

Abstract
Cell line-derived xenografts (CDX) or patient-derived xenografts (PDX), in immune deficient mice, have revolutionized our understanding of normal and malignant human hematopoiesis. Transgenic approaches further improved in vivo hematological research, allowing the development of human cytokine producing mice, which show superior human cell engraftment. The most popular mouse strains used in research, the NOG (NOD.Cg-Prkdcscid Il2rγtm1Sug/Jic), and the NSG (NOD/SCID-IL2Rγ−/−, NOD.Cg-PrkdcscidIl2rγtm1Wjl/SzJ) mouse and their human cytokine-producing (IL-3, GM-CSF, and SCF) counterparts (huNOG and NSGS), rely partly on a mutation in the DNA repair protein PRKDC causing a severe combined immune deficiency phenotype rendering the mice less tolerant to DNA damaging therapeutics, thereby limiting their usefulness in the investigation of novel AML therapeutics. NRG (NOD/RAG1/2−/−IL2Rγ−/−) mice show equivalent immune ablation through a defective recombination activation gene (RAG) leaving DNA damage repair intact and human cytokine producing NRGS mice were generated, improving myeloid engraftment. Our findings indicate that unconditioned NRG and NRGS mice can harbor established AML CDX, and can tolerate aggressive induction chemotherapy at higher doses than NSG mice without overt toxicity. However, unconditioned NRGS mice developed less clinically relevant disease with CDXs forming solid tumors throughout the body, while unconditioned NRG mice were incapable of efficiently supporting PDX or human hematopoietic stem cell engraftment. These findings emphasize the contextually dependent utility of each of these powerful new strains in the study of normal and malignant human hematopoiesis. Thus, the choice of mouse strain cannot be random, but must be based on the experimental outcomes and questions to be addressed.
INTRODUCTION
Immune deficient mice have revolutionized biomedical research, including the study of both normal human hematopoiesis and leukemogenesis.[1–3] Capable of harboring both normal and malignant human xenografts without rejection, highly immune deficient mice are indispensable in hematological research allowing differentiation and proliferation of these cells in vivo.[4] Xenograft mouse models consistently better predict the success of experimental chemotherapeutics in clinical trials[5], likely due to the complex and dynamic interaction between the bone marrow (BM) microenvironment and heterogeneous populations of leukemic cells.[6] Seminal xenograft studies showed that only the most primitive fraction of patient leukemic cells (Lin-,CD34+), but not mature blasts, were capable of transferring disease to primary and secondary immune deficient mice.[7, 8] These cells, dubbed leukemic stem cells (LSCs), exist at the top of a hierarchy similar to normal hematopoiesis, in which a small population of slowly proliferating, self-renewing LSCs gives rise to a large population of clonally expanded blasts.[8] Furthermore, current evidence supports the notion that specific chemotherapeutic protected niches exist within the BM microenvironment providing sanctuary for LSCs, eventually causing lethal refractory relapse.[9, 10] Thus, it becomes clear that xenograft models are the most reliable preclinical method to model the highly complex interplay between leukemic populations, BM microenvironment, clinical treatment outcomes, and by extension pharmacological efficacy of novel investigational therapeutics.[11–13]
Discovered in 1962, nude mice were the first immune deficient mice capable of accepting human cells and tissues, and since then there has been continued effort to further increase immune ablation and improve patient sample engraftment.[3, 14] Nude mice were followed by severe combined immune deficiency (SCID) mice[15], which lack the ability to mount and maintain adaptive immune responses due to a recessive mutation resulting in loss of enzyme DNA-dependent protein kinase catalytic subunit (Prkdc) activity, an enzyme critically important for DNA damage repair. This enzyme is required for V(D)J recombination, and thus both T and B lymphocyte maturation is blocked.[16, 17] Further immune ablation was achieved by cross breeding with non-obese diabetic (NOD) mice[18] and backcrossing to introduce a mutated IL-2rγ resulting in two of the most commonly used strains in hematopoietic research, NOG and NSG (NOD/SCID IL-2Rγnull).[19] These mice in addition to lacking T and B lymphocytes also lack natural killer cells, exhibit decreased macrophage function, and have impaired eosinophil function thereby allowing for efficient engraftment of human tissues.[20–22] Despite these advances in murine leukemic xenograft models, most samples of acute myeloid leukemia (AML) fail to sufficiently engraft, likely due to several factors including: 1.) Homing to BM, 2.) Lack of stromal support, 3.) AML sample intrinsic factors, 4.) Absence of human cytokines, and 5.) Residual innate mouse immunity.[23]
Transgenic approaches have allowed researchers to develop several iterations of NOD/SCID IL-2Rynull mice (NSGS, NSS, etc.) expressing several human myeloid cytokines (IL-3, GM-CSF, and SCF), dramatically increasing engraftment of human AML.[20, 21, 24, 25] Human cytokine expression supports myelopoiesis and engraftment of a variety of patient leukemia, but also the engraftment and multilineage expansion of mature human hematopoietic cells from CD34+ hematopoietic stem cells (HSCs).[26, 27] While these advancements are indeed significant, SCID mediated immune deficiency makes the mice much less tolerant to SOC DNA damaging AML therapeutics like anthracyclines and cytarabine.[5, 23] Thus, it is difficult to model xenograft response to standard of care (SOC) therapy, a necessary component of a truly physiologically and clinically relevant preclinical model. In 2003 Shultz et. Al., developed another strain of mice called NRG (NOD/Rag1/2null), whose lymphocyte developmental phenotype is maintained through a defective recombination activation gene (RAG), leaving DNA damage repair unimpaired, with comparable immune ablation.[28]
Recently in 2014, NRG mice were crossed with NSGS mice (NSG-hIL3, h-GM-CSF, hSCF) and the SCID mutation removed through selective crossbreeding with NRG mice.[29] This new strain of mice called NRGS (NRG-SGM3) have a comparable degree of immune ablation compared to SCID mice, express human myeloid cytokines, and can likely tolerate higher doses of SOC therapy in a dosing regimen more similar to clinical practice.[23] These mice show consistent highly efficient engraftment, model the influence of the BM microenvironment, human cytokine signaling, and therapeutic outcomes. Given the recent development of these strains and their relative lack of use in AML research, the goal of this study was to provide an in-depth analysis of the comparative utility of unconditioned NRG and NRGS mice in the investigation of: 1.) Cell line-derived xenografts (CDX), 2.) Patient-derived xenografts (PDX), 3.) Response to SOC chemotherapeutics, 4.) Humanization potential.
