

Abstract
Members of the Wee family of kinases negatively regulate the cell cycle via phosphorylation of CDK1 and are considered potential drug targets. Herein, we investigated the structure–function relationship of human Wee1, Wee2, and Myt1 (PKMYT1). Purified recombinant full-length proteins and kinase domain constructs differed substantially in phosphorylation states and catalytic competency, suggesting complex mechanisms of activation. A series of crystal structures reveal unique features that distinguish Wee1 and Wee2 from Myt1 and establish the structural basis of differential inhibition by the widely used Wee1 inhibitor MK-1775. Kinome profiling and cellular studies demonstrate that, in addition to Wee1 and Wee2, MK-1775 is an equally potent inhibitor of the polo-like kinase PLK1. Several previously unrecognized inhibitors of Wee kinases were discovered and characterized. Combined, the data provide a comprehensive view on the catalytic and structural properties of Wee kinases and a framework for the rational design of novel inhibitors thereof.
Graphical Abstract
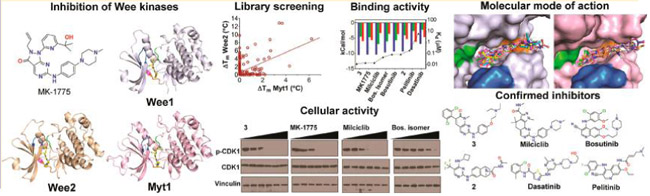
INTRODUCTION
Members of the Wee family of protein kinases, Wee1 (Wee1A), Wee2 (Wee1B), and Myt1 (PKMYT1), function as negative regulators of the cell cycle by inhibiting the CDK1-cyclin B complex. Wee1 is a tyrosine kinase first identified in fission yeast1 which inactivates the CDK1-cyclin B complex through phosphorylation of Tyr15, thereby preventing entry into mitosis at the G2-M transition.2,3 Normal cells repair DNA during G1 arrest, whereas cancer cells with aberrant G1 checkpoint activity depend on the ability of Wee1 to temporarily inhibit CDK1, thereby permitting a G2-M arrest for DNA repair and avoiding premature entry into mitosis to prevent mitotic catastrophe.4–6 Therefore, inhibition of Wee1 by small molecule drugs has emerged as an anticancer therapeutic strategy, particularly in combination with DNA-damaging agents.7–10 A different function of Wee1 occurs in postmitotic neurons during the establishment of distinct axonal and dendritic compartments, an essential step during neuronal development.11 Wee1 has also been reported to phosphorylate histone H2B at Tyr37 which suppresses histone transcription in the late S phase,12 providing a direct link between epigenetics and cell-cycle progression.13
Unlike the ubiquitously expressed Wee1, Wee2 expression is germ-cell-specific and inhibits meiosis by phosphorylating Tyr15 of the CDK1-cyclin B complex.14–16 In mouse oocytes, Wee2 is essential for metaphase II exit17 and is therefore considered a potential nonhormonal female contraceptive target. Myt1 is a dual specificity protein kinase localized to the ER-Golgi complex which in vitro inhibits CDK1-cyclin B by phosphorylating Thr14 and Tyr15.18,19 In Drosophila loss of both Myt1 and Wee1 resulted in synthetic lethality, indicating that they are functionally redundant during fly development.20 In mammalian somatic cells Myt1 is essential for Golgi and ER assembly at the end of mitosis through its ability to suppress both CDK1-cyclin B1 and CDK1-cyclin B2 activities.21
Recently it was demonstrated that in human fetal neural stem cells Myt1 acts redundantly with Wee1 to promote timely completion of mitosis, while in glioblastoma stemlike cells this redundancy is broken resulting in the essential requirement for Myt1 functionality.22
Previous drug discovery efforts have focused on Wee1 rather than Wee2 or Myt1 as prospective drug targets. MK-1775 (aka AZD1775) was the first selective Wee1 inhibitor reported23,24 and is currently in clinical trials for the treatment of diverse solid tumors.25,26 Structural information on Wee kinases was limited to the Wee1 kinase domain liganded with pyrrolo-carbazole containing inhibitors27,28 and of the unliganded Myt1 kinase domain (PDB entry 3P1A, no associated citation). Here we investigated the structure–function relationship of the entire human Wee family of kinases using biochemical, biophysical, and cell biological methods. Purified recombinant full-length proteins and kinase domain constructs differed substantially in phosphorylation states and catalytic competency toward CDK1-Tyr15. A series of novel crystal structures were determined, revealing unique structural features that distinguish Wee1 and Wee2 from Myt1 and providing the structural basis of differential inhibition by MK-1775 for the first time. Kinome profiling and cellular studies established that, in addition to Wee1 and Wee2, MK-1775 is an equally potent inhibitor of other kinases including the polo-like kinase PLK1. Several previously unrecognized Wee kinase inhibitors were discovered and fully characterized, including the pan-CDK inhibitor milciclib, which is in clinical trials for the treatment of solid tumors.29 Combined, the data provide a comprehensive view on the catalytic and structural properties of the Wee family of kinases and a new framework for the rational design of novel inhibitors thereof.
RESULTS AND DISCUSSION
Wee Kinases Differ Substantially in Catalytic Activity and Phosphorylation State.
Upon recombinant overexpression in E. coli the kinase domains of human Wee1, Wee2, and Myt1 indicated in Figure 1A showed good solubility and yielded crystallization-grade proteins using affinity, anion exchange, and size exclusion chromatography. While the kinase domain of Myt1 was able to phosphorylate Tyr15 of CDK1, both Wee1 and Wee2 kinase domains were catalytically inactive (Figure 1B). By contrast, full-length Wee1 and Wee2, recombinantly overexpressed in mammalian Expi293 cells and purified to high homogeneity, were enzymatically active. Full-length Wee1 showed 30-fold and 10-fold increased specific activity over Wee2 and Myt1, respectively. For the purified kinase domains, mass spectrometric analysis revealed multiple serine, threonine, and tyrosine phosphorylation sites in Myt1, whereas the Wee1 lacked phosphorylated residues and Wee2 showed only a single phosphorylated residue (Tyr227) (Figure 1A, Supporting Information Table S1). As the kinase domains were expressed in E. coli, phosphorylated residues are likely the result of autophosphorylation. Thus, the kinase domain of Myt1 shows an unusual ability to indiscriminately autophos-phorylate multiple serine, threonine, and tyrosine residues, while autophosphorylation and activation of Wee1 and Wee2 kinases domains appear to be impaired. By contrast, purified full-length Wee1 was phosphorylated at 25 residues, 10 of which are located in the kinase domain (Figure 1A). Purified full-length Wee2 was phosphorylated at five residues with only the autophosphorylation site Tyr227 located in the kinase domain. The phosphorylation sites are significantly different between all three family members, perhaps reflecting the differential catalytic activity toward CDK1 phosphorylation (Figure 1B). By sequence alignment none of the phosphorylation sites are conserved across all three family members. Wee1 and Wee2 share a single strictly conserved phosphorylated residue (Ser642 and Ser557, respectively), while Wee1 and Myt1 share two phosphorylation sites, a strictly conserved residue (Tyr325 and Tyr136, respectively) and a conserved residue (Ser472 and Thr260, respectively). Ser642 of Wee1 has previously been reported as a phosphorylation site of the brain-selective kinases BRSK1 and BRSK2, leading to the down-regulation of Wee1 activity in polarized neurons.11,30 The finding that the equivalent residue in Wee2 is also a phosphorylation site suggests an important role of the very carboxyl terminus in regulating the functionality of Wee kinases. However, certain sites found to be phosphorylated might not be functionally relevant due to the recombinant overexpression of these proteins in host cells.
