

Abstract
In addition to playing a role in adhesion, desmoglein 2 (Dsg2) is an important regulator of growth and survival signaling pathways, cell proliferation, migration and invasion, and oncogenesis. While low-level Dsg2 expression is observed in basal keratinocytes and is downregulated in non-healing venous ulcers, overexpression has been observed in both melanomas and non-melanoma malignancies. Here, we show that transgenic mice overexpressing Dsg2 in basal keratinocytes primed the activation of mitogenic pathways, but did not induce dramatic epidermal changes or susceptibility to chemical-induced tumor development. Interestingly, acceleration of full-thickness wound closure and increased wound-adjacent keratinocyte proliferation was observed in these mice. As epidermal cytokines and their receptors play critical roles in wound healing, Dsg2-induced secretome alterations were assessed with an antibody profiler array and revealed increased release and proteolytic processing of the urokinase-type plasminogen activator receptor (uPAR). Dsg2 induced uPAR expression in the skin of transgenic compared to wild-type mice. Wound healing further enhanced uPAR in both epidermis and dermis with concomitant increase in the pro-healing laminin-332, a major component of the basement membrane zone, in transgenic mice. This study demonstrates that Dsg2 induces epidermal activation of various signaling cascades and accelerates cutaneous wound healing, in part, through uPAR-related signaling cascades.
Keywords: Carcinogenesis, Desmoglein 2, Exosomes, Keratinocyte, uPAR, Wound healing
INTRODUCTION
Epidermal keratinocytes undergo orchestrated proliferation and differentiation that is intricately regulated by crosstalk between the dermis and epidermis (Dhouailly and Sawyer, 1984). Disruption of homeostasis during wound healing requires coordinated activation of signaling cascades to facilitate. Stem-like basal keratinocytes divide asymmetrically into one differentiating and one basal/proliferative keratinocyte to maintain the stratification of the self-renewing epidermis (Lechler and Fuchs, 2005, Roshan et al., 2016). Keratinocytes adjacent to the wounds express basal-like and ‘activated’ cytokeratins and stem-like transcription factors, including Slug (Aomatsu et al., 2012, McGowan and Coulombe, 1998). It is paramount to the intrinsic self-renewal capabilities of the skin to maintain basal keratinocytes in a pro-proliferative state and responsive to migratory signals.
Signaling mediated by adhesion proteins is emerging as a key player in maintaining epidermal homeostasis and function (Kowalczyk and Green, 2013). The desmosomal cadherin desmoglein 2 (Dsg2) has been shown to play a critical role in cell proliferation and migration. Embryonic lethal Dsg2−/− mice illustrate that Dsg2 is essential for developmental cell survival and growth (Eshkind et al., 2002). Transgenic overexpression of Dsg2 in suprabasal epidermis enhances activation of mitogenic signaling pathways and increases keratinocyte proliferation, resulting in epidermal hyperplasia and enhanced susceptibility to tumor growth (Brennan et al., 2007). How Dsg2 modulates mitogenic signaling may involve its localization to lipid rafts and interaction with caveolin-1, an integral membrane component and signal regulator of lipid rafts (Brennan et al., 2012). Dsg2 also modulates EGFR levels and activation, promoting increased cell proliferation, migration, and invasion (Klessner et al., 2009, Overmiller et al., 2016). Furthermore, Dsg2 upregulates Gli1 and Ptch1, target genes of the Hh signaling pathway, and compound Dsg2/Ptc1+/lacZ mice have accelerated development of basal and squamous cell carcinoma (BCC and SCC) and tumorigenesis in response to chemical carcinogens (Brennan-Crispi et al., 2015). Dsg2 modulates exosome release from keratinocytes and induces pro-proliferative activity in dermal fibroblasts (Overmiller et al., 2017). These results indicate that Dsg2 can profoundly affect the homeostasis of the epidermis and facilitate skin carcinogenesis.
In the epidermis, Dsg2 expression is restricted to the basal layer and decreases with age (Brennan and Mahoney, 2009, Schäfer et al., 1994). Mutations in the human DSG2 gene are associated with fibrosis and cardiomyopathy with no obvious skin phenotype (Awad et al., 2006, Pilichou et al., 2006). Dsg2 is downregulated in non-healing venous ulcers, though other desmosomal cadherins, including Dsc2 and Dsg3, are upregulated (Stojadinovic et al., 2008). Given that the proliferative and differentiating populations of keratinocytes in the epidermis are functionally distinct, we sought to determine the effects of Dsg2 overexpression in basal keratinocytes through K14-driven expression (K14-Dsg2). The effect of basal Dsg2 on epidermal homeostasis, malignant transformation, and wound healing was investigated, and the results illustrate that basal keratinocytes overexpressing Dsg2 are ‘primed’ to maintain the epidermal architecture following tissue damage without inducing hyperplasticity in the skin.
RESULTS
Establishment and characterization of K14-Dsg2 transgenic mice
In the interfollicular epidermis, Dsg2 is barely detectable, and loss-of-function Dsg2lo/lo mice develop fibrotic lesions in the heart without skin morphological changes (Ebert et al., 2016). To assess the role of Dsg2 in the epidermis, we generated a K14-Dsg2 construct by subcloning the C-terminal Flag-tagged mouse Dsg2 cDNA downstream of the human keratin 14 (KRT14) promoter (Figure S1a) (Brennan et al., 2007). K14-Dsg2 transgenic mice did not exhibit any gross developmental, morphological, or life span defects compared to wild-type mice and litter sizes were normal (between 6-10 pups per litter). We confirmed the expression of Dsg2 in the skin of these mice by immunoblotting (Figure 1a) and immunofluorescence for the Flag tag (Figure S1b). The staining pattern of Flag-tagged Dsg2 was not continuous along the interfollicular epidermis and occasionally appeared to be cytoplasmic, similar to that of endogenous Dsg2 (Brennan and Mahoney, 2009). As suprabasal expression of Dsg2 induced epidermal hyperplasia at all ages, the epidermis of K14-Dsg2 mice was evaluated. Surprisingly, K14-driven expression of Dsg2 did not induce any apparent changes in epidermal thickness (Figures 1b and S1c). Furthermore, basal-Dsg2 did not alter keratinocyte proliferation, as measured by PCNA immunofluorescence (Figures S1d, e). In summary, K14-driven expression of Dsg2 in the basal epidermis had no detectable gross morphological effects on mouse development, fecundity, or viability, and did not alter the overall architecture of the skin.
Figure 1. Dsg2 enhances activation of mitogenic signaling species.