MATERIALS AND METHODS
Cell Culture and Patient Samples
Established human AML cell lines K562, MV411, and U937 were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in RPMI medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C with 5% CO2. The patient derived xenograft models (CCHMC-23, CCHMC-7, CCHMC-9, and CCHMC-35), were established at Cincinnati Children’s Hospital Medical Center from specimens acquired under an IRB-approved protocol (#2008–0021). These primary specimens were established and expanded in NSGS mice at CCHMC and BM aspirates obtained from leukemic mice were frozen at −80°C in RPMI with 10% FBS and 10% DMSO until xenograft. Primary AML blasts were isolated from de-identified diluted whole marrow aspirate acquired from JGBCC biorepository with informed consent and under IRB guidelines (IRB#13.0188), by Histopaque gradient and centrifugation at 500 × g for 20 minutes at room temperature. Human CD34+ hematopoietic stem cells purified from umbilical cord blood were obtained from AllCells, Alameda, California, USA).
In vivo studies
NRG (NOD/RAG1/2−/−IL2Rγ−/−, stock no: 007799) and NRGS (NOD/RAG1/2−/−IL2Rγ−/−Tg[CMV-IL3,CSF2,KITLG]1Eav/J, stock no: 024099) mice producing 24 ng/ml of human IL-3, GM-CSF, and SCF[28] were obtained from Jackson laboratories, and bred and maintained under standard conditions in the University of Louisville Rodent Research Facility (RRF) on a 12-hour light/12-hour dark cycle with food and water provided ad libitum. Animal procedures were approved by the Institutional Animal Care and Use Committee. For all cell line derived xenograft studies and the human HSC study mice received 5 × 10^5 cells, and for all passaged patient derived xenograft studies 1.25 × 10^5 human cells suspended in 200μl PBS per mouse by bolus intravenous injection. Similarly, mice engrafted with the primary patient sample JGB-AML1 received 2 × 10^6 cells in 200 μl per mouse by intravenous bolus. Mouse health was monitored for characteristic signs of leukemia like scruffiness, and hind-limb paralysis, at which time mice were euthanized. Analysis of human cell engraftment was conducted on a case-by-case basis when mice became moribund and humane endpoints were reached.
5+3 Induction Chemotherapy Regimen
Using previously published data we established a similar 5+3 induction therapy regimen using intravenous bolus injection of doxorubicin (3 mg/kg, Days 1–3) combined with cytarabine (75 mg/kg, Days 1–5). Mice began treatment seven (or twenty five) days after transplantation of human leukemic cells with all mice receiving injections balanced to a final volume of 200ul PBS. Patients receiving standard 7+3 induction therapy (60 mg/m2 doxorubicin and 100 mg/m2 cytarabine) get approximately 1.5 mg/kg doxorubicin and 2.5 mg/kg cytarabine. Therefore, similar to previously published work, our dose of doxorubicin was doubled and the cytarabine concentration increased by 20-fold in an attempt to achieve necessary plasma concentrations of the drugs using a bolus injection as compared to continuous infusion given in clinical care.[23]
Chemotherapeutics
Clinical formulations of both doxorubicin and cytarabine were obtained from the James Graham Brown Cancer Center as self-sealing vials containing 20 mg/10 ml or 2g/20ml respectively dissolved in saline, manufactured by APP a division of Fresenius Kabi USA LLC (Lake Zurich, IL).
FACS Analysis
The spleen and BM cells were analyzed by FACS as previously described.[31] Briefly cells were isolated from tissues, red blood cells were lysed and blocked for 10 mins at 4°C with Fc Block (#553142 BD Biosciences, Miami, FL, USA). The samples were then stained with the appropriate fluorophore conjugated anti-human antibody (Alexa 700-CD45, APC-CD11b, APCCD19, PE-CD3, PE-Cy.5-CD34, and PE-Cy.5-CD33) from BioLegend (San Diego, CA) for 20 mins at 4° C and then analyzed on a Becton Dickinson FACScan with FlowJo.
Statistical analysis
All statistics were performed using GraphPad Prism 6 software. Unless specified below significance was determined by one-way ANOVA, followed by Tukey tests, using a cut off of P < 0.05. For all survival curves the log rank (Mantel-Cox) test was used, with a cut off of P < 0.05.