Figure 1.

Phosphorylation state, catalytic activity, and thermodynamics of Wee kinases. (A) Diagram of the amino acid sequences of Wee kinases. Full-length Wee1 and Wee2 were expressed in mammalian Expi293 cells. The boxes indicate the kinase domain constructs expressed in E. coli. The highlighted residues indicate the phosphorylation sites detected by mass spectrometry (see Supporting Information Table S1 for original data). (B) Specific activity of full-length (FL) and kinase domain (KD) of Wee kinases toward Tyr15 phosphorylation of CDK1 as determined by ELISA assay. (C) ITC of MK-1775 interaction with the kinase domains of Wee kinases. The left panel shows the titration peaks, the middle panel the fitted data, and the right panel the resulting thermodynamic signatures. For experimental conditions and original titration data see Supporting Information Figures S1–S3 and Table S2.
Structural Basis of Wee Kinases Inhibition by MK-1775.
Although the kinase domains of Wee1 and Wee2 are devoid of enzymatic activity, direct binding studies by isothermal titration calorimetry (ITC) established high binding affinity for MK-1775, a phase II clinical trial small-molecule Wee1 inhibitor, with Kd values of 13 and 27 nM, respectively (Figure 1C, Supporting Information Figures S1–S3 and Table S2). Complex formation was characterized by favorable enthalpy and entropy contributions for Wee1 and Wee2. By contrast, the interaction of MK-1775 with Myt1 showed unfavorable entropy changes resulting in significantly weaker binding affinity (Kd = 320 nM). Previously known X-ray crystal structures of Wee kinases were those of Wee1 liganded with one series of pyrrolo-carbazole inhibitors27,28 and of unliganded Myt1 (PDB entry 3P1A). We therefore determined high-resolution X-ray cocrystal structures of the kinase domains of all three Wee kinases with MK-1775 (Figure 2, Supporting Information Figures S4–S6 and Tables S3–S4). As expected, MK-1775 is a type I inhibitor interacting primarily with the main chain atoms of a conserved cysteine residue of the hinge region. An additional H-bond exists between the pyrazolo-pyrimidinone oxygen of MK-1775 and the side chain of the gatekeeper residue, which is an asparagine residue in Wee1 and Wee2 and a threonine residue in Myt1 (Figure 2B). Several hydrophobic interactions stabilize MK-1775 in the respective ATP site (Figure 2C). The overall structures of the three MK-1775-kinase complexes are highly similar to root-mean-square deviation (rmsd) values of 0.66 Å between Wee2 and Wee1 and of 1.2 Å between Myt1 and Wee1. Major structural changes around the ATP site occur in the P-loop (Wee1 residues 307–312) which is in a “closed” conformation in Wee2 and in the C-helix (Wee1 residues 338–353) which adopts a different conformation in Myt1 away from the ATP site. Residues comprising the ATP site are strictly conserved between Wee1 and Wee2 with the exception of Asp386Wee1-to-Ala substitution in the solvent exposed front specificity pocket. Myt1 differs in five residues of the ATP site of which the Pro192Myt1 for Gly382Wee1 substitution appears to reduce the shape complementarity toward MK-1775 binding (Figure 2D). A conspicuous structural element in both Wee1 and Wee2 kinase domains is a large flexible loop preceding the DLG motif and connecting an antiparallel β-sheet next to the hinge region (Figure 2A, Figure 2E). In Myt1 and most other kinases this 22–26 residue long loop is reduced to a regular three-residue β-turn. The loops are acidic (pI = 4.0 and 4.1), rich in glutamate and serine residues, and predicted to contain a potential PEST proteolytic cleavage site31 in Wee1 but not in Wee2 (******emboss.bioinformatics.nl/cgi-bin/emboss/epestfind). Wee1 contains three additional PEST motifs in the N-terminus (residues 24–126), Wee2 contains a single potential PEST site in the N-terminus (residues 90–101), while Myt1 lacks PEST motifs. The larger number of potential proteolytic cleavage sites in Wee1 may ensure tight control of its intracellular half-life through multiple built-in venues toward enzyme inactivation and degradation.
Figure 2.

Crystal structures of Wee kinases in complex with MK-1775. (A) Overall structures of the kinase domains with MK-1775 (yellow) liganded in the ATP site. (B) Detailed binding interactions between MK-1775 and protein residues with hydrogen bonding and hydrophobic interactions indicated as black and green dotted lines, respectively. Stereoviews with Fo – Fc omit density maps are shown in Supporting Information Figures S4–S6. Crystallization conditions are listed in Supporting Information Table S3 and crystallographic data collection and refinement statistics in Supporting Information Table S4. The gatekeeper residue is shown in magenta, the hinge region is in orange, residues of the DLG/DFG motifs are in green, residues of the P-loop are in blue, and water molecules are indicated as cyan spheres. (C) Schematics of MK-1775 interactions in the respective ATP sites. (D) Superimposition of Wee1 (cyan) and Myt1 (pink) emphasizing the amino acid differences in the vicinity of MK-1775 (center). (E) Sequence alignments emphasizing the large loops preceding the DLG motifs characteristic of Wee1 and Wee2.
MK-1775 Is a Dual Wee-PLK Inhibitor.
Having determined the precise binding mode of MK-1775 in all Wee family members, we next asked if this inhibitor is uniquely selective for Wee kinases. Profiling of MK-1775 for binding activity across the human kinome (403 wild-type and 65 mutant kinases) revealed good selectivity with scores of S(1) = 0.017 and S(10) = 0.032 for kinases of less than 1% and 10% remaining activity, respectively (Figure 3A). Statistical evaluation of cumulative inhibition32 yielded a Gini coefficient of 0.63. In addition to Wee1 and Wee2, other kinases targeted by MK-1775 were PLK1, −2 and −3, JAK2 and −3, and mutant forms of ABL1, FLT3, and GCN2 (Figure 3A, Supporting Information Table S5). Follow-up qPCR-based binding studies established that MK-1775 is an equipotent ligand of Wee1, Wee2, and PLK1 with Kd values of 3.2, 3.9, and 3.0 nM, respectively (Figure 3B). The Kd values for other kinases within the top 5% were between 7.5 and 110 nM, with FLT3 being the most sensitive and JAK2 the least sensitive target of MK-1775. The high binding potential of MK-1775 toward PLK1 was unexpected as this inhibitor was previously thought to be selective for Wee1 and is widely utilized as a chemical probe to study Wee1 function in cancer models.7,8 However, in a recent independent study using chemical proteomics PLK1 was also identified as a prime target of MK-1775 in H322 non-small-cell lung cancer cells.33 Molecular modeling using the well-known structure of the PLK1 kinase domain indicates a good fit of MK-1775 in the ATP site through two H-bonds with the main chain atoms of Cys133 of the hinge region (Figure 3C), the same principle binding mode as observed in the Wee kinases (Figure 2).
