

S. canis Cas9 is a natural CRISPR enzyme that uses two motif insertions to enable flexible targeting of DNA sequences.
Abstract
RNA-guided DNA endonucleases of the CRISPR-Cas system are widely used for genome engineering and thus have numerous applications in a wide variety of fields. CRISPR endonucleases, however, require a specific protospacer adjacent motif (PAM) flanking the target site, thus constraining their targetable sequence space. In this study, we demonstrate the natural PAM plasticity of a highly similar, yet previously uncharacterized, Cas9 from Streptococcus canis (ScCas9) through rational manipulation of distinguishing motif insertions. To this end, we report affinity to minimal 5′-NNG-3′ PAM sequences and demonstrate the accurate editing capabilities of the ortholog in both bacterial and human cells. Last, we build an automated bioinformatics pipeline, the Search for PAMs by ALignment Of Targets (SPAMALOT), which further explores the microbial PAM diversity of otherwise overlooked Streptococcus Cas9 orthologs. Our results establish that ScCas9 can be used both as an alternative genome editing tool and as a functional platform to discover novel Streptococcus PAM specificities.
INTRODUCTION
RNA-guided endonucleases of the CRISPR-Cas system, such as Cas9 (1) and Cpf1 (also known as Cas12a) (2), have proven to be versatile tools for genome editing and regulation (3), which have numerous implications in medicine, agriculture, bioenergy, food security, nanotechnology, and beyond (4). The range of targetable sequences is limited, however, by the need for a specific protospacer adjacent motif (PAM), which is determined by DNA-protein interactions, to immediately follow the DNA sequence specified by the single-guide RNA (sgRNA) (1, 5–8). For example, the most widely used variant, Streptococcus pyogenes Cas9 (SpCas9), requires a 5′-NGG-3′ motif downstream of its RNA-programmed DNA target (1, 4–8). To relax this constraint, additional Cas9 and Cpf1 variants with distinct PAM requirements have been either discovered (9–13) or engineered (12–15) to diversify the range of targetable DNA sequences. In total, these studies have provided only a handful of CRISPR effectors with minimal PAM requirements that enable wide targeting capabilities.
To help augment this list, we characterize an orthologous Cas9 protein from S. canis (ScCas9; UniProt I7QXF2), having 89.2% sequence similarity to SpCas9. We find that, despite this homology, ScCas9 prefers a more minimal 5′-NNG-3′ PAM. To explain this divergence, we identify two notable insertions within its open reading frame (ORF) that differentiate ScCas9 from SpCas9 and contribute to its PAM-recognition flexibility. We show that ScCas9 can efficiently and accurately edit genomic DNA in mammalian cells and investigate possible explanations for PAM divergence between Streptococcus orthologs. Last, we construct a bioinformatics pipeline to explore the PAM specificities of other Streptococcus orthologs.
RESULTS
Identification of SpCas9 homologs
While numerous Cas9 homologs have been sequenced, only a handful of Streptococcus orthologs have been characterized or functionally validated. To explore this space, we curated all Streptococcus Cas9 protein sequences from UniProt (16), performed global pairwise alignments using the BLOSUM62 scoring matrix (17), and calculated percent sequence homology to SpCas9. From them, ScCas9 stood out, not only because of its remarkable sequence homology (89.2%) to SpCas9 but also due to a positive-charged insertion of 10 amino acids within the highly conserved REC3 domain, in positions 367 to 376 (Fig. 1A). Exploiting both of these properties, we modeled the insertion within the corresponding domain of PDB 4OO8 (18) and, when viewed in PyMOL, noticed that it formed a “loop”-like structure, of which several of its positive-charged residues come in close proximity with the target DNA near the PAM (Fig. 1B). We further identified an additional insertion of two amino acids (KQ) immediately upstream of the two critical arginine residues necessary for PAM binding (19) in positions 1337 and 1338 (Fig. 1A). We thus hypothesized that these insertions may affect the PAM specificity of this enzyme. To support this prediction, we computationally characterized the PAM for ScCas9 by first mapping spacer sequences from the Cas9-associated type II CRISPR loci in the S. canis genome (20) to viral and plasmid genomes using BLAST (21), extracting the sequences 3′ to the mapped protospacers, and subsequently generating a WebLogo (22) representation of the aligned PAM sequences. Our analysis suggested 5′-NNGTT-3′ PAM (Fig. 1C). Intrigued by these novel motifs and motivated by the potentially reduced specificity at position 2 of the PAM sequence, we selected ScCas9 as a candidate for further PAM characterization and engineering.
Fig. 1. In silico characterization of ScCas9.
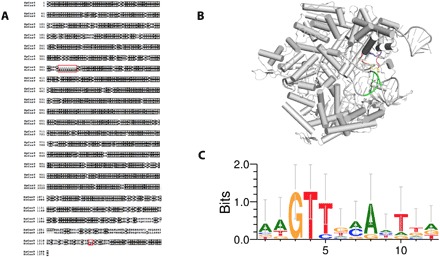
(A) Global pairwise sequence alignment of SpCas9 and ScCas9. Despite sharing 89.2% sequence homology to SpCas9, ScCas9 contains two notable insertions, one positive-charged insertion in the REC domain (367 to 376) and another KQ insertion in the PAM-interacting domain (PID; 1337 and 1338), as indicated. (B) Insertion of novel REC motif into PDB 4OO8 (18). The 367 to 376 insertion demonstrates a loop-like structure (red). Several of its positive-charged residues (yellow) come in close proximity to the target DNA near the PAM (green). (C) WebLogo (22) for sequences found at the 3′ end of protospacer targets identified in plasmid and viral genomes using type II spacer sequences within S. canis as BLAST (21) queries.
