

Abstract
Food and Drug Administration (FDA) and Centers for Medicare and Medicaid Services (CMS) rely on evidence from clinical trials when approving a therapeutic for marketing and insurance coverage in the US, respectively. No study has compared the quality and quantity of evidence examined by these agencies.
To characterize evidence used by FDA and CMS to support marketing approval and National Coverage Determinations (NCDs), respectively, of novel therapeutics reviewed for CMS coverage from 2005 through 2016.
A cross-sectional study of clinical trials described in FDA approval documents and CMS NCD memoranda. We compared the number of clinical trials used by each agency as well as the following characteristics among original clinical trials: study size, randomization, double-blinding, and control arm.
Twelve medical products met our inclusion criteria. FDA approvals of these products were based on 22 pivotal trials. CMS NCDs were based on 27 original clinical trials; 14 clinical trials were used by both agencies. Between FDA pivotal and CMS original clinical trials, there was no significant difference in study size (P = .53), use of randomization (P = .75), double-blinding (P = .55), or control arm (P = .54). There was no statistically significant difference in median age between participants in trials reviewed by CMS versus those reviewed by FDA (62 vs 59 years, P = .26). The median time from FDA approval to publication of CMS NCD memorandum was 17 (interquartile range, 13–36) months.
FDA approvals and CMS NCDs are based on a similar number and quality of trials, although trial participants are not reflective of the Medicare population, and the process of finalizing coverage determinations requires an additional 17 months.
Keywords: Centers for Medicare and Medicaid Services, Food and Drug Administration, national coverage determination
1. Introduction
Marketing approval for medical devices, drugs, and biologics in the United States is granted by the Food and Drug Administration (FDA). FDA's final decision for high-risk devices, drugs, and biologics is based on several factors that ensure the products are “safe and effective” for their intended use, including the strength of supporting evidence, the risks and benefits of other available therapies, the need for post-market data collection, the likelihood that effects seen in trials will apply to broader populations, and the nature and severity of the condition the product is intended to treat.[1] An exception is moderate-risk devices, which generally receive clearance by demonstrating “substantial equivalence” to a predicate device.[2]
After regulatory approval, a product requires third-party payer coverage for clinical adoption; in the US, the largest is the Centers for Medicare and Medicaid Services (CMS) and Medicare alone covers 56.8 million Americans.[3] By statute, products covered by CMS must be “reasonable and necessary for the diagnosis or treatment of an illness or injury.’”[4] Whereas most coverage decisions are made by regional contractors, controversial medical products, or those predicted to have a large impact on the health of Medicare beneficiaries are reviewed by CMS through the National Coverage Determination (NCD) process.[5–7]
FDA relies primarily on pivotal clinical trials to determine whether a product is “safe and effective.”[8,9] CMS also relies, in part, on clinical trial data when determining coverage, stipulating that randomization, blinding, contemporaneous controls, and sufficient study size constitute a stronger evidence base.[7,10] In addition, CMS may consider other sources of evidence and information for NCDs, such as recommendations from the Medicare Evidence Development and Coverage Advisory Committee (MEDCAC), an advisory committee consisting of members from patient groups, industry, and the scientific community.[5,11] Despite differences in the process for FDA approval and CMS NCDs, no study has formally evaluated the type and quality of evidence reviewed by FDA and CMS to support their decisions. Understanding this question is important because making beneficial treatments available to patients requires continuity between both approval and coverage processes.
Therefore, we conducted a cross-sectional study to characterize the evidence supporting FDA approval and CMS coverage for drugs, biologics, and moderate- and high-risk medical devices that received NCDs from 2005 through 2016. Results from this study will provide information on the strength of evidence required for FDA approval and national CMS coverage and on the characteristics of participants evaluated in clinical trials, with the goal of helping understand how data generation can be aligned for future approval and coverage determinations.
2. Methods
2.1. Data sources: CMS NCDs
The Medicare Coverage Database (https://www.cms.gov/medicare-coverage-database/) offers public access to local and NCD memoranda and other documents germane to the Medicare coverage determination process. NCDs are published memoranda that include the following information: the coverage decision (covered, covered with evidence development, or not covered), a concise background of the disease intended to be diagnosed or treated, the history of Medicare coverage, a timeline of recent activities, FDA status of the medical product, general principles of Medicare's evidence review, the evidence base evaluated by CMS through both internal and external technology assessments, reports of MEDCAC meetings, and professional society position statements or guidelines that informed the final coverage decision.