Figure 3.

Selectivity of MK-1775 across the human kinome. (A) Kinome profiling results of MK-1775 (0.5 μM) showing prominent activity against kinases other than Wee1 and Wee2. Large circles indicate high inhibitory potential. The complete data set is presented in Supporting Information Table S5. (B) Dissociation constants of MK-1775 interaction with selected kinases determined by qPCR based assay. (C) Molecular docking model of MK-1775 bound to the ATP site of PLK1. (D) Cell growth inhibitory activity of known PLK1 inhibitors in comparison with MK-1775 upon 72 h drug treatment. (E) MK-1775, but not BI-2536 or 1, shows dual Wee1-PLK1 activity by simultaneously attenuating the levels of phosphorylated CDK1 and TCTP in synchronized noncancerous HEK293T and breast cancer MDA-MB-231 cells upon 6 h drug treatment.
Cellular activity of dual Wee1-PLK1 inhibition by MK-1775 was compared with that of the known PLK1 inhibitors BI-253634 and (R)-5-(6-((4-methylpiperazin-1-yl)methyl)-1H-benzo[d]imidazol-1-yl)-3-(1-(2-(trifluoromethyl)phenyl)-ethoxy)thiophene-2-carboxamide (GSK-46136435) using the adherent cell lines HEK293T (noncancerous kidney) and MDA-MB-231 (breast cancer), as well as the liquid tumor cell line MM1.S (multiple myeloma) (Figure 3D). Although the reported inhibitory activities of BI-2536 (IC50 = 0.83 nM) and 1 (GSK-461364) (Ki = 2.2 nM) against PLK1 are similar to the Kd value of 3.0 nM for the PLK1-MK-1775 complex, they were 2 orders of magnitude more potent than MK-1775 against cell viability (IC50 values between 1 and 3.5 nM for BI-2536 and 1 and between 290 and 310 nM for MK-1775). Immunoblotting confirmed the dual Wee1-PLK1 inhibitory activity of MK-1775 by simultaneously reducing the levels of phosphorylated CDK1 (Wee substrate) and of translationally controlled tumor protein (TCTP, PLK1 substrate) (Figure 3E). BI-2536 and 1 had no effect on CDK1 phosphorylation but were highly effective at reducing p-TCTP levels with half-maximal concentrations of <0.1 μM, while MK-1775 reduced the phosphorylation levels of CDK1 and PLK1 about 10 times less effectively, perhaps caused by limited cell permeability.
Discovery and Characterization of Other Wee Kinase Inhibitors.
In an attempt to identify chemical scaffolds other than MK-1775 capable of inhibiting Wee kinases, we used DSF to screen a library of 278 diverse kinase inhibitors from Selleck Chemicals against Wee2 and Myt1. MK-1775 served as a positive control, inducing large increases in melting temperature (Tm) for Wee1 and Wee2 (ΔTm = 15 and 12 °C, respectively) and a moderate increase for Myt1 (ΔTm = 3 °C) (Table 1). The top 5% of compounds screened showed ΔTm values between 2 and 6 °C for Myt1 and between 5 and 13 °C for Wee2 (Figure 4A). As expected from the similar architecture of the target sites, the two data sets showed good correlation with a Pearson’s coefficient of 0.48, both enzymes sharing five of nine compounds characterized in more detail (Figure 4B). Among the most selective inhibitors was the designated Aurora kinase inhibitor 2 (PF-0381473536), which showed no binding potential for Myt1, and the multitargeted inhibitor PP-12137 showing only weak binding potential for Wee2. Selected hit compounds supplemented with the previously reported Wee1 inhibitor 3 (PD-16628538,39) and an isomer of bosutinib40 were further characterized by ITC (Figure 4C–E). The resulting thermodynamic signatures established 3, the pan-CDK inhibitor milciclib (aka PHA-848125),41 and bosutinib isomer as ligands with highest binding affinity for both Wee1 and Wee2, with only bosutinib isomer showing increased binding potential for Wee2. These were followed by bosutinib, 2, pelitinib, and dasatinib with Kd values between 0.078 and 5.7 μM, with dasatinib showing 11-fold higher binding potential for Wee2. Compounds with strongest binding potential for Myt1 were dasatinib, 3, and bosutinib with Kd values between 150 and 320 nM, with only dasatinib showing increased selectivity for Myt1. For all three enzymes the dissociation constants of the respective protein–inhibitor complexes as determined by ITC were logarithmically proportional to the increase in melting temperatures as determined by DSF (Figure 4F). The most potent compounds were further characterized for cellular activity, with 3 and MK-1775 showing the highest growth inhibitory potential against HEK293T, MM1.S, and MDA-MB-231 cells with IC50 values between 0.16 and 0.52 μM (Table 2, Figure 5). Milciclib was slightly less effective, while bosutinib isomer was the least active compound with IC50 values between 1.6 and 3.9 μM. Growth inhibitory activity was paralleled by a dose-dependent reduction of phospho-CDK1 levels, indicating on-target inhibition of Wee1 in these cells (Figure 5). However, differences in kinase selectivity properties may also account for the observed differential cellular activity of these compounds. Notably milciclib, which is in clinical trials for the treatment of solid tumors,29 was previously not reported to also potently inhibit Wee1 and Wee2.
Table 1.
Binding Potential of Identified Wee Kinase Inhibitors
| Wee1 |
Wee2 |
Myt1 |
||||
|---|---|---|---|---|---|---|
| compd | Kd (nM)a | ΔTm (°C)b | Kd (nM) | ΔTm (°C) | Kd (nM) | ΔTm (°C) |
| 3 | 11.0 ± 1.6 | 16.5 ± 1.10 | 5.0 ± 0.8 | 14.2 ± 0.80 | 247 ± 58.1 | 6.0 ±0.51 |
| MK-1775 | 13.4 ± 1.2 | 15.4 ± 0.83 | 27 ± 3.6 | 11.6 ± 0.32 | 324 ± 27.3 | 3.3 ±0.09 |
| milciclib | 13.6 ± 2.0 | 13.4 ± 0.0 | 26.6 ± 4.2 | 11.5 ± 0.0 | 990 ± 55.2 | 3.6 ±0.0 |
| bosutinib isomer | 43.7 ± 10.0 | 10.0 ± 0.0 | 4.7 ± 2.3 | 12.3 ± 0.0 | 444 ± 41.0 | 3.0 ± 0.0 |
| bosutinib | 77.5 ± 6.0 | 7.2 ± 0.48 | 106 ± 4.3 | 9.7 ± 0.24 | 270 ± 17.2 | 3.9 ± 0.0 |
| 2 | 78.7 ± 8.3 | 7.9 ± 0.0 | 128 ± 17.3 | 8.5 ± 0.0 | NBc | −0.1 ± 0.0 |
| pelitinib | 172 ± 22.3 | 6.4 ± 0.88 | 498 ± 48.3 | 5.6 ± 0.71 | 1105 ± 82.9 | 2.4 ± 0.02 |
| dasatinib | 5700 ± 314 | 5.0 ± 0.35 | 481 ± 30.5 | 9.2 ± 0.25 | 150 ± 9.1 | 6.0 ± 0.35 |
| saracatinib | NBc | 1.2 ± 0.06 | NBc | 0.65 ± 0.14 | 2817 ± 144.4 | 2.1 ± 0.32 |
ITC, single measurement and associated standard error of data fit.