RESULTS
Cell line derived xenograft (CDX) in unconditioned NRG and NRGS mice
First, we wanted to examine differential engraftment efficiency of several established, commonly used human leukemic cell lines (K562, MV411, and U937) in NRG and NRGS mice. All cell lines showed high rates of engraftment in both strains resulting in disease progression and eventual euthanasia upon achievement of humane endpoints (Fig 1A). However, the formation of large intraperitoneal (IP) tumors developed selectively in NRGS mice engrafted with all established cell lines tested, while NRG mice developed clinically similar diffuse disease with expected signs of disease progression like splenomegaly. Necropsy revealed that most of the NRGS mice engrafted with MV411 cells had the occurrence of masses, found both intraperitoneally and lymphatically (Fig 1B, bottom right). K562 cells had a high propensity to form tumors in the kidneys (Fig 1B, bottom left), while U937 cells formed solid tumors in the spleens of only NRGS mice (Fig 1B, bottom middle). All mice engrafted with any of these cell lines showed pronounced leukemic infiltration of hematopoietic organs (Fig 2A, 2B), but NRG mice selectively failed to develop IP masses or solid tumors, and show a more clinically relevant disease presentation. Thus, it is evident that NRG mice, but not NRGS mice, are well suited for studies involving CDXs.
Figure 1. Engraftment of established human AML cell lines in NRG and NRGS mice.
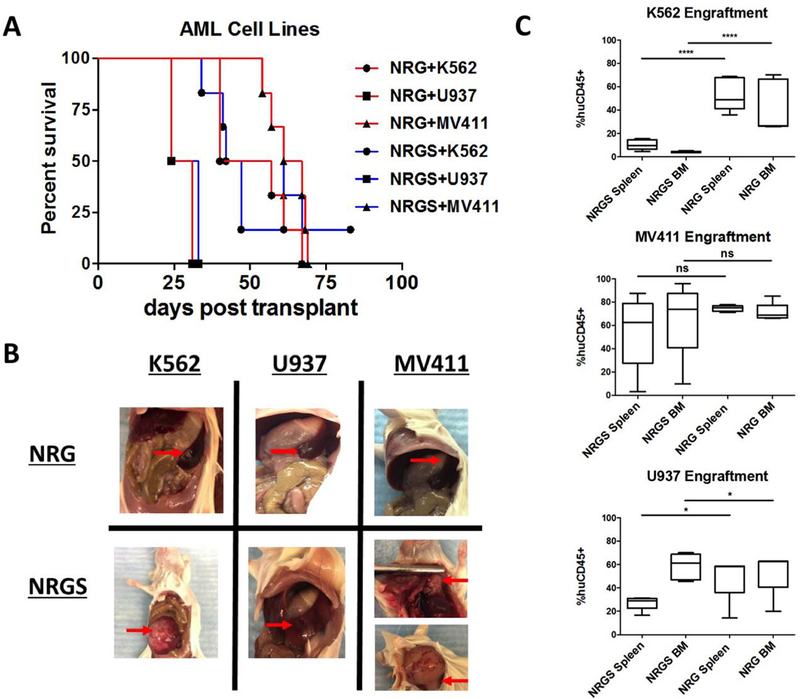
(A) Survival curves of NRG and NRGS mice post intravenous xenograft of 5 × 10^5 of each respective cell line K562, U937, and MV411. (B) NRG mice transplanted with human AML cell lines showed normal leukemic progression with diffuse disease and splenomegaly (as depicted), while NRGS mice developed solid splenic tumors (U937), solid intraperitoneal tumors (K562, MV411), and solid lymphatic tumors (MV411). (C) Box-and-whisker plots showing the percent of detectable human cells in the spleens and bone marrow of NRG (n=5) and NRGS (n=5) mice, as quantified by flow cytometry for human CD45.
Figure 2. H&E staining of spleen sections from NRG and NRGS mice xenografted with established human cell lines.
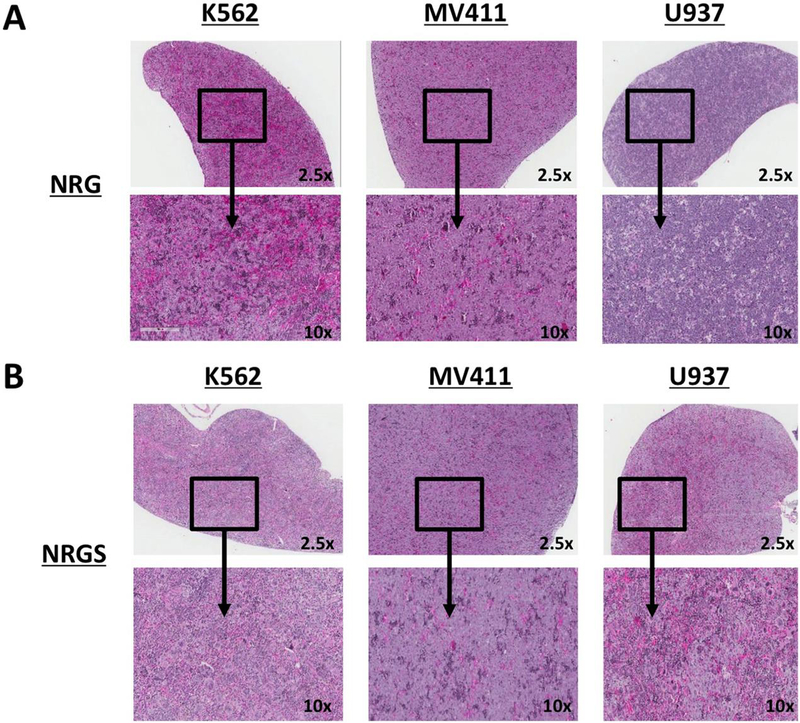
(A) H&E stained sections of representative formalin fixed spleens spleens from NRG mice intravenously xenografted with 5 × 10^5 K562, MV411, or U937 cells per mouse suspended in 200ul of PBS. (B) Representative spleens from NRGS mice intravenously xenografted with 5 × 10^5 K562, MV411, or U937 cells per mouse suspended in 200ul of PBS.