In December 2016 and January 2017 we downloaded all NCD memoranda and external technology assessments referenced in the memoranda.
2.2. Data sources: FDA approval documents
For moderate-risk devices, 510(k) summaries were downloaded from the FDA website. In brief, 510(k) summaries provide evidence of substantial equivalence to another legally US marketed device.[2]
Links to FDA Summaries of Safety and Effectiveness Data (SSEDs) for all high-risk medical devices were included in the corresponding Medicare determination memoranda. The SSED is a document whose goal is to provide “a reasoned, objective, and balanced critique of the scientific evidence which served as the basis of the decision to approve or deny the premarket approval (PMA) [application].’”[12] Approval of a PMA application is “based on a determination by FDA that the PMA contains sufficient valid scientific evidence to assure that [a high-risk device] is safe and effective for its intended use(s).”[12]
Approval documents for pharmaceuticals and biologics were obtained through the Drugs@FDA on-line database and the FDA site for approved products under Vaccines, Blood, and Biologics, respectively, in December 2016 and January 2017.
2.3. Study sample
We included all NCD memoranda published from January 1, 2005, through December 31, 2016, that pertained to high-risk medical devices, moderate-risk medical devices, pharmaceuticals, or biologics, including both NCDs that resulted in coverage and those that did not. We excluded NCDs for diagnostic technologies, non-medical services, or surgical procedures that did not include discussion of a specific therapeutic product (Fig. 1).
Figure 1.
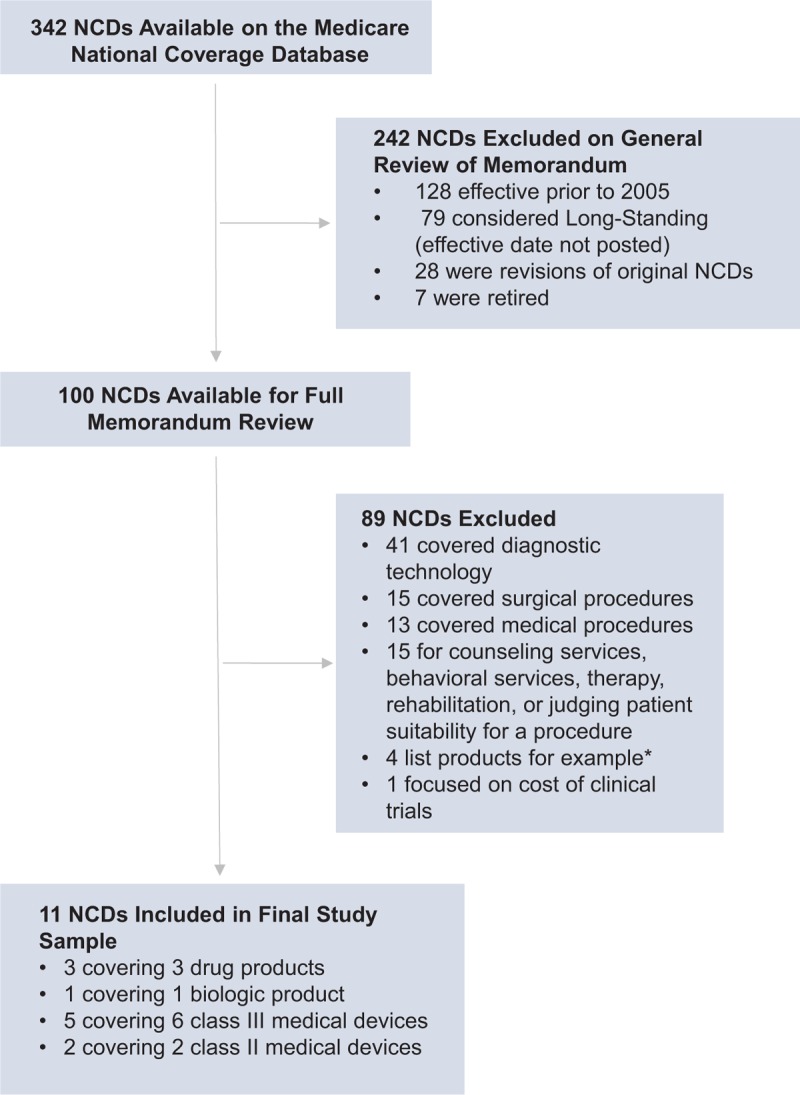
Selection of NCDs issued by Medicare from 2005 through 2016, covering medical devices, drugs, and biologics. ∗The products were listed in the context of a therapeutic procedure, but were not the focus of the NCD. NCD = national coverage determination.