Determination of PAM sequences recognized by ScCas9
Because of the relatively low number of protospacer targets, we validated the PAM binding sequence of ScCas9 using an existent positive selection bacterial screen based on green fluorescent protein (GFP) expression conditioned on PAM binding, termed PAM-SCANR (23). A plasmid library containing the target sequence, followed by a randomized 5′-NNNNNNNN-3′ (8N) PAM sequence, was bound by a nuclease-deficient ScCas9 (and dSpCas9 as a control) and an sgRNA both specific to the target sequence and general for SpCas9 and ScCas9, allowing for the repression of lacI and expression of GFP. Plasmid DNA from fluorescence-activated cell sorting (FACS)–sorted GFP-positive cells and presorted cells were extracted and amplified, and enriched PAM sequences were identified by Sanger sequencing and visualized using DNA chromatograms. Our results provide initial evidence that ScCas9 can bind to the minimal 5′-NNG-3′ PAM, distinct to that of SpCas9’s 5′-NGG-3′ (Fig. 2A).
Fig. 2. PAM determination of engineered ScCas9 variants.
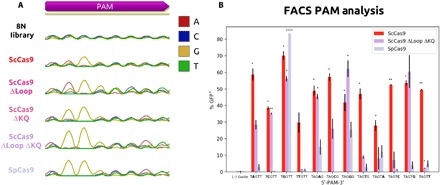
(A) PAM binding enrichment on a 5′-NNNNNNNN-3′ (8N) PAM library. PAM profiles are represented by Sanger sequencing chromatograms via amplification of PAM region following plasmid extraction of GFP+ E. coli cells. (B) Examination of PAM preference for ScCas9. For individual PAMs, all four bases were iterated at a single position (2, 4, 5). Each PAM-containing plasmid was electroporated in duplicates, subjected to FACS analysis, and gated for GFP expression. Subsequently, GFP expression levels were averaged. SD was used to calculate error bars, and statistical significance analysis was conducted using a two-tailed Student’s t test as compared to the negative control.
We hypothesized that the previously described insertions may contribute to this flexibility, and thus engineered ScCas9 to remove either insertion or both, and subjected these variants to the same screen. Only removing the loop (ScCas9 Δ367-376 or ScCas9 ΔLoop) extended the PAM of ScCas9 to 5′-NAG-3′, with reduced specificity for C and G at position 2, while only removing the KQ insertion (ScCas9 Δ1337-1338 or ScCas9 ΔKQ) reverted its specificity to a more 5′-NGG-3′–like PAM, with reduced specificity for A at position 2 (Fig. 2A). Last, the most SpCas9-like variant, where both insertions are removed (ScCas9 Δ367-376 Δ1337-1338 or ScCas9 ΔLoop ΔKQ), expectedly reverts its specificity back to 5′-NGG-3′ (Fig. 2A). Thus, from a functional perspective, these insertions operate in tandem to reduce the specificity of ScCas9 from the canonical 5′-NGG-3′ PAM to a more minimal 5′-NNG-3′.
To confirm the results of the library assay and to rule out limiting downstream requirements, we decided to elucidate the minimal PAM requirements of ScCas9 by using fixed PAM sequences. We replaced the PAM library with individual PAM sequences, which were varied at positions 2, 4, and 5, to test each possible base. Our results demonstrate that, while ScCas9 exhibits no clear additional base dependence with activity for all base iterations at each position, ScCas9 ΔLoop ΔKQ demonstrates significant binding at 5′-NGG-3′ PAM sequences and at some, but not all, 5′-NNGNN-3′ motifs, indicating an intermediate PAM specificity between that of SpCas9 and ScCas9 (Fig. 2B).
Assessment of ScCas9 PAM specificity in human cells
We compared the PAM specificity of ScCas9 to SpCas9 in human cells by cotransfecting human embryonic kidney–293T (HEK293T) cells with plasmids expressing these variants along with sgRNAs directed to a native genomic locus (VEGFA) with varying PAM sequences (table S1). We first tested editing efficiency at a site containing an overlapping PAM (5′-GGGT-3′). After 48 hours posttransfection, gene modification rates as detected by the T7E1 assay demonstrated comparable editing activities of SpCas9, ScCas9, and ScCas9 ΔLoop ΔKQ (Fig. 3, A and B). In addition, we constructed sgRNAs to sites with various nonoverlapping 5′-NNGN-3′ PAM sequences. While SpCas9’s cleavage activity was impaired at other non–5′-NGG-3′ sequences (Fig. 3) (24), ScCas9 maintained comparable activity to that of SpCas9 on its 5′-NGG-3′ target across all tested targets with 5′-NNGN-3′ PAM sequences (Fig. 3). Consistent with our bacterial data, ScCas9 ΔLoop ΔKQ was able to cleave at the 5′-NGG-3′ target, along with significant activity on the 5′-NNGA-3′ target, with reduced gene modification levels at all other 5′-NNGN-3′ targets (Fig. 3). Overall, these results verify that ScCas9 can serve as an effective alternative to SpCas9 for genome editing in mammalian cells, both at overlapping 5′-NGG-3′ and more minimal 5′-NNGN-3′ PAM sequences.
Fig. 3. ScCas9 PAM specificity in human cells.
