

Abstract
The disruption of meiotic sex chromosome inactivation (MSCI) has been proposed to be a major developmental mechanism underlying the rapid evolution of hybrid male sterility. We tested this idea by analyzing cell-specific gene expression across spermatogenesis in two lineages of house mice and their sterile and fertile reciprocal hybrids. We found pervasive disruption of sex chromosome gene expression in sterile hybrids at every stage of spermatogenesis. Failure of MSCI was developmentally preceded by increased silencing of autosomal genes, supporting the hypothesis that divergence at the hybrid incompatibility gene, Prdm9, results in increased rates of autosomal asynapsis which in turn triggers widespread silencing of unsynapsed chromatin. We also detected opposite patterns of postmeiotic overexpression or hyper-repression of the sex chromosomes in reciprocal hybrids, supporting the hypothesis that genomic conflict has driven functional divergence that leads to deleterious X–Y dosage imbalances in hybrids. Our developmental timeline also exposed more subtle patterns of mitotic misregulation on the X chromosome, a previously undocumented stage of spermatogenic disruption in this cross. These results indicate that multiple hybrid incompatibilities have converged on a common regulatory phenotype, the disrupted expression of the sex chromosomes during spermatogenesis. Collectively, these data reveal a composite regulatory basis to hybrid male sterility in mice that helps resolve the mechanistic underpinnings of the well-documented large X-effect in mice speciation. We propose that the inherent sensitivity of spermatogenesis to X-linked regulatory disruption has the potential to be a major driver of reproductive isolation in species with chromosomal sex determination.
Keywords: speciation, sexual conflict, hybrid male sterility, gene expression, fluorescence activated cell sorting, meiotic sex chromosome inactivation, postmeiotic sex chromosome repression
Introduction
Proper regulation of the sex chromosomes is an essential component of normal male fertility in mammals (Handel and Schimenti 2010; Royo et al. 2010). The X and Y chromosomes are transcriptionally silenced early in meiosis (meiotic sex chromosome inactivation or MSCI; Handel 2004; Turner et al. 2004) and remain repressed for the duration of spermatogenesis (postmeiotic sex chromosome repression or PSCR) save a relatively small subset of postmeiotically expressed genes (Namekawa et al. 2006). Spermatogenesis has more stringent cellular and molecular checkpoints than oogenesis (Morelli and Cohen 2005; Burgoyne et al. 2009), both MSCI and PSCR are necessary for normal sperm development (Forejt 1985; Ellis et al. 2005; Homolka et al. 2007; Cocquet et al. 2009; Royo et al. 2010), and failure of meiotic X-inactivation is thought to be an important cause of male sterility in humans (Turner 2007; Zamudio et al. 2008). The sex chromosomes also play a large role in speciation (Haldane 1922; Coyne and Orr 1989), often through the rapid evolution of sex-linked hybrid male sterility (Tao et al. 2003; Masly and Presgraves 2007). These parallel observations raise the intriguing possibility that the recurrent evolution of hybrid male sterility—one of the most general and rapidly evolving causes of speciation—may have a common developmental basis.
Does disruption of MSCI, or related sex-linked regulatory processes, provide a common developmental mechanism for the rapid evolution of hybrid male sterility? Nearly a half century ago Lifschytz and Lindsley (1972) proposed that divergence in the regulation of MSCI could contribute to the pervasiveness of hybrid male sterility in species with XY sex determination (i.e., Haldane’s rule; Haldane 1922). This mechanistic hypothesis was later expanded to the more general idea that spermatogenesis is inherently sensitive and prone to disruption in hybrids (Jablonka and Lamb 1991; Wu and Davis 1993). In addition to Haldane’s Rule, an inherent sensitivity of MSCI could also explain the disproportionately large role that X chromosomes plays in the accumulation of hybrid incompatibilities (i.e., the large X-effect; Coyne and Orr 1989; Masly and Presgraves 2007). This compelling hypothesis remains largely untested and only a few studies have linked disrupted sex chromosome regulation to reproductive failure in hybrids (Good et al. 2010; Bhattacharyya et al. 2013; Campbell et al. 2013; Oka and Shiroishi 2014; Davis et al. 2015). MSCI has been best characterized in mammals where it appears to be a special case of meiotic silencing of unsynapsed chromatin (MSUC; Turner 2007). However, while MSCI-like processes have independently evolved in diverse animal lineages (Sin and Namekawa 2013), the underlying molecular details remain unresolved in most species (Landeen et al. 2016). Moreover, sterility usually results in systematic differences in testis cellular composition that can confound genome-wide patterns of expression in the absence of a strong developmental framework (Good et al. 2010), making the MSCI hypothesis difficult to test.
Spermatogenesis and MSCI have been thoroughly studied in the house mouse (Forejt 1985; Homolka et al. 2007; Turner 2007; Zamudio et al. 2008), providing a powerful model to test the role of sex chromosome gene regulation in the evolution of hybrid male sterility. The closely related subspecies Mus musculus musculus and Mus musculus domesticus (hereafter musculus and domesticus) hybridize in a well-characterized zone (Janousek et al. 2012) where hybrid males often have reduced fertility (Turner et al. 2012). There is extensive evidence for a large X-effect for reproductive isolation between musculus and domesticus based on patterns of gene flow across the hybrid zone (Tucker, Sage, et al. 1992b; Payseur et al. 2004; Janousek et al. 2012), the genetic architecture of male sterility (White et al. 2011; Dzur-Gejdosova et al. 2012; Turner et al. 2014), and analyses of chromosome substitution strains (Oka et al. 2004; Storchová et al. 2004; Oka et al. 2007; Good, Dean, et al. 2008; Gregorova et al. 2008). In laboratory crosses, F1 hybrid males with a musculus mother are usually sterile, whereas the reciprocal cross typically yields males with higher fertility (Good, Handel, et al. 2008). The asymmetry is partly due to deleterious epistatic interactions involving the musculus X chromosome (White et al. 2011; Bhattacharyya et al. 2014; Turner et al. 2014) and divergence at the PRDM9 DNA-binding protein (Mihola et al. 2009). PRDM9 directs the location of double-strand breaks (DSBs) by binding specific DNA sequences that are systematically eroded when DSBs are repaired from the unbroken sister chromatid (Baker et al. 2015). This process drives divergence at PRDM9 binding sites and leads to asymmetric PRDM9 binding in hybrids, which impedes DSB repair (Davies et al. 2016), increases rates of autosomal asynapsis during early pachytene (Bhattacharyya et al. 2013, 2014; Davies et al. 2016), and appears to subsequently disrupt MSCI (Good et al. 2010; Bhattacharyya et al. 2013; Campbell et al. 2013). It is still unclear how this process is modulated through interacting loci (Mihola et al. 2009; Dzur-Gejdosova et al. 2012; Flachs et al. 2012; Bhattacharyya et al. 2014) or how disrupted X chromosome inactivation alters autosomal gene networks during spermatogenesis (Turner et al. 2014).
Divergence at PRDM9 binding sites and epistatic disruption of MSCI provide a clear pathway to one aspect of hybrid sterility, but these incompatibilities alone cannot explain the large X-effect for reproductive isolation between musculus and domesticus. Sex-linked hybrid sterility occurs across a broad range of genetic architectures and reproductive phenotypes (Good, Handel, et al. 2008; Oka et al. 2010; White et al. 2011; Flachs et al. 2012; White et al. 2012; Oka et al. 2014) and has been predicted to have other drivers, such as genetic conflict between the X and Y chromosomes (Macholán et al. 2008; Ellis et al. 2011; Cocquet et al. 2012; Campbell and Nachman 2014). The ampliconic sex-linked genes Slx/Slx1 and Sly show dosage-dependent interactions with antagonistic effects on the regulation of PSCR (Cocquet et al. 2009, 2010, 2012). Sly-deficient mice overexpress sex-linked genes resulting in severely abnormal sperm morphology, increased X transmission, and female-biased litters. In contrast, Slx/Slxl1-deficient mice show greater sex-linked repression resulting in moderate sperm defects, increased transmission of the Y, and male-biased sex ratios (Cocquet et al. 2009, 2010, 2012). This conflict is thought to be mediated through relative gene copy numbers, which have independently driven rapid ampliconic expansions of Slx and Sly in musculus (100 Slx/80 Sly) and domesticus (50 Slx/50 Sly) (Ellis et al. 2011; see also Scavetta and Tautz 2010). As a result, reciprocal F1 hybrids are predicted to be either Slx-deficient (fertileDxM: 50 Slx/80 Sly) or Sly-deficient (sterileMxD 100 Slx/50 Sly). This model of X-Y imbalance is not testable in hybrid crosses involving some classic laboratory strains of domesticus (e.g., C57BL, Bhattacharyya et al. 2013; Davies et al. 2016; Wang et al. 2016) because they carry an introgressed musculus Y chromosome (Tucker, Lee, et al. 1992). However, studies using other strains (Campbell et al. 2012; Campbell and Nachman 2014; Case et al. 2015) have shown that interactions between the musculus X and the domesticus Y result in abnormal sperm morphology. Unfortunately, evidence for disruption of PSCR has been at least partially confounded by disruption of MSCI in the same cross (Good et al. 2010; Campbell et al. 2013) and this alternative model of sex-linked regulatory disruption in hybrid mice remains untested.