2.4. Identification of original clinical trials reviewed by CMS
Each NCD was reviewed by 1 investigator (ACR) who identified original clinical trials used by CMS to support its NCD. Included trial evidence was summarized within the memoranda under the ‘Internal Technology Assessment’ section, which summarizes CMS’ evidence assessment. Original trials (e.g., excluding follow-up studies, interim-analyses, and pooled studies) reviewed by CMS were compared with pivotal trials used to support FDA approval. Uncertainty about trial inclusion was resolved by consensus between 2 investigators (ACR and JSR). We also recorded those additional studies that were based either on the original clinical trials reviewed by CMS or on FDA pivotal trials. These studies were not included in the sample of original clinical trials and included follow-up studies, pooled analyses, or interim analyses.
2.5. Identification of FDA pivotal efficacy trials
One investigator (ACR) identified trials labeled “pivotal” or “primary efficacy and safety” in FDA Medical Reviews for each included pharmaceutical or biologic and in SSEDs for high-risk devices. Of note, some pivotal trials in SSEDs were identified with a preceding statement such as “data from these clinical studies were the basis of the PMA approval decision.” Trials with narrative summaries within 510(k) documents for moderate-risk devices were classified as pivotal trials. Any uncertainty on identification of a pivotal trial was resolved by consensus among all study investigators.
2.6. Data extraction
For each NCD, the following data were extracted from the decision memorandum: date of NCD initiation, date of NCD posting by CMS, individual or agency requesting the NCD, number and type of products specified for coverage, whether MEDCAC was convened, whether an external technology assessment was requested (either from the Agency for Healthcare Research and Quality [AHRQ] or another external health technology assessment body), whether the referenced external technology assessment discussed cost-effectiveness, and the final coverage decision (covered, covered with evidence development [CED], or not covered). Links to NCD decision memoranda are provided on the CMS website for each NCD. The FDA approval date was determined from the NCD decision memoranda or from primary FDA approval documents.
We collected the following data to characterize all original trials evaluated by CMS and FDA: study size, use of randomization, use of double-blinding, inclusion of a control arm (placebo or active control), age of trial participants, proportion of female trial participants, proportion of trials incorporating clinical outcome(s) as a primary efficacy measure, proportion of trials including a US study location, and proportion of trials with multiple enrollment sites. Four of these variables (study size, use of randomization, double-blinding, and inclusion of a control arm) were considered primary trial characteristics based on the Cochrane Study Quality Guide and prior studies examining quality of evidence reviewed in CMS NCDs.[13–15]
2.7. Statistical analysis
2.7.1. Characterization of NCDs
Descriptive statistics were used to evaluate mean CMS review time (from initiation of NCD request to publication of decision memorandum); proportion of NCDs initiated internally by CMS; proportion of memoranda that referenced an advisory report from MEDCAC; proportion of memoranda that referenced an external technology assessment; proportion of external technology assessments that included a discussion of cost-effectiveness; median number of public comments in each of the 2 public comment periods (an initial 30-day period after initiation of the NCD request, and a second 30-day period after publication of the proposed decision memorandum); and proportion of NCDs referencing professional society statements, clinical practice guidelines, FDA approval documents, or expert opinion. We also determined the proportion of CMS evaluations leading to a positive coverage decision, including CED. All calculations were performed in Microsoft Excel v. 15.31 by 1 investigator (ACR) with verification by JSR and SSD.