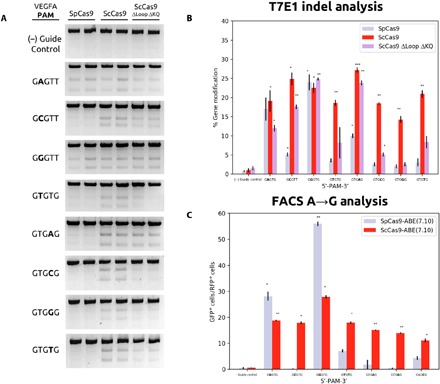
(A) T7E1 analysis of indels produced at VEGFA loci with indicated PAM sequences. The Cas9 used is indicated above each lane. All samples were performed in biological duplicates. As a background control, SpCas9, ScCas9, and ScCas9 ΔLoop ΔKQ were transfected without targeting guide RNA vectors [(−) guide control]. (B) Quantitative analysis of T7E1 products. Unprocessed gel images were quantified by line scan analysis using Fiji (41), the total intensity of cleaved bands were calculated as a fraction of total product, and percent gene modification was calculated. All samples were performed in duplicates, and quantified modification values were averaged. SD was used to calculate error bars, and statistical significance analysis was conducted using a two-tailed Student’s t test as compared to the negative control. (C) ScCas9-mediated A→G base editing. GFP+ cells were calculated as a percentage of mCherry+ (RFP+) cells for indicated PAM sequences using the TLR (25) with an early stop codon. All samples were performed in duplicates, and quantified percentages were averaged. SD was used to calculate error bars, and statistical significance analysis was conducted using a two-tailed Student’s t test. RFP+, red fluorescent protein–positive.
We further assessed the PAM specificity of ScCas9 base editors using a synthetic traffic light reporter (TLR) (25) plasmid, containing an early stop codon upstream of a GFP ORF and downstream of an mCherry ORF. Successful A→G base editing using the ABE(7.10) architecture, as described in Gaudelli et al. (26), converts an early, in-frame TAG stop codon to a TGG tryptophan codon, thus restoring GFP expression. After gating cells based on mCherry expression, we observed significant base-editing efficiency at all 5′-NNGN-3′ target PAM sequences for ScCas9-ABE(7.10), as compared to the SpCas9-ABE(7.10) architecture, which only demonstrates significant A→G conversion on the standard 5′-NGG-3′ and tolerated 5′-NAG-3′ motifs in this assay (Fig. 3C).
Off-target analysis of ScCas9
We next sought to evaluate the accuracy of this enzyme in comparison to SpCas9. We used previous genome-wide analysis of SpCas9 targeting accuracy to select three genomic targets (VEGFA site 3, FANCF site 2, and DNMT1 site 4) that have multiple off-target sites on which SpCas9 demonstrates activity (27). Each of these three sites additionally has a single off-target that has been particularly difficult to mediate via engineering of high-fidelity Cas9 variants (28–30). We first analyzed ScCas9’s activity on these off-targets. After cotransfection of sgRNAs to the three aforementioned sites alongside both SpCas9 and ScCas9, we amplified genomic DNA flanking, both the on-target and difficult off-target sequences, to assess their genome modification activities. Consistent with previously reported data (30), SpCas9 demonstrated high off-to-on targeting on all three examined targets (Fig. 4A). ScCas9 demonstrated comparable on-target activities for the three targets, but exhibited negligible activity on the VEGFA site 3 and DNMT1 site 4 off-targets, and a nearly 1.5-fold decrease in off-to-on target ratio for FANCF site 2 (Fig. 4A), suggesting improved accuracy over SpCas9 on overlapping 5′-NGG-3′ targets.
Fig. 4. ScCas9 performance as a genome editing tool.
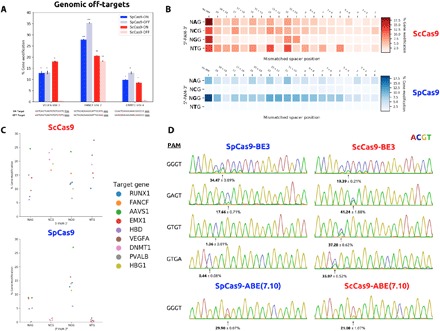
(A) Quantitative analysis of T7E1 products for indicated genomic on- and off-target editing. All samples were performed in duplicates, and quantified modification values were averaged. SD was used to calculate error bars, and statistical significance analysis was conducted using a two-tailed Student’s t test as compared to each negative control (table S2). Mismatched positions within the spacer sequence are highlighted in red. (B) Efficiency heatmap of mismatch tolerance assay. Quantified modification efficiencies, as assessed by the T7E1 assay, are exhibited for each labeled single or double mismatch in the sgRNA sequence for each indicated PAM (table S1). (C) Dot plot of on-target modification percentages at various gene targets for indicated PAM, as assessed by the T7E1 assay. Duplicate modification percentages were averaged (table S2). (D) Genomic base-editing characterization. For each indicated PAM, a representative Sanger sequencing chromatogram is shown, demonstrating the most efficiently edited base in the target sequence. Percent edited values, as quantified by BEEP in comparison to an unedited negative control, were averaged, and SD was subsequently calculated.
To examine ScCas9’s accuracy across its wider PAM targeting range, we used a mismatch tolerance assay (30) on target sequences with 5′-NAG-3′, 5′-NCG-3′, 5′-NGG-3′, and 5′-NTG-3′ PAMs. We generated sgRNAs containing both single and adjacent double mismatches at every other base across each of the four on-target sequences (table S1) and subsequently measured the genome modification efficiencies for these mismatched sgRNAs. Our results demonstrate that ScCas9 generally tolerates single mismatches better than double mismatches for each analyzed spacer position and is similarly less likely to tolerate mismatches within the seed region of the crRNA, though with greater sensitivity than SpCas9 (Fig. 4B). Across all of the four PAM targets, ScCas9 does, however, tolerate mismatches within the middle of the crRNA sequence, with highest efficiencies reported for the 5′-NTG-3′ target. SpCas9 expectedly demonstrates negligible genome modification activity on the 5′-NCG-3′ and 5′-NTG-3′ targets but weakly tolerates single and double mismatches across the entire crRNA sequence, with reduced tolerance in the seed region for the standard 5′-NGG-3′ target, corroborating previous mismatch tolerance studies (30). Last, ScCas9 exhibits a mismatch tolerance profile similar to SpCas9 on the 5′-NAG-3′ target, albeit with a higher reported on-target efficiency (Fig. 4B).