DSF, mean value of 2–4 measurements and associated standard deviation.
NB: no binding.
Figure 4.

Discovery of inhibitors of Wee kinases using DSF. (A) Distribution of compounds from the kinase inhibitor library of Selleck Chemicals able to increase the melting temperatures of Wee2 and Myt1 kinase domains. (B) Correlation of the two data sets with selective and unselective hit compounds highlighted. (C–E) Thermodynamic signatures of compound interactions with Wee1, Wee2, and Myt1. (F) Logarithmic proportionality between temperature shifts and dissociation constants.
Table 2.
Cell Growth Inhibitory Potential of Wee1/2 Inhibitors
| cell growth inhibition, IC50 (μM) |
|||
|---|---|---|---|
| compd | HEK293T | MM1.S | MDA-MB-231 |
| 3 | 0.29 ± 0.013 | 0.52 ± 0.044 | 0.16 ± 0.008 |
| MK-1775 | 0.29 ± 0.046 | 0.31 ± 0.008 | 0.26 ± 0.016 |
| milciclib | 1.5 ± 0.037 | 0.72 ± 0.086 | 0.31 ± 0.052 |
| bosutinib isomer | 1.6 ± 0.19 | 3.9 ± 0.50 | 1.8 ± 0.061 |
Figure 5.

Cellular activity of Wee1 inhibitors. The most potent biochemical inhibitors of Wee1 and Wee2 were evaluated for growth inhibitory activity (left panel, cell titer blue assay upon 72 h drug treatment) and the ability to reduce phospho-CDK1 (Tyr15) levels (right panel, immunoblotting upon 6 h drug treatment) of cell lines HEK293T (A), MM1.S (B), and MDA-MB-231 (C). IC50 values are shown in Table 2.
To establish a framework for the structure-based drug design of novel Wee kinases inhibitors, high-resolution crystal structures were determined for five Wee1-inhibitor complexes (Figure 6) and six Myt1-inhibitor complexes (Figure 7). All compounds are type I inhibitors with similar binding poses in the ATP site, establishing single or dual H-bonds with the main chain atoms of Cys379Wee1 or Cys190Myt1. Superimposition of the Wee1 and Myt1 inhibitor complexes demonstrates the high similarity of the inhibitor binding poses in both enzymes (Figure 6B, Figure 7B). All inhibitors assume a mainly planar conformation in the ATP site with noticeable rotational freedom for functional groups directed toward the DFG motif and the solvent exposed front specificity pocket on opposite sides of the inhibitor. A notable difference in the ATP sites of Wee kinases is the narrow front specificity pocket in Myt1, which likely causes the generally weaker binding potential of kinase inhibitors toward Myt1. The Wee1-inhibitor complexes differ mostly in the N-terminal domain, particularly in the P-loop and the C-helix with rmsd values of up to 4 Å, while the Myt1-inhibitor complexes show largest structural differences in the activation loop with rmsd values of up to 8 Å (Suppoting Information Figure S7).
Figure 6.

Cocrystal structures of Wee1 with diverse inhibitors. (A) Chemical structures and binding interactions of compounds in the ATP site. Orange highlighted atoms directly interact with Cys379 of the hinge region. (B) Superimposition of all inhibitors in the ATP site. The color code is the same as in Figure 2B. Stereopresentations including Fo – Fc electron density maps are shown in Supporting Information Figure S4. Crystallization conditions and data/refinement statistics are listed in Supporting Information Tables S3 and S4.
Figure 7.

Cocrystal structures of Myt1 with diverse kinase inhibitors. (A) Chemical structures and binding interactions of compounds in the ATP site. Orange highlighted atoms directly interact with Cys190 of the hinge region. (B) Superimposition of all inhibitors in the ATP site. The color code is the same as in Figure 2B. Stereopresentations including Fo – Fc electron density maps are shown in Supporting Information Figure S5. Crystallization conditions and data/refinement statistics are listed in Supporting Information Tables S3 and S4.
CONCLUSIONS
Despite their critical role in mitosis and meiosis, relatively little is known about the structure–function relationship of Wee kinases and inhibitors thereof. The comparative analysis of this work demonstrates that Wee1 and Wee2 differ significantly from Myt1 in structural properties and catalytic competency, suggesting that regions outside the kinase domains of Wee1 and Wee2 regulate enzymatic activity through currently unknown mechanisms. Crystal structures of all three Wee kinases liganded with MK-1775 provide the structural basis of inhibition by this cancer drug candidate for the first time. However, the finding that MK-1775 is an equipotent inhibitor of PLK1 limits its usefulness as a chemical probe to study Wee1 or Wee2 biology. While several kinase inhibitors showed high binding potential for Wee1 and Wee2, it appears that the narrower ATP site in Myt1 confers increased tolerance to kinase inhibitors in general. The precise knowledge of the binding interactions of chemically diverse compounds with Wee kinases provides a new framework for the rational design of novel inhibitors with improved selectivity profiles, for example, by applying scaffold hopping approaches.42
EXPERIMENTAL SECTION
Reagents and Compounds.
Reagents and compounds for biochemical and crystallographic experiments were purchased from Sigma-Aldrich (St. Louis, MO) and Hampton Research (Aliso Viejo, CA) unless otherwise indicated. The L1200 kinase inhibitor library and individual kinase inhibitors were purchased from Selleck Chemicals (Houston, TX) except 3 and bosutinib isomer, which were from Sigma-Aldrich and Santa Cruz Biotechnology (Dallas, TX), respectively. 3 was ≥98% pure and all other inhibitors were ≥99% pure according to the certificates of analysis provided by the vendors. The concentration of purified protein was determined by A280 molar absorbance using a Nanodrop ND-2000c spectrophotometer (Nanodrop Technologies).
Cloning and Expression.
The genes encoding human full-length Wee1 (Wee1-FL, residues 1–646) and full-length Wee2 (Wee2-FL, residues 1–567) were synthesized by GeneArt (Life Technologies, Carlsbad, CA) and subcloned into pcDNA3.3-TOPO providing an N-terminal His8 tag. To generate full-length recombinant Wee1 and Wee2, human Expi293F cells were propagated in Erlenmeyer flasks at 37 °C and 5% CO2 and shaken at 125 rpm in a New Brunswick shaker/incubator S41i. On the day of transfection, an amount of 850 mL of cells was adjusted to a cell density of ~3 × 106 cells per mL. 1000 μg of plasmid DNA in 50 mL of Opti-MEM and 2.66 mL of ExpiFectamine in 50 mL of Opti-MEM was incubated separately for 5 min and then incubated together for another 20 min. 100 mL of the DNA/transfection mixture was then added to the 850 mL culture. After 20 h in the incubator, 5 mL of transfection enhancer 1 and 50 mL of transfection enhancer 2 were added to the culture. Cells were harvested 2 days post-transfection, washed in PBS, and stored at −80 °C until further processed.