2.7.2. Characterization of trials evaluated by the FDA and CMS
For original clinical trials, we used descriptive statistics to evaluate the aforementioned trial characteristics. The proportion of secondary analyses (i.e., interim-studies, follow-up analyses, pooled studies) reporting data on patients with an average age of at least 65 years was also determined. We used the Fisher Exact Test to compare the following characteristics among trials reviewed by FDA and CMS: use of randomization; double-blinding; inclusion of a control arm (active or placebo); inclusion of a US study location; enrollment from multiple study sites; and inclusion of a clinical outcome in primary efficacy analyses. The Wilcoxon Signed-Rank Test for paired samples was used to compare the total number of trials evaluated by each agency, the age of participants across original trials, the proportion of female trial participants, and the sample size across original clinical trials. Of note, the proportion of female trial participants was only evaluated for 10 of the 12 products because 2 products pertained to the treatment of prostate cancer (PROVENGE and Plenaxis). All tests were 2-tailed and used a type I error rate of 0.05. Analyses were conducted using Excel v. 15.31 by 1 investigator (ACR.), with subsequent verification by JSR and SSD.
Because this study made use of publicly-available documents and materials and did not use human subject data, it was exempt from review by the Yale Institutional Review Board.
3. Results
3.1. Characteristics of NCDs
From 2005 through 2016, 11 CMS NCDs covering 12 products (1 NCD included coverage for 2 FDA approved products) met our inclusion criteria. These included 3 pharmaceuticals, 1 biologic, 6 high-risk medical devices, and 2 moderate-risk devices. CMS initiated 4 NCD requests, 3 were requested by product manufacturers, 2 by medical societies, 1 by a Medicare beneficiary, and 1 by a physician.
Median CMS review time was 263 (IQR, 248–272) days. Only 1 (9.1%) NCD included MEDCAC review. Five (45%) NCDs reviewed an external technology assessment, 2 of which included information on cost-effectiveness (Table 1). Eight (73%) NCDs considered professional society statements, and 7 (64%) referenced clinical practice guidelines. Nine (82%) NCDs specifically stated that FDA approval documents were taken into consideration during the coverage determination process. Six NCDs (54%) referenced expert opinion.
Table 1.
Characteristics of trials used by the FDA for approval decisions and CMS for national coverage determinations.
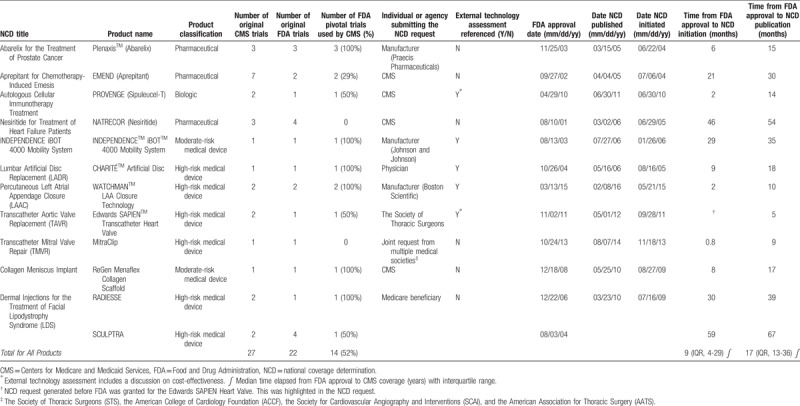
The median number of public comments during the first period for all NCDs was 43 (IQR 19-139), and for the second comment period was 76 (IQR 38-164).
A total of 8 (73%) NCDs were positive coverage determinations, with 3 (38%) of those requiring coverage with evidence development. The remaining 3 NCDs were determinations by CMS to not provide coverage. For more detailed information on each NCD, please see Appendix A.