ScCas9 genome editing capabilities
Finally, to establish ScCas9 as a useful genome editing tool, we evaluated its ability to modify a variety of gene targets for a handful of different PAM sequences. We constructed sgRNAs to 24 targets within nine endogenous genes in HEK293T cells and evaluated on-target gene modification using the T7E1 assay. Our results demonstrate that ScCas9 maintains efficiencies comparable to that of SpCas9 on 5′-NGG-3′ sequences and on selected 5′-NNG-3′ PAM targets (Fig. 4C), supporting our previous findings (Fig. 3B). SpCas9 expectedly performs efficiently on 5′-NGG-3′ and weakly on 5′-NAG-3′ targets but demonstrates negligible editing capabilities on 5′-NCG-3′ and 5′-NTG-3′ PAM sequences (Fig. 4C), as previously demonstrated. Notably, ScCas9 performed less effectively on selected target sequences in the hemoglobin subunit δ (HBD) gene while demonstrating higher efficiencies on 5′-NNG-3′ sequences in VEGFA and DNMT1, for example. This variation in efficiency within each PAM group and across different genes indicates that proper target selection within specified genomic regions is critical for successful ScCas9-mediated gene modification (Fig. 4C).
We subsequently measured the efficacy of ScCas9 integrated within the BE3 (31) and ABE(7.10) (26) base-editing architectures on endogenous genomic loci. To evaluate the efficiency of base-editing activities, we developed a simple, easy-to-use Python program, termed the Base Editing Evaluation Program (BEEP), that takes as input both a negative control ab1 Sanger sequencing file and the edited sample ab1 file and outputs the efficiency of an indicated base conversion at a specific position (read from 5′ to 3′) along the target sequence. BEEP analysis on ab1 files, following transfection of ScCas9 base editors, genomic amplification, and subsequent Sanger sequencing, demonstrates that ScCas9 is capable of mediating C→T and A→G base conversion at both overlapping 5′-NGG-3′ and nonoverlapping 5′-NNG-3′ PAM sequences (Fig. 4D). While ScCas9 base editors perform efficiently on the non–5′-NGG-3′ targets, as compared to SpCas9 (Figs. 3C and 4D), ScCas9 is less effective at editing 5′-NGG-3′ genomic targets than SpCas9 for both architectures (Fig. 4D), indicating that further development is necessary for broad usage of ScCas9 base editors.
Investigation of sequence conservation between S. canis and other streptococcus Cas9 orthologs
To further investigate the distinguishing motif insertions in ScCas9, we inserted the loop (SpCas9 ::Loop), the KQ motif (SpCas9 ::KQ), or both (SpCas9 ::Loop ::KQ) into the SpCas9 ORF and analyzed binding on the 8N library using PAM-SCANR. Of these variants, only SpCas9 ::KQ showed target binding affinity in the PAM-SCANR assay. Sequencing on enriched GFP-expressing cells demonstrated an unaffected preference for 5′-NGG-3′ (Fig. 5A). FACS analysis on a fixed 5′-TGG-3′ PAM confirmed these binding profiles, with SpCas9 ::KQ yielding half the fraction of GFP-positive cells compared to SpCas9 (Fig. 5B). These data, in conjunction with the binding profiles of ScCas9 variants (Fig. 2), suggest that, while these insertions within ScCas9 do distinguish its PAM preference from SpCas9, other sequence features of ScCas9 also contribute to its divergence.
Fig. 5. Relationship of ScCas9 to other Streptococcus orthologs.
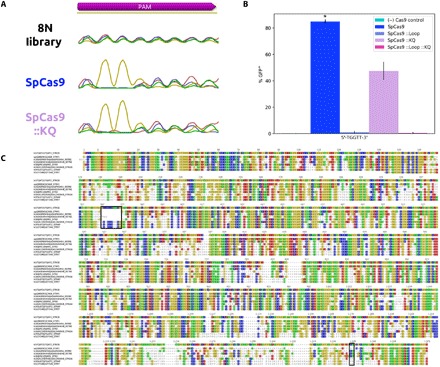
(A) PAM binding enrichment on a 5′-NNNNNNNN-3′ PAM library of ScCas9-like SpCas9 variants. The PAM-SCANR screen (23) was applied to variants of SpCas9 containing the loop, KQ insertions, or both. SpCas9 ::Loop and SpCas9 ::Loop ::KQ failed to demonstrate PAM binding and thus GFP expression. (B) FACS analysis of binding at 5′-NGG-3′ PAM. All samples were performed in duplicates and averaged. SD was used to calculate error bars. (C) Sequence conservation of Streptococcus orthologs with ScCas9 as a reference. Each ortholog is referred to by its UniProt ID (16). The loop (367 to 376) and KQ (1337 and 1338) insertion alignments are indicated.
S. canis has been reported to infect dogs, cats, cows, and humans and has been implicated as an adjacent evolutionary neighbor of S. pyogenes, as evidenced by various phylogenetic analyses (20, 32, 33). In addition to sharing common hosts, we identified S. canis CRISPR spacers that map to phage lysogens in S. pyogenes genomes, which suggests that they are overlapping viral hosts as well. This close evolutionary relationship has manifested itself in the sequence homology of ScCas9 and SpCas9, among other orthologous genes, predicted to be a result of lateral gene transfer (LGT) (20, 32, 33). Nonetheless, from the alignment of SpCas9 and ScCas9, the first 1240 positions score with 93.5% similarity and the last 144 positions score with 52.8%. To account for the exceptional divergence in the PID at the C terminus of ScCas9 and the positive-charged inserted loop, we focused on alignment of the distinguishing sequences of ScCas9 to other Streptococcus Cas9 orthologs (Fig. 5C). Notably, the loop motif is present in certain orthologs, such as those from S. gordonii, S. anginosus, and S. intermedius, while the ScCas9 PID is mostly composed of disjoint sequences from other orthologs, such as those from S. phocae, S. varani, and S. equinis. Additional LGT events between these orthologs, as opposed to isolated divergence, more likely explain the differences between ScCas9 and SpCas9. Our demonstration that two insertion motifs in ScCas9 alter PAM preferences, yet do not abolish PAM binding when removed, suggests other functional evolutionary intermediates in the formation of effective PAM preferences.