Patient derived xenografts (PDX) in unconditioned NRG and NRGS mice
We next examined the differential engraftment efficiency of several patient-derived samples in both NRG and NRGS mice. All patient derived samples were acquired from pediatric patients upon relapse, and three of four displayed MLL-translocations, while the final sample was cytogenetically normal (Fig 3A). Disease progression required euthanasia upon reaching humane endpoints in all of the NRGS mice irrespective of sample origin, while all NRG mice survived till the 100 day study endpoint without overt signs of disease (Fig 3B). As expected efficient engraftment occurred with all four patient derived samples in NRGS mice, leading to leukemic infiltration of the hematopoietic compartment, while three of the four patient samples tested (CCHMC-7, CCHMC-23, CCHMC-35) showed virtually no engraftment of human cells (<5%) in NRG mice (Fig 3C). Furthermore, no intraperitoneal or lymphatic solid tumors formed in the NRGS mice. Interestingly, large numbers of human cells were detected in the hematopoietic organs of NRG mice engrafted with CCHC-9 cells, but without symptomatic disease presentation
Figure 3. Engraftment of patient derived AML cells in NRG and NRGS mice.
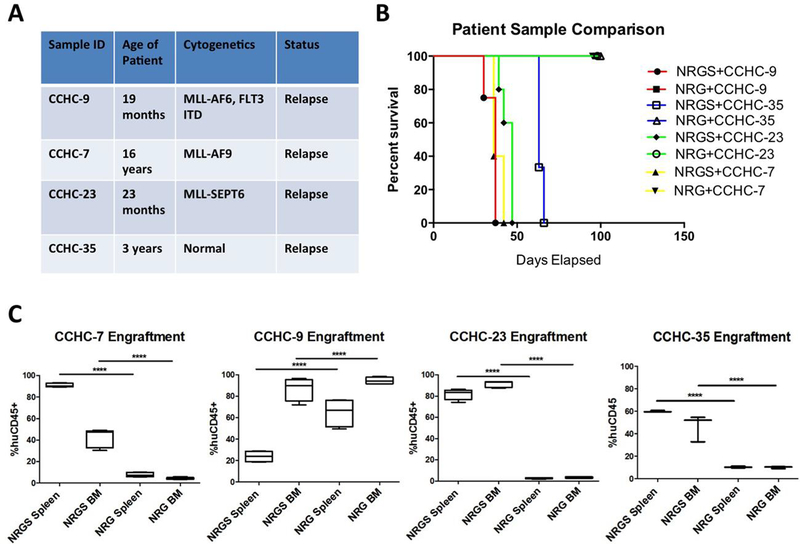
(A) All patient samples were initially obtained from pediatric patients upon relapse, and passaged once through NSGS mice. All xenograft experiments performed in these studies utilized the bone marrow of NSGS mice harboring high levels of each respective patient leukemic cells, as quantified by flow cytometry for human CD45. (B) Survival curves of NRG and NRGS mice post intravenous xenograft of 1.25 × 10^5 human cells of each respective patient sample. Mice showing no signs of disease were euthanized 100 days post xenograft, the end-point of the study. (C) Box-and-whisker plots showing the percent of detectable human cells in the spleens and bone marrow of NRG (n=5) and NRGS (n=5) mice, as quantified by flow cytometry for human CD45.
CD34+ engraftment potential in unconditioned NRG and NRGS mice
Given the recent push for the development of humanized mice, bearing functional human immunity, we next examined the engraftment potential and multilineage expansion of human CD34+ HSCs. Purified CD34+ cells from umbilical cord blood (Fig 4A) were transplanted into NRG and NRGS mice, but only NRGS mice showed significant, durable engraftment of human hematopoietic cells 105 days post xenograft. Human cells were still detectable in the hematopoietic organs of NRG mice, but at an extremely low percentage. H&E staining of spleen sections from NRGS revealed distinct patches of cells absent in NRG mice, likely human HSCs undergoing extrameduallary hematopoiesis (Fig 4B). Multilineage expansion of human pre-B cells, myeloid cells, and T-cells was observed only in NRGS mice as characterized by expression of human CD19, CD33, and CD3 respectively (Fig 4C). In contrast to published work indicating that human cytokine expression is counterproductive to human HSC engraftment, we show that unconditioned NRGS mice retain high levels of human HSCs (huCD45+/huCD34+) in both the spleen (Fig 5A) and the BM (Fig 5B) even 105 days post xenotransplant.
Figure 4. Comparative humanization potential of NRG and NRGS mice, using human umbilical cord derived CD34+ HSCs.