Here we use fluorescence activated cell sorting (FACS) to developmentally stage spermatogenic gene expression in musculus, domesticus, and their reciprocal sterile and fertile F1 hybrids. We detected disrupted X chromosome expression at every major stage of spermatogenesis, each of which likely reflects distinct mechanistic and genetic bases. We show that both MSCI and PSCR are disrupted in sterile males, resulting in dramatic over-expression of the X chromosome. Early in meiosis, we found a correlation between local gene silencing and rates of asymmetric PRDM9 binding, indicating the widespread initiation of MSUC immediately preceding the disruption of MSCI in sterile males. In postmeiotic cells, we identified opposite patterns of overexpression or hyper-repression of the sex chromosomes in reciprocal hybrids, supporting the hypothesis that X–Y genomic conflict causes deleterious dosage imbalances in hybrid males. Our developmental timeline also exposed early mitotic misregulation of the X chromosome, a previously undocumented stage of spermatogenic disruption in this cross. Finally, we found a strong tendency for autosomal and sex-linked genes to be upregulated in sterile males, indicating that regulatory incompatibilities tend to result in a loss of repression. Collectively, we demonstrate that disrupted sex chromosome expression plays a central role in the evolution of hybrid male sterility and that the large X-effect in mouse speciation has a composite regulatory basis.
Results
Cell-Specific Gene Expression across Spermatogenesis
Spermatogenesis is a complex and asynchronous developmental process, encompassing numerous transitions in cellular composition and cell-specific patterns of gene expression. In addition, testis cellular composition can be strongly impacted by genetic perturbations (e.g., sterility) or evolutionary divergence (Firman et al. 2015). Thus, whole testis expression profiles may confound real expression differences with cellular artifacts (Good et al. 2010; Saglican et al. 2014), impeding general tests for disrupted MSCI or other regulatory mechanisms in sterile hybrids (Good et al. 2010; Turner et al. 2014; Davis et al. 2015; Mack et al. 2016). To overcome these issues, we utilized FACS (Getun et al. 2011) to isolate four highly enriched cell populations spanning three phases of spermatogenesis: mitosis (spermatogonia), early meiosis prior to MSCI (leptotene/zygotene spermatocytes), meiosis after the onset of MSCI (diplotene spermatocytes), and postmeiotic cells (round spermatids, supplementary fig. S1A, Supplementary Material online).
We isolated and sequenced RNA from three individuals for each of these four cell types in domesticus, musculus, and their reciprocal F1 hybrids (n = 48, 4 cell types × 3 replicates × 4 crosses). For each subspecies, we generated interstrain F1s using wild-derived inbred strains to reduce the impacts of inbreeding depression on spermatogenesis (domesticus: WSB/EiJ females x LEWES/EiJ males; musculus: CZECHII/EiJ females × PWK/PhJ males). We previously reported FACS-based transcriptome data from these subspecific strains as part of a broader examination of molecular evolution across mouse spermatogenesis (Larson et al. 2016). Here we focused on the regulatory dynamics of reciprocal F1 hybrid males between two strains (PWK and LEWES) using FACS-based transcriptome data. We confirmed previous studies that the severity of male reproductive deficits is asymmetrical in these hybrids (Good, Handel, et al. 2008; Campbell et al. 2012). F1 hybrids with musculus mothers had smaller testes, lower sperm counts and a high proportion of abnormal sperm morphologies compared with both subspecies (table 1). Although they can produce some sperm and are not completely sterile (Good, Handel, et al. 2008), we will refer to these males as sterileMxD to reflect their severe reproductive deficits. In contrast, males from the reciprocal cross had slightly smaller testes, lower sperm counts, and more abnormal sperm morphology compared with domesticus but were within the range of fertile musculus males (hereafter fertileDxM, table 1). Hybrid mice from these strains have been previously evaluated using cell specific expression for a handful of loci (Campbell et al. 2013) and whole-testes transcriptomes (Good et al. 2010; Mack et al. 2016). This is the first cell-specific quantification of genome-wide expression in F1 hybrids, allowing for a detailed and systematic test of the developmental timeline of disrupted sex chromosome expression in this powerful model system.
Table 1.
Male Reproductive Traits of Domesticus, Musculus and Their F1 Hybrids.
| SterileMxD | FertileDxM | Musculus | Domesticus | |
|---|---|---|---|---|
| Number of males | 14 | 10 | 7 | 49 |
| Body weight (g) | 16.8 ± 0.6 | 19.5 ± 0.6 | 18.1 ± 0.7 | 17.8 ± 0.2 |
| Seminal vesicle weight (mg/g)1 | 4.9 ± 0.3 | 5.8 ± 0.5 | 6.0 ± 0.9 | 5.3 ± 0.2 |
| Testis weight (mg/g)1 | 7.1 ± 0.2▼▽ | 9.3 ± 0.2▼ | 9.7 ± 0.5 | 11.6 ± 0.1 |
| Sperm count (1x106) | 3.4 ± 0.7▼▽ | 12.8 ± 1.5▼ | 16.1 ± 2.3 | 21.4 ± 1.3 |
| Proportion motile sperm | 0.68 ± 0.08 | 0.71 ± 0.07 | 0.75 ± 0.05 | 0.75 ± 0.02 |
| Sperm head morphology index | 0.03 ± 0.01▼▽ | 0.95 ± 0.02▼ | 0.97 ± 0.02 | 0.98 ± 0.00 |
Note.—Values are mean and standard error. Arrows indicate significantly lower values in F1 hybrids compared with domesticus (closed) and musculus (open) based on Wilcoxon rank sum test with FDR correction (Benjamini and Hochberg 1995).
Paired tissue weight (mg) per gram of body weight.
For each cross, we generated between 21 and 30 million uniquely mapped paired reads per cell type (428 million paired reads; supplementary table S1, Supplementary Material online) using reciprocal mapping to remove reference bias (Huang et al. 2014). In addition, we counted multiply mapped fragments for 252 multicopy X-linked genes (Mueller et al. 2013) and all Y-linked genes. After filtering, we retained 14,887 protein-coding genes expressed in at least one cell type. To validate cell population purity, we confirmed cell-specific expression of select candidate genes (supplementary fig. S1B, Supplementary Material online) and clean partitioning of gene expression profiles by cell type (supplementary fig. S1C, Supplementary Material online). As expected, cell types partitioned by cross, with each subspecies forming a distinct cluster and F1 hybrids mid-way between musculus and domesticus (supplementary fig. S1D, Supplementary Material online). In both subspecies, the X chromosome was inactivated in diplotene spermatocytes and repressed in round spermatids (supplementary fig. S2, Supplementary Material online), consistent with previously described patterns of X-linked expression (Namekawa et al. 2006; Larson et al. 2016). Altogether, these results verified that our FACS enrichment yielded highly purified cell populations.
Widespread Overexpression of the X Chromosome across Spermatogenesis
We observed three striking patterns in comparisons between sterile hybrids and all fertile males (i.e., the fertile hybrid and both subspecies). First, the X chromosome was highly enriched for differentially expressed (DE) genes across spermatogenesis (fig. 1 and supplementary fig. S3, Supplementary Material online). At each stage, the X chromosome had as many or more DE genes than all of the autosomes combined (table 2). These results were robust to differences in expression thresholds and handling of multicopy genes on both the autosomes and the sex chromosomes (supplementary table S2, Supplementary Material online). This is opposite to what we previously observed in evolutionary contrasts between musculus and domesticus, where X-linked genes showed similar (spermatogonia) or less expression divergence (round spermatids) than autosomal genes expressed in the same cells (Larson et al. 2016).
Fig. 1.

X chromosome gene expression is disrupted in sterile hybrids at every stage of spermatogenesis. (A) Timeline of spermatogenesis highlighting FACS-enriched cell populations, which span major transitions in gene expression. The sex chromosomes are transcriptionally silenced in early prophase (MSCI) and are repressed during postmeiotic development (PSCR). (B) Cell-specific differential expression (DE) between sterileMxD hybrids and all fertile males across spermatogenesis. Gray lines are the difference in normalized FPKM for DE genes, plotted along each chromosome (top is the centromere). At every cell stage the X chromosome was enriched for DE genes and enriched for genes overexpressed in sterileMxD males. All enrichment tests are based on FDR corrected chromosome-wise hypergeometric tests. (C) The proportion of DE genes for the autosomes and the X chromosome. Significant differences are based on FDR corrected Pearson’s chi-squared test. FPKM values are normalized so that the sum of squares equals 1.
Table 2.