Genus-wide prediction of divergent Streptococcus Cas9 PAMs
Demonstrations of efficient genome editing by Cas9 nucleases with distinct PAM specificity from several Streptococcus species, including S. canis, motivated us to develop a bioinformatics pipeline for discovering additional Cas9 proteins with novel PAM requirements in the Streptococcus genus. We call this method the Search for PAMs by ALignment Of Targets (SPAMALOT). Briefly, we mapped a 20-nt portion of spacers flanked by known Streptococcus repeat sequences to candidate protospacers that align with no more than two mismatches in phages associated with the genus (34). We grouped 12-nt protospacer 3′-adjacent sequences from each alignment by genome and CRISPR repeat and then generated group WebLogos (22) to compute presumed PAM features. Figure 6A shows that resulting WebLogos accurately reflect the known PAM specificities of ScCas9 (this work), S. pyogenes, S. thermophilus, and S. mutans (7, 35, 36). We identified a notable diversity in the WebLogo plots derived from various S. thermophilus cassettes with common repeat sequences (Fig. 6B), each of which could originate from any other such S. thermophilus WebLogo upon subtle specificity changes that traverse intermediate WebLogos among them. We observe a similar relationship between two S. oralis WebLogos that also share this repeat and unique putative PAM specificities associated with CRISPR cassettes containing S. mutans–like repeats from the S. oralis, S. equinis, and S. pseudopneumoniae genomes (Fig. 6C).
Fig. 6. SPAMALOT PAM predictions for Streptococcus Cas9 orthologs.
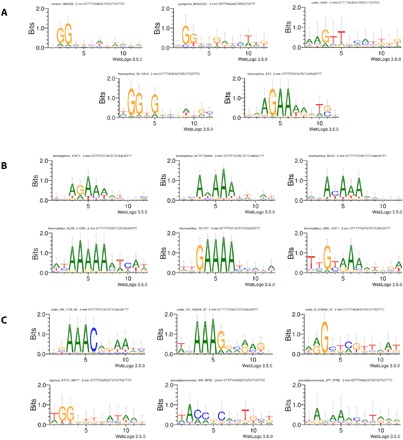
Spacer sequences found within the type II CRISPR cassettes associated with Cas9 ORFs from specified Streptococcus genomes were aligned to S. phage genomes to generate spacer-protospacer mappings. WebLogos (22), labeled with the relevant species, genome, and CRISPR repeat, were generated for sequences found at the 3′ end of candidate protospacer targets with no more than two mismatches (2 mm). (A) PAM predictions for experimentally validated Cas9 PAM sequences in previous studies. (B) Novel PAM predictions of alternate S. thermophilus Cas9 orthologs with putative divergent specificities. (C) Novel PAM predictions of uncharacterized Streptococcus orthologs with distinct specificities.
DISCUSSION
As the growth and development of CRISPR technologies continue, the range of targetable sequences remains limited by the requirement for a PAM sequence flanking a given target site. While significant discovery and engineering efforts have been undertaken to expand this range (9–15), there are still only a handful of CRISPR endonucleases with minimal specificity requirements. Here, we have developed an analogous platform for genome editing using ScCas9, a highly similar SpCas9 ortholog with affinity to minimal 5′-NNG-3′ PAM sequences.
Established PAM engineering methods, such as random mutagenesis and directed evolution, can only generate substitution mutations in protein coding sequences. During the preparation of this manuscript, another group used phage-assisted continuous evolution (37) to evolve an SpCas9 variant, xCas9(3.7), with preference for various 5′-NG-3′ PAM sequences (38). An alternative approach consists of inserting or removing motifs with specific properties, which may provide a sequence search space that more common mutagenic techniques cannot directly access. Here, we demonstrate an evolutionary example of this method with ScCas9, whose sequence disparities with SpCas9 include two divergent motifs that contribute to its minimal PAM sequence. Engineered variants lacking these motifs exhibit more stringent PAM specificities in our PAM determination assays, and the removal of both motifs reverts its PAM specificity back to a more 5′-NGG-3′–like preference. While minimal inconsistencies in PAM preference between the used assays may arise from PAM-dependent allosteric changes that drive DNA cleavage (19), the PAM flexibility of ScCas9, as compared to SpCas9, remains consistent in all tested contexts.
To date, there are limited open-source tools or platforms specifically for the prediction of PAM sequences, though prior studies have conducted internal bioinformatics-based characterizations before experimental validation (9–11, 13, 30, 39). Here, we have established SPAMALOT as an accessible resource that we share with the community for application to CRISPR cassettes from other genera. Future development will include broadening the scope of candidate targets beyond genus-associated phage to capture additional sequences that could be beneficial targets, such as lysogens in species that host the same phage. We hope that this pipeline can be used to more efficiently validate and engineer PAM specificities that expand the targeting range of CRISPR, especially for strictly PAM-constrained technologies such as base editing (26, 31) and homology repair induction (40).