3.2. Number of trials evaluated by the FDA and CMS
CMS NCDs for the 12 medical products were based on 27 original clinical trials, and 11 secondary analyses of the original clinical trials. FDA approval of these same 12 products was based on review of 22 pivotal trials. Fourteen (52%) of the 27 original clinical trials evaluated by CMS were the same pivotal trials examined by the FDA (Table 1). Eight (4%) of the pivotal trials evaluated by FDA were not included in CMS NCDs. Of these 8 trials, 4 were for a pharmaceutical (Natrecor), 3 for 1 high-risk medical device (SCULPTRA), and 1 for a different high-risk medical device (MitraClip). The NCD for Natrecor did include a brief result summary from the FDA pivotal trials, along with reference to a meta-analysis that included data from clinical trials used to support FDA approval. The NCD for dermal injections of facial lipodystrophy syndrome, which included SCULPTRA, included results from a 24-month extension of 1 of the original FDA pivotal trials. Of the remaining 2 trials supporting SCULPTRA's FDA approval, 1 was a randomized, controlled trial conducted in the US and the other was a single-arm trial conducted in the UK. Neither was specifically mentioned in the CMS NCD. The NCD for transcatheter mitral valve repair (MitraClip) includes a discussion of the FDA pivotal trial in CMS’ internal technology assessment.
There was no significant difference in the median number of clinical trials evaluated by the FDA and CMS for all products (FDA 1 [IQR, 1-2] versus CMS 2 [IQR, 1-2]; P = .59).
3.3. Characteristics of trials evaluated by the FDA and CMS
There were no significant differences in the characteristics of trials used to support FDA approval and CMS NCDs. Specifically, the following characteristics in original and pivotal clinical trials were similar in both groups: randomization (FDA: 15/22 [68%] versus CMS: 20/27 [74%]; P = .75), use of either placebo or active control (FDA 14/22 [64%] versus CMS: 20/27 [74%]; P = .54), double-blinding (FDA: 6/22 [27%] versus CMS: 10/27 [37%]; P = .55), and median number of enrolled study participants (FDA: 279 [IQR, 120–345] versus CMS: 291 [IQR, 174–328]; P = .53) (Table 2).
Table 2.
Comparison of trial design between the FDA and CMS.
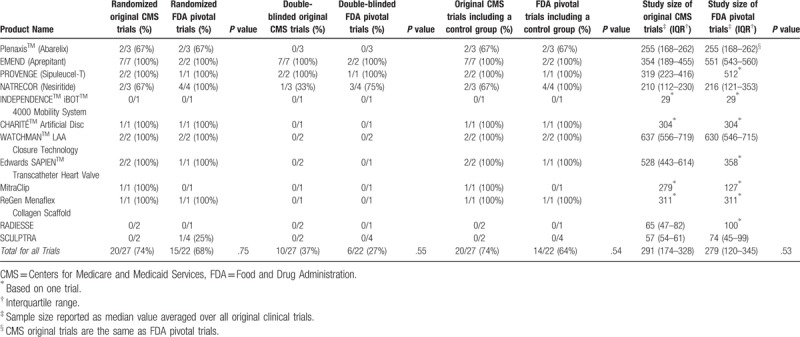
The number of CMS clinical trials including a US study location was approximately equal to the number of FDA clinical trials including a US study location (FDA: 18/19 [95%] versus CMS: 22/27 [81%]; P = .63). Of note, study location was unavailable for 3 FDA pivotal trials. There was no significant difference in the number of multi-center clinical trials evaluated by FDA and CMS (FDA: 17/22 [77%] versus CMS: 22/27 [81%]; P = .74). There was no significant difference in the use of a clinical outcome measure as a primary efficacy endpoint (FDA: 18/22 [82%] versus CMS: 22/27 [81%]; P = 1.00).
There was no significant difference in overall trial participant age (FDA: 59 years [IQR, 45–73] versus CMS: 62 years [IQR, 48–72]; P = .26). Of the original clinical trials reviewed by CMS, 4 (15%) included a subgroup analysis for patients older than 65 years of age. Of the 11 secondary analyses reviewed by CMS based on the original trials, 4 (36%) included subgroup data on patients older than 65 years. There was no significant difference in the proportion of female participants in FDA and CMS clinical trials (FDA: 29% [IQR, 20–44] versus CMS: 32% [24–29]; P = 1.00).
3.4. Time from FDA approval to initiation and publication of a CMS NCD
The median time from FDA approval to initiation of an NCD was 9 (IQR, 4–29) months. The median time from FDA approval to publication of a CMS NCD memorandum was 17 (IQR, 13–36) months.