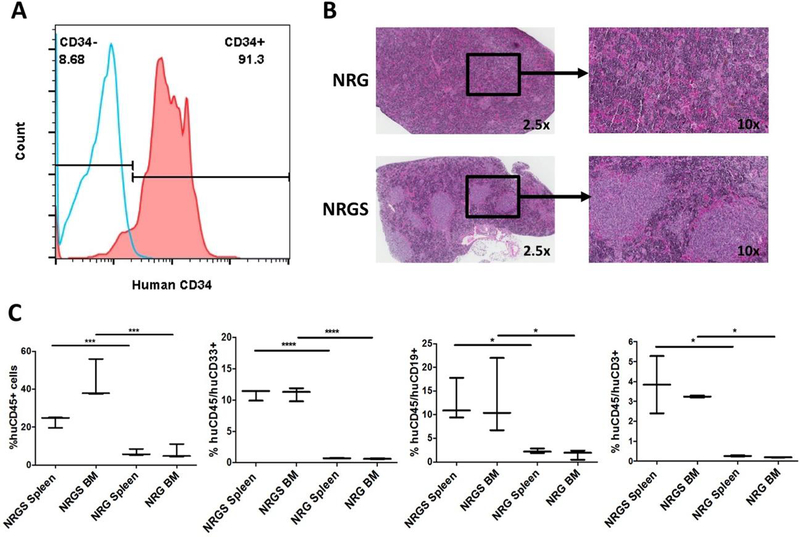
(A) Histogram showing the percent human CD34+ cells purified from umbilical cord blood before xenotransplantation, blue representing unstained cells and red representing cells stained with anti-human CD34-PE/Cy5. (B) Immunohistochemistry analysis with H&E staining of representative NRG and NRGS spleens, where distinct patches of human cells are clearly visible only in NRGS spleens. (C) Box-and-whisker plots showing the percent human cells in the spleens and bone marrow of NRG (n=3) and NRGS (n=3) mice 105 days post transplantation of 5 × 10^5 human CD34+ cells, and lineage analysis by FACS staining using anti-human CD45-Alexa 700 (human pan-hematopoietic), CD19-APC (pan-B cell), and CD33-PE/Cy5 (pan-myeloid).
Figure 5. Comparison of long term engraftment of mature and stem human hematopoietic cells in unconditioned NRG and NRGS mice.
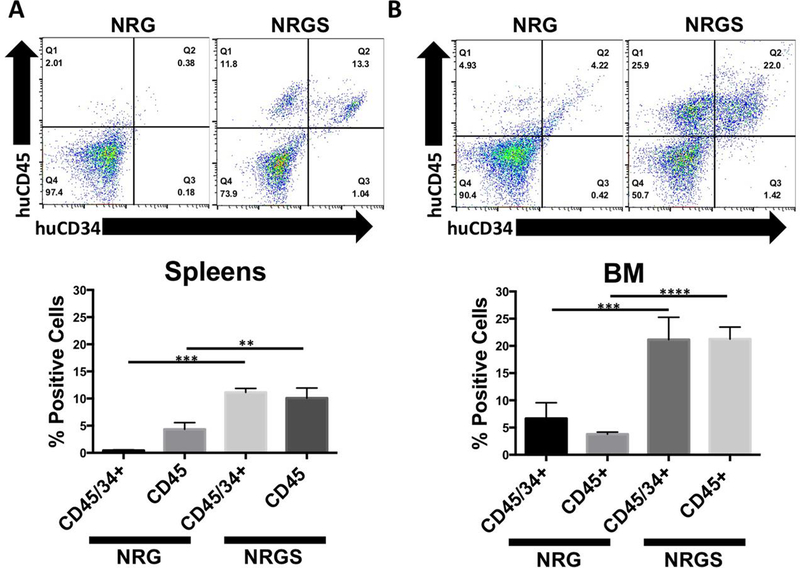
(A) Representative flow plots and relative percentages of human stem (huCD45+/huCD34+) and mature (huCD45+/huCD34-) hematopoietic cells in the spleens of unconditioned NRG (n=5) and NRGS (n=5) mice 105 days post transplantation of 5 × 10^5 human CD34+ UBC cells, indicating significantly higher engraftment of both stem and mature human hematopoietic cells. (B) Representative flow plots and relative percentages of mature and stem human hematopoietic cells detected in the bone marrow of unconditioned NRG and NRGS mice, showing highly significant enrichment of both stem and mature human hematopoietic cells selectively in NRGS mice.