The genes encoding Wee1 kinase domain (Wee1-KD, residues 291–575) and Wee2 kinase domain (Wee2-KD, residues 202–492) were amplified by PCR and subcloned into a modified pET28a plasmid to provide cleavable MBP fusion protein with a N-terminal His6 tag. 43 The gene encoding human Myt1 kinase domain (Myt1-KD, residues 75–361) cloned in-frame with an N-terminal His6 tag was received in a pNIC28-Bsa4 vector from Addgene (plasmid 39061). The gene encoding full-length Escherichia phage lambda serine/threonine-protein phosphatase (LAMBD, residues 1–221) was synthesized by GeneArt with optimized codon usage for E. coli protein expression and subcloned into pGEX-6p-2 vector with N-terminal GST tag. Wee1-KD and Wee2-KD in modified pET28a as well as plasmid 39061 were subsequently transformed into competent E. coli strain BL21(DE3) CodonPlus RIPL (Agilent) for protein expression. LAMBD plasmid was transformed into competent E. coli strain BL21(DE3) for protein expression. Bacterial cultures were grown for 2–3 h at 37 °C until OD600 reached 0.5, at which time the temperature was decreased to 16 °C prior to induction with 0.1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Cultures were grown for an additional 20 h at 16 °C and then harvested by centrifugation (8000g for 15 min, 4 °C).
Protein Purification.
All protein purification steps were performed by fast protein liquid chromatography (FPLC) at 4 °C. Harvested bacterial cell pellets for Wee1-KD, Wee2-KD, and Myt1-KD were resuspended in 50 mM Na/K phosphate buffer (pH 7.4) containing 300 mM NaCl, 10 mM imidazole, and 0.01% Triton X-100 at 4 °C for 1 h. After being lysed by homogenizer and centrifugation (30 000g for 60 min, 4 °C), the supernatant was purified by immobilized Ni-affinity chromatography (Qiagen, Hilden, Germany) using a linear gradient of 20–250 mM imidazole. Following incubation of peak fractions with His-TEV protease (for Wee1-KD) or PreScission protease (for Wee2-KD) at 4 °C, cleaved His-MBP tag was removed by a second Ni-affinity column. Wee1-KD or Wee2-KD were further purified by a Mono Q 10/100 GL column (anion exchange, GE Healthcare Life Sciences, Pittsburgh, PA) using 50 mM Tris (pH 8.0) buffer containing 1 mM DTT and a linear gradient of 0–500 mM NaCl. Peak fractions of Wee1-KD and Wee2-KD were loaded onto a Superdex 75 (26/60) column (GE Healthcare Life Sciences) and eluted with 50 mM Tris buffer (pH 8.0) containing 150 mM NaCl and 1 mM DTT. Peak fractions of His6-tagged Myt1-KD from Ni-affinity column were concentrated and loaded onto a Superdex 75 (26/60) column and eluted with 50 mM HEPES buffer (pH 7.5) containing 300 mM NaCl and 1 mM DTT. Purified Wee1 KD, Wee2 KD, and Myt1 were concentrated to 5–7 mg mL−1 and stored at −80 °C.
For full-length Wee1 and Wee2 enzymes, harvested Expi293F cells were resuspended in 50 mM Na/K phosphate buffer (pH 7.4) containing 300 mM NaCl, 20 mM imidazole, and Pierce protease inhibitor (EDTA-free, Thermo Scientific) at 4 °C for 0.5 h. Cells were lysed using a homogenizer, the lysate was centrifuged at 30 000g for 60 min, and the supernatant was subjected to immobilized Ni-affinity chromatography. Peak fractions were further purified by a Mono Q 10/100 GL column using 50 mM Tris (pH 8.0) buffer containing 1 mM DTT and a linear gradient of 0–500 mM NaCl.
Dephosphorylation of Myt1 by LAMBD.
E. coli cells overexpressing LAMBD phosphatase were resuspended in 50 mM Tris buffer (pH 8.0) containing 150 mM NaCl, 5 mM MnCl2, 1 mM DTT, and 0.01% Triton X-100 at 4 °C for 1 h. After lysis by homogenizer and centrifugation (30 000g for 60 min, 4 °C), the supernatant was loaded onto a GST-affinity column (GE Healthcare Life Sciences) in 50 mM Tris (pH 8.0), 150 mM NaCl, 5 mM MnCl2, 1 mM DTT, and a step gradient of 0–10 mM reduced glutathione. Peak fractions of GST-LAMBD were exchanged into 50 mM Tris buffer (pH 8.0) containing 150 mM NaCl, 1 mM MnCl2, 1 mM DTT by rapid gel filtration. Purified GST-LAMBD was concentrated to 5 mg/mL and stored at −80 °C. Specific activity of purified GST-LAMBD was assessed together with commercially available LAMBD (catalog P0753S, New England Biolabs) following the manufacturer’s protocol. Purified Myt1 (30 mg) was treated with 4 mg of GST-LAMBD at 30 °C for 1 h in 50 mM Tris buffer (pH 8.0) containing 150 mM NaCl, 1 mM MnCl2, 1 mM DTT. Then the mixture was loaded onto a Ni-affinity column and dephosphorylated Myt1 was eluted with a step elution of 0–250 mM imidazole. Peak fractions were exchanged into 50 mM Tris buffer (pH 8.0) containing 150 mM NaCl, and 1 mM DTT by rapid gel filtration and concentrated to 5 mg mL−1. Dephosphorylation of Myt1 was confirmed by immunoblotting using phosphotyrosine (Cell Signaling no. 9416), phosphothreonine (Cell Signaling no. 9381), and phosphoserine antibodies (Abcam no. 9332).
ELISA Assay.
Enzymatic activity of purified Wee1-FL (60 ng per well), Wee1-KD (1 μg per well), Wee2-FL (250 ng per well), Wee2-KD (1 μg per well), Myt1-KD (250 ng per well) was assessed by CycLex Wee1 kinase assay (MBL International Corp.) following the manufacturer’s instructions and using commercially available Wee1 (40 mU, MBL International Corp.) as a positive control. Stop solution was added to each well following incubation with substrate reagent for 8.5 min, and absorbance was measured in a SpectraMax 340PC384 plate reader (Molecular Devices) at dual wavelengths of 450 and 540 nm.
Differential Scanning Fluorimetry (DSF).