Summary of DE Genes. There were a total of 14,887 genes expressed in at least one cell stage in the testis. Differential expression was compared between reciprocal hybrids (sterileMxD vs. fertileDxM) and between sterileMxD hybrids and fertile males (sterileMxD vs. musculus, domesticus and fertileDxM). ▼ and ▲ refer to genes that have lower or higher expression in the sterileMxD hybrid (e.g., sterileMxD – fertileDxM). Significance differences in the proportion of under/overexpressed genes were evaluated using a χ2 test: P-values: * ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001.
| Spermatogonia |
Leptotene/zygotenespermatocytes |
Diplotenespermatocytes |
Round spermatids |
||||||
|---|---|---|---|---|---|---|---|---|---|
| ▼ | ▲ | ▼ | ▲ | ▼ | ▲ | ▼ | ▲ | ||
| Expressed genes sterileMxD vs, fertileDxM | Auto | 6581*** | 5092 | 5598** | 5307 | 5700*** | 4385 | 5287 | 5460 |
| X | 235 | 248 | 196 | 211 | 6 | 145*** | 32 | 314*** | |
| Y | 12 | 6 | 13 | 6 | 1 | 1 | 63 | 58 | |
| DE genes sterileMxD vs. fertileDxM | Auto | 32 | 55* | 45* | 24 | 31 | 78*** | 54 | 152*** |
| X | 30 | 36 | 15 | 23 | 1 | 67*** | 13 | 178*** | |
| Y | 9* | 1 | 9* | 1 | 0 | 1 | 8 | 39*** | |
| DE genes sterileMxD vs. all fertile | Auto | 3 | 15** | 7 | 7 | 7 | 18* | 11 | 36*** |
| X | 1 | 19*** | 0 | 6* | 0 | 56*** | 0 | 74*** | |
| Y | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 4 | |
Second, transgressive X-linked gene expression in comparisons between sterileMxD hybrids and all fertile males (i.e., genes with expression outside the range of fertile males) were due to higher expression in sterileMxD hybrids. This striking asymmetry persisted when we controlled for evolutionary divergence by directly comparing expression of the musculus X in sterileMxD hybrids versus the X chromosome in musculus males (fig. 2). Thus, the musculus X chromosome was overexpressed in the sterileMxD hybrid background across all developmental stages, though the extent and magnitude varied across different stages of spermatogenesis (table 2). There are specific developmental mechanisms (i.e., disruption of MSCI and PSCR) that can explain the overexpression of the X chromosome late in spermatogenesis (discussed below). However, we also detected a strong enrichment of overexpressed X-linked genes in spermatogonia of sterileMxD hybrids, prior to MSCI. In spermatogonial cells, sterileMxD hybrids had a higher proportion of DE genes on the X chromosome than all of the autosomes combined (X chromosome: 4.14%, autosomes: 0.15%, fig. 1C) and nearly all X-linked DE genes were overexpressed in sterileMxD hybrids compared with all fertile males (table 2) and in comparisons only with musculus males (fig. 2). Importantly, there was little overlap among DE genes expressed at each time point (supplementary fig. S4, Supplementary Material online), indicating the presence of transcripts from X-linked genes in the later stages was not simply due to incomplete FACS isolation or persistence of transcripts from earlier cell stages.
Fig. 2.

Sex chromosome expression is disrupted in hybrids relative to their conspecific chromosome. The proportion of expressed genes that are DE genes between hybrids and a single parent (musculus or domesticus). Positive and negative logFC indicate the proportion of expressed genes that were over or underexpressed in hybrids. Differences in the proportion of over/underexpressed DE genes and differences in the total proportion of DE genes (brackets) are indicated above each contrast (Pearson’s chi square test, FDR corrected P-values: *≤0.05, **≤0.01, ***≤0.001). SP = spermatogonia, LZ = leptotene/zygotene spermatocytes, DIP = diplotene spermatocytes, RS = round spermatids.
Third, while we found few autosomal differences associated with hybrid male sterility overall, most autosomal DE genes were also overexpressed in sterileMxD hybrids in all but leptotene/zygotene spermatocytes (table 2). This asymmetry occurred only in a small subset of genes and cannot be explained by genome-wide asymmetries in our data (supplementary fig. S5, Supplementary Material online). In fact, when we considered all genes (i.e., even those that were not DE), the autosomes tended to be slightly underexpressed in sterileMxD hybrids (table 2) and this result was robust to different library normalization methods. These results suggest that disrupted expression in hybrids usually involves a loss of repression on the autosomes and the sex chromosomes.
Disruption of MSCI and Meiotic Silencing of Unsynapsed Chromatin
We found chromosome-wide disruption of MSCI in sterileMxD hybrids (fig. 1B). A total of 151 X-linked genes were expressed in diplotene spermatocytes of F1 hybrids. Of these, 145 had higher expression in sterileMxD hybrids (table 2) and only 44 were expressed in fertileDxM hybrids. That means more than three times as many genes were expressed in diplotene spermatocytes of sterileMxD hybrids compared with fertileDxM hybrids. When compared with all fertile males, 56 of the expressed X-linked genes were significantly overexpressed in sterileMxD hybrids. This was substantially more X-linked DE genes than on all of the autosomes combined (table 2) and a stronger chromosome-wide signal of MSCI disruption than was apparent in previous studies (Good et al. 2010; Bhattacharyya et al. 2013; Turner et al. 2014). However, sterileMxD hybrids still had relatively low diplotene X-linked expression compared with other cell types (supplementary fig. S2, Supplementary Material online), suggesting some persistence of an overall repressive regulatory environment at this stage. This may reflect cell-to-cell variation in the occurrence and/or magnitude of disrupted MSCI. We also observed meiotic inactivation of the Y chromosome (supplementary fig. S6, Supplementary Material online), but there were too few Y-linked genes expressed prior to MSCI (18–19 genes) to evaluate patterns of misexpression in diplotene spermatocytes (table 2).
Previous studies have shown a strong association between PRDM9 binding asymmetry, levels of autosomal asynapsis, and disruption of MSCI in hybrids (Bhattacharyya et al. 2013; Davies et al. 2016; see above). However, it has remained unclear how these phenomena impact the global landscape of sex-linked and autosomal gene expression in sterile males (Good et al. 2010; Turner et al. 2014). We tested the prediction that elevated autosomal asynapsis triggers the MSUC response in late zygotene cells (Bhattacharyya et al. 2013; Davies et al. 2016), causing localized signatures of reduced gene expression in sterile hybrids. We used a sliding gene window to test for local enrichment of underexpressed autosomal genes in leptotene/zygotene cells of sterileMxD hybrids compared with fertileDxM hybrids. We found that chromosomes 4, 7, 17, and 19 had regions enriched for genes underexpressed in sterileMxD hybrids (fig. 3A). Notably, a strongly underexpressed region on chromosome 7 was coincident with a locus shown to interact with Meir1, a major X chromosome modifier of differences in global recombination rates between musculus and domesticus (Dumont and Payseur 2011; Balcova et al. 2016). Patterns of underexpression were strongest for chromosomes 17 and 19, where at least half of all gene windows were underexpressed in sterileMxD hybrids.
Fig. 3.

Spatial patterns of expression reveal regions of potential autosomal asynapsis. (A) Counts of genes in gene windows (250 genes) that are underexpressed in sterileMxD hybrids compared with fertileDxM hybrids for leptotene/zygotene spermatocytes. Highlighted regions on chromosomes 4, 7, 17, and 19 fall outside of the 99th quantile modeled with a Poisson distribution. (B) Chromosome-wide expression immediately prior to MSCI (leptotene/zygotene spermatocytes) correlates with DMC1 signals, an estimate of the degree of asymmetric PRDM9 binding (data from Davis et al. 2016). Chromosomes with higher rates of asymmetric PRDM9 binding (i.e., larger DMC1 signal ratios) have overall lower expression in sterileMxD hybrids, consistent with autosomal silencing. Chromosomes in bold were evaluated in Bhattacharyya et al. (2013).
Chromosomes 17 and 19 also showed the highest rates of meiotic chromosomal asynapsis in sterile musculus and domesticus F1 hybrids in a previous survey of five autosomes (17: 32.1% and 19: 46.6% of cells, Bhattacharyya et al. 2013). While we do not currently have estimates of autosomal asynapsis rates for the strains used in this study, Davies et al. (2016) found that the rate of asymmetric PRDM9 binding in musculus × domesticus hybrids provides a good proxy for chromosome-specific rates of asynapsis. Davies et al. (2016) quantified PRDM9 binding asymmetry based on the proportion of cells marked at a given locus with DMC1 (a protein that binds to single-stranded 3′ DNA sequence following DSBs). We found the per chromosome median difference in expression (logFC) between sterileMxD and fertileDxM hybrids in leptotene/zygotene cells was negatively correlated with DMC1-based estimates of asynapsis [i.e., the ratio of DMC1 heat values in sterile versus fertile males from Davies et al. (2016); fig. 3B]. Chromosomes with lower overall expression in sterileMxD hybrids had the highest DMC1-estimated rates of autosomal asynapsis associated with asymmetrical Prdm9 binding. There was also a strong negative correlation between lower expression in sterileMxD hybrids and DMC1 ratios in diplotene spermatocytes (r2 = 0.334, P = 0.005), indicating that silencing of these chromosomes persists after synapsis is complete. These variables were weakly correlated in spermatogonia (r2 = 0.144, P = 0.060) and showed no relationship in round spermatids (r2 = −0.006, P = 0.357). Overall, these patterns strongly support the hypothesis that chromosome-specific variation in the extent of asymmetric Prdm9 binding in sterileMxD hybrids influences rates of autosomal asynapsis, which triggers MSUC followed by disrupted MSCI.