Comparison of induction therapy
Useful AML xenograft mouse models also require the ability to model standard of care(SOC) therapeutic outcomes, which in current clinical practice rely heavily on the cytotoxic combination of an anthracycline and cytarabine. Therefore, we wanted to examine both the tolerability and the efficacy of an adapted 5+3 induction regimen (five consecutive days of cytarabine with doxorubicin given for the first three days) in both NRG and NRGS mice xenografted with human leukemia. Established human cell lines K562 and MV411 formed large solid tumors intraperitoneally and lymphatically selectively in NRGS mice upon xenotransplant. Mice injected with U937 cells show similar survival trajectories and engraftment efficiency in both strains, and NRGS solid tumor formation is restricted to the spleen. Further, given the finding that PDX samples fail to engraft efficiently in NRG mice, we chose U937 cells to examine comparative SOC therapeutic outcomes between NRG and NRGS mice. Mice from both strains were able to tolerate our adapted 5+3 bolus IV induction regimen, and all treated mice showed a highly significant enhancement in survival compared to vehicle injected controls (Fig 6A). These findings indicate both strains are tolerant of a clinically similar induction regimen, and therapy is efficacious in treating disease (Fig 6B). Comparing patient xenograft induction sensitivity in each strain proved a challenge as none of the four patient samples (CCHC-7, CCHC-9, CCHC-23, CCHC-35) gave rise to leukemia in NRG mice at the timepoints examined. Although the inherent limitations of NRG mice prevented us from examining comparative SOC therapeutic outcomes with patient-derived AML xenografts, we still felt it was important to examine the efficacy of our regimen against patient derived AML. As such, NRG and NRGS mice were engrafted with the patient sample CCHC-23, and 5+3 induction therapy was initiated seven days post xenograft. Again, we were able significantly prolong the survival of all treated NRGS mice as compared to vehicle-injected controls (Fig 6C), and therapy reduced the leukemic burden (Fig 6C). NRG mice in this study failed to develop disease and survived till the experimental endpoint. Similarly, our induction therapy regimen was efficacious in significantly prolonging the survival of NRGS mice harboring either CCHC-7 or CCHC-9 PDX (Fig 6D). These findings indicate that our 5+3 induction regimen is efficacious against several patient derived AML of varied etiology, at rates comparable to those observed in the aforementioned study with the established U937 cell line. This data is crucial to establish the feasibility and efficacy of modeling SOC treatment outcomes in mice xenografted with patient derived AML.
Figure 6. Comparison of standard of care chemotherapeutic response using 5+3 induction regimen in NRG and NRGS mice harboring cell line or patient derived leukemia.
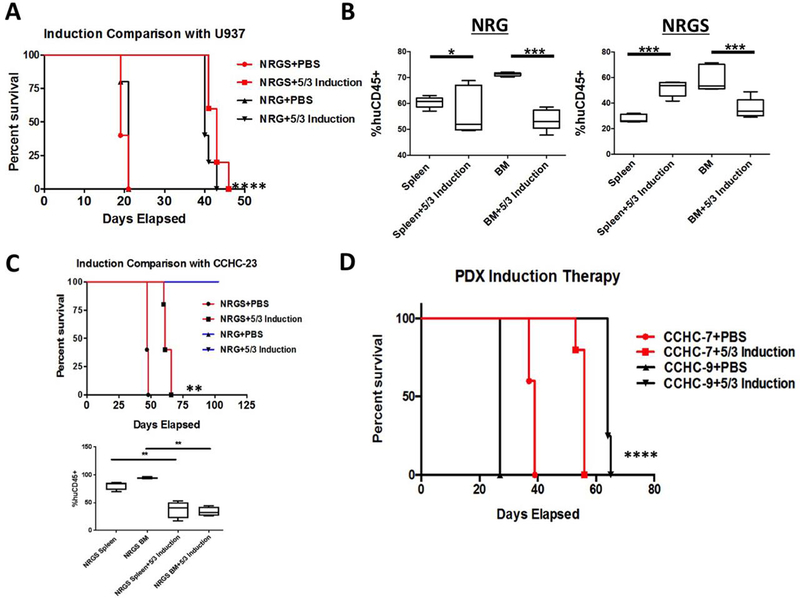
(A) Survival curves for NRG (n=5) and NRGS (n=5) mice engrafted with 5 × 10^5 U937 cells and subsequently treated with a 5+3 induction regimen [3 mg/kg doxorubicin (days 1–3) and 75 mg/kg cytarabine (all 5 days)] or the vehicle (PBS) by intravenous injection 7 days post xenograft. (B) Box-and-whisker plots showing significant differences in the percent human cells detected in the spleens and bone marrow of the NRG or NRGS mice from the study in panel A. (C) Survival curves and box-and-whisker plots showing percent human cells of NRG (n=5 per group) and NRGS (n=5 per group) mice xenografted with 1.25 × 10^5 CCHC-23 cells and subsequently treated with 5+3 induction or the vehicle 7 days post xenograft. NRG mice failed to develop disease and survived until the endpoint of the study with or without induction therapy. (D) Survival curves showing significantly prolonged survival of NRGS mice treated with 5+3 induction using two additional patient xenografts CCHC-7 and CCHC-9 to confirm therapeutic efficacy in multiple PDX samples.
Escalation and Modulation of Induction Therapy
Once established that our model strains were tolerant of our aggressive 5+3 induction regimen, we wanted to test the limits of our system by doubling the doses of both agents in NRGS mice transplanted with the patient sample CCHC-23. Comparable to human patients, the dose-limiting agent in this study was determined to be doxorubicin and groups of mice receiving double the dose of doxorubicin with or without increased cytarabine showed widespread toxicity and mortality (7 of 10 mice). Interestingly, mice receiving double the dose of cytarabine showed no overt increase in signs of toxicity, and the survival trajectory showed no change when compared to our standard 5+3 regimen (Fig 7A). Perhaps most intriguing the few mice that survived treatment with double the dosage of doxorubicin (6 mg/kg) showed no signs of leukemia at the endpoint of the study. This data is highly significant as it demonstrates that with proper optimization of drug concentration and dosing schedule it is possible to achieve a “remission like” phenotype. This would be of great importance in accurately modeling patient disease as up to 75% of patients with de novo AML experience complete remission upon the first cycle of induction therapy, with the majority of mortality being driven by refractory relapsed disease.[31] Finally, in an effort to faithfully model clinical treatment of AML we examined the therapeutic efficacy of induction therapy 7 days and 25 days post-xenograft. Concurrent with previously published reports, we found survival was significantly different between the cohorts of mice receiving induction 7 days or 25 days post xenograft, with mice surviving longer with earlier therapy (Fig 7B).[23] Thus, further investigation and optimization of treatment schedules may allow the development of a patient xenograft AML model that can accurately recapitulate clinical treatment outcomes, including minimal residual disease as well as relapse.