Last, because ScCas9 does not require any alterations to the sgRNA of SpCas9, and because of its significant sequence homology with SpCas9, we presume that identical modifications from previous studies (28–30) can be made to increase the accuracy and efficiency of the endonuclease and its variants, although it already demonstrates potentially improved on-to-off activity as compared to the standard SpCas9 on 5′-NGG-3′ targets. In addition, while we have exhaustively evaluated the PAM specificity of ScCas9 on multiple targets in a variety of genome editing contexts, we do not rule out the possibility that there may exist untested 5′-NNG-3′ genomic targets on which ScCas9 does not have significant activity. When used together with SpCas9 and xCas9(3.7), however, ScCas9 expands the target range of currently used Cas9 enzymes for genome editing purposes. With further development, we anticipate that this broadened Streptococcus Cas9 toolkit, containing both ScCas9 and additional, uncharacterized orthologs with expanded targeting range, will enhance the current set of CRISPR technologies.
MATERIALS AND METHODS
Identification of Cas9 homologs and generation of plasmids
The UniProt database (16) was mined for all Streptococcus Cas9 protein sequences, which were used as inputs to either the BioPython pairwise2 module or Geneious to conduct global pairwise alignments with SpCas9, using the BLOSUM62 scoring matrix (17) and subsequently calculate percent homology. The ScCas9 was codon optimized for Escherichia coli, ordered as multiple gBlocks from Integrated DNA Technologies and assembled using Golden Gate Assembly. The pSF-EF1-Alpha-Cas9WT-EMCV-Puro (OG3569) plasmid for human expression of SpCas9 was purchased from Oxford Genetics, and the ORFs of Cas9 variants were individually amplified by polymerase chain reaction (PCR) to generate 35–base pair (bp) extensions for subsequent Gibson Assembly into the OG3569 backbone. Engineering of the coding sequence of ScCas9 and SpCas9 for removal or insertion of motifs was conducted using either the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) or Gibson Assembly. To generate ScCas9 base-editing plasmids, pCMV-ABE(7.10) (Addgene plasmid no. 102919) and pCMV-BE3 (Addgene plasmid no. 73021) were received as gifts from D. Liu. Similarly, the ORF of the ScCas9 D10A nickase was amplified by PCR to generate 35-bp extensions for subsequent Gibson Assembly into each base-editing architecture backbone. sgRNA plasmids were constructed by annealing oligonucleotides coding for crRNA sequences (table S1) and 4-bp overhangs and subsequently performing a T4 DNA ligase–mediated ligation reaction into a plasmid backbone immediately downstream of the human U6 promoter sequence. Assembled constructs were transformed into 50 μl of NEB Turbo Competent E. coli cells and plated onto Luria broth (LB) agar supplemented with the appropriate antibiotic for subsequent sequence verification of colonies and plasmid purification.
PAM-SCANR assay
Plasmids for the SpCas9 sgRNA and PAM-SCANR genetic circuit, as well as BW25113 ΔlacI cells, were provided by the Beisel Lab (North Carolina State University). Plasmid libraries containing the target sequence followed by either a fully randomized 8-bp 5′-NNNNNNNN-3′ library or fixed PAM sequences were constructed by conducting site-directed mutagenesis on the PAM-SCANR plasmid flanking the protospacer sequence. Nuclease-deficient mutations (D10A and H850A) were introduced to the ScCas9 variants using Gibson Assembly. The provided BW25113 cells were made electrocompetent using standard glycerol wash and resuspension protocols. The PAM library and sgRNA plasmids, with resistance to kanamycin (Kan) and carbenicillin (Crb), respectively, were coelectroporated into the electrocompetent cells at 2.4 kV, outgrown, and recovered in Kan + Crb LB medium overnight. The outgrowth was diluted 1:100, grown to ABS600 of 0.6 in Kan + Crb LB liquid medium, and made electrocompetent. Indicated Cas9 plasmids, with resistance to chloramphenicol (Chl), were electroporated in duplicates into the electrocompetent cells harboring both the dCas9 and sgRNA plasmids, outgrown, and collected in 5 ml of Kan + Crb + Chl LB medium. Overnight cultures were diluted to an ABS600 of 0.01 and cultured to an OD600 (optical density at 600 nm) of 0.2. Cultures were analyzed and sorted on a FACSAria machine (Becton Dickinson). Events were gated on the basis of forward scatter and side scatter, and fluorescence was measured in the fluorescein isothiocyanate (FITC) channel (488-nm laser excitation, 530/30 filter for detection), with at least 30,000 gated events for data analysis. Sorted GFP-positive cells were grown to sufficient density, and plasmids from the presorted and sorted populations were then isolated, and the region flanking the nucleotide library was PCR-amplified and submitted for Sanger sequencing (Genewiz). Bacteria harboring nonlibrary PAM plasmids, performed in duplicates, were analyzed by FACS following electroporation and overnight incubation and represented as the percentage of GFP-positive cells in the population using SD to calculate error bars. The PAM-SCANR spacer sequence is 5′-CGAAAGGTTTTGCACTCGAC-3′. Additional details on the PAM-SCANR assay can be found in Leenay et al. (23).
Cell culture and gene modification analysis
HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 100 U/ml penicillin, 100 U/ml streptomycin, and 10% fetal bovine serum. sgRNA plasmid (500 ng) and effector [nuclease, BE3, or ABE(7.10)] plasmid (500 ng) were transfected into cells as duplicates (2 × 105 per well in a 24-well plate) with Lipofectamine 2000 (Invitrogen) in Opti-MEM (Gibco). After 48 hours posttransfection, genomic DNA was extracted using QuickExtract Solution (Epicentre), and genomic loci were amplified by PCR using the KAPA HiFi HotStart ReadyMix (Kapa Biosystems). For base-editing analysis, amplicons were purified and submitted for Sanger sequencing (Genewiz). For indel analysis, the T7E1 reaction was conducted according to the manufacturer’s instructions, and equal volumes of products were analyzed on a 2% agarose gel stained with SYBR Safe (Thermo Fisher Scientific). Unprocessed gel image files were analyzed in Fiji (41). The cleaved bands of interest were isolated using the rectangle tool, and the areas under the corresponding peaks were measured and calculated as the fraction cleaved of the total product. Percent gene modification was calculated as follows (42)
All samples were performed in duplicates, and percent gene modifications were averaged. SD was used to calculate error bars.