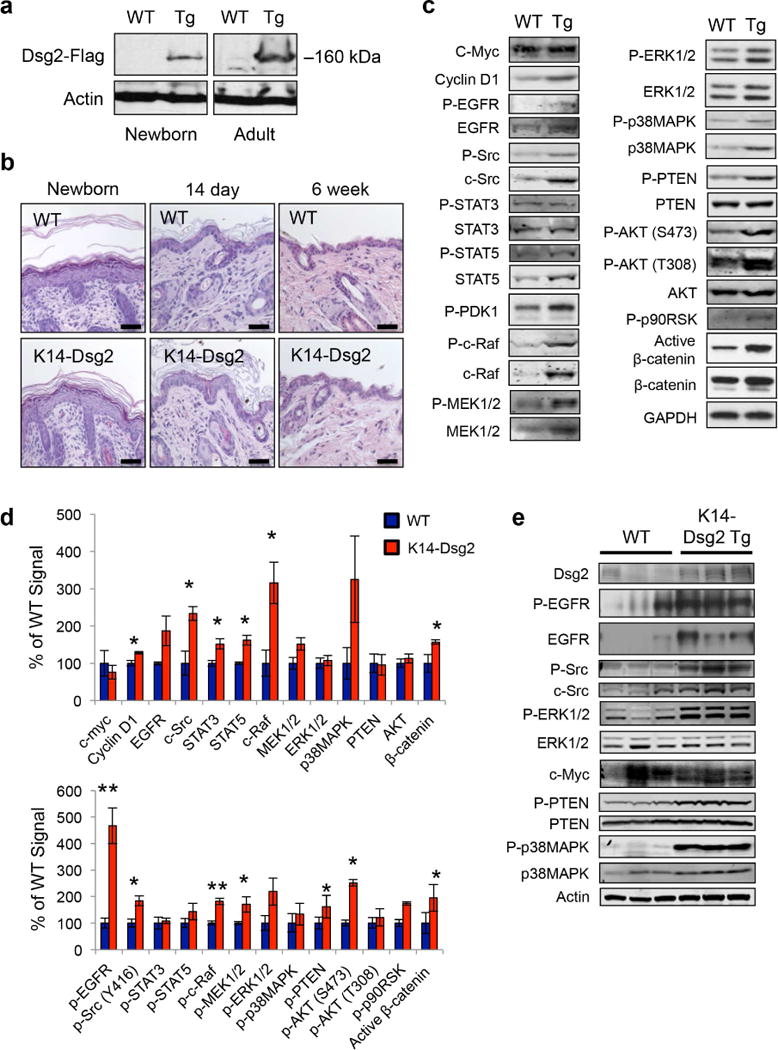
(a) Skin from wild-type and K14-Dsg2 transgenic mice was lysed and immunoblotted for Flag. (b) Representative hematoxylin and eosin staining of newborn, 14 day, and 6 week old dorsal skin of wild-type and transgenic mice. (c) Dorsal skin from wild-type and transgenic mice was lysed and proteins were immunoblotted for phosphorylated or total signaling proteins. Representative results from one of three independent experiments shown. (d) The signals were quantified and unphosphorylated proteins were normalized to GAPDH loading control while phosphorylated proteins were normalized to respective total proteins. (e) Wild-type and K14-Dsg2 mouse epidermis was immunoblotted for phosphorylated and total proteins. Data are depicted as percentage of wild-type and average ± SEM. N=3.
Basal Dsg2 activates mitogenic and survival signaling pathways but does not increase susceptibility to chemical-induced carcinogenesis
Dsg2 had been previously shown to positively regulate activation of the EGFR/MAPK signaling and other factors including c-myc, PI3K/AKT, c-Src, and STAT3 (Brennan et al., 2007, Overmiller et al., 2016). While the total levels of many signaling proteins including EGFR, STAT3, c-Raf, c-Src, and Cyclin D1 were increased in the transgenic mice, the level of c-Myc, a transcription factor that plays a critical role in keratinocyte proliferation was not affected (Figure 1c) (Hirning et al., 1991, Mugrauer et al., 1988). Next, we examined the phosphorylation of various mitogenic signaling proteins in the skin of K14-Dsg2 transgenic mice (Figure 1d). Remarkably, the K14-Dsg2 mice showed enhanced levels of phosphorylated EGFR, Src, c-Raf, AKT and PTEN. Furthermore, enhanced stabilization of activated β-catenin (Ctnnb1) and increased expression of the positive cell-cycle regulator Cyclin D1 (Ccnd1) was observed in K14-Dsg2 skin. The increased expression of some of these factors in K14-Dsg2 epidermis was confirmed by specifically harvesting intact epidermal fractions from juvenile mice, as the epidermis is several cell layers thick and permissible to epidermal-dermal separation in young mice (Figure 1e). Signaling cascades, such as those downstream of EGFR, are activated in the developing skin as they are required for epidermal morphogenesis in perinatal mice—i.e. keratinocyte differentiation and hair follicle development/cycling (Schneider et al., 2008, Sotiropoulou and Blanpain, 2012). Though juvenile epidermis is mitotically active and undergoing rapid alterations, overexpression of Dsg2 potentiated the activation of various signaling cascades, but was unable to drive hyperplasia, even in this context (Figures 1b, S1c). Thus the results so far support our previous findings that Dsg2 can enhance mitogenic signaling, but appear to be insufficient to promote cell proliferation in the basal epidermis.
Activated signaling in the K14-Dsg2 mice suggests that the skin is maintained in a ‘primed’ homeostatic state. A similar phenomenon was observed in the epidermis of caveolin-1 knockout mice which had no overt epidermal dysplasia, but were highly susceptible to DMBA-TPA-induced tumorigenesis (Capozza et al., 2003). As many of these pathways drive early tumor growth and are positively regulated by Dsg2, we next investigated tumor formation in K14-Dsg2 mice. No sporadic epidermal lesions were observed in the epidermis of older transgenic mice (up to 9 months of age). DMBA/TPA can drive the growth of mostly benign papillomas and, rarely, advanced squamous cell carcinomas after prolonged treatment (Filler et al., 2007). Wild-type and transgenic mice, following the DMBA/TPA treatment protocol, developed comparable hyperplasia in the dorsal epidermis and histopathologically similar tumors (Figure S2a, b). Both wild-type and transgenic mice developed an equal number of tumors larger than 2 mm (Figure S2c), which grew at the same rate (Figure S2d). These data illustrate that hyperactivation of signaling cascades in the skin of transgenic mice is insufficient to drive spontaneous and chemical carcinogenesis.
Dsg2 overexpression accelerates cutaneous wound healing
Aberrations in keratinocyte activation can dramatically disrupt proper wound healing (Barrientos et al., 2008). Therefore, we asked whether Dsg2 influences keratinocyte dynamics after wounding. Full-thickness wounds were inflicted on the dorsal skin of wild-type and K14-Dsg2 transgenic mice and allowed to heal for up to four days (Figure 2a). The rate of wound closure in the transgenic mice was enhanced compared to the wild-type littermates (Figure 2b, c). Histologically, both wild-type and transgenic mouse wounds appeared similar with a dense lymphocytic infiltrate after wounding and similar reorganization of the dermis in regenerating tissue (Figures 2b and S3a). Keratinocyte proliferation is an important aspect of cutaneous wound healing, generally occurring at a later stage of the healing process (Pastar et al., 2014). Wounds from the K14-Dsg2 mice had more actively proliferating keratinocytes (Figure 2d). K14 expression in keratinocytes flanking and re-epithelializing the wound bed is a feature of normal cutaneous wound healing while K6 is a marker for activated keratinocytes, also highly expressed in wound-adjacent keratinocytes (Freedberg et al., 2001, Schafer and Werner, 2008). K6 and K14 expression was observed 24 hours post-wounding in both wild-type and transgenic mice, but the expression of both keratins and Dsg2 itself was enhanced in the K14-Dsg2 wound-adjacent keratinocytes (Figure S3b-d). These data illustrate that basal overexpression of Dsg2 activates several pro-growth and cell division pathways that prime the skin for enhanced wound healing.