The binding potential of compounds against Wee1-KD, Wee2-KD, and Myt1-KD was assessed by DSF using a StepOnePlus real-time PCR system (Applied Biosystems, Grand Island, NY). Protein (2 μM final concentration in 50 mM HEPES (pH 7.5), 150 mM NaCl, and 1 mM DTT were assayed in a 96-well plate in quadruplicate. Inhibitors were added to a final concentration of 50 μM (with Wee1-KD and Wee2-KD) or 100 μM (with Myt1-KD) and 2% DMSO. Protein thermal shift dye (1:1000; Applied Biosystems) was used as the fluorescent probe and fluorescence was measured using the ROX reporter channel (620 nm). Protein stability was investigated by programing the thermocycler to increase the temperature from 25 to 99 °C using 0.2 °C increments and 10 s incubations per increment. The inflection point of the transition curve/melting temperature (Tm) was calculated using the Boltzmann equation within the Protein Thermal Shift software (version 1.2) (Applied Biosystems, Grand Island, NY). The ΔTm values were calculated using DMSO control as a reference.
Isothermal Titration Calorimetry (ITC).
Binding of small molecules to Wee1-KD, Wee2-KD, and Myt1-KD was analyzed using a MicroCal iTC200 (Malvern) at 15 °C, 25 °C, and 25 °C, respectively. Wee1 was exchanged into 50 mM Na/K phosphate (pH 6.9) buffer, while Wee2 and Myt1 were exchanged into 50 mM HEPES (pH 7.5) buffer containing 150 mM NaCl. After an initial 0.2 μL injection, 29 aliquots (1.35 μL each) of the syringe solution (200–750 μM ligands) were titrated into 200 μL of the protein solution (20–30 μM) in the cell, which was stirred constantly at 1000 rpm. Data were recorded for 120–180 s between injections. Generation of heat due to dilution was determined in a separate experiment by diluting protein into buffer and subtracting these as blank values for each injection. Corrected heat values were fitted using a nonlinear least-squares curve-fitting algorithm (Microcal Origin 7.0) to obtain the stoichiometry (n), binding constants (Ka, Kd), and change in enthalpy for each enzyme–ligand interaction (ΔH). Additional experimental procedures are described in Supporting Information (Supporting Information Table S2).
X-ray Crystallography.
Purified kinase domains of Wee1, Wee2, and Myt1 were exchanged into crystallization buffer via PD-10 columns (GE Healthcare Life Sciences), concentrated, and crystallized by the hanging-drop vapor diffusion method using a 1:1 (v/v) ratio of protein–ligand solution and precipitant at 18 °C (Supporting Information Table S3). Crystals were harvested in cryoprotectant (reservoir containing 10–25% (v/v) ethylene glycol) and flash frozen in a stream of nitrogen gas. X-ray diffraction data were recorded at −180 °C at the beamlines 22-ID and 22-BM, SER-CAT, Advanced Photon Source, Argonne National Laboratories and in the Moffitt Structural Biology Core using Cu Kα X-rays generated by a Rigaku Micro-Max 007-HF X-ray generator, focused by mirror optics and equipped with a Rigaku CCD Saturn 944 system. Data were reduced and scaled with XDS;44 PHENIX45 was employed for phasing and refinement, and model building was performed using Coot.46 PDB entries 1X8B and 3P1A were used as the search models for Wee1/Wee2 and Myt1, respectively. Initial models for the small molecule ligands were generated using MarvinSketch (ChemAxon, Cambridge, MA) with ligand restraints from eLBOW of the PHENIX suite. All structures were validated by MolProbity and phenix.model_vs_data.47 Figures were prepared using PyMOL (Schrödinger, LLC).
Kinase Inhibition/Binding Assays and Profiling.
The half maximal inhibitory concentration (IC50) of each compound against WEE1 and PLK1 was determined in dose–response by Reaction Biology Corp. using a 33P-ATP radiolabeled assay (10 inhibitor doses from 0.5 nM to 10 μM) in the presence of 10 μM ATP. Residual enzymatic activity (in % of DMSO controls) was determined in duplicate. Profiling of MK-1775 across a panel of 468 human kinases using a single inhibitor concentration of 500 nM and dissociation constants of MK-1775 interaction with selected kinases was determined by Discoverx Corp. The amount of kinase captured on an immobilized ligand in the presence or absence of compound was measured using a quantitative real-time polymerase chain reaction (qPCR) method that detects the associated DNA label tagged to the kinase. The results are reported as
Mass Spectrometric Identification of Phosphorylation Sites.
Mass spectrometric studies were performed by MS Bioworks, LLC (Ann Arbor, MI). 2.4 μg of Wee1-FL, Wee1-KD, Wee2-FL, Wee2-KD, and Myt1-KD was processed by SDS–PAGE using a 4–12% Bis-Tris NuPage Mini-gel (Invitrogen) with the MOPS buffer system. The target protein was excised for further processing. In-gel digestion with trypsin digestion was performed on the excised gel band using a ProGest robot (DigiLab) with the following protocol: (1) washed with 25 mM ammonium bicarbonate followed by acetonitrile; (2) reduced with 10 mM dithiothreitol at 60 °C followed by alkylation with 50 mM iodoacetamide at room temperature; (3) digested with sequencing grade trypsin (Promega) at 37 °C for 4 h; (4) quenched with formic acid, and the supernatant was analyzed directly without further processing. The gel digest was analyzed by nano LC-MS/MS with a Waters NanoAcquity HPLC system interfaced to a ThermoFisher Q Exactive. Peptides were loaded on a trapping column and eluted over a 75 μm analytical column at 350 nL/min; both columns were packed with Luna resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 60 000 fwhm and 17 500 fwhm for MS and MS/MS respectively. The 15 most abundant ions were selected for MS/MS. Data were processed with Mascot Distiller and searched using a local copy of Mascot against the UniProt Human appended with the custom sequence.
Cellular Studies.
Human MM1.S, MDA-MB-231, and HEK293T cells were purchased from ATCC and passaged in the laboratory for fewer than 6 months after receipt or resuscitation. MM1.S cells were maintained in RPMI-1640 medium, and the other two cell lines were maintained in DMEM (Life Technologies). All cells were grown and maintained at 37 °C in a humidified atmosphere containing 5% CO2. The following antibodies were purchased from Cell Signaling: phospho-CDK1(Y15) (no. 9111), CDK1 (no. 9116), phospho-TCTP(S46) (no. 5251), TCTP (no. 5128). Vinculin antibody was purchased from Sigma-Aldrich (no. V9131). Peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch. For viability assays, cells were seeded in 96-well plates at approximately 3000 adherent or 20 000 suspension cells per well (0.1 mL). Adherent cells were allowed to attach overnight, and suspension cells were incubated for 1 h prior to dosing. Cells were incubated with the compounds indicated in the figure legends ranging from 1 nM to 10 μM in the presence of vehicle (0.1% DMSO) for 72 h, with 6 replicates per concentration. After drug treatment, 15 μL of CellTiter Blue reagent (Promega) was added to each well, followed by vigorous orbital shaking for 5 min and incubation for 3 h at 37 °C. Plates were placed in a Wallac EnVision 2103 multilabel reader (PerkinElmer), and fluorescence was determined using excitation and emission filters of 570 and 615 nm, respectively. Growth inhibition data were analyzed with the Prism 6 software (GraphPad).