Base-editing analysis with TLR
HEK293T cells were maintained, as previously described, and transfected with the corresponding sgRNA plasmids (333 ng), ABE7.10 plasmids (333 ng), and synthetically constructed TLR plasmids (333 ng) into cells as duplicates (2 × 105 per well in a 24-well plate) with Lipofectamine 2000 (Invitrogen) in Opti-MEM (Gibco). After 5 days posttransfection, cells were harvested and analyzed on a FACSCelesta machine (Becton Dickinson) for mCherry (561-nm laser excitation, 610/20 filter for detection) and GFP (488-nm laser excitation, 530/30 filter for detection) fluorescence. Cells expressing mCherry were gated, and percent GFP+ calculation of the subset were calculated. All samples were performed in duplicates, and percentage values were averaged. SD was used to calculate error bars. The TLR spacer sequence is 5′-TTCTGTAGTCGACGGTACCG-3′.
Base-editing evaluation program (BEEP)
The BEEP software was written in Python, using the pandas data manipulation library and BioPython package. As inputs, the program requires a sample ab1 file, a negative control ab1 file, a target sequence, and the position of the specified base conversion, handled as a.csv file either for multiple sample analysis or for individual samples on the command line. Briefly, the provided target sequences are aligned to the base calls of each input ab1 file to determine the absolute position of the target within the file. Subsequently, the peak values for each base at the indicated position in the spacer are obtained, and the editing percentage of the specified base conversion is calculated. Last, a separate function normalizes the editing percentage to that of the negative control ab1 file to account for background signals of each base. The final base conversion percentage is outputted to the same .csv file for downstream analysis. The BEEP software can be downloaded at https://github.com/mitmedialab/BEEP.
SPAMALOT pipeline
All 11,440 Streptococcus bacterial and 53 Streptococcus-associated phage genomes were downloaded from the National Center for Biotechnology Information (NCBI). CRISPR repeats cataloged for the genus were downloaded from CRISPRdb hosted by the University of Paris-Sud (43). For each genome, spacers upstream of a specific repeat sequence were collected with a toolchain consisting of the fast and memory-efficient Bowtie 2 alignment (44). Each genome and repeat-type–specific collection of spacers were then matched to all phage genomes using the original Bowtie short-sequence alignment tool (45) to identify candidate protospacers with at most one, two, or no mismatches. Unique candidates were input into the WebLogo 3 (22) command line tool for prediction of PAM features. The SPAMALOT software can be downloaded at https://github.com/mitmedialab/SPAMALOT.
Statistical analysis
Data are shown as means ± SD unless stated otherwise. Statistical analysis was performed using the two-tailed Student’s t test, using the SciPy software package. Calculated P values, as compared to the negative control, are represented as follows: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001. Data were plotted using Matplotlib.
Supplementary Material
Acknowledgments
We are very grateful to R. Leenay and C. Beisel for sharing PAM-SCANR plasmids and cells and for providing guidance in implementing the PAM-SCANR protocol. We thank L. Nip for technical assistance and T. Karydis for critical assistance in modeling PDB domain insertions. We also thank E. Boyden for access to cell culture, in addition to N. Gershenfeld and S. Zhang for shared laboratory equipment. Author contributions: P.C. and N.J. conceived study and designed experiments. P.C. and N.J. carried out experiments. P.C. and N.J. conducted data analyses. P.C. and N.J. designed and implemented bioinformatics workflows. P.C. and N.J. wrote the paper. P.C., N.J., and J.M.J. reviewed the paper. J.M.J. supervised the project. Competing interests: P.C., N.J., and J.M.J. are inventors on a provisional patent related to this work (no. 62/560,630; filed 19 September 2017). All authors declare that they have no additional competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. BEEP and SPAMALOT code, as well as WebLogos, are hosted on GitHub. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau0766/DC1
Table S1. Protein and nucleotide sequences used in this study, grouped by corresponding figure.
Table S2. Extended data containing raw and calculated values acquired in this study, grouped by corresponding figure.