Figure 2. Dsg2 promotes cutaneous wound healing.
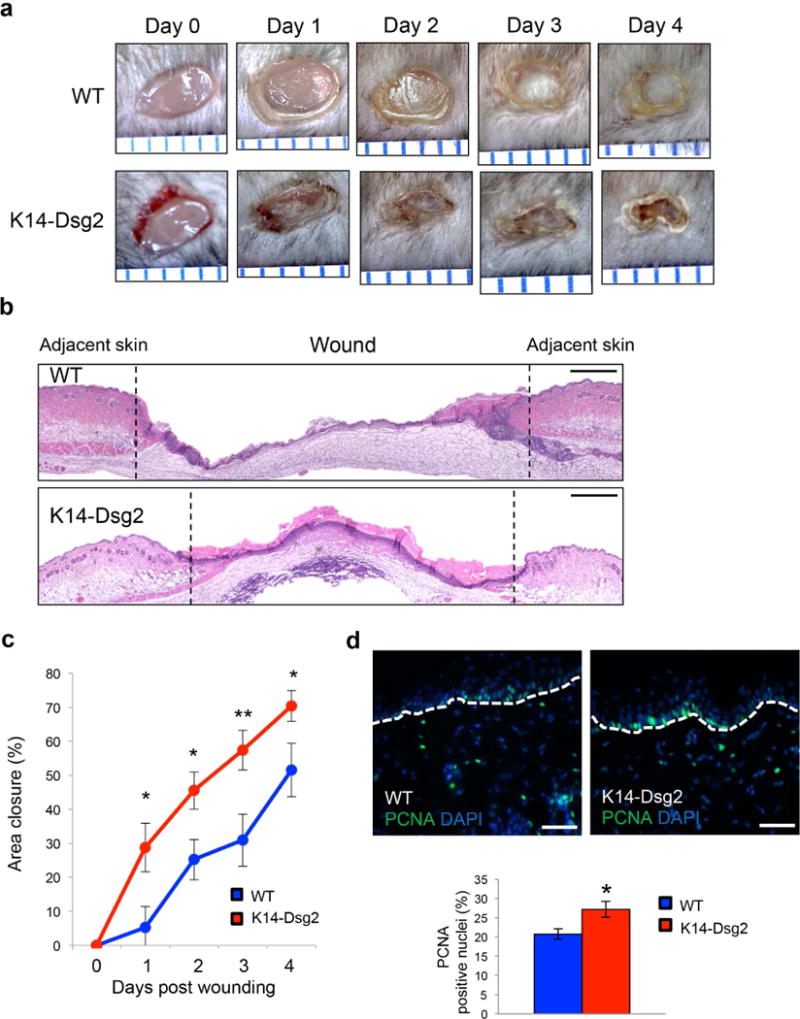
(a) Full-thickness wounds were inflicted on the dorsal skin of wild-type and K14-Dsg2 transgenic mice and photographed up to 4 days post-wounding. Space between blue ruler lines is 1 mm. (b) H&E staining of 4-day old wild-type and transgenic wounds. Dashed lines demarcate wound edge. Scale bar=100 μm (c) Wound area of non-epithelialized tissue was measured in ImageJ (mm2) and normalized to the area of fresh wounds. Data are expressed as average relative area of re-epithelialized tissue covering the initial wound (mm2) ± SEM. N=13 wild-type; 16 transgenic. (d) Representative immunofluorescence of wound-adjacent wild-type and transgenic mouse skin 4 days post wounding for PCNA (green) and DAPI (blue). Scale bars=50 μm.
Dsg2 alters secretion of wound healing-associated cytokines and chemokines
Keratinocytes are exposed to and release soluble cytokines, chemokines, and growth factors secreted into the wound milieu to promote their migration and proliferation (Landen et al., 2016, Reinke and Sorg, 2012). To determine if Dsg2 could modulate the secretome of keratinocytes, we analyzed the conditioned media from HaCaT keratinocytes with a secretome antibody array. Dsg2 overexpression in HaCaTs altered the keratinocyte secretome with increases in the secretion of interleukin 6 receptor (IL-6R), urokinase plasminogen activator receptor (uPAR), and tumor necrosis factor receptor (TNFRI) (Figures 3a, b, S4a, and Table 1), cytokines that are critical for modulating the rate of wound repair by fibroblasts and keratinocytes (Behm et al., 2012).
Figure 3. Cytokine profiling identifies uPAR as a key molecule upregulated in response to Dsg2.
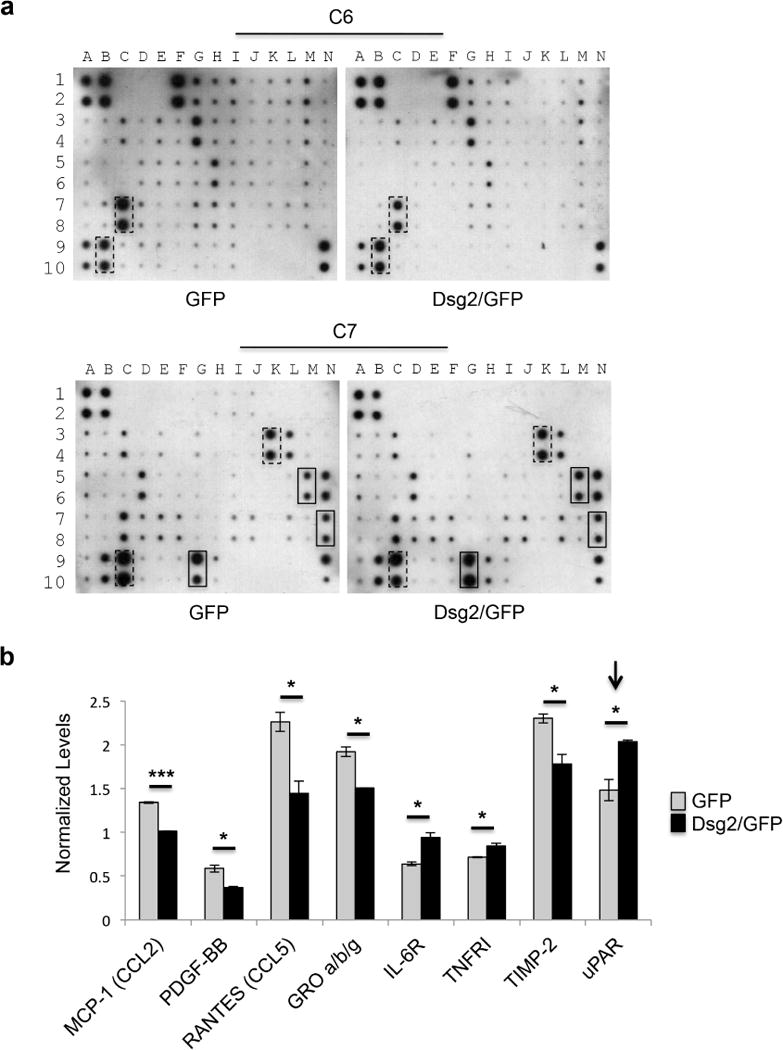
(a) Human Cytokine Antibody Arrays C6 and C7 were assayed with conditioned medium from HaCaT keratinocytes overexpressing GFP or Dsg2/GFP. Boxes indicate cytokines that showed statistically significant changes (dashed lines: downregulated; solid lines: upregulated). (b) Densitometry was performed of each cytokine by ImageJ software and histogram bars represent the relative intensity value of each cytokine secreted (white: HaCaT-GFP; black: HaCaT-Dsg2/GFP). Data are normalized to on-membrane positive control and depicted as relative abundancy in samples ± SEM. N=2.