X–Y Conflict and Disruption of PSCR
Next we focused on patterns of sex-linked expression in postmeiotic round spermatids to test the model that dosage-dependent interactions between Slx and Sly have antagonistic effects on the regulation of PSCR (Cocquet et al. 2009, 2010, 2012). Our working model assumed copy number estimates for musculus (100 Slx/80 Sly) and domesticus (50 Slx/50 Sly) derived from Ellis et al. (2011) analysis of inbred strains from each lineage. Quantitative estimates of Slx/Sly copy numbers vary by study and genotype (Scavetta and Tautz 2010; Ellis et al. 2011; Case et al. 2015). However, differences in relative copy numbers between musculus and domesticus are qualitatively consistent across studies and have been predicted to cause dosage imbalances between SLX and SLY in F1 hybrids (Ellis et al. 2011; Cocquet et al. 2012). Under this model, PSCR should be disrupted in Sly-deficient hybrids (sterileMxD) causing sex-linked genes to be overexpressed, whereas Slx-deficient hybrids (fertileDxM) should show some decreased sex-linked expression (i.e., hyper-repression). To test this we used a method developed to provide more accurate expression estimates for multicopy genes (Bray et al. 2016) to quantify transcript-level expression of Slx and Sly. As we would predict based on copy number, there was higher expression of Slx and Sly in musculus compared with domesticus. Consistent with a model of Slx-Sly imbalance, Sly-deficient hybrids overexpressed Slx relative to musculus. Sly expression in hybrids corresponded to species-specific differences in copy numbers, and was not over/underexpressed relative to their conspecific chromosome (fig. 4A). Overall patterns of sex-linked expression were also consistent with the Slx–Sly imbalance model (Cocquet et al. 2012). There was dramatically higher X and Y-linked expression in round spermatids of sterileMxD hybrids compared with the musculus X and the domesticus Y (figs. 2 and 4B). Genes that are normally silenced in postmeiotic cells were expressed and genes that escape PSCR had higher expression levels (supplementary fig. S2, Supplementary Material online). We saw the opposite pattern in fertileDxM hybrids (Slx-deficient), which had lower X-linked expression compared with the domesticus X (figs. 2 and 4B). Importantly, we observed a strong negative correlation in gene expression between reciprocal hybrids in round spermatids (Spearman’s ρ = −0.168, P = 0.003). Genes that were highly expressed in sterileMxD hybrids tended to show lower expression in fertileDxM hybrids relative to species-specific controls. There was no evidence for a negative correlation between the reciprocal hybrids in any other cell type (supplementary fig. S7, Supplementary Material online).
Fig. 4.
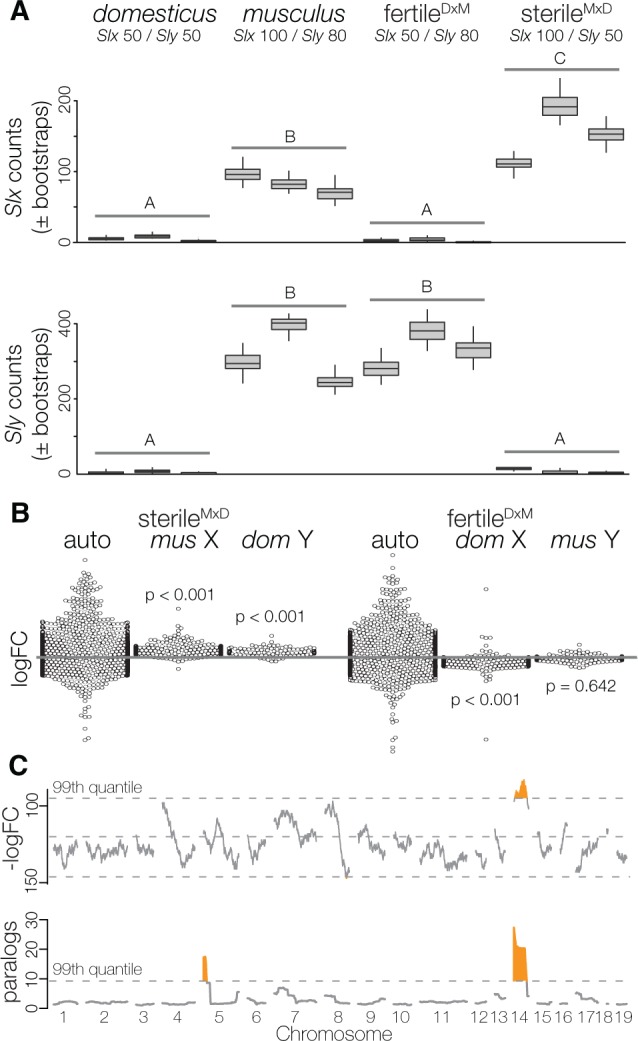
Signals of genomic conflict in sex-linked gene expression in round spermatids. (A) Expression levels of Slx and Sly in round spermatids, estimated using transcript-level counts based on pseudoalignments in kallisto. Letters represent significant differences using likelihood ratio tests in the R package sleuth. (B) Expression differences in rounds spermatids (all genes) between hybrids and both subspecies (mus = musculus, dom = domesticus). Each point represents an observed logFC, with point densities “wrapped” at edges. P-values are differences in the median logFC between autosomes and the sex chromosomes (Wilcoxon test). (C) Sliding-gene windows (250 genes) for counts of underexpressed genes (top) and total number of paralogs (bottom) expressed in round spermatids of sterileMxD hybrids relative to fertileDxM hybrids. Highlighted regions fall outside of the 99th quantile modeled with a Poisson distribution. Chromosome 14 had a run of genes overexpressed in sterileMxD hybrids (Wald–Wolfowitz runs test, FDR corrected P-value < 0.01), and both chromosomes 5 and 14 are enriched for paralogs.
Slx and Sly primarily regulate sex-linked expression through antagonistic effects on PSCR, but there is also evidence for co-regulation of several other genes (Cocquet et al. 2009, 2012; Comptour et al. 2014) including some ampliconic clusters of autosomal genes expressed in round spermatids. To test for similar effects in our reciprocal hybrids, we conducted a sliding gene-window analysis to identify genomic regions with clusters of over- or under-expressed genes between reciprocal hybrids in postmeiotic cells. One large region on chromosome 14 had elevated expression in Sly-deficient hybrids (fig. 4C). This region was not identified in prior studies that have looked for autosomal co-regulation with Slx or Sly (Cocquet et al. 2009). Interestingly, we found this region of chromosome 14 was also highly enriched for gene paralogs expressed in round spermatids (fig. 4C), suggesting that these multicopy gene families may be co-regulated with Slx and/or Sly.
Discussion
When Lifschytz and Lindsley (1972) first articulated the general model for meiotic inactivation of the X chromosome in males, they hypothesized a link between regulatory disruption at this key developmental stage and the evolution of hybrid male sterility. Here we show that the X chromosome is misregulated in sterile hybrid males at every major stage of spermatogenesis, demonstrating a recurrent regulatory syndrome that is broader than MSCI and likely plays a key role in the large X-effect for hybrid male sterility in mice. Below we discuss evidence that these patterns reflect the disruption of MSCI, as well as distinct mitotic and postmeiotic developmental processes that involve independent genetic mechanisms. Collectively, these patterns extend our mechanistic understanding of the predominant role that the X chromosome plays in mouse speciation and reveal that spermatogenic sex chromosome regulation is an inherently sensitive process that is prone to disruption with evolutionary divergence.
The Composite Developmental Basis of X-Linked Hybrid Male Sterility
Our developmental timeline provides insight into the disrupted regulatory landscape of early meiosis and, when combined with other recent findings, presents a detailed genetic model for MSCI and Prdm9-related hybrid male sterility in house mice. Biased gene conversion has driven rapid functional divergence of PRDM9 binding sites between closely-related mouse lineages (Mihola et al. 2009; Baker et al. 2015), which in turn leads to asymmetric DSBs in F1 hybrids between musculus and domesticus (Davies et al. 2016) and a predisposition to autosomal asynapsis (Mihola et al. 2009; Bhattacharyya et al. 2013). The extent of Prdm9 binding site divergence varies across autosomes leading to chromosomal variation in rates of asynapsis (Davies et al. 2016). Building on this foundation, we showed a chromosome-level association between the extent of PRDM9 binding site divergence (Davies et al. 2016) and the down-regulation of autosomal genes that is consistent with the induction of MSUC in response to asynapsis. As predicted (Homolka et al. 2007; Turner 2007; Burgoyne et al. 2009), this global regulatory response developmentally precedes the disruption of MSCI in sterile males (fig. 3).