REFERENCES AND NOTES
- 1.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E., A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zetsche B., Gootenberg J. S., Abudayyeh O. O., Slaymaker I. M., Makarova K. S., Essletzbichler P., Volz S. E., Joung J., van der Oost J., Regev A., Koonin E. V., Zhang F., Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qi L. S., Larson M. H., Gilbert L. A., Doudna J. A., Weissman J. S., Arkin A. P., Lim W. A., Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrangou R., Horvath P., A decade of discovery: CRISPR functions and applications. Nat. Microbiol. 2, 17092 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Mojica F. J., Díez-Villaseñor C., García-Martínez J., Almendros C., Short motif sequences determine the targets of the prokaryotic CRISPR defense system. Microbiology 155, 733–740 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Shah S. A., Erdmann S., Mojica F. J., Garrett R. A., Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biol. 10, 891–899 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sternberg S. H., Redding S., Jinek M., Greene E. C., Doudna J. A., DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang F., Zhou K., Ma L., Gressel S., Doudna J. A., A Cas9-guide RNA complex preorganized for target DNA recognition. Science 384, 1477–1481 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Ran F. A., Cong L., Yan W. X., Scott D. A., Gootenberg J. S., Kriz A. J., Zetsche B., Shalem O., Wu X., Makarova K. S., Koonin E. V., Sharp P. A., Zhang F., In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esvelt K. M., Mali P., Braff J. L., Moosburner M., Yaung S. J.,Church G. M., Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 10, 1116–1121 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim E., Koo T., Park S. W., Kim D., Kim K., Cho H. Y., Song D. W., Lee K. J., Jung M. H., Kim S., Kim J. H., Kim J. H., Kim J. S., In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 8, 14500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirano H., Gootenberg J. S., Horii T., Abudayyeh O. O., Kimura M., Hsu P. D., Nakane T., Ishitani R., Hatada I., Zhang F., Nishimasu H., Nureki O., Structure and engineering of Francisella novicida Cas9. Cell 164, 950–961 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrington L. B., Paez-Espino D., Staahl B. T., Chen J. S., Ma E., Kyrpides N. C., Doudna J. A., A thermostable Cas9 with increased lifetime in human plasma. Nat. Commun. 8, 1424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleinstiver B. P., Prew M. S., Tsai S. Q., Topkar V. V., Nguyen N. T., Zheng Z., Gonzales A. P., Li Z., Peterson R. T., Yeh J. R., Aryee M. J., Joung J. K., Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao L., Cox D. B. T., Yan W. X., Manteiga J. C., Schneider M. W., Cox D. B. T., Yan W. X., Manteiga J. C., Schneider M. W., Yamano T., Nishimasu H., Nureki O., Crosetto N., Zhang F., Engineered Cpf1 variants with altered PAM specificities. Nat. Biotechnol. 35, 789–792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The UniProt Consortium , UniProt: The universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henikoff S., Henikoff J. G., Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 89, 10915–10919 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishimasu H., Ran F. A., Hsu P. D., Konermann S., Shehata S. I., Dohmae N., Ishitani R., Zhang F., Nureki O., Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anders C., Bargsten K., Jinek M., Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol. Cell 61, 895–902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lefébure T., Richards V. P., Lang P., Pavinski-Bitar P., Stanhope M. J., Gene repertoire evolution of Streptococcus pyogenes inferred from phylogenomic analysis with Streptococcus canis and Streptococcus dysgalactiae. PLOS ONE 7, e37607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. Jour. of Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 22.Crooks G. E., Hon G., Chandonia J. M., Brenner S. E., WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leenay R. T., Maksimchuk K. R., Slotkowski R. A., Agrawal R. N., Gomaa A. A., Briner A. E., Barrangou R., Beisel C. L., Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol. Cell 62, 137–147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., Zhang F., DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Certo M. T., Ryu B. Y., Annis J. E., Garibov M., Jarjour J., Rawlings D. J., Scharenberg A. M., Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 8, 671–676 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaudelli N. M., Komor A. C., Rees H. A., Packer M. S., Badran A. H.,Liu D. R., Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai S. Q., Zheng Z., Nguyen N. T., Liebers M., Topkar V. V., Wyvekens N., Khayter C., Iafrate A. J., Le L. P., Aryee M. J., Joung J. K., GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slaymaker I. M., Gao L., Zetsche B., Scott D. A., Yan W. X., Zhang F., Rationally engineered Cas9 Nucleases with improved specificity. Science 351, 84–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleinstiver B. P., Pattanayak V., Prew M. S., Tsai S. Q., Nguyen N. T., Zheng Z., Joung J. K., High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J. S., Dagdas Y. S., Kleinstiver B. P., Welch M. M., Sousa A. A., Harrington L. B., Sternberg S. H., Joung J. K., Yildiz A., Doudna J. A., Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komor A. C., Kim Y. B., Packer M. S., Zuris J. A., Liu D. R., Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richards V. P., Zadoks R. N., Pavinski Bitar P. D., Lefébure T., Lang P., Werner B., Tikofsky L., Moroni P., Stanhope M. J., Genome characterization and population genetic structure of the zoonotic pathogen, Streptococcus canis. BMC Microbiol. 12, 293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards V. P., Palmer S. R., Pavinski Bitar P. D., Qin X., Weinstock G. M., Weinstock G. M., Highlander S. K., Town C. D., Burne R. A., Stanhope M. J., Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol. Evol. 6, 741–753 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shmakov S. A., Sitnik V., Makarova K. S., Wolf Y. I., Severinov K. V., Koonin E. V., The CRISPR spacer space is dominated by sequences from species-specific mobilomes. MBio 8, e01397–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller M., Lee C. M., Gasiunas G., Davis T. H., Cradick T. J., Siksnys V., Bao G., Cathomen T., Mussolino C., Streptococcus thermophilus CRISPR-Cas9 systems enable specific editing of the human genome. Mol. Ther. 24, 636–644 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fonfara I., Rhun A. L., Chylinski K., Makarova K. S., Lécrivain A. L., Bzdrenga J., Koonin E. V., Charpentier E., Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 42, 2577–2590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esvelt K. M., Carlson J. C., Liu D. R., A system for the continuous directed evolution of biomolecules. Nature 472, 499–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu J. H., Miller S. M., Geurts M. H., Tang W., Chen L., Sun N., Zeina C. M., Gao X., Rees H. A., Lin Z., Liu D. R., Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hidalgo-Cantabrana C., Crawley A. B., Sanchez B., Barrangou R., Characterization and exploitation of CRISPR Loci in Bifidobacterium longum. Front. Microbiol. 8, 1851 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richardson C. D., Ray G. J., DeWitt M. A., Curie G. L., Corn J. E., Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34, 339–344 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guschin D. Y., Waite A. J., Katibah G. E., Miller J. C., Holmes M. C., Rebar E. J., A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 649, 247–256 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Grissa I., Vergnaud G., Pourcel C., The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 8, 172 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau0766/DC1
Table S1. Protein and nucleotide sequences used in this study, grouped by corresponding figure.
Table S2. Extended data containing raw and calculated values acquired in this study, grouped by corresponding figure.