Table 1.
Cytokines altered in keratinocytes in response to Dsg2 (HaCaT-Dsg2/GFP/HaCaT-GFP)
| Cytokines | Fold change | P-value |
|---|---|---|
| IL-6R | 1.48 | * |
| uPAR | 1.37 | * |
| TNFRI | 1.18 | * |
| TIMP-2 | −1.29 | * |
| GRO α/β/γ | −1.28 | * |
| MCP-1 (CCL2) | −1.32 | *** |
| RANTES (CCL5) | −1.57 | * |
| PDGF-BB | −1.59 | * |
HaCaT: spontaneously transformed immortal keratinocytes; Dsg2: desmoglein 2; GFP: green fluorescence protein; IL-6R: interleukin-6 receptor; uPAR: urokinase-type plasminogen activator receptor; TNFRI: tumor necrosis factor receptor I; TIMP-2: tissue inhibitor of metalloproteinase 2; GRO a/b/g: growth-related oncogene; MCP-1/CCL2: monocyte chemoattractant protein 1/C-C motif chemokine 2; RANTES: regulated on activation, normal T cell expressed and secreted; PDGF-BB: Platelet-derived growth factor subunit B.
P< 0.05,
P < 0.001
uPAR plays a central role in cell surface-associated plasminogen activation leading to degradation and remodeling of the extracellular matrix (Ellis et al., 1989, Stephens et al., 1989). Loss of uPAR function in knockout mice showed delayed wound-healing response, and decreased keratinocytes proliferation and migration (D’Alessio et al., 2008). uPAR is a highly glycosylated, GPI-anchored receptor with 3 extracellular domains (D1-3). Proteolytic cleavage can occur in the linker region between D1 and D2 to yield the uPARD2D3 fragment, which can undergo further cleavage of the GPI-linker releasing the soluble form uPARD2D3 (Sidenius et al., 2000). Immunoblotting of lysates and immunohistochemistry of skin from wild-type and K14-Dsg2 transgenic mouse epidermis showed a significant increase in glycosylated (~55-70 kDa) uPAR, soluble uPARD2D3 (~ 37 kDa) and full-length uPAR (~45 kDa) in response to Dsg2 overexpression (Figure 4a and b). HaCaTs overexpressing Dsg2 had enhanced expression of uPAR, and the dermis of transgenic mice had significant increases in the uPAR fragments (Figure S4b, c). Lysates from the day 1 wounds had increased uPAR expression and processing in the transgenic mice, corroborating uPAR immunostaining in wild-type and K14-Dsg2 transgenic wounds (Figure S5a, b). The source of dermal uPAR could be from soluble full-length uPAR or uPARD2D3 from the epidermis, although we cannot rule out the release of uPAR from peri-wound dermal fibroblasts.
Figure 4. Dsg2 enhances epidermal and dermal uPAR in wounded tissues.
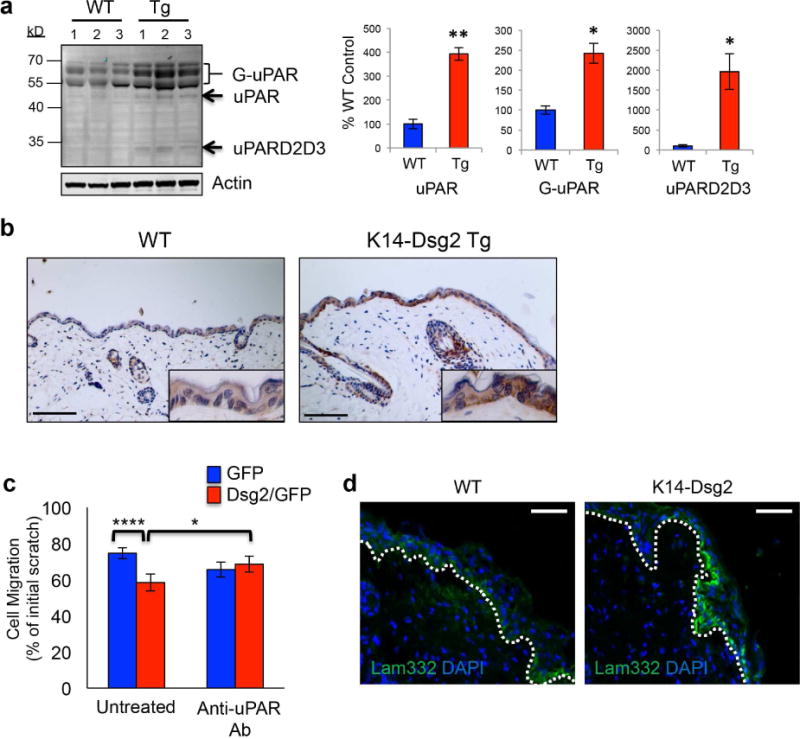
(a) Wild-type and K14-Dsg2 transgenic mouse epidermis (N=3) was immunoblotted for uPAR and actin. Signals were quantified, normalized to actin, and expressed as % of wild-type (right). G-uPAR: glycosylated uPAR; uPAR: non-glycosylated uPAR; uPARD2D3: D2D3 domains of uPAR. (b) Wild-type and K14-Dsg2 transgenic mice skins were immunostained for uPAR. (c) HaCaT+GFP or +Dsg2/GFP were treated with 1μg/mL anti-uPAR antibody and allowed to migrate for 24 h following scratch. Data are normalized to initial wound width of respective conditions and expressed as % distance remaining to be re-epithelialized. N=3. (d) Wild-type and K14-Dsg2 wounds (day 1) were immunostained for Laminin-332. Dashed line: regenerating basement membrane. Scale bars=50 μm.
Dsg2 enhances SCC cell migration in vitro in an EGFR- and c-Src-dependent fashion (Overmiller et al., 2016). HaCaTs stably overexpressing Dsg2 had similarly enhanced migration in an in vitro scratch assay, which was attenuated by blocking uPAR activity in the cells with an anti-uPAR antibody (Figure 4c). uPAR signaling induces downstream expression of laminin-332 in keratinocytes in an EGFR-dependent manner (D’Alessio et al., 2008). Laminin-332 is a critical structural component of the basement membrane in healthy skin and is secreted by keratinocytes to promote cellular migration in re-epithelializing tissue (Iorio et al., 2015). HaCaTs overexpressing Dsg2 had increased laminin-332 (Figure S4b). The basal expression of laminin-332 in healthy basement membranes was similar between wild-type and transgenic mice. Intriguingly, the transgenic wounds had more laminin-332 expression in the migrating keratinocytes and subsequently deposited in the regenerating basement membrane in a pattern similar to that seen in other mouse models of cutaneous wound repair (Figures 4d and S5c) (Jackow et al., 2016, Krampert et al., 2004, Veit et al., 2011). uPAR can be secreted through exosomes, cell-derived vesicles of approximately 30-150 nm (Jung et al., 2009, Mu et al., 2013). We have previously shown Dsg2 potentiates the secretion of exosomes from both HaCaTs and malignant SCC cells (Overmiller et al., 2017). Indeed, both HaCaT+GFP and +Dsg2/GFP cells secreted exosomes equally enriched in a ~70kDa fraction of uPAR, likely corresponding to the full-length, highly glycosylated protein (Figure S5d). These results suggest that Dsg2 may facilitate wound repair through uPAR.
DISCUSSION
Differentiation-dependent expression of various desmosomal cadherins in the epidermis may be an important factor for maintaining strata-specific cell-cell adhesion (Delva et al., 2009). However disruption of this expression pattern expression has revealed additional biological functions. For example, forced expression of Dsg2 in the suprabasal epidermis of the Inv-Dsg2 mice altered cell signaling and promoted cell growth and proliferation (Brennan et al., 2007). Interestingly, suprabasal expression of Dsg3 also induced epidermal hyperplasia and abnormal differentiation (Elias et al., 2001, Merritt et al., 2002). In this report, overexpression of Dsg2 in the basal cell compartment of the epidermis was unable to disrupt tissue homeostasis.
Various mitogenic signaling pathways were significantly activated in the skin of K14-Dsg2 transgenic mice, including ERK1/2, AKT, and β-catenin. These pathways, when abnormally activated in the skin, typically drive epidermal hyperplasia and susceptibility to cutaneous malignancy (Dhillon et al., 2007, Hafner et al., 2010, Sherwood and Leigh, 2016). Furthermore, PTEN inactivation is a hallmark of various cancer types and is mutated in multiple hamartoma syndrome (Ming and He, 2009). PTEN phosphorylation is associated with cytoplasmic sequestration and inhibition of its negative regulator role in the PI3K/AKT pathway (Das et al., 2003). Though basal expression of Cyclin D1 was increased in our transgenic mice, knockout studies demonstrated that cyclin D1 alone is not sufficient to activate cell replication (Robles et al., 1998). Similarly, while oncogenes such as STAT3 or c-Src play a critical role during cancer development, their role in the epidermis during normal homeostasis is poorly defined. Interestingly, c-Myc expression was unaltered in the skin of K14-Dsg2 mice, in contrast to Inv-Dsg2 mice (Brennan et al., 2007). Suprabasal overexpression of c-myc in mice resulted in hyperplasia while basal overexpression led to increased keratinocyte proliferation, but attenuated migration during wound healing, due to β1 integrin transcriptional repression (Waikel et al., 2001, Waikel et al., 1999). It is possible that c-Myc may be the important driver of Dsg2-induced epidermal hyperplasia that is not upregulated in K14-Dsg2 skin (Figure 5). Inducible knockout of c-Myc from basal keratinocytes delayed cutaneous wound healing and depleted the population of epidermal stem cells in a time-dependent manner (Zanet et al., 2005). Though overexpression of Dsg2 did not promote hyperplasia or tumor growth in response to DMBA/TPA treatment, Dsg2 in the basal keratinocytes accelerated cutaneous wound healing. Healing and regeneration in skin is an well-orchestrated event requiring keratinocyte proliferation and migration to completely cover and regenerate the wounded skin, with molecular signals such as cytokines and chemokines playing an essential role for restoration of tissue integrity (Gurtner et al., 2008). Though keratin patterning in the healthy epidermis is typified by basal keratins K5/K14 and differentiation keratins K1/K10, wound-adjacent keratinocytes express high levels of the activation marker K6 (Freedberg et al., 2001). The K14-Dsg2 transgenic mice had enhanced expression of K6 following cutaneous injury suggesting that the cells, indolent in healthy skin, are indeed primed for activation and healing.
Figure 5. Dsg2 potentiates the wound healing response.
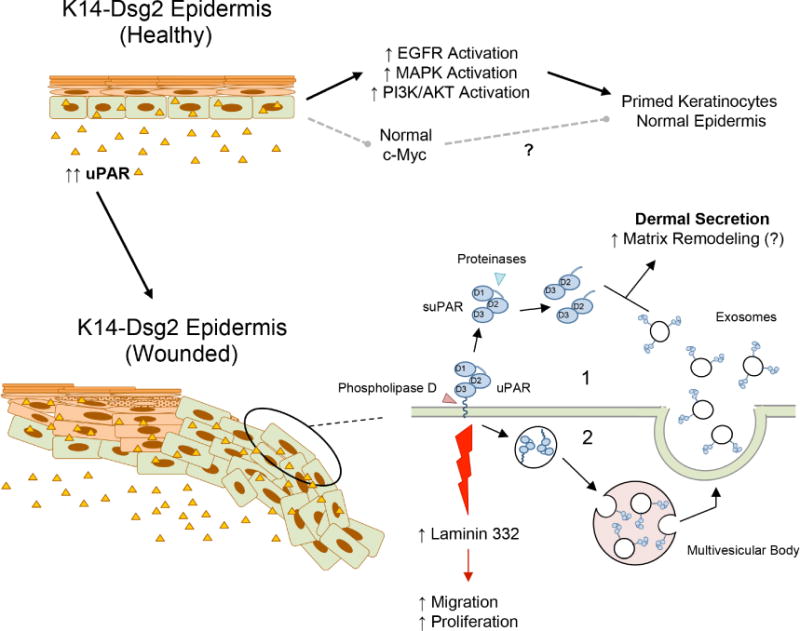
(Top) Basal overexpression of Dsg2 drives activation of various signaling cascade, but not overexpression of c-Myc, presumably leaving the epidermis primed, but unperturbed. Additionally, there is increased epidermal and dermal uPAR expression. (Bottom) During wound healing, uPAR can be shed and processed from the keratinocytes membranes as soluble uPAR (suPAR) and the D2D3 fragment, respectively. uPAR can also be shed via exosomes after shuttling through the endosomal sorting pathway; both mechanisms increase dermal uPAR concentration and promote matrix remodeling and wound resolution. Through uPAR-mediated signaling, Dsg2 potentiates expression of pro-migratory laminin-332 in the regenerating basement membrane.