For immunoblotting, cells were seeded in 6-well plates (2 × 105 adherent or 1 × 106 suspension cells per well) and incubated for 6 h with increasing inhibitor concentrations. For synchronization in prometaphase, HEK293T or MDA-MB-231 cells were seeded at 4 × 106 cell density in a T75 flask 16 h before drug treatment with 0.2 or 0.1 μg/mL nocodazole, respectively, then incubated for additional 6 h with inhibitors. Cells were harvested by centrifugation at 300g for 5 min and resuspended in CelLytic M cell lysis reagent (Sigma-Aldrich) containing Halt protease and phosphatase inhibitor cocktail (Thermo Scientific) and 5 mM EDTA at 4 °C. Protein concentration was determined with the Bio-Rad protein assay reagent, and samples were diluted with ⅓ volume 4× SDS sample buffer and heated at 85 °C for 5 min. Samples were subjected to 12.5% SDS–PAGE and transferred to PVDF membranes. Western blots were developed with the appropriate pairs of primary and secondary antibodies, and signals were visualized using HyGLO ChemiluminescentSignalFire ECL reagent (Denville ScientificCell Signaling, no. 6883).
Supplementary Material
ACKNOWLEDGMENTS
We thank the Moffitt Chemical Biology Core for use of the protein crystallography facility (National Cancer Institute Grant P30-CA076292) and the Southeast Regional Collaborative Access Team (SER-CAT, University of Georgia) for synchrotron data collection. We also thank William Goodheart and Paula Cranfill (Moffitt Cancer Center) for assistance with large scale protein production. This work was supported by the National Institute for Child Health & Human Development (NICHD) Grants U01-HD076542, U54-HD055744, and HHSN275201300017C.
ABBREVIATIONS USED
- DSF
differential scanning fluorimetry
- ITC
isothermal titration calorimetry
- TCTP
translationally controlled tumor protein
- KD
kinase domain
- FL
full-length
- PLK
polo-like kinase
- LAMBD
Escherichia phage lambda serine/threonine-protein phosphatase
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.jmedchem.7b00996.
Tables of phosphorylated residues, detailed experimental conditions for ITC studies and original titration data, crystallization conditions, stereofigures including omit electron density maps, rmsd analysis graphs (PDF) X-ray data collection and refinement statistics, kinome profiling results (XLSX)
Molecular formula strings (CSV)
Accession Codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 5V5Y, 5VC5, 5VC6, 5VC4, 5VC3, 5VD2, 5VDK, 5VCV, 5VCW, 5VCX, 5VD0, 5VD1, 5VCZ, 5VCY, and 5VD3.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Russell P; Nurse P Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987, 49, 559–567. [DOI] [PubMed] [Google Scholar]
- (2).Lundgren K; Walworth N; Booher R; Dembski M; Kirschner M; Beach D Mik1 and Wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell 1991, 64, 1111–1122. [DOI] [PubMed] [Google Scholar]
- (3).Parker LL; Piwnica-Worms H Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [DOI] [PubMed] [Google Scholar]
- (4).Kastan MB; Bartek J Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [DOI] [PubMed] [Google Scholar]
- (5).Bucher N; Britten CD G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chen T; Stephens PA; Middleton FK; Curtin NJ Targeting the S and G2 checkpoint to treat cancer. Drug Discovery Today 2012, 17, 194–202. [DOI] [PubMed] [Google Scholar]
- (7).Do K; Doroshow JH; Kummar S Wee1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3159–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Matheson CJ; Backos DS; Reigan P Targeting WEE1 kinase in cancer. Trends Pharmacol. Sci 2016, 37, 872–881. [DOI] [PubMed] [Google Scholar]
- (9).Matheson CJ; Venkataraman S; Amani V; Harris PS; Backos DS; Donson AM; Wempe MF; Foreman NK; Vibhakar R; Reigan P A WEE1 inhibitor analog of AZD1775 maintains synergy with cisplatin and demonstrates reduced single-agent cytotoxicity in medulloblastoma cells. ACS Chem. Biol 2016, 11, 921–930. [DOI] [PubMed] [Google Scholar]
- (10).Jhuraney A; Woods NT; Wright G; Rix L; Kinose F; Kroeger JL; Remily-Wood E; Cress WD; Koomen JM; Brantley SG; Gray JE; Haura EB; Rix U; Monteiro AN PAXIP1 potentiates the combination of WEE1 inhibitor AZD1775 and platinum agents in lung cancer. Mol. Cancer Ther 2016, 15, 1669–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Muller M; Lutter D; Puschel AW Persistence of the cell-cycle checkpoint kinase Wee1 in SadA- and SadB-deficient neurons disrupts neuronal polarity. J. Cell Sci 2010, 123, 286–294. [DOI] [PubMed] [Google Scholar]
- (12).Mahajan K; Fang B; Koomen JM; Mahajan NP H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat. Struct. Mol. Biol 2012, 19, 930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mahajan K; Mahajan NP WEE1 tyrosine kinase, a novel epigenetic modifier. Trends Genet 2013, 29, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nakanishi M; Ando H; Watanabe N; Kitamura K; Ito K; Okayama H; Miyamoto T; Agui T; Sasaki M Identification and characterization of human Wee1B, a new member of the Wee1 family of Cdk-inhibitory kinases. Genes Cells 2000, 5, 839–847. [DOI] [PubMed] [Google Scholar]
- (15).Han SJ; Chen R; Paronetto MP; Conti M Wee1B is an oocyte-specific kinase involved in the control of meiotic arrest in the mouse. Curr. Biol 2005, 15, 1670–1676. [DOI] [PubMed] [Google Scholar]
- (16).Hanna CB; Yao S; Patta MC; Jensen JT; Wu X WEE2 is an oocyte-specific meiosis inhibitor in rhesus macaque monkeys. Biol. Reprod 2010, 82, 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Oh JS; Susor A; Conti M Protein tyrosine kinase Wee1B is essential for metaphase II exit in mouse oocytes. Science 2011, 332, 462–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Booher RN; Holman PS; Fattaey A Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J. Biol. Chem 1997, 272, 22300–22306. [DOI] [PubMed] [Google Scholar]