Figure 7. Escalation of 5+3 induction chemotherapy and modulation of timing of chemotherapy initiation.
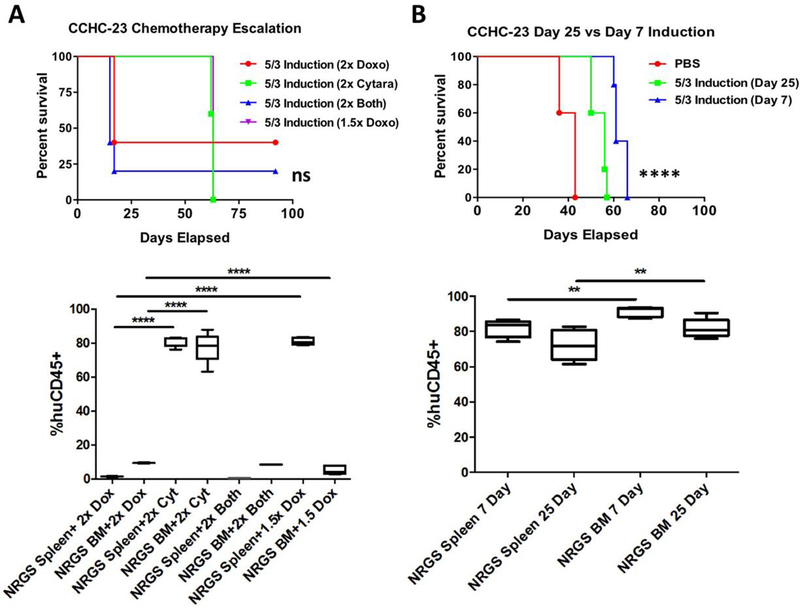
(A) Survival curves of NRGS mice (n=5 per group) xenografted with 1.25 × 10^5 CCHC-23 cells and subsequently treated with 5+3 induction therapy this time with either double the dose of both drugs (6 mg/kg doxorubicin, 150 mg/kg cytarabine), double the dose of each drug singly without manipulation of concentration of the other, or 1.5× the standard dose of doxorubicin (4.5 mg/kg, 75 mg/kg cytarabine). Box-and-whisker plots showing the percent of human cells detected in the spleen or bone marrow in each treatment escalation group. (B) Survival curves of NRGS mice (n=5 per group) xenografted with 1.25 × 10^5 CCHC-23 cells and treated with 5+3 induction 7 days post xenotransplant or 25 days post xenotransplant. Box-and-whisker plots showing percent human cells detected in the spleens and bone marrow of NRGS mice upon disease progression and euthanasia following 5+3 induction therapy initiated either 7 days or 25 days post xenograft.
Primary patient sample engraftment
Our final line of investigation was to determine the differential engraftment efficiency of an un-passaged primary patient AML sample (detailed description Fig 8A) obtained from a BM aspirate from the James Graham Brown Cancer Center (Fig 8B). Unconditioned NRG and NRGS mice were xenografted with 2 × 10^6 nucleated cells obtained from leukemic marrow aspirate. Mice were euthanized 100 days post injection at which time the spleens and BM were analyzed for human CD45 expression by flow cytometry. On average NRGS mice had consistently higher rates of both spleen and BM engraftment as compared to NRG mice (Fig 8C). Interestingly, splenic engraftment rates were low in both strains of mice (NRG average %huCD45= 3.07, NRGS average %huCD45= 5.87), with NRGS mice harboring approximately twice as many spleen resident human cells. H&E staining of spleen sections revealed a higher degree of leukemic infiltration in NRGS mice as compared to NRG mice (Fig 8D).
Figure 8. Engraftment of primary patient derived AML in NRG and NRGS mice.