Keratinocytes have long been recognized as a major source of growth factors and cytokines that are critical during wound healing, including uPA and uPAR (Werner and Grose, 2003). There was increased uPAR expression in the epidermis and secretion into the dermis of the K14-Dsg2 mice as well as the actively proliferating and migrating keratinocytes adjacent to the wounded tissue. Overexpression of uPAR constitutively activates EGFR and c-Src to promote cell motility through α5β1 integrin, while loss of uPAR attenuates EGFR-dependent laminin-332 secretion without affecting integrin expression or signaling (D’Alessio et al., 2008, Tarui et al., 2003). While EGFR and Src are wholly capable of promoting hyperproliferation and motility in keratinocytes independently, synergy with uPAR-related signaling could further stimulate this signaling cascade. Dsg2 enhances the release of uPAR into the dermis and wound area by two potential mechanisms (Figure 5). Phospholipase D (PLD), which is activated in wounded keratinocytes, cleaves the GPI anchor to release soluble uPAR (Arun et al., 2013). Proteinases further cleave within the linker region between domains D1 and D2. uPAR can also undergo endocytosis into endosomes and multivesicular bodies and, when fused with the plasma membrane, release uPAR-loaded exosomes. Though Dsg2 does not enhance the sorting of uPAR to exosomes, we have previously shown that both malignant and non-malignant keratinocytes overexpressing Dsg2 release more exosomes per cell (Overmiller et al., 2017). The cleaved uPAR in the extracellular milieu potentially promotes additional fibroblast activation and matrix remodeling in the healing dermis.
Loss of uPAR in knockout mice disrupted wound repair by reducing laminin-332 expression in an EGFR-dependent manner (D’Alessio et al., 2008). We observed increased laminin-332 in the epithelial tongue of the healing wounds of transgenic mice. In the healthy epidermis, both EGFR and Src were hyperactivated in the transgenic mice, corroborating our previous in vitro findings (Overmiller et al., 2016). We postulate that uPAR may enhance cell growth and migration through its interaction with integrins and associated downstream signaling pathways including EGFR and c-Src (Ghosh et al., 2000, Xue et al., 1997). Indeed, these results suggest that EGFR/c-Src act in conjunction or synergize with the uPAR signaling axis to promote enhanced wound healing in our transgenic mouse model. c-Src itself is an upstream regulator of uPAR transcription, which may be an important intermediary for upregulating uPAR expression in the K14-Dsg2 mouse keratinocytes (Okan et al., 2001). In summary, these data demonstrate a broad effect of Dsg2 in priming basal keratinocytes mitogenic and pro-migratory cellular signaling following trauma, at least in part by increased secretion of a panel of soluble factors that can enhance the repair program.
MATERIALS AND METHODS
Details of protocols and reagents utilized can be found in the Supplementary Materials and Methods online.
Ethics statement on animal research
Animal experiments were facilitated within the guidelines provided within the Guide for the Care and Use of Laboratory Animals of the NIH and in accordance with Laboratory Animal Services at TJU. The animal usage protocol was approved by IACUC at TJU after review by the staff veterinarian.
Generating transgenic mice and genotyping
The Dsg2.Flag cDNA was subcloned into a targeting construct containing ~2,300 bp of the human cytokeratin KRT14 promoter and a polyadenylation tail (kind gift from Dr. Elaine Fuchs, Rockefeller University) (Brennan et al., 2007). Founder mice were backcrossed at least 5 generations to the C57BL/6J inbred strain background for further analysis.
Immunofluorescence and immunohistochemistry
Sections (4 μm) of OCT (Tissue-Tek; Fisher) frozen tissues were immunostained for Flag, pEGFR (Tyr845), pSrc (Tyr416), Laminin-332, and Dsg2 and FFPE tissues for PCNA, K14, K6, and uPAR (Brennan et al., 2007).
Keratinocyte proliferation
Proliferation was assayed by immunostaining mouse skin for PCNA and DAPI. The number of PCNA+ cells was counted in at least 3 random high-powered (40x) fields or a single, encompassing medium-powered (20x) field for the normal and wounded tissue, respectively.
DMBA-TPA-induced skin carcinogenesis
Wild-type and K14-Dsg2 transgenic mice (6-8 weeks; male and female) were treated with DMBA (7,12-dimethylbenz[a]antracene) and biweekly applications of TPA (12-O-tetradecanoylphorbol 13-acetate) to promote skin tumor growth (Brennan et al., 2007, Filler et al., 2007). Tumors were measured biweekly using digital calipers.
Full-thickness skin wounding
Punch biopsies (4 mm) were used to generate wounds in wild-type and K14-Dsg2 transgenic mice (6-8 weeks; male and female) (Wang et al., 2013). Wounds were photographed daily by digital microscope (Plugable USB 2.0 Digital Microscope).
Cytokine antibody array
Cytokine profiles of conditioned media (48 h) from HaCaT-GFP and HaCaT-Dsg2/GFP cells were analyzed using a Human Cytokine Antibody Array C1000 (#AAH-CYT-1000-2; Raybiotech, Norcross, GA). Quantification of dot intensity was performed in ImageJ following background subtraction, and the raw data normalized to the array-specific positive control dots.
Scratch Assay
Confluent HaCaT+GFP and +Dsg2/GFP sheets were scratched with a P200 pipet tip and incubated 24 hr in DMEM ± 1 μg/mL anti-uPAR antibody. Images were taken immediately post-scratch and 24 hr later; data expressed as % of initial width between epithelial sheets remaining.
Exosome isolation
Exosomes were purified from conditioned medium of HaCaT keratinocytes by sequential ultracentrifugation (Greening et al., 2015, Overmiller et al., 2017). Medium was centrifuged at 300 × g, 2,000 × g, and 10,000 × g to remove live and dead cells, and microvesicles. Supernatant was centrifuged at 110,000 × g (Beckman 45Ti) to pellet exosomes.
Statistical analysis
Data are expressed average value ± SEM. Student’s t test, unless otherwise noted in Supplemental Materials and Methods * p<0.05; ** p<0.01; *** p<0.001.
Supplementary Material
Acknowledgments
Research reported here was supported by the NIH (Mahoney, AR056067), the Sidney Kimmel Cancer Center (Mahoney), and the Thomas Jefferson University Dean’s Transformational Science Award (Mahoney).
Abbreviations
- Dsg2
Desmoglein 2
- SCC
Squamous cell carcinomas
- MMP
Matrix metalloproteinase
- ADAM
A disintegrin and metalloproteinase
- EGFR
Epidermal growth factor receptor
- FFPE
Formalin-fixed paraffin-embedded
- DMBA
7,12-dimethylbenz[a]anthracene
- TPA
12-O-tetradecanoylphorbol-13-acetate
- H&E
Haematoxylin and Eosin
- uPAR
Urokinase-type plasminogen activator receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ORCID
My Mahoney: 0000-0003-0203-9171
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
AUTHOR CONTRIBUTIONS
FC, AMO, DMB, MGM were involved in the study design and concept. LDS, JKW, and MGM provided reagents. FC, AMO, AL, DMB, KPM, MRM, LDS and MGM were involved in data acquisition. FC, AMO, DMB, KPM, MRM, NAR-D, JKW and MGM were involved in data analysis and interpretation. AMO, FC and MGM drafted the manuscript. FC, AMO, DMB, NAR-D, LDS, JKW, and MGM edited the manuscript.