- (19).Liu F; Stanton JJ; Wu Z; Piwnica-Worms H The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol. Cell. Biol 1997, 17, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jin Z; Homola E; Tiong S; Campbell SD Drosophila myt1 is the major cdk1 inhibitory kinase for wing imaginal disc development. Genetics 2008, 180, 2123–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nakajima H; Yonemura S; Murata M; Nakamura N; Piwnica-Worms H; Nishida E Myt1 protein kinase is essential for Golgi and ER assembly during mitotic exit. J. Cell Biol 2008, 181, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Toledo CM; Ding Y; Hoellerbauer P; Davis RJ; Basom R; Girard EJ; Lee E; Corrin P; Hart T; Bolouri H; Davison J; Zhang Q; Hardcastle J; Aronow BJ; Plaisier CL; Baliga NS; Moffat J; Lin Q; Li XN; Nam DH; Lee J; Pollard SM; Zhu J; Delrow JJ; Clurman BE; Olson JM; Paddison PJ Genomewide CRISPR-Cas9 screens reveal loss of redundancy between PKMYT1 and WEE1 in glioblastoma stem-like cells. Cell Rep 2015, 13, 2425–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hirai H; Iwasawa Y; Okada M; Arai T; Nishibata T; Kobayashi M; Kimura T; Kaneko N; Ohtani J; Yamanaka K; Itadani H; Takahashi-Suzuki I; Fukasawa K; Oki H; Nambu T; Jiang J; Sakai T; Arakawa H; Sakamoto T; Sagara T; Yoshizumi T; Mizuarai S; Kotani H Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther 2009, 8, 2992–3000. [DOI] [PubMed] [Google Scholar]
- (24).Hirai H; Arai T; Okada M; Nishibata T; Kobayashi M; Sakai N; Imagaki K; Ohtani J; Sakai T; Yoshizumi T; Mizuarai S; Iwasawa Y; Kotani H MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther 2010, 9, 514–522. [DOI] [PubMed] [Google Scholar]
- (25).Do K; Wilsker D; Ji J; Zlott J; Freshwater T; Kinders RJ; Collins J; Chen AP; Doroshow JH; Kummar S Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J. Clin. Oncol 2015, 33, 3409–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Leijen S; van Geel RM; Pavlick AC; Tibes R; Rosen L; Razak AR; Lam R; Demuth T; Rose S; Lee MA; Freshwater T; Shumway S; Liang LW; Oza AM; Schellens JH; Shapiro GI Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J. Clin. Oncol 2016, 34, 4371–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Squire CJ; Dickson JM; Ivanovic I; Baker EN Structure and inhibition of the human cell cycle checkpoint kinase, Wee1A kinase: an atypical tyrosine kinase with a key role in CDK1 regulation. Structure 2005, 13, 541–550. [DOI] [PubMed] [Google Scholar]
- (28).Smaill JB; Lee HH; Palmer BD; Thompson AM; Squire CJ; Baker EN; Booth RJ; Kraker A; Hook K; Denny WA Synthesis and structure-activity relationships of soluble 8-substituted 4-(2-chlorophenyl)-9-hydroxypyrrolo[3,4-c]carbazole-1,3-(2H,6H)-diones as inhibitors of the Wee1 and Chk1 checkpoint kinases. Bioorg Med Chem. Lett 2008, 18, 929–933. [DOI] [PubMed] [Google Scholar]
- (29).Aspeslagh S; Shailubhai K; Bahleda R; Gazzah A; Varga A; Hollebecque A; Massard C; Spreafico A; Reni M; Soria JC Phase I dose-escalation study of milciclib in combination with gemcitabine in patients with refractory solid tumors. Cancer Chemother. Pharmacol 2017, 79, 1257–1265. [DOI] [PubMed] [Google Scholar]
- (30).Lu R; Niida H; Nakanishi M Human SAD1 kinase is involved in UV-induced DNA damage checkpoint function. J. Biol. Chem 2004, 279, 31164–31170. [DOI] [PubMed] [Google Scholar]
- (31).Rogers S; Wells R; Rechsteiner M Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 1986, 234, 364–368. [DOI] [PubMed] [Google Scholar]
- (32).Graczyk PP Gini coefficient: a new way to express selectivity of kinase inhibitors against a family of kinases. J. Med. Chem 2007, 50, 5773–5779. [DOI] [PubMed] [Google Scholar]
- (33).Wright G; Golubeva V; Remsing Rix LL; Berndt N; Luo Y; Ward GA; Gray JE; Schonbrunn E; Lawrence HR; Monteiro ANA; Rix U Dual targeting of WEE1 and PLK1 by AZD1775 elicits single agent cellular anticancer activity. ACS Chem. Biol 2017, 12, 1883–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Steegmaier M; Hoffmann M; Baum A; Lenart P; Petronczki M; Krssak M; Gurtler U; Garin-Chesa P; Lieb S; Quant J; Grauert M; Adolf GR; Kraut N; Peters JM; Rettig WJ BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol 2007, 17, 316–322. [DOI] [PubMed] [Google Scholar]
- (35).Gilmartin AG; Bleam MR; Richter MC; Erskine SG; Kruger RG; Madden L; Hassler DF; Smith GK; Gontarek RR; Courtney MP; Sutton D; Diamond MA; Jackson JR; Laquerre SG Distinct concentration-dependent effects of the pololike kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res 2009, 69, 6969–6977. [DOI] [PubMed] [Google Scholar]
- (36).Jani JP; Arcari J; Bernardo V; Bhattacharya SK; Briere D; Cohen BD; Coleman K; Christensen JG; Emerson EO; Jakowski A; Hook K; Los G; Moyer JD; Pruimboom-Brees I; Pustilnik L; Rossi AM; Steyn SJ; Su C; Tsaparikos K; Wishka D; Yoon K; Jakubczak JL PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol. Cancer Ther 2010, 9, 883–894. [DOI] [PubMed] [Google Scholar]
- (37).Apsel B; Blair JA; Gonzalez B; Nazif TM; Feldman ME; Aizenstein B; Hoffman R; Williams RL; Shokat KM; Knight ZA Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol 2008, 4, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang Y; Li J; Booher RN; Kraker A; Lawrence T; Leopold WR; Sun Y Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res 2001, 61, 8211–8217. [PubMed] [Google Scholar]
- (39).Panek RL; Lu GH; Klutchko SR; Batley BL; Dahring TK; Hamby JM; Hallak H; Doherty AM; Keiser JA In vitro pharmacological characterization of PD 166285, a new nanomolar potent and broadly active protein tyrosine kinase inhibitor. J. Pharmacol. Exp. Ther 1997, 283, 1433–1444. [PubMed] [Google Scholar]
- (40).Levinson NM; Boxer SG Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS One 2012, 7, e29828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Brasca MG; Amboldi N; Ballinari D; Cameron A; Casale E; Cervi G; Colombo M; Colotta F; Croci V; D’Alessio R; Fiorentini F; Isacchi A; Mercurio C; Moretti W; Panzeri A; Pastori W; Pevarello P; Quartieri F; Roletto F; Traquandi G; Vianello P; Vulpetti A; Ciomei M Identification of N,1,4,4-tetramethyl-8-{[4-(4-methylpiperazin-1-yl)phenyl]amino}-4,5-dihydro-1H-py razolo[4,3-h]quinazoline-3-carboxamide (PHA-848125), a potent, orally available cyclin dependent kinase inhibitor. J. Med. Chem 2009, 52, 5152–5163. [DOI] [PubMed] [Google Scholar]
- (42).Sun H; Tawa G; Wallqvist A Classification of scaffoldhopping approaches. Drug Discovery Today 2012, 17, 310–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Olesen SH; Zhu JY; Martin MP; Schonbrunn E Discovery of diverse small-molecule inhibitors of mammalian Sterile20-like kinase 3 (MST3). ChemMedChem 2016, 11, 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kabsch W Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Afonine PV; Grosse-Kunstleve RW; Chen VB; Headd JJ; Moriarty NW; Richardson JS; Richardson DC; Urzhumtsev A; Zwart PH; Adams PD phenix.model_vs_data: a high-level tool for the calculation of crystallographic model and data statistics. J. Appl. Crystallogr 2010, 43, 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.