4. Discussion
We characterized clinical trials used to support FDA approval and CMS NCDs for novel medical products (pharmaceuticals, biologics, moderate and high-risk medical devices) that were issued NCDs from 2005 through 2016, focusing on characteristics reflective of high-quality evidence.[15] There were no significant differences in use of randomization, double-blinding, control arm, or study size between original clinical trials evaluated by CMS and pivotal trials reviewed by FDA. Over half of original clinical trials reviewed by CMS were the same pivotal trials evaluated by FDA. This finding is of particular interest given differences in mandates for FDA approval and CMS coverage.
FDA's goal is to ensure that pivotal trials demonstrate safety and efficacy of the medical product, whereas CMS must ensure that trials show that a product is reasonable and necessary for diagnosis or treatment of an illness or injury in the Medicare population.[16] It is, therefore, reasonable to expect differences in trial characteristics. For example, FDA might be more likely to rely on a greater proportion of randomized, double-blind, placebo-controlled trials. CMS would be expected to prioritize trials that generalize to Medicare beneficiaries, including larger proportions of female participants over 65 years of age and that utilize active controls to determine if there is a benefit to covering the newer therapeutic over available alternatives. However, our results show that FDA and CMS not only rely on many of the same trials but even when other clinical trials are considered in Medicare's review, there is no overall difference in the quality of trials evaluated by each agency. Variation in the objectives of evidence review by FDA and CMS does not reflect variation in the trials—and their characteristics—evaluated of product approval/coverage.
Participant demographics in trials reviewed by FDA and CMS were also similar. Females constituted fewer than 50% of CMS trial participants, which is not significantly different from the median percent of female participants in FDA pivotal trials. This is important because more than half (54%) of Medicare beneficiaries are women.[17] In addition, neither CMS nor FDA trials had a median participant age above 65 years. However, CMS did include additional studies in its evaluation that frequently incorporated sub-group analyses of the original clinical trials for patients older than 65 years. CMS may use the time from initiation to publication of an NCD to acquire these additional data. This is unlikely to affect the time gap from FDA approval to CMS NCD because the CMS has a standard time period allotted for review of NCDs. However, knowing the data specifications requested by CMS in advance might potentially help reduce the time lag from FDA approval to NCD initiation. While the data from our study does not provide enough power to conduct such analyses, this may be a potential point of consideration for regulators and manufacturers. This is the goal of the Parallel Review program that was piloted starting in 2011 and officially established in 2016 for medical devices.[18]
Parallel review is an FDA–CMS collaboration designed to reduce the time between FDA review and CMS evaluation and to help manufacturers understand and address the data requirements of both agencies.[16] Through the program, manufacturers can request initiation of a CMS NCD while the product is still under FDA review.[16] For example, CMS initiated their coverage review for the Edwards SAPIEN transcatheter heart valve more than 1 month (9/28/11) before FDA approved the device (11/02/11). Whereas the SAPIEN transcatheter heart valve was not officially part of the parallel review program, this was 1 instance in which the manufacturer engaged both FDA and CMS before starting its pivotal clinical trial.[19] Early review obviated the median 9-month delay from FDA approval to initiation of a CMS NCD that we found. Therefore, parallel review may substantially decrease the delay in FDA approval to CMS coverage for medical devices.
Though parallel review could eliminate the need for CMS to acquire additional trial data in the Medicare population after FDA approval, other factors must be considered when merging FDA and CMS review processes. CMS, unlike FDA, holds 2 30-day public comment periods for each NCD and its decision memorandum includes a summary of, and response to, comments received. Public comments are designed to enhance the “quality of agency decision making” and can encompass contributions ranging from medical society recommendations to patient narratives.[20]
Furthermore, CMS considers evidence-based guidelines and the opinions of members of the medical or scientific community (labeled “expert opinion” in NCD memoranda) and may consider MEDCAC reports. Since many of these additional reports and guidelines reflect direct patient and physician experiences with the medical product once it is on the market, it will be difficult to merge these processes through parallel review. An additional barrier to the parallel review program may involve the relationship between manufacturers and the national CMS agency. Because of specific requirements for CPT coding, manufacturers may receive lower reimbursement rates from CMS once an NCD is issued. In addition, anecdotally, it has been suggested that manufacturers may have the potential to exert more influence in the local coverage determination (LCD) process, although this has not been systematically studied.