References
- Aomatsu K, Arao T, Abe K, Kodama A, Sugioka K, Matsumoto K, et al. Slug is upregulated during wound healing and regulates cellular phenotypes in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2012;53(2):751–6. doi: 10.1167/iovs.11-8222. [DOI] [PubMed] [Google Scholar]
- Arun SN, Xie D, Howard AC, Zhong Q, Zhong X, McNeil PL, et al. Cell wounding activates phospholipase D in primary mouse keratinocytes. J Lipid Res. 2013;54(3):581–91. doi: 10.1194/jlr.M027060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am j hum genet. 2006;79(1):136–42. doi: 10.1086/504393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- Behm B, Babilas P, Landthaler M, Schreml S. Cytokines, chemokines and growth factors in wound healing. J Eur Acad Dermatol Venereol. 2012;26(7):812–20. doi: 10.1111/j.1468-3083.2011.04415.x. [DOI] [PubMed] [Google Scholar]
- Brennan-Crispi DM, Hossain C, Sahu J, Brady M, Riobo NA, Mahoney MG. Crosstalk between Desmoglein 2 and Patched 1 accelerates chemical-induced skin tumorigenesis. Oncotarget. 2015;6(11):8593–605. doi: 10.18632/oncotarget.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan D, Hu Y, Joubeh S, Choi YW, Whitaker-Menezes D, O’Brien T, et al. Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J Cell Science. 2007;120(5):758–71. doi: 10.1242/jcs.03392. [DOI] [PubMed] [Google Scholar]
- Brennan D, Mahoney MG. Increased expression of Dsg2 in malignant skin carcinomas: A tissue-microarray based study. Cell Adh Migr. 2009;3(2):148–54. doi: 10.4161/cam.3.2.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan D, Peltonen S, Dowling A, Medhat W, Green KJ, Wahl JK, et al. A role for caveolin-1 in desmoglein binding and desmosome dynamics. Oncogene. 2012;31(13):1636–48. doi: 10.1038/onc.2011.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, et al. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am J Pathol. 2003;162(6):2029–39. doi: 10.1016/S0002-9440(10)64335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio S, Gerasi L, Blasi F. uPAR-deficient mouse keratinocytes fail to produce EGFR-dependent laminin-5, affecting migration in vivo and in vitro. J Cell Sci. 2008;121(Pt 23):3922–32. doi: 10.1242/jcs.037549. [DOI] [PubMed] [Google Scholar]
- Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A. 2003;100(13):7491–6. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delva E, Tucker DK, Kowalczyk AP. The desmosome. Cold Spring Harb Perspect Biol. 2009;1(2):a002543. doi: 10.1101/cshperspect.a002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Dhouailly D, Sawyer RH. Avian scale development. XI. Initial appearance of the dermal defect in scaleless skin. Dev Biol. 1984;105(2):343–50. doi: 10.1016/0012-1606(84)90291-4. [DOI] [PubMed] [Google Scholar]
- Elias PM, Matsuyoshi N, Wu H, Lin C, Wang ZH, Brown BE, et al. Desmoglein isoform distribution affects stratum corneum structure and function. J Cell Biol. 2001;153(2):243–9. doi: 10.1083/jcb.153.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis V, Scully MF, Kakkar VV. Plasminogen activation initiated by single-chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J Biol Chem. 1989;264(4):2185–8. [PubMed] [Google Scholar]
- Eshkind L, Tian Q, Schmidt A, Franke WW, Windoffer R, Leube RE. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur J Cell Biol. 2002;81:592–8. doi: 10.1078/0171-9335-00278. [DOI] [PubMed] [Google Scholar]
- Filler RB, Roberts SJ, Girardi M. Cutaneous two-stage chemical carcinogenesis. CSH Protoc. 2007;2007 doi: 10.1101/pdb.prot4837. pdb prot4837. [DOI] [PubMed] [Google Scholar]
- Freedberg IM, Tomic-Canic M, Komine M, Blumenberg M. Keratins and the keratinocyte activation cycle. J Invest Dermatol. 2001;116(5):633–40. doi: 10.1046/j.1523-1747.2001.01327.x. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Brown R, Jones JC, Ellerbroek SM, Stack MS. Urinary-type plasminogen activator (uPA) expression and uPA receptor localization are regulated by alpha 3beta 1 integrin in oral keratinocytes. J Biol Chem. 2000;275(31):23869–76. doi: 10.1074/jbc.M000935200. [DOI] [PubMed] [Google Scholar]
- Greening DW, Xu R, Ji H, Tauro BJ, Simpson RJ. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol Biol. 2015;1295:179–209. doi: 10.1007/978-1-4939-2550-6_15. [DOI] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–21. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Hafner C, Landthaler M, Vogt T. Activation of the PI3K/AKT signalling pathway in non-melanoma skin cancer is not mediated by oncogenic PIK3CA and AKT1 hotspot mutations. Exp Dermatol. 2010;19(8):e222–7. doi: 10.1111/j.1600-0625.2009.01056.x. [DOI] [PubMed] [Google Scholar]
- Hirning U, Schmid P, Schulz WA, Rettenberger G, Hameister H. A comparative analysis of N-myc and c-myc expression and cellular proliferation in mouse organogenesis. Mech Dev. 1991;33(2):119–25. doi: 10.1016/0925-4773(91)90078-k. [DOI] [PubMed] [Google Scholar]
- Iorio V, Troughton LD, Hamill KJ. Laminins: Roles and Utility in Wound Repair. Adv Wound Care (New Rochelle) 2015;4(4):250–63. doi: 10.1089/wound.2014.0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackow J, Loffek S, Nystrom A, Bruckner-Tuderman L, Franzke CW. Collagen XVII Shedding Suppresses Re-Epithelialization by Directing Keratinocyte Migration and Dampening mTOR Signaling. J Invest Dermatol. 2016;136(5):1031–41. doi: 10.1016/j.jid.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Jung T, Castellana D, Klingbeil P, Cuesta Hernandez I, Vitacolonna M, Orlicky DJ, et al. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia. 2009;11(10):1093–105. doi: 10.1593/neo.09822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klessner JL, Desai BV, Amargo EV, Getsios S, Green KJ. EGFR and ADAMs cooperate to regulate shedding and endocytic trafficking of the desmosomal cadherin desmoglein 2. Mol Biol Cell. 2009;20(1):328–37. doi: 10.1091/mbc.E08-04-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk AP, Green KJ. Structure, function, and regulation of desmosomes. Prog Mol Biol Transl Sci. 2013;116:95–118. doi: 10.1016/B978-0-12-394311-8.00005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampert M, Bloch W, Sasaki T, Bugnon P, Rulicke T, Wolf E, et al. Activities of the matrix metalloproteinase stromelysin-2 (MMP-10) in matrix degradation and keratinocyte organization in wounded skin. Mol Biol Cell. 2004;15(12):5242–54. doi: 10.1091/mbc.E04-02-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen NX, Li D, Stahle M. Transition from inflammation to proliferation: a critical step during wound healing. Cell Mol Life Sci. 2016;73(20):3861–85. doi: 10.1007/s00018-016-2268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437(7056):275–80. doi: 10.1038/nature03922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan K, Coulombe P. In: The wound repair-associated keratins 6, 16, and 17 Insights into the role of intermediate filaments in specifying keratinocyte cytoarchitecture. Herrmann H, Harris JR, editors. New York: Plenum Press; 1998. pp. 173–204. [PubMed] [Google Scholar]