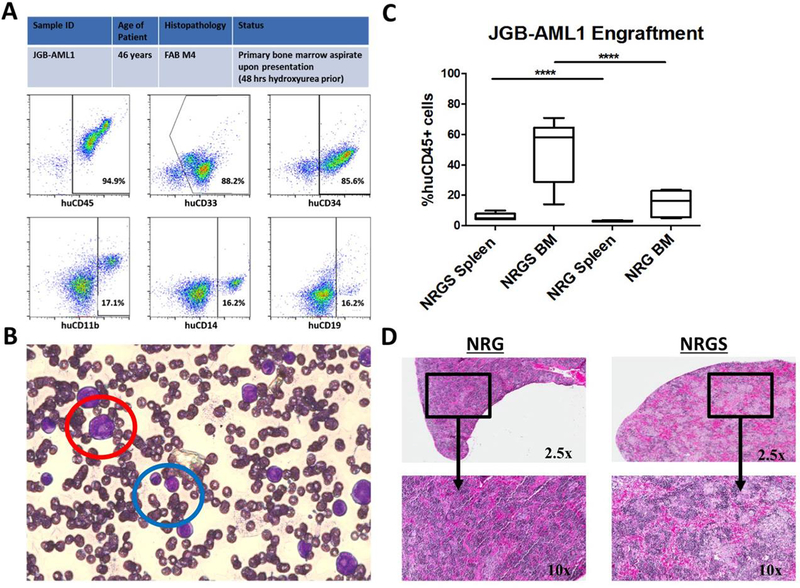
(A) Initial characterization of primary patient AML sample upon acquisition of bone marrow aspirate and subsequent Ficoll gradient separation of mononucleated cells, by flow cytometry for human CD45, CD34, CD33, CD19, CD14, and CD11b. (B) Bone marrow aspirate slide smear stained with Protocol© Hema 3 showing both leukemic myeloblasts (red) and monoblasts (blue) (photo acquired at 40× on Zeiss Observer AX10 microscope connected to Sony Nex-5n HD camera), typical for M4 subtype AML. (C) Box-and-whisker plot showing the percent human cells detected in the spleens and bone marrow of NRG (n=5) and NRGS (n=5) mice 100 days post xenograft of 2 × 10^6 mononucleated cells. (D) Immunohistochemistry of representative formalin fixed, H&E stained, NRG and NRGS spleens xenografted with 2 × 10^6 mononuclear cells obtained from Ficoll gradient separation of clinical bone marrow aspirate.
Discussion
The understanding of normal and malignant hematopoiesis in vivo has been revolutionized by the use of immune deficient xenograft mouse models, which continue to improve in both accurate recapitulation of patient disease and scope of use. This holds especially true for the study of AML, which exhibits a vast heterogeneity unique to individual patients in terms of prognoses and chemotherapeutic sensitivity.[32, 33] Thus, preclinical investigation of novel AML therapeutics with a high likelihood of success in clinical trials, require the development of translational mouse models capable of modeling patient disease and SOC outcomes. This poses a significant challenge as most currently used models rely on SCID mediated immune deficient mice after costly, difficult, and time-consuming myeloablative conditioning by radiation or chemotherapeutics, rendering the mice much less tolerant to DNA damaging SOC chemotherapeutics. New, under-utilized alternative model systems reliant on RAG deficiency, like NRG and NRGS mice, show equivalent comparative immune ablation with the additional advantage of increased tolerance to relevant doses of SOC induction therapeutics making them better model systems for examining treatment outcomes or investigating novel AML therapeutics. Comparative analysis of our data highlights the importance of selecting the appropriate mouse strain, especially in effective preclinical development of novel AML therapeutics. All tested established human AML cell lines were capable of engraftment at comparable efficiencies in both unconditioned strains, and both exhibited similar survival trajectories. These findings are in line with published studies showing that conditioning of mice by irradiation or busulfan does not change the xenotransplantation efficiency of AML cell lines in NSG mice[35], and may suggest that conditioning is unnecessary for CDX establishment in NRG or NRGS mice. It is important to note that a majority of only NRGS mice developed solid tumors when transplanted with all cell lines. Conversely, the NRG mice followed expected disease progression with all cell lines colonizing the BM and subsequently expanding into the other hematopoietic compartments resulting in splenomegaly, paralysis, and diminished survival. The solid tumors in the NRGS mice were mostly intraperitoneal or in the lymphatic system (except U937, formed solid splenic tumors), likely due to the whole-body constitutive expression of human myeloid cytokines. Xenotransplanted cells receive constant cytokine signaling in NRGS mice, promoting their survival and proliferation wherever they end up upon injection. Interestingly, there was a high propensity of solid tumor formation in the kidneys of NRGS mice transplanted only with K562 cells (Fig 1C lower left), which are the largest of all the cell lines studied, indicating that cell size may be of important consideration. Cells that are too large to fit through mouse microvasculature likely get lodged in renal capillary beds where they undergo cytokine mediated proliferation. These findings are of significant importance as they indicate that NRGS mice are not ideal for CDX studies as they develop clinically dissimilar disease.
Conversely, we found the exact opposite to be true for patient derived leukemia, which engraft with classical disease presentation and kinetics selectively in NRGS mice, not in NRG mice. Two of the three patient samples tested failed completely to engraft in NRG mice, and while one patient sample (CCHC-9) was capable of engraftment, mice failed to develop symptomatic disease even 100 days post xenograft. Unlike cell lines, there was no solid tumor formation in any of the NRGS mice transplanted with any of the patient samples, and all mice developed lethal leukemia with classical symptomatic presentation including scruffiness, splenomegaly, and hind-limb paralysis. It is important to note that all PDX samples engrafted with high efficiency in NRGS mice without any myeloablative conditioning, an additional advantage compared to currently used AML PDX models in terms of throughput, ease, and cost. These findings indicate that NRGS mice should exclusively be used for studies involving patient derived leukemia, while NRG mice should be used for studies with established human cell lines.
Using a modified version of a previously published protocol, we next examined the comparative chemotherapeutic response of both strains xenografted with U937 cells. These cells were chosen as they display highly similar engraftment efficiency in both strains, with solid tumor formation in NRGS mice restricted to the spleen. Not only were both strains capable of tolerating aggressive chemotherapy at clinically relevant doses, but treatment was also effective in significantly enhancing survival compared to vehicle treated mice. These findings are especially important, as modeling SOC therapeutic outcomes as better experimental controls, will be crucial in the discovery of novel efficacious AML therapeutics. Finally, we wanted to examine if dose escalation would be feasible, and if it would be possible to induce a remission like phