laparotomy, showing a trabecular pattern with nuclear atypia and absence of portal tract (low magnification, ×10).
Abbreviations: HCC, hepatocellular carcinoma; DEN, diethylnitrosamine.
An overall mortality rate of 38.8% (26/67) was observed at week 17, with only 41 of 67 animals alive at the end of the 4-week therapeutic phase. Mortality was significantly higher in animals receiving chemotherapy (21/37) compared with the DMSO controls (5/30) (56.7% vs 16.6%, respectively, P=0.001). However, mortality did not differ in relation to the chemotherapeutic regimens (P=0.322), and deaths were homogenously distributed over the entire period of treatment.
In order to assess the efficacy of salirasib alone or in combination with sorafenib in our HCC-model, the number of suspect lesions (whitish nodules >2 mm) and tumor burden in all surviving animals (n=41) were evaluated at sacrifice. The mean number of tumors per liver was 7 (range: 4–14). The number of tumors in control and treated groups (irrespective of the chemotherapeutic regimen) was not statistically different (data not shown; P=0.213). Because the absolute number of tumors might only partially reflect the antitumor treatment response, we also determined the overall tumor burden, as defined in the Materials and methods section. Interestingly, tumor burden was significantly lower in rats receiving chemotherapy than in DMSO control group (P=0.044), suggesting some response of HCC to the different treatment regimens. However, no significant differences could be found between the different types of treatment (sorafenib alone at low or high doses, and salirasib alone or in combination with sorafenib) (Figure 4).
Figure 4.

Median (range) values of tumor burden (Y-axis) at sacrifice. Tumor burden (assessed as length×width for each target lesion, and sum of the value of each tumor) was significantly decreased in treated rats compared with DMSO control group (P=0.044).
Abbreviation: DMSO, dimethyl sulfoxide.
Pathological and immunohistological assessment of tumor lesions
All macroscopically suspect liver lesions were dissected at sacrifice and subjected to conventional histology (H&E staining). Histology revealed well-differentiated or moderately differentiated pure HCC in 19 of the 25 (76%) surviving animals of the DMSO group. The histological diagnosis was confirmed by immunohistochemistry, since tumors only expressed AFP (Figure 5A and B), but neither CK7 nor CK19. In the six remaining DMSO rats, five had combined HCC/CHC tumor, with CK7 and CK19 positivity on immunostaining in at least one tumoral nodule; in the last DMSO animal, histology was suggestive of an angiosarcoma. By contrast, only a minority of animals (6/16) treated with salirasib and/or sorafenib showed a histologically and immunohistochemically proven pure HCC in all examined tumors. The proportion of HCC/CHC combined tumors was significantly higher in animals receiving chemotherapy than in controls (P=0.018). Indeed, 62.5% (10/16) presented at least two tumors that were either negative or weakly positive for AFP but strongly expressing CK7 and/or CK19, highly suggestive of HCC/CHC mixed type tumor (Figure 5C–F).
Figure 5.

Histology and immunohistochemistry of tumors.
Notes: (A) Conventional histology (H&E) of pure HCC in DMSO rat, with thick trabecular growth pattern composed of tumor cells showing eosinophilic cytoplasm with irregular nuclear and distinct nucleoli, with massively positive AFP immunolabeling; (B) conventional histology (H&E) showing a mixed type HCC/CHC in treated rat with two components; (C) thick trabecular/solid structure and (E) clearly glandular structures and positive CK-19 (D) and CK-7 (F) immunolabeling (×20 magnification).
Abbreviations: AFP, alpha-fetoprotein; DMSO, dimethyl sulfoxide; HCC, hepatocellular carcinoma; HCC/CHC, hepatocholangiocellular carcinoma.
Assessment of the overall proliferation–apoptosis balance in tumor nodules
The impact of chemotherapy on the apoptosis–proliferation balance in tumor nodules was also evaluated, aiming to analyze whether variations of this balance could at least partially explain the appearance of a different tumor phenotype. As a marker of cell proliferation, immunolabeling for Ki67 on tumoral and nontumoral tissue was performed in a total of 28 animals, including treated (n=12) or untreated (DMSO control group, n=16) rats. In the control group, as well as in treated animals whatever the type of treatment, Ki67 index was higher in tumoral tissue compared with the surrounding nontumoral cirrhotic tissue (Figure 6). The comparison between treated (n=12) and untreated (n=16) tumors revealed a significantly lower Ki67 expression in the treated groups than in control group (Figure 7; P=0.001). A similar, but nonsignificant trend toward lower Ki67 expression was also found in nontumoral tissue of treated rats when compared with cirrhotic, nontumoral liver in DMSO group. These observations suggest an antiproliferative effect of chemotherapies mainly directed to tumor cells. Nevertheless, in treated animals, no significant difference in Ki67 expression could be found when pure HCC were compared with combined HCC/CHC tumors (20.5% vs 16.8%; P=0.6). Caspase-3 activity was assessed as a surrogate marker for apoptosis in 22 animals (six in DMSO group, 16 in treated groups). A trend toward lower caspase-3 activity in tumoral and nontumoral tissues in the treated groups compared with tumoral and nontumoral tissues in DMSO group was observed (Figure 8). However, the difference did not reach statistical significance (P=0.156).
Figure 6.

Representative immunohistochemistry of Ki67 in tumoral and nontumoral tissue of DMSO control group and in treated animals (magnification ×20).
Abbreviation: DMSO, dimethyl sulfoxide.
Figure 7.
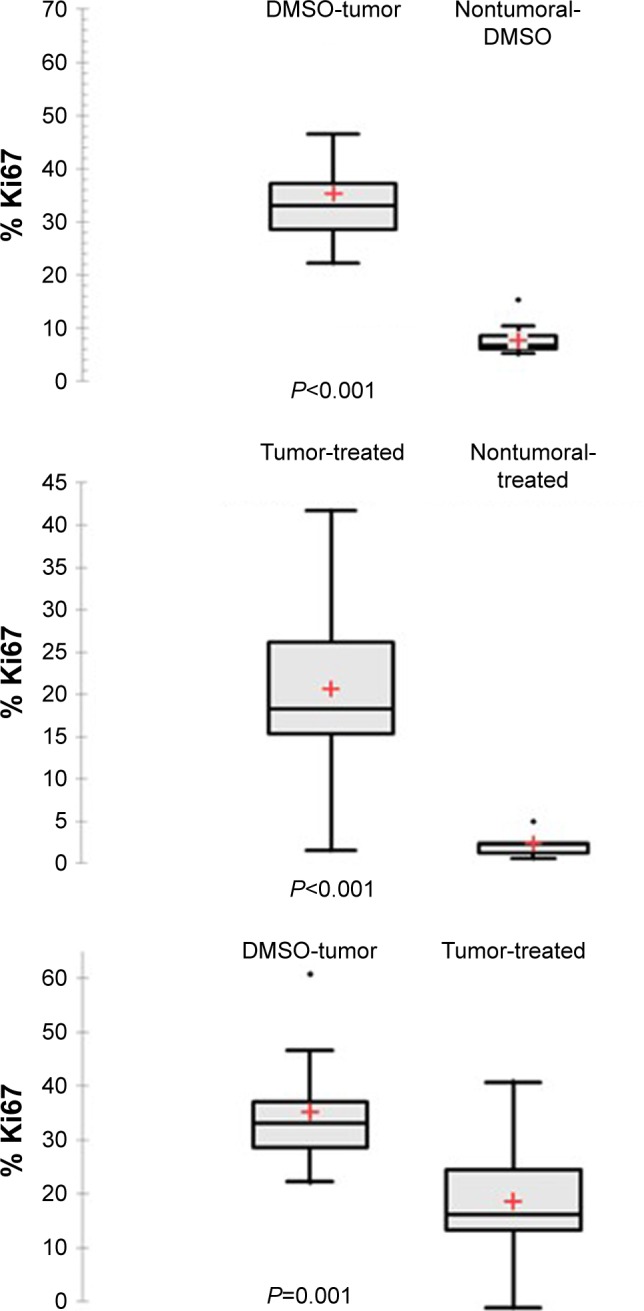
Ki67 labeling index after immunohistochemical staining Ki67 expression was significantly higher in tumoral than in nontumoral tissue in DMSO group as well as in treated animals (P<0.001).
Notes: A statistically significant difference was also found between tumoral tissue in DMSO group (n=16) compared with treated tumor (n=12) (P=0.001). +Mean values.
Abbreviation: DMSO, dimethyl sulfoxide.
Figure 8.

Caspase-3 activity in tumoral and nontumoral tissue.
Notes: Caspase-3 activity was lower in tumoral and nontumoral tissues in the treated groups; as shown, a slight but nonsignificant reduction in caspase-3 activity was found in tumor of animals after administration of salirasib and/or sorafenib (n=16) compared with DMSO control group (n=6) (P=0.154; NS).
Abbreviations: DMSO, dimethyl sulfoxide; NS, not significant.
Discussion
More than 80% of HCC develop on cirrhotic liver, and accordingly, a curative approach must take into account the cancer as well as the underlying liver disease. This explains why partial surgical resection is not considered as an option in the majority of patients who present with advanced liver impairment at the time of tumor diagnosis. In order to better stratify therapeutic strategies, preclinical models coupling cirrhosis and HCC should be designed and used. Several models of chemically induced HCC on liver fibrosis and cirrhosis have already been published.22 The alkylating agent DEN is frequently used in rodents as it induces cirrhosis as well as liver tumors that are molecularly similar to human HCC with poor prognosis.23 This model has already been used by our group, DEN being administered orally during 6 weeks or intraperitoneally during various time intervals.24,25 In the present study, DEN was administered orally for 9 weeks, inducing cirrhosis and tumor development in all animals, as confirmed by macroscopic, histological, and immunohistological analysis.
Current knowledge on molecular mechanisms involved in hepatic carcinogenesis has led to the development of targeted therapies, specific to several pathogenic pathways.26 In particular, VEGF-related tumor angiogenesis, as well as proliferation and cell survival dependent on m-TOR and Ras signaling pathways, may constitute targets of interest for treatment in HCC. Sorafenib represents the only therapy currently approved as first-line VEGF and Ras inhibitor in patients with nonresectable HCC, but with only a modest benefit in terms of patient survival.10
Conceptually, targeted combination therapies should reduce the size of tumors to make them resectable or to consider liver transplantation within Milan or San Francisco criteria.3,27 Several combination chemotherapies have been proposed in Phase III drug trial.28 Nevertheless, the results obtained up to now with combined chemotherapies remain unsatisfactory in terms of tumor control and patient survival, with, in addition, a significant toxicity.29 Our group has previously shown the preventive effect of salirasib in the development of tumor foci in DEN-induced HCC model.8 Moreover, Charette et al14 showed the antitumoral efficacy of salirasib in a xenograft model where Hep G2 cell line derived from human hepatoblastoma was implanted subcutaneously in athymic mice.9 However, evidence upon efficacy of salirasib once a tumor has developed (curative purpose) is lacking. The choice to combine sorafenib and salirasib in the present study comes from the clinical use of the former and the interesting experimental results obtained so far by Charette et al for the latter. Accordingly, our study aimed to assess both toxicity and antitumoral efficacy of such combination administered at different doses after tumor development. In the present experiment, a very high mortality rate was found in all treated groups (56.7%), which could be related to a poor tolerance to these compounds in cirrhotic livers with a high tumor burden. However, no evidence of increased toxicity expressed by creatinine, bilirubin, and transaminases was demonstrated when comparing DMSO and treated rats (data not shown). From an antitumoral efficiency perspective, this study showed significantly decreased tumor burden in the treated groups, irrespective of the treatment, compared with the control group (P=0.044). However, our data could not confirm a synergistic therapeutic effect of salirasib and sorafenib, but this observation should be definitively confirmed in a larger cohort of animals. The doses of salirasib and sorafenib used in our experiment were chosen on the basis of other published works and after having carried out an in vitro study with FTO-2B hepatoma cells of rats. Based on this study and data in the literature, we suspected that the use of lower doses of salirasib (eg, 10 mg/kg) and sorafenib (eg, 7.5 mg/kg) would no longer be effective. However, we cannot formally exclude that in vivo in this rat model, using smaller doses of these two molecules may still have a cytotoxic antitumor effect with a better tolerance.
Nevertheless, this regimen should be mentioned with extreme caution and might be proposed only if other alternatives may not be applied.
Intriguingly, a significant percentage of surviving rats treated with salirasib and/or sorafenib showed tumoral phenotypic changes, as evidenced by the development of combined HCC/CHC in 62.5% of treated animals vs 20% in the control/DMSO group (P=0.018). Mixed HCC/CHC was defined according to the 2010 WHO classification, as tumor showing small monotonous glands with antler-like intersection patterns and hepatocytes with pronounced nuclei.30 In addition, immunohistochemistry confirmed the histological diagnosis, because immunostaining for CK7 and CK19 was positive in these instances. This phenomenon of phenotypic tumoral change has recently been reported by several authors after locoregional treatment, such as transarterial chemoembolization (TACE), administered in patients with HCC on cirrhosis.5,6 Preliminary clinical data from our group confirmed that combined HCC/CHC were found on the native liver specimen at transplantation in 14% of patients (6/43) receiving at least two locoregional treatments prior to liver transplantation, while a phenotypic tumoral change was never found in patients transplanted without TACE (0/18). Beside its therapeutic effect, these data suggest that TACE may also induce phenotypic changes in HCC similar to those observed in our animals after targeted therapy. Therefore, a phenotypic change of tumors under high selection pressure (eg, TACE or combined targeted therapies) could potentially trigger some tumor escape mechanism, with the selection of cellular variants resistant to the treatment administered. The mechanism involved in such tumor escape process may involve increased cell proliferation leading to increased mutation rate, enhanced angiogenesis, or reduced apoptosis, but it remain poorly understood. In the present study, there was no relation between the phenotypic change and increased cellular proliferation in the mixed tumor type. Ki67 expression was significantly lower in tumor nodules of treated animals (regardless of the tumor phenotype) compared with HCC tumors in the DMSO group. This contributes to reinforce the initial observation of an antitumor, antiproliferative effect of the treatment regimens. Our results on caspase-3 activity also showed that apoptosis was not significantly increased after administration of salirasib and/or sorafenib. We cannot formally exclude that a potential resistance to apoptosis could have contributed to the tumoral phenotypic change. An additional explanation might be epithelial-to-mesenchymal transition (EMT). From a histological point of view, we had no clear sign of a morphological change of tumor cells into fusiform cells, which is typical in EMT. EMT is also characterized by a loss of E-cadherin and gain of mesenchymal markers such as vimentin. In our study, E-cadherin and vimentin immunohistochemistry did not produce any convincing results in favor of EMT (data not shown). Clearly, additional mechanistic studies (eg, analyzing angiogenesis) are needed to better understand this tumor escape process.
The development of mixed tumors with a cholangiocellular component could have important clinical implications in HCC management. Such HCC/CHC constitutes a rare form of primary liver cancer (0.4%–14%) and is clinically associated with poor prognosis.31,32 Vascular invasion, regional organ involvement, nodal and distant metastases seem to play a role in tumor aggressiveness.32 These recent observations could lead to modify the therapeutic algorithm used until now for HCC in humans. In particular, a tumoral downstaging by iterative TACE in patients with HCC awaiting liver transplantation could theoretically lead to the development of mixed HCC/CHC tumors, with a poorer prognosis. Clinicians should remain cautious before administrating iterative TACE and targeted therapies to patients potentially eligible for liver transplantation, unless further studies are available to validate the efficacy of these treatments in terms of better prognosis and survival.
In conclusion, the present study provided the first preclinical model of targeted chemotherapy-induced change of HCC phenotype and advocated caution before using iteratively this type of therapy in HCC patients. Furthermore, this model could be useful to further study the mechanistic phenomena underlying phenotypic changes in tumors exposed to a high selection pressure and to define the useful ways that might help to counteract this process.
Acknowledgments
The abstract of this paper was presented at the 18th Congress of the European Society for Organ Transplantation as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in Transplant International, 2017;30(S2):390–576 (https://doi.org/10.1111/tri.13049).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Wallace MC, Preen D, Jeffrey GP, Adams LA. The evolving epidemiology of hepatocellular carcinoma: a global perspective. Expert Rev Gastroenterol Hepatol. 2015;9(6):765–779. doi: 10.1586/17474124.2015.1028363. [DOI] [PubMed] [Google Scholar]
- 3.Mazzaferro V, Regalia E, Doci R, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med. 1996;334(11):693–700. doi: 10.1056/NEJM199603143341104. [DOI] [PubMed] [Google Scholar]
- 4.Ciccarelli O, Lai Q, Goffette P, et al. Liver transplantation for hepatocellular cancer: UCL experience in 137 adult cirrhotic patients. Alpha-foetoprotein level and locoregional treatment as refined selection criteria. Transpl Int. 2012;25(8):867–875. doi: 10.1111/j.1432-2277.2012.01512.x. [DOI] [PubMed] [Google Scholar]
- 5.Nishihara Y, Aishima S, Kuroda Y, et al. Biliary phenotype of hepatocellular carcinoma after preoperative transcatheter arterial chemoembolization. J Gastroenterol Hepatol. 2008;23(12):1860–1868. doi: 10.1111/j.1440-1746.2008.05601.x. [DOI] [PubMed] [Google Scholar]
- 6.Zen C, Zen Y, Mitry RR, et al. Mixed phenotype hepatocellular carcinoma after transarterial chemoembolization and liver transplantation. Liver Transpl. 2011;17(8):943–954. doi: 10.1002/lt.22314. [DOI] [PubMed] [Google Scholar]
- 7.Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol. 2013;87(2):227–247. doi: 10.1007/s00204-012-0931-2. [DOI] [PubMed] [Google Scholar]
- 8.Villanueva A, Chiang DY, Newell P, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135(6):1972–1983. doi: 10.1053/j.gastro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Shi Y, Jiang CY, Wei LX, Wang YL, Dai GH. Expression and prognostic role of pan-Ras, Raf-1, pMEK1 and pERK1/2 in patients with hepatocellular carcinoma. Eur J Surg Oncol. 2011;37(6):513–520. doi: 10.1016/j.ejso.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 10.Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130(4):1117–1128. doi: 10.1053/j.gastro.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Haklai R, Weisz MG, Elad G, et al. Dislodgment and accelerated degradation of Ras. Biochemistry. 1998;37(5):1306–1314. doi: 10.1021/bi972032d. [DOI] [PubMed] [Google Scholar]
- 12.Marom M, Haklai R, Ben-Baruch G, Marciano D, Egozi Y, Kloog Y. Selective inhibition of Rasdependent cell growth by farnesylthiosalisylic acid. J Biol Chem. 1995;270(38):22263–22270. doi: 10.1074/jbc.270.38.22263. [DOI] [PubMed] [Google Scholar]
- 13.Schneider-Merck T, Borbath I, Charette N, et al. The Ras inhibitor farnesylthiosalicyclic acid (FTS) prevents nodule formation and development of preneoplastic foci of altered hepatocytes in rats. Eur J Cancer. 2009;45(11):2050–2060. doi: 10.1016/j.ejca.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Charette N, de Saeger C, Lannoy V, Horsmans Y, Leclercq I, Stärkel P. Salirasib inhibits the growth of hepatocarcinoma cell lines in vitro and tumor growth in vivo through ras and mTOR inhibition. Mol Cancer. 2010;9:256. doi: 10.1186/1476-4598-9-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 16.Ha WS, Kim CK, Song SH, Kang CB. Study on mechanism of multistep hepatotumorigenesis in rat: development of hepatotumorigenesis. J Vet Sci. 2001;2(1):53–58. [PubMed] [Google Scholar]
- 17.Zvibel I, Fiorino AS, Brill S, Reid LM. Phenotypic characterization of rat hepatoma cell lines and lineage-specific regulation of gene expression by differentiation agents. Differentiation. 1998;63(4):215–223. doi: 10.1111/j.1432-0436.1998.00215.x. [DOI] [PubMed] [Google Scholar]
- 18.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 19.Schlageter M, Terracciano LM, D’Angelo S, Sorrentino P. Histopathology of hepatocellular carcinoma. World J Gastroenterol. 2014;20(43):15955–15964. doi: 10.3748/wjg.v20.i43.15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Hul NK, Abarca-Quinones J, Sempoux C, Horsmans Y, Leclercq IA. Relation between liver progenitor cell expansion and extracellular matrix deposition in a CDE-induced murine model of chronic liver injury. Hepatology. 2009;49(5):1625–1635. doi: 10.1002/hep.22820. [DOI] [PubMed] [Google Scholar]
- 21.Picard C, Starkel P, Sempoux C, Saliez A, Lebrun V, Horsmans Y. Molecular mechanisms of apoptosis in the liver of rats after portal branch ligation with and without retrorsine. Lab Invest. 2004;84(5):618–628. doi: 10.1038/labinvest.3700085. [DOI] [PubMed] [Google Scholar]
- 22.Delire B, Stärkel P, Leclercq I. Animal models for fibrotic liver diseases: what we have, what we need, and what is under development. J Clin Transl Hepatol. 2015;3(1):53–66. doi: 10.14218/JCTH.2014.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36(12):1306–1311. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- 24.Borbath I, Leclercq IA, Sempoux C, Abarca-Quinones J, Desaeger C, Horsmans Y. Efficacy of lanreotide in preventing the occurrence of chemically induced hepatocellular carcinoma in rats. Chem Biol Interact. 2010;183(1):238–248. doi: 10.1016/j.cbi.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Loeppen S, Schneider D, Gaunitz F, et al. Overexpression of glutamine synthetase is associated with beta-catenin-mutations in mouse liver tumors during promotion of hepatocarcinogenesis by phenobarbital. Cancer Res. 2002;62(20):5685–5688. [PubMed] [Google Scholar]
- 26.Desai JR, Ochoa S, Prins PA, He AR. Systemic therapy for advanced hepatocellular carcinoma: an update. J Gastrointest Oncol. 2017;8(2):243–255. doi: 10.21037/jgo.2017.02.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao FY. Liver transplantation for hepatocellular carcinoma: beyond the Milan criteria. Am J Transplant. 2008;8(10):1982–1989. doi: 10.1111/j.1600-6143.2008.02351.x. [DOI] [PubMed] [Google Scholar]
- 28.Wu Z, Martinez-Fong D, Trédaniel J, Forgez P. Neurotensin and its high affinity receptor 1 as a potential pharmacological target in cancer therapy. Front Endocrinol. 2012;3:184. doi: 10.3389/fendo.2012.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chau I, Peck-Radosavljevic M, Borg C, et al. Ramucirumab as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib: patient-focused outcome results from the randomised phase III REACH study. Eur J Cancer. 2017;81:17–25. doi: 10.1016/j.ejca.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Akiba J, Nakashima O, Hattori S, et al. Clinicopathologic analysis of combined hepatocellular-cholangiocarcinoma according to the latest WHO classification. Am J Surg Pathol. 2013;37(4):496–505. doi: 10.1097/PAS.0b013e31827332b0. [DOI] [PubMed] [Google Scholar]
- 31.Kassahun WT, Hauss J. Management of combined hepatocellular and cholangiocarcinoma. Int J Clin Pract. 2008;62(8):1271–1278. doi: 10.1111/j.1742-1241.2007.01694.x. [DOI] [PubMed] [Google Scholar]
- 32.Lin G, Toh CH, Wu RC, et al. Combined hepatocellular cho-langiocarcinoma: prognostic factors investigated by computed tomography/magnetic resonance imaging. Int J Clin Pract. 2008;62(8):1199–1205. doi: 10.1111/j.1742-1241.2007.01291.x. [DOI] [PubMed] [Google Scholar]