- Merritt AJ, Berika MY, Zhai W, Kirk SE, Ji B, Hardman MJ, et al. Suprabasal desmoglein 3 expression in the epidermis of transgenic mice results in hyperproliferation and abnormal differentiation. Mol Cell Biol. 2002;22(16):5846–58. doi: 10.1128/MCB.22.16.5846-5858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming M, He YY. PTEN: new insights into its regulation and function in skin cancer. J Invest Dermatol. 2009;129(9):2109–12. doi: 10.1038/jid.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu W, Rana S, Zoller M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia. 2013;15(8):875–87. doi: 10.1593/neo.13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugrauer G, Alt FW, Ekblom P. N-myc proto-oncogene expression during organogenesis in the developing mouse as revealed by in situ hybridization. J Cell Biol. 1988;107(4):1325–35. doi: 10.1083/jcb.107.4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okan E, Drewett V, Shaw PE, Jones P. The small-GTPase RalA activates transcription of the urokinase plasminogen activator receptor (uPAR) gene via an AP1-dependent mechanism. Oncogene. 2001;20(15):1816–24. doi: 10.1038/sj.onc.1204260. [DOI] [PubMed] [Google Scholar]
- Overmiller AM, McGuinn KP, Roberts BJ, Cooper F, Brennan-Crispi DM, Deguchi T, et al. c-Src/Cav1-dependent activation of the EGFR by Dsg2. Oncotarget. 2016;7(25):37536–55. doi: 10.18632/oncotarget.7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmiller AM, Pierluissi JA, Wermuth PJ, Sauma S, Martinez-Outschoorn U, Tuluc M, et al. Desmoglein 2 modulates extracellular vesicle release from squamous cell carcinoma keratinocytes. FASEB J. 2017;31(8):3412–24. doi: 10.1096/fj.201601138RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG, Sawaya A, et al. Epithelialization in Wound Healing: A Comprehensive Review. Adv Wound Care (New Rochelle) 2014;3(7):445–64. doi: 10.1089/wound.2013.0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113(9):1171–9. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. 2012;49(1):35–43. doi: 10.1159/000339613. [DOI] [PubMed] [Google Scholar]
- Robles AI, Rodriguez-Puebla ML, Glick AB, Trempus C, Hansen L, Sicinski P, et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12(16):2469–74. doi: 10.1101/gad.12.16.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshan A, Murai K, Fowler J, Simons BD, Nikolaidou-Neokosmidou V, Jones PH. Human keratinocytes have two interconvertible modes of proliferation. Nat Cell Biol. 2016;18(2):145–56. doi: 10.1038/ncb3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9(8):628–38. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- Schäfer S, Koch PJ, Franke WW. Identification of the ubiquitous human desmoglein, Dsg2, and the expression catalogue of the desmoglein subfamily of desmosomal cadherins. Exp Cell Res. 1994;211:391–9. doi: 10.1006/excr.1994.1103. [DOI] [PubMed] [Google Scholar]
- Schneider MR, Werner S, Paus R, Wolf E. Beyond wavy hairs: the epidermal growth factor receptor and its ligands in skin biology and pathology. Am J Pathol. 2008;173(1):14–24. doi: 10.2353/ajpath.2008.070942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood V, Leigh IM. WNT Signaling in Cutaneous Squamous Cell Carcinoma: A Future Treatment Strategy? J Invest Dermatol. 2016;136(9):1760–7. doi: 10.1016/j.jid.2016.05.108. [DOI] [PubMed] [Google Scholar]
- Sidenius N, Sier CF, Blasi F. Shedding and cleavage of the urokinase receptor (uPAR): identification and characterisation of uPAR fragments in vitro and in vivo. FEBS Lett. 2000;475(1):52–6. doi: 10.1016/s0014-5793(00)01624-0. [DOI] [PubMed] [Google Scholar]
- Sotiropoulou PA, Blanpain C. Development and homeostasis of the skin epidermis. Cold Spring Harb Perspect Biol. 2012;4(7):a008383. doi: 10.1101/cshperspect.a008383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens RW, Pollanen J, Tapiovaara H, Leung KC, Sim PS, Salonen EM, et al. Activation of pro-urokinase and plasminogen on human sarcoma cells: a proteolytic system with surface-bound reactants. J Cell Biol. 1989;108(5):1987–95. doi: 10.1083/jcb.108.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojadinovic O, Pastar I, Vukelic S, Mahoney MG, Brennan D, Krzyzanowska A, et al. Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. J Cell Mol Med. 2008;12(6B):2675–90. doi: 10.1111/j.1582-4934.2008.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarui T, Andronicos N, Czekay RP, Mazar AP, Bdeir K, Parry GC, et al. Critical role of integrin alpha 5 beta 1 in urokinase (uPA)/urokinase receptor (uPAR, CD87) signaling. J Biol Chem. 2003;278(32):29863–72. doi: 10.1074/jbc.M304694200. [DOI] [PubMed] [Google Scholar]
- Veit G, Zwolanek D, Eckes B, Niland S, Kapyla J, Zweers MC, et al. Collagen XXIII, novel ligand for integrin alpha2beta1 in the epidermis. J Biol Chem. 2011;286(31):27804–13. doi: 10.1074/jbc.M111.220046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet. 2001;28(2):165–8. doi: 10.1038/88889. [DOI] [PubMed] [Google Scholar]
- Waikel RL, Wang XJ, Roop DR. Targeted expression of c-Myc in the epidermis alters normal proliferation, differentiation and UV-B induced apoptosis. Oncogene. 1999;18:4870–8. doi: 10.1038/sj.onc.1203040. [DOI] [PubMed] [Google Scholar]
- Wang X, Ge J, Tredget EE, Wu Y. The mouse excisional wound splinting model, including applications for stem cell transplantation. Nat Protoc. 2013;8(2):302–9. doi: 10.1038/nprot.2013.002. [DOI] [PubMed] [Google Scholar]
- Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83(3):835–70. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- Xue W, Mizukami I, Todd RF, 3rd, Petty HR. Urokinase-type plasminogen activator receptors associate with beta1 and beta3 integrins of fibrosarcoma cells: dependence on extracellular matrix components. Cancer Res. 1997;57(9):1682–9. [PubMed] [Google Scholar]
- Zanet J, Pibre S, Jacquet C, Ramirez A, de Alboran IM, Gandarillas A. Endogenous Myc controls mammalian epidermal cell size, hyperproliferation, endoreplication and stem cell amplification. J Cell Sci. 2005;118(8):1693–704. doi: 10.1242/jcs.02298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.