Limitations of our study include a small number of total reviewed NCDs; only 10 to 15 are issued annually by CMS,[13] whereas the majority of coverage decisions are made by regional contractors, whose determinations are not made publicly available. The recently enacted 21st Century Cures Act institutes new requirements mandating greater transparency of these regional coverage decisions, including public availability of documents summarizing evidence that supported development of a local coverage determination.[21] However, since NCDs are for the most controversial technologies likely to have a significant impact on the Medicare population and apply to all contractors, they are among the most important to study. In the future, it would be interesting to gather information from additional NCDs as they accrue over the years and conduct additional subgroup analyses on trial characteristics, such as time of follow-up and use of an active or placebo comparison group. In addition, CMS and FDA documents occasionally had missing data. For example, information on study site was unavailable for 3 FDA pivotal trials.
In conclusion, FDA approval and CMS NCDs of novel therapeutics often rely on the same clinical trial evidence and on trials of similar quality. However, the process of finalizing coverage determination requires an additional 17 months. FDA and CMS should continue to work together to ensure timely coverage decisions after FDA approval, perhaps by encouraging manufacturers to include larger proportions of older and female participants in their trials supporting FDA approval.
Author contributions
Conceptualization: Joseph Ross.
Data curation: Aliya C. Roginiel.
Formal analysis: Aliya C. Roginiel, Sanket Dhruva.
Funding acquisition: Aliya C. Roginiel, Joseph Ross.
Investigation: Aliya C. Roginiel, Sanket Dhruva, Joseph Ross.
Methodology: Aliya C. Roginiel, Sanket Dhruva, Joseph Ross.
Supervision: Joseph Ross.
Validation: Sanket Dhruva.
Writing – original draft: Aliya C. Roginiel.
Writing – review & editing: Sanket Dhruva, Joseph Ross.
Footnotes
Abbreviations: CMS = Centers for Medicare and Medicaid Services, FDA = Food and Drug Administration, MEDCAC = Medicare Evidence Development and Coverage Advisory Committee, NCD = national coverage determination, PMA = premarket approval, SSED = Summaries of Safety and Effectiveness Data.
Ms. Roginiel received a student research grant provided by the Yale School of Medicine Office of Student Research under NIH training grant award T35DK104689. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors assume full responsibility for the accuracy and completeness of the ideas presented.
Ms. Roginiel has no conflicts of interest to report. Over the past 24 months, Dr. Dhruva has been a fellow in the Robert Wood Johnson Foundation Clinical Scholars program at Yale, supported in part by the Department of Veterans Affairs. Over the past 36 months, Dr. Ross received support through Yale University from Johnson and Johnson to develop methods of clinical trial data sharing, from Medtronic, Inc. and the Food and Drug Administration (FDA) to develop methods for postmarket surveillance of medical devices, from the Food and Drug Administration as part of the Centers for Excellence in Regulatory Science and Innovation (CERSI) program, from the Blue Cross Blue Shield Association to better understand medical technology evaluation, from the Centers of Medicare and Medicaid Services (CMS) to develop and maintain performance measures that are used for public reporting, from the Agency for Healthcare Research and Quality to examine community predictors of healthcare quality, and from the Laura and John Arnold Foundation, which established the Collaboration for Research Integrity and Transparency (CRIT) at Yale University.