The exact mechanistic links between autosomal asynapsis and disruption of MSCI remain unclear. Disruption of MSCI is associated with elevated autosomal asynapsis (Burgoyne et al. 2009), possibly due to the exhaustion of limited essential MSUC proteins necessary to silence the sex chromosomes (Turner 2015) or intrusion of unsynapsed autosomes on the nuclear domain where inactivated sex chromosomes are sequestered (Forejt 1985; Bhattacharyya et al. 2013). None of the genes known to be involved in the initiation or progression of MSUC and MSCI (Turner 2007, 2015) showed disrupted expression in sterileMxD hybrids (supplementary table S3, Supplementary Material online), suggesting that the gene networks involved in silencing unpaired chromosomes are functional even when MSCI is disrupted. Regardless of the mechanistic causes, Prdm9-related asynapsis in mouse hybrids depends on an epistatic interaction with the musculus X chromosome (Forejt 1996; Bhattacharyya et al. 2014; Turner et al. 2014). The musculus X-linked gene(s) underlying F1 hybrid male sterility and the associated disruption of MSCI have been localized to a relatively small interval (Hstx2, Bhattacharyya et al. 2014) coincident with a major locus (Meir1) that influences differences in global recombination rates between musculus and domesticus (Dumont and Payseur 2011; Balcova et al. 2016). This suggests a close relationship between autosomal asynapsis, MSCI, X-linked meiotic control of recombination, and the evolution of hybrid male sterility in mice (Balcova et al. 2016). These results raise the intriguing possibility that divergence in the control of meiotic recombination may be an important driver in the evolution of reproductive isolation (Payseur 2016). In mice, this divergence is partially driven by rapid evolution of PRDM9 binding sites that impairs DSB repair (Davies et al. 2016); a mechanism that could contribute more broadly to the evolution of hybrid male sterility in mammals given the conserved role of Prdm9 in recombination (Oliver et al. 2009). Similarly, other processes that disrupt autosomal synapsis (e.g., chromosomal translocations and rearrangements) could also trigger MSUC and subsequent failure of MSCI (Homolka et al. 2007), but it is unclear if these other forms of autosomal divergence would also involve interactions with the X chromosome (i.e., be relevant to the large X-effect).
The ongoing genetic resolution of Prdm9/MSCI-related hybrid sterility is exciting, but our study demonstrates that this is just one regulatory aspect of the large X-effect for hybrid male sterility in mice. Alleles underlying sterile interactions at Prdm9 are polymorphic in musculus and domesticus (Piálek et al. 2008; Good et al. 2010; Flachs et al. 2012), and sterility only occurs when a hybrid male with a musculus X chromosome is heterozygous for two sterile alleles (Flachs et al. 2012; Bhattacharyya et al. 2014; Flachs et al. 2014). In principle, this complex genetic architecture (i.e., epistatic, polymorphic, underdominant, and asymmetric) should be rare in nature. For example, the musculus–domesticus hybrid zone is dominated by advanced generation hybrids (Turner and Harr 2014) where heterozygous tracks of divergent PRDM9 binding sites should be relatively rare, making extensive asymmetric Prdm9 binding less likely to be an effective reproductive barrier between subspecies. In reality, Prdm9 does not show restricted gene flow across the mouse hybrid zone, whereas both sex chromosomes have strongly restricted gene flow between musculus and domesticus (Macholán et al. 2011; Janousek et al. 2012). It is also clear from genetic mapping studies that the large X-effect for reproductive isolation has a complex genetic and phenotypic basis in mice (Storchová et al. 2004; Good, Dean, et al. 2008; White et al. 2011; Campbell et al. 2012; Campbell and Nachman 2014; Turner and Harr 2014; Turner et al. 2014). Different regions of the genome contribute to sterility in different hybrid crosses, while some regions (i.e., chromosome 17 and the X chromosome) are repeatedly found to play a large role in sterility (fig. 5). The genetic and developmental bases of these complex sterility polymorphisms and their association with X-linked incompatibilities have remained elusive.
Fig. 5.

Genomic regions with disrupted expression in hybrids. Genome plot showing the overlap among disrupted genomic regions identified in this study and evaluations of gene expression and sterility phenotypes in other studies. Highlighted chromosomes (17 and X) are hotspots for disrupted gene regulation, with overlap among this study and at least three others. 1Turner et al. (2014), 2Turner and Harr (2014), 3White et al. (2011).
Our data provide strong evidence that reciprocal hybrid males show directional disruption of PSCR (fig. 2) as predicted under the model of genomic conflict between Sly and Slx (Cocquet et al. 2012). Sly is a key regulator of the epigenetic marks that repress sex chromosome expression in round spermatids, while Slx/Slxl1 promote postmeiotic sex-linked expression (Cocquet et al. 2012). These antagonistic effects, combined with copy number divergence between musculus and domesticus (Scavetta and Tautz 2010; Ellis et al. 2011), set the stage for hybrid disruption of PSCR due to copy number imbalances. Alternatively, loss of PSCR in sterileMxD hybrids could simply reflect the downstream effects of disrupted MSCI (Campbell et al. 2013) because PSCR is partially dependent on the transmission of the repressive epigenetic environment established at MSCI (Turner et al. 2006). Previous genome-wide expression studies on whole testis have detected up-regulation of the X chromosome in these same crosses (Good et al. 2010; Campbell et al. 2013; Mack et al. 2016), but could not differentiate between MSCI and PSCR . Campbell et al. (2013) used quantitative PCR to show that both MSCI and PSCR appear disrupted in sterileMxD hybrids (seven genes, FACS-enriched cells) and that disrupted X-linked expression (presumably disruption of MSCI) can be detected in whole testis independent of Y chromosome genotype (12 genes). None of these previous studies directly tested the reciprocal predictions of Sly/Slx genomic conflict model (Cocquet et al. 2012).
Here we detected both massive overexpression of the sex chromosomes in Sly-deficient sterileMxD hybrids and under-expression of X-linked genes in Slx-deficient fertileDxM hybrids (figs. 2 and 4). Indeed, we found a strong negative correlation in expression levels between reciprocal hybrids in round spermatids, while there was a weak (non-significant) positive trend coincident with the disruption of MSCI in diplotene spermatocytes (supplementary fig. S7, Supplementary Material online). We also detected overexpression of a highly repetitive region of chromosome 14 in Sly-deficient sterileMxD hybrids, suggesting that these autosomal genes may be co-regulated with Slx through the repressive effects of Sly. These reciprocal patterns of hypo- and hyper-repression and autosomal co-regulation fit well with data from SLY/SLX knockdowns (Cocquet et al. 2009, 2010, 2012) and would not be expected if postmeiotic disruptions were entirely a consequence of disrupted MSCI. Genetic conflict during spermatogenesis is thought to be a widespread phenomenon (Meiklejohn and Tao 2010; Ellegren 2011; Sin and Namekawa 2013). Our results provide some of the strongest evidence to date of hybrid male sterility evolving as a direct consequence of antagonistic co-evolution between the sex chromosomes.
Early in spermatogenesis, prior to MSCI and PSCR, we detected a strong enrichment of X-linked DE genes (fig. 1) and the majority of X-linked spermatogonia genes were overexpressed in sterileMxD hybrids (table 2). This unexpected asymmetry suggests disrupted expression of the X chromosome manifests prior to entering meiosis. It is unclear what regulatory phenomena could lead to such strong directional effects in spermatogonia, but a qualitatively similar X-linked regulatory effect was observed in sterile consomic lines between the hybrid lineage M. m. molossinus (a late generation hybrid of musculus x M. m. castaneus) and classic mouse inbred strains (primarily domesticus) (Oka et al. 2014). In these mice, there were more X-linked DE genes during the first wave of spermatogenesis (whole testis, 5–7 days postpartum), possibly due to the misregulation of X-linked genes in some early cell types (stem cells, spermatogonia, or sertoli cells) and differences in testis histology due to delayed fertility or mitotic arrest (Oka et al. 2014). Together with our finding of disrupted X-linked expression early in musculus and domesticus hybrids, this suggests that misregulation of the X chromosome early in spermatogenesis could underlie cryptic sterility phenotypes that contribute to the cumulative disruption of spermatogenesis.
General Insights into Hybrid Regulatory Incompatibilities
Transgressive autosomal and X-linked expression in sterileMxD hybrids was strongly associated with upregulation across the developmental timeline, suggesting that disrupted gene regulation often reflects loss of gene repression. Transgressive expression in hybrids has previously been found to be bi-directional or underexpressed (reviewed in Ortíz-Barrientos et al. 2006; Oka and Shiroishi 2014). However, there are notable exceptions that have found overexpressed autosomal genes in sterile F1 hybrids (copepods, Barreto et al. 2015; mice, Mack et al. 2016) or in interspecific introgression lines (Drosophila, Meiklejohn et al. 2014; tomato, Guerrero et al. 2016). Studies focusing on whole animal or tissue level expression have limited ability to detect asymmetrical transgressive expression, since relative expression is highly sensitive to differences in cellular composition. Nevertheless, late-generation introgression lines suggest that divergence in trans-regulatory factors can disrupt interactions between gene regulators that suppress expression and their targets (Meiklejohn et al. 2014; Guerrero et al. 2016). Additional cell-specific data are needed to determine the generality of overexpression in disrupted hybrid regulatory networks.
Spermatogenic Sex Chromosome Regulation Is Broadly Sensitive to Evolutionary Divergence
The predominance of hybrid male sterility in XY systems (Laurie 1997; Presgraves 2008; Delph and Demuth 2016) indicates that having distinct sex chromosomes renders spermatogenesis unusually prone to hybrid breakdown. Numerous evolutionary or developmental theories that have been proposed to explain these patterns, including faster X-linked evolution (Charlesworth et al. 1987), genomic conflict via sex-chromosome meiotic drive (Frank 1991; Hurst and Pomiankowski 1991; Meiklejohn and Tao 2010), intense sexual selection on male-biased genes (Wu and Davis 1993), X-autosome gene movement (Moyle et al. 2010), and an inherent sensitivity of spermatogenesis (Wu and Davis 1993) mediated by disruption of MSCI (Lifschytz and Lindsley 1972; Jablonka and Lamb 1991). There is some empirical support for all of these evolutionary (reviewed in Masly and Presgraves 2007; Meiklejohn and Tao 2010; Moyle et al. 2010; Larson et al. 2016) and developmental phenomena (Turner 2015), but their direct contributions to the evolution of reproductive isolation have remained unclear.
We and others have previously linked disruption of MSCI to the evolution of hybrid male sterility (Good et al. 2010; Bhattacharyya et al. 2013; Campbell et al. 2013; see also Davis et al. 2015). Here we have shown recurrent disruption of X-linked expression across spermatogenesis (fig. 1), indicating that sex-linked gene regulation is inherently sensitive to divergence and that this large X-regulatory effect extends well beyond MSCI. Consistent with this, X-linked gene expression and DNA methylation divergence between mouse lineages is most highly constrained in postmeiotic cells (Larson et al. 2016); the same stage of spermatogenesis where we see the greatest X-linked disruption in sterile hybrids. Nonetheless, this large X-regulatory effect in mouse hybrids is associated with rapid evolution, either through rapid divergence of recombination machinery (e.g., Prdm9-binding site evolution) or intense genomic conflict between the sex chromosomes (e.g., rapidly co-evolving Slx/Sly amplicons). Given these patterns, we propose that the evolution of hybrid male sterility is ultimately driven by interactions between rapidly evolving components of sex chromosome regulation and the inherent sensitivity of spermatogenesis to changes in XY expression. If generally true, this inherent developmental vulnerability has the potential to be a major driver of Haldane’s rule (Haldane 1922) and the large-X effect in XY genetic systems.
Materials and methods
Crosses and Male Sterility Phenotypes
We used wild-derived inbred strains of two subspecies of house mice: M. m. domesticus (WSB/EiJ, LEWES/EiJ) and M. m. musculus (PWK/PhJ, CZECHII/EiJ). We generated inter-strain F1s for each subspecies (WSB females × LEWES males, CZII females × PWK males) to reduce the impacts of inbreeding depression on spermatogenesis following previous reproductive studies on these mice (Good, Dean, et al. 2008; Good et al. 2010; Campbell et al. 2013). For intersubspecific hybrid crosses we utilized two of these strains, LEWES and PWK, in reciprocal crosses to generate sterile (sterileMxD) and fertile (fertileDxM) hybrids. Experimental mice were obtained from breeding colonies established with mice purchased from the Jackson Laboratory (http://jaxmice.jax.org) and maintained at the University of Montana (UM) Department of Laboratory Animal Resources (IACUC protocol 002-13). Males were weaned at ∼21 days after birth, housed in same-sex sibling groups until they were isolated at 45 days. Males were euthanized between 60 and 90 days using CO2 followed by cervical dislocation. We quantified male reproductive traits as described previously (Good, Dean, et al. 2008; Good, Handel, et al. 2008).
Testicular Cell Sorting and RNAseq Libraries
We used FACS to isolate enriched testicular cell populations as described in Getun et al. (2011) and Larson et al. (2016). Testes were decapsulated, washed twice in 1 mg/mL collagenase (Worthington Biochemical) and GBSS (Sigma) and disassociated in 1 mg/mL trypsin (Worthington Biochemical). Trypsin was inactivated with 0.16 mg/mL fetal calf serum (Sigma) and cells were stained in 0.36 mg/mL of Hoechst 33343 (Invitrogen) and 0.002 mg/mL propidium iodide. At each step, solutions were placed in a VWR minishaker at 120 rpm at 33 °C for 15 min and 0.004 mg/mL DNase was added to eliminate clumps. Cells were filtered twice through a 40 μm cell strainer and kept on ice prior to sorting. Cell sorting was performed on a FACSAria IIu cell sorter (BD Biosciences) at the UM Center for Environmental Health Sciences Fluorescence Cytometry Core. FACS isolates cells based on size, granularity, and fluorescence. Enriched cell-populations were collected in 15 μL beta mercaptoethanol (Sigma) per mL of RLT lysis buffer (Qiagen). Our experimental protocol was optimized for cell purity using both gene expression metrics and microscopy (see supplementary fig. S1, Supplementary Material online). RNA was extracted from each cell type using the Qiagen RNeasy kit and quantified on a Bioanalzyer 2000 (Aglient). Samples with RNA integrity (RIN) ≥ 8 were prepared for sequencing using the Illumina Truseq Sample Prep Kit v2 using a design that avoided batch effects between cell populations and genotypes. Libraries were sequenced (paired end, 100 bp) on Illumina HiSeq 2000 at the QB3 Vincent J. Coates Genomics Sequencing Laboratory at University of California Berkley and the Utah Microarray Core at the University of Utah.
Read Mapping
We removed Illumina adaptors and low quality bases from reads using Trimmomatic v0.32 (Bolger et al. 2014) and mapped reads using Tophat v2.0.10 (Kim et al. 2013) to published strain-specific pseudo-references for M. m. musculus (PWK/PhJ) and M. m. domesticus (WSB/EiJ) that incorporated all known single nucleotide variants, indels, and structural variants relative to the Genome Reference Consortium mouse build 38 (GRCm38, Ensembl release 75) (Huang et al. 2014). This approach minimizes mapping bias to the mouse reference genome, which is predominantly derived from M. m. domesticus (Yang et al. 2011). We translated reads back into the GRCm38 coordinates using Lapels v1.0.5 and merged alignments using Suspenders v0.2.4 (Huang et al. 2014). We used featureCounts v1.4.4 (Liao et al. 2013) to assign uniquely mapped paired reads (Q 20, C, paired) to annotated genes (Ensembl release 78). We counted multiply-mapped reads for X-linked genes that are multicopy (108 genes) or ampliconic (144 genes) (Mueller et al. 2013) and all Y-linked genes (183 genes). We also counted multiply mapped reads for the large multicopy Speer family on chromosome 5, which is potentially co-regulated with Sly (Cocquet et al. 2009). To validate our estimates of Slx/Sly expression, we estimated transcript level read counts using a pseudoalignment approach, which has been proposed to be more robust to counts of multiply mapped reads, implemented in kallisto v0.43.0 (Bray et al. 2016), with differential expression evaluated in the R package sleuth v 0.28.1 (Pimentel et al. 2016).
Differential Expression
All gene expression analyses were conducted using the Bioconductor v3.0 package edgeR v3.6.8 (Robinson et al. 2010) in R v3.1.1, with false discovery rates of 5% (Benjamini and Hochberg 1995). We normalized our data using the scaling factor method in edgeR and restricted our analysis to protein-coding genes with a minimum expression of FPKM > 1 in 3/48 samples. For all analyses, we tested a range of expression thresholds (FPKM > 1–10), different ways of handling multiply mapped reads, and an alternative normalization method (i.e., weighted trimmed mean of M-values). We fit our data with negative binomial generalized linear models with Cox-Reid tagwise dispersion estimates (McCarthy et al. 2012). Our model included cross type and cell type as a single factor and we constructed a design matrix that contrasted each unique combination (e.g., sterileMxD vs. fertileDxM for each cell type). To evaluate differential expression, we used likelihood ratio tests, dropping one coefficient from the design matrix (i.e., the “null” model) and comparing that to the full model. For all analyses, we restrict our results to genes that are expressed in the focal cell type (FPKM > 1 in 3/6 samples). We defined genes as having disrupted expression for a given cell type in sterileMxD hybrids when expression was higher or lower in the sterile males compared with all fertile males (fertileDxM, musculus and domesticus).
Data Accessibility
The data reported in this paper are available through the National Center for Biotechnology Information under accession number SRP065082.
Supplementary Material
Supplementary tables S1–S3 and figures S1–S7 are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Pamela Shaw, Irina Getun, and Bivian Torres for assistance with FACS; members of the Good lab, Polly Campbell, Peter Ellis, Andrew Morgan, and three anonymous reviewers for helpful feedback and the staff of the UM Laboratory Animal Research facility. We thank the University of Montana Fluorescence Cytometry Core, supported by the National Institute of General Medicine Sciences of the National Institutes of Health (P30GM103338), the UM Genomics Core, supported by a grant from the M.J. Murdock Charitable Trust, and the Vincent J. Coates Genomics Sequencing Laboratory at University of California Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303. This work was funded by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health (R01-HD073439 to JMG) and the National Institute of General Medical Sciences (R01-GM098536 to MDD).
References
- Baker CL, Kajita S, Walker M, Saxl RL, Raghupathy N, Choi K, Petkov PM, Paigen K. 2015. PRDM9 drives evolutionary erosion of hotspots in Mus musculus through haplotype-specific initiation of meiotic recombination. PLoS Genet. 11:e1004916.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcova M, Faltusova B, Gergelits V, Bhattacharyya T, Mihola O, Trachtulec Z, Knopf C, Fotopulosova V, Chvatalova I, Gregorova S, et al. 2016. Hybrid sterility locus on chromosome X controls meiotic recombination rate in mouse. PLoS Genet. 12:e1005906–e1005916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto FS, Pereira RJ, Burton RS. 2015. Hybrid dysfunction and physiological compensation in gene expression. Mol Biol Evol. 32:613–622. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 57:289–300. [Google Scholar]
- Bhattacharyya T, Gregorova S, Mihola O, Anger M, Sebestova J, Denny P, Simecek P, Forejt J. 2013. Mechanistic basis of infertility of mouse intersubspecific hybrids. Proc Natl Acad Sci U S A. 110:E468–E477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya T, Reifova R, Gregorova S, Simecek P, Gergelits V, Mistrik M, Martincova I, Piálek J, Forejt J. 2014. X chromosome control of meiotic chromosome synapsis in mouse inter-subspecific hybrids. PLoS Genet. 10:e1004088.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 34:525–527. [DOI] [PubMed] [Google Scholar]
- Burgoyne PS, Mahadevaiah SK, Turner JMA. 2009. The consequences of asynapsis for mammalian meiosis. Nat Rev Genet. 10:207–216. [DOI] [PubMed] [Google Scholar]
- Campbell P, Good JM, Dean MD, Tucker PK, Nachman MW. 2012. The contribution of the Y chromosome to hybrid male sterility in house mice. Genetics 191:1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P, Good JM, Nachman MW. 2013. Meiotic sex chromosome inactivation is disrupted in sterile hybrid male house mice. Genetics 193:819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P, Nachman MW. 2014. X-Y interactions underlie sperm head abnormality in hybrid male house mice. Genetics 196:1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case LK, Wall EH, Osmanski EE, Dragon JA, Saligrama N, Zachary JF, Lemos B, Blankenhorn EP, Teuscher C. 2015. Copy number variation in Y chromosome multicopy genes is linked to a paternal parent-of-origin effect on CNS autoimmune disease in female offspring. Genome Biol. 16:R28.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH. 1987. The relative rates of evolution of sex chromosomes and autosomes. Am Nat. 130:113–146. [Google Scholar]
- Cocquet J, Ellis PJI, Mahadevaiah SK, Affara NA, Vaiman D, Burgoyne PS. 2012. A genetic basis for a postmeiotic X versus Y chromosome intragenomic conflict in the mouse. PLoS Genet. 8:e1002900.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquet J, Ellis PJI, Yamauchi Y, Mahadevaiah SK, Affara NA, Ward MA, Burgoyne PS. 2009. The multicopy gene Sly represses the sex chromosomes in the male mouse germline after meiosis. PLoS Biol. 7:e1000244.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquet J, Ellis PJI, Yamauchi Y, Riel JM, Karacs TPS, Rattigan A, Ojarikre OA, Affara NA, Ward MA, Burgoyne PS. 2010. Deficiency in the multicopy Sycp3-like X-linked genes Slx and Slxl1 causes major defects in spermatid differentiation. Mol Biol Cell. 21:3497–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comptour A, Moretti C, Serrentino M-E, Auer J, Ialy-Radio C, Ward MA, Touré A, Vaiman D, Cocquet J. 2014. SSTY proteins co-localize with the post-meiotic sex chromatin and interact with regulators of its expression. FEBS J. 281:1571–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. 1989. Two rules of speciation In: Otte D, Endler JA, editors. Speciation and its consequences. Sunderland, MA: Sinauer; p. 180–207. [Google Scholar]
- Davies B, Hatton E, Altemose N, Hussin JG, Pratto F, Zhang G, Hinch AG, Moralli D, Biggs D, Diaz R, et al. 2016. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530:171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BW, Seabury CM, Brashear WA, Li G, Roelke-Parker M, Murphy WJ. 2015. Mechanisms underlying mammalian hybrid sterility in two feline interspecies models. Mol Biol Evol. 32:2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delph LF, Demuth JP. 2016. Haldane’s rule: genetic bases and their empirical support. J Hered. 107:383–391. [DOI] [PubMed] [Google Scholar]
- Dumont BL, Payseur BA. 2011. Genetic analysis of genome-scale recombination rate evolution in house mice. PLoS Genet. 7:e1002116.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzur-Gejdosova M, Simecek P, Gregorova S, Bhattacharyya T, Forejt J. 2012. Dissecting the genetic architecture of F1 hybrid sterility in house mice. Evolution 66:3321–3335. [DOI] [PubMed] [Google Scholar]
- Ellegren H. 2011. Sex-chromosome evolution: recent progress and the influence of male and female heterogamety. Nat Rev Genet. 12:157–166. [DOI] [PubMed] [Google Scholar]
- Ellis PJI, Bacon J, Affara NA. 2011. Association of Sly with sex-linked gene amplification during mouse evolution: a side effect of genomic conflict in spermatids?. Hum Mol Genet. 20:3010–3021. [DOI] [PubMed] [Google Scholar]
- Ellis PJI, Clemente EJ, Ball P, Touré A, Ferguson L, Turner JMA, Loveland KL, Affara NA, Burgoyne PS. 2005. Deletions on mouse Yq lead to upregulation of multiple X- and Y-linked transcripts in spermatids. Hum Mol Genet. 14:2705–2715. [DOI] [PubMed] [Google Scholar]
- Firman RC, Garcia-Gonzalez F, Thyer E, Wheeler S, Yamin Z, Yuan M, Simmons LW. 2015. Evolutionary change in testes tissue composition among experimental populations of house mice. Evolution 69:848–855. [DOI] [PubMed] [Google Scholar]
- Flachs P, Bhattacharyya T, Mihola O, Piálek J, Forejt J, Trachtulec Z. 2014. Prdm9 incompatibility controls oligospermia and delayed fertility but no selfish transmission in mouse intersubspecific hybrids. PLoS One 9:e95806.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachs P, Mihola O, Simecek P, Gregorova S, Schimenti JC, Matsui Y, Baudat F, de Massy B, Piálek J, Forejt J, et al. 2012. Interallelic and intergenic incompatibilities of the Prdm9 (Hst1) gene in mouse hybrid sterility. PLoS Genet. 8:e1003044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forejt J. 1985. Chromosomal and genic sterility of hybrid type in mice and men. Exp Clin Immunogenet. 2:106–119. [PubMed] [Google Scholar]
- Forejt J. 1996. Hybrid sterility in the mouse. Trends Genet. 12:412–417. [DOI] [PubMed] [Google Scholar]
- Frank SA. 1991. Divergence of meiotic drive-suppression systems as an explanation for sex-biased hybrid sterility and inviability. Evolution 45:262–267. [DOI] [PubMed] [Google Scholar]
- Getun IV, Torres B, Bois PRJ. 2011. Flow cytometry purification of mouse meiotic cells. JoVE 50:e2602.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Dean MD, Nachman MW. 2008. A complex genetic basis to X-linked hybrid male sterility between two species of house mice. Genetics 179:2213–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Giger T, Dean MD, Nachman MW. 2010. Widespread over-expression of the X chromosome in sterile F1 hybrid mice. PLoS Genet. 6:e1001148.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Handel MA, Nachman MW. 2008. Asymmetry and polymorphism of hybrid male sterility during the early stages of speciation in house mice. Evolution 62:50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorova S, Divina P, Storchová R, Trachtulec Z, Fotopulosova V, Svenson KL, Donahue LR, Paigen B, Forejt J. 2008. Mouse consomic strains: exploiting genetic divergence between Mus m. musculus and Mus m. domesticus subspecies. Genome Res. 18:509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero RF, Posto AL, Moyle LC, Hahn MW. 2016. Genome-wide patterns of regulatory divergence revealed by introgression lines. Evolution 70:696–706. [DOI] [PubMed] [Google Scholar]
- Haldane JBS. 1922. Sex ratio and unisexual sterility in hybrid animals. J Genet. 12:101–109. [Google Scholar]
- Handel M. 2004. The XY body: a specialized meiotic chromatin domain. Exp Cell Res. 296:57–63. [DOI] [PubMed] [Google Scholar]
- Handel MA, Schimenti JC. 2010. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat Rev Genet. 11:124–136. [DOI] [PubMed] [Google Scholar]
- Homolka D, Ivanek R, Capkova J, Jansa P, Forejt J. 2007. Chromosomal rearrangement interferes with meiotic X chromosome inactivation. Genome Res. 17:1431–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Holt J, Kao C-Y, McMillan L, Wang W. 2014. A novel multi-alignment pipeline for high-throughput sequencing data. Database 2014:bau057.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Pomiankowski A. 1991. Causes of sex ratio bias may account for unisexual sterility in hybrids: a new explanation of Haldane’s rule and related phenomena. Genetics 128:841–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka E, Lamb MJ. 1991. Sex chromosomes and speciation. Proc Biol Sci. 243:203–208. [DOI] [PubMed] [Google Scholar]
- Janousek V, Wang L, Luzynski K, Dufkova P, Vyskocilova MM, Nachman MW, Munclinger P, Macholán M, Piálek J, Tucker PK. 2012. Genome-wide architecture of reproductive isolation in a naturally occurring hybrid zone between Mus musculus musculus and M. m. domesticus. Mol Ecol. 21:3032–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeen EL, Muirhead CA, Wright L, Meiklejohn CD, Presgraves DC. 2016. Sex chromosome-wide transcriptional suppression and compensatory cis-regulatory evolution mediate gene expression in the Drosophila male Germline. PLoS Biol. 14:e1002499–e1002429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EL, Vanderpool D, Keeble S, Zhou M, Sarver BAJ, Smith AD, Dean MD, Good JM. 2016. Contrasting levels of molecular evolution on the mouse X chromosome. Genetics 203:1841–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC. 1997. The weaker sex is heterogametic: 75 years of Haldane’s rule. Genetics 147:937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2013. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. [DOI] [PubMed]
- Lifschytz E, Lindsley DL. 1972. The role of X-chromosome inactivation during spermatogenesis. Proc Natl Acad Sci U S A 69:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macholán M, Baird SJ, Munclinger P, Dufkova P, Bímová B, Piálek J. 2008. Genetic conflict outweighs heterogametic incompatibility in the mouse hybrid zone? BMC Evol Biol. 8:271.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macholán M, Baird SJE, Dufkova P, Munclinger P, Bímová B, Piálek J. 2011. Assessing multilocus introgression patterns: a case study on the mouse X chromosome in central Europe. Evolution 65:1428–1446. [DOI] [PubMed] [Google Scholar]
- Mack KL, Campbell P, Nachman MW. 2016. Gene regulation and speciation in house mice. Genome Res. 26:451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masly JP, Presgraves DC. 2007. High-resolution genome-wide dissection of the two rules of speciation in Drosophila. PLoS Biol. 5:e243.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40:4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Coolon JD, Hartl DL, Wittkopp PJ. 2014. The roles of cis- and trans-regulation in the evolution of regulatory incompatibilities and sexually dimorphic gene expression. Genome Res. 24:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Tao Y. 2010. Genetic conflict and sex chromosome evolution. Trends Ecol Evol. 25:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihola O, Trachtulec Z, Vlcek C, Schimenti JC, Forejt J. 2009. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 323:373–375. [DOI] [PubMed] [Google Scholar]
- Morelli MA, Cohen PE. 2005. Not all germ cells are created equal: aspects of sexual dimorphism in mammalian meiosis. Reproduction 130:761–781. [DOI] [PubMed] [Google Scholar]
- Moyle LC, Muir CD, Han MV, Hahn MW. 2010. The contribution of gene movement to the "Two rules of speciation". Evolution 64:1541–1557. [DOI] [PubMed] [Google Scholar]
- Mueller JL, Skaletsky H, Brown LG, Zaghlul S, Rock S, Graves T, Auger K, Warren WC, Wilson RK, Page DC. 2013. Independent specialization of the human and mouse X chromosomes for the male germ line. Nat Genet. 45:1083–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Park PJ, Zhang L-F, Shima JE, McCarrey JR, Griswold MD, Lee JT. 2006. Postmeiotic sex chromatin in the male germline of mice. Curr Biol. 16:660–667. [DOI] [PubMed] [Google Scholar]
- Oka A, Aoto T, Totsuka Y, Takahashi R, Ueda M, Mita A, Sakurai-Yamatani N, Yamamoto H, Kuriki S, Takagi N, et al. 2007. Disruption of genetic interaction between two autosomal regions and the X chromosome causes reproductive isolation between mouse strains derived from different subspecies. Genetics 175:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Mita A, Sakurai-Yamatani N, Yamamoto H, Takagi N, Takano-Shimizu T, Toshimori K, Moriwaki K, Shiroishi T. 2004. Hybrid breakdown caused by substitution of the X chromosome between two mouse subspecies. Genetics 166:913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Mita A, Takada Y, Koseki H, Shiroishi T. 2010. Reproductive isolation in hybrid mice due to spermatogenesis defects at three meiotic stages. Genetics 186:339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Shiroishi T. 2014. Regulatory divergence of X-linked genes and hybrid male sterility in mice. Genes Genet Syst. 89:99–108. [DOI] [PubMed] [Google Scholar]
- Oka A, Takada T, Fujisawa H, Shiroishi T. 2014. Evolutionarily diverged regulation of X-chromosomal genes as a primal event in mouse reproductive isolation. PLoS Genet. 10:e1004301.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, Phadnis N, Beatson SA, Lunter G, Malik HS, Ponting CP. 2009. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 5:e1000753–e1000714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortíz-Barrientos D, Counterman BA, Noor MAF. 2006. Gene expression divergence and the origin of hybrid dysfunctions. Genetica 129:71–81. [DOI] [PubMed] [Google Scholar]
- Payseur BA, Krenz JG, Nachman MW. 2004. Differential patterns of introgression across the X chromosome in a hybrid zone between two species of house mice. Evolution 58:2064–2078. [DOI] [PubMed] [Google Scholar]
- Payseur BA. 2016. Genetic links between recombination and speciation. PLoS Genet. 12:e1006066–e1006064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piálek J, Vyskocilová M, Bímová B, Havelková D, Piálková J, Dufkova P, Bencová V, Ďureje L, Albrecht T, Hauffe HC, et al. 2008. Development of unique house mouse resources suitable for evolutionary studies of speciation. J Hered. 99:34–44. [DOI] [PubMed] [Google Scholar]
- Pimentel HJ, Bray N, Puente S, Melsted P, Pachter L. 2016. Differential analysis of RNA-Seq incorporating quantification uncertainty. bioRxiv:http:dx.doi.org–10.1101–058164. [DOI] [PubMed]
- Presgraves DC. 2008. Sex chromosomes and speciation in Drosophila. Trends Genet. 24:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royo H, Polikiewicz G, Mahadevaiah SK, Prosser H, Mitchell M, Bradley A, de Rooij DG, Burgoyne PS, Turner JMA. 2010. Evidence that meiotic sex chromosome inactivation is essential for male fertility. Curr Biol. 20:2117–2123. [DOI] [PubMed] [Google Scholar]
- Saglican E, Ozkurt E, Hu H, Erdem B, Khaitovich P. 2014. Heterochrony explains convergent testis evolution in primates. bioRxiv:010553.
- Scavetta RJ, Tautz D. 2010. Copy number changes of CNV regions in intersubspecific crosses of the house mouse. Mol Biol Evol. 27:1845–1856. [DOI] [PubMed] [Google Scholar]
- Sin H-S, Namekawa SH. 2013. The great escape. Epigenetics 8:887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchová R, Gregorová S, Buckiová D, Kyselová V, Divina P, Forejt J. 2004. Genetic analysis of X-linked hybrid sterility in the house mouse. Mamm Genome 13:515–524. [DOI] [PubMed] [Google Scholar]
- Tao Y, Chen S, Hartl DL, Laurie CC. 2003. Genetic dissection of hybrid incompatibilities between Drosophila simulans and D. mauritiana. I. Differential accumulation of hybrid male sterility effects on the X and autosomes. Genetics 164:1383–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker PK, Lee BK, Lundrigan BL, Eicher EM. 1992. Geographic origin of the Y chromosomes in "old" inbred strains of mice. Mamm Genome 3:254–261. [DOI] [PubMed] [Google Scholar]
- Tucker PK, Sage RD, Warner J, Wilson AC, Eicher EM. 1992. Abrupt cline for sex chromosomes in a hybrid zone between two species of mice. Evolution 46:1146–1163. [DOI] [PubMed] [Google Scholar]
- Turner JMA, Aprelikova O, Xu X, Wang R, Kim S, Chandramouli GVR, Barrett JC, Burgoyne PS, Deng C-X. 2004. BRCA1, histone H2AX phosphorylation, and male meiotic sex chromosome inactivation. Curr Biol. 14:2135–2142. [DOI] [PubMed] [Google Scholar]
- Turner JMA, Mahadevaiah SK, Ellis PJI, Mitchell MJ, Burgoyne PS. 2006. Pachytene asynapsis drives meiotic sex chromosome inactivation and leads to substantial postmeiotic repression in spermatids. Dev Cell 10:521–529. [DOI] [PubMed] [Google Scholar]
- Turner JMA. 2007. Meiotic sex chromosome inactivation. Development 134:1823–1831. [DOI] [PubMed] [Google Scholar]
- Turner JMA. 2015. Meiotic silencing in mammals. Annu Rev Genet. 49:395–412. [DOI] [PubMed] [Google Scholar]
- Turner LM, Harr B. 2014. Genome-wide mapping in a house mouse hybrid zone reveals hybrid sterility loci and Dobzhansky-Muller interactions. eLife 3:eLife.02504. [DOI] [PMC free article] [PubMed]
- Turner LM, Schwahn DJ, Harr B. 2012. Reduced male fertility is common but highly variable in form and severity in a natural house mouse hybrid zone. Evolution 66:443–458. [DOI] [PubMed] [Google Scholar]
- Turner LM, White MA, Tautz D, Payseur BA. 2014. Genomic networks of hybrid sterility. PLoS Genet. 10:e1004162.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Guo Y, Liu W, Zhao W, Song G, Zhou T, Huang H, Guo X, Sun F. 2016. Proteomic analysis of pachytene spermatocytes of sterile hybrid male mice. Biol Reprod. 95:52.. [DOI] [PubMed] [Google Scholar]
- White MA, Steffy B, Wiltshire T, Payseur BA. 2011. Genetic dissection of a key reproductive barrier between nascent species of house mice. Genetics 189:289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Stubbings M, Dumont BL, Payseur BA. 2012. Genetics and evolution of hybrid male sterility in house mice. Genetics 191:917–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-I, Davis AW. 1993. Evolution of postmating reproductive isolation: the composite nature of Haldane’s rule and its genetic bases. Am Nat. 142:187–212. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang JR, Didion JP, Buus RJ, Bell TA, Welsh CE, Bonhomme F, Yu AH-T, Nachman MW, Piálek J, et al. 2011. Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet. 43:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio NM, Chong S, O’Bryan MK. 2008. Epigenetic regulation in male germ cells. Reproduction 136:131–146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data reported in this paper are available through the National Center for Biotechnology Information under accession number SRP065082.