References
- [1].Califf RM. Benefit-risk assessments at the US food and drug administration: finding the balance. JAMA 2017;3177:693–4. [DOI] [PubMed] [Google Scholar]
- [2].U.S. Food and Drug Administration Division of Industry and Consumer Education. Premarket Notification 510(k) 2016. Available at: https://www.fda.gov/medicaldevices/deviceregulationandguidance/howtomarketyourdevice/premarketsubmissions/premarketnotification510k/. Accessed August 12, 2017. [Google Scholar]
- [3].Mnuchin ST, Acosta RA, Price TE, et al. The 2017 Annual Report of the Boards of Trustees of the Federal Hospital Insurance and Federal Supplementary Medical Insurance Trust Funds. Available at: https://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/ReportsTrustFunds/Downloads/TR2017.pdf. Accessed December 2017. [Google Scholar]
- [4].The Social Security Administration. Compilation of The Social Security Laws: Social Security Act. Available at: https://www.ssa.gov/OP_Home/ssact/ssact-toc.htm. Accessed August 12, 2017. [Google Scholar]
- [5].Sebelius K, Tavenner M. Medicare program; revised process for making national coverage determinations. Dep Health Hum Serv Cent Medicare Medicaid Services Federal Register 2013;78152:48164–9. [Google Scholar]
- [6].Chambers JD, Morris S, Neumann PJ, et al. Factors predicting medicare national coverage: an empirical analysis. Med Care 2012;50:249–56. [DOI] [PubMed] [Google Scholar]
- [7].Chambers JD, Chenoweth M, Cangelosi MJ, et al. Medicare is scrutinizing evidence more tightly for national coverage determinations. Health Aff Millwood 2015;34:253–60. [DOI] [PubMed] [Google Scholar]
- [8].Rathi VK, Krumholz HM, Masoudi FA, et al. Characteristics of clinical studies conducted over the total product life cycle of high-risk therapeutic medical devices receiving fda premarket approval in 2010 and 2011. JAMA 2015;314:604–12. [DOI] [PubMed] [Google Scholar]
- [9].Downing NS, Aminawung JA, Shah ND, et al. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005–2012. JAMA 2014;311:368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Phurrough S, Jacques L, Truong E, et al. Decision memo for abarelix for the treatment of prostate cancer. Centers Medicare Medicaid Serv 2005;1–30. [Google Scholar]
- [11].Lavertu S, Walters DE, Weimer DL. Scientific expertise and the balance of political interests: MEDCAC and medicare coverage decisions. J-PART 2011;55–81. [Google Scholar]
- [12].The U.S. Food and Drug Administration. PMA Application Contents 2017. Available at: https://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/PremarketApprovalPMA/ucm050289.htm. Accessed August 12, 2017. [Google Scholar]
- [13].Neumann PJ, Kamae MS, Palmer JA. Medicare's national coverage decisions for technologies, 1999–2007. Health Aff Millwood 2008;27:1620–31. [DOI] [PubMed] [Google Scholar]
- [14].Neumann PJ, Divi N, Beinfeld MT, et al. Medicare's national coverage decisions, 1999–2003: quality of evidence and review times. Health Aff Millwood 2005;24:243–54. [DOI] [PubMed] [Google Scholar]
- [15].Ryan R. Cochrane Consumers and Communication Review Group Study Quality Guide. Pgs. 1–48. Available at: https://cccrg.cochrane.org/sites/cccrg.cochrane.org/files/public/uploads/StudyQualityGuide_May%202013.pdf. Accessed December 2017. [Google Scholar]
- [16].Richardson L. Health policy brief: aligning FDA and CMS review. Health Aff 2015;1–5. [Google Scholar]
- [17].Centers for Medicare and Medicaid Services, Office of Enterprise Data and Analytics. 2016 CMS Statistics. Available at: https://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/CMS-Statistics-Reference-Booklet/Downloads/2016_CMS_Stats.pdf. Accessed August 2017. [Google Scholar]
- [18].The U.S. Food and Drug Administration. Payer Communication Task Force (PCTF): Parallel Review. 2017. Available at: https://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cdrh/cdrhinnovation/ucm456149.htm. Accessed August 12, 2017. [Google Scholar]
- [19].Jim Pomager. Bridging the FDA-CMS Divide: An Optimized Route to Market for Medical Devices, Part 1. 2014. Available at: https://www.meddeviceonline.com/doc/bridging-the-fda-cms-divide-an-optimized-route-to-market-for-medical-devices-part-1-0001. Accessed August 24, 2017. [Google Scholar]
- [20].The Centers for Medicare and Medicaid Services. Coverage Information Exchange: Public Comments. 2014. Available at: https://www.cms.gov/Medicare/Coverage/InfoExchange/publiccomments.html. Accessed August 12, 2017. [Google Scholar]
- [21].114th Congress of the United States of America. Text of House Amendment to the Senate Amendment to H.R. 34, Tsunami Warning, Education, and Research Act of 2015 [Showing the text of the 21st Century Cures Act]. Available at: https://docs.house.gov/billsthisweek/20161128/CPRT-114-HPRT-RU00-SAHR34.xml. Accessed December 2017. [Google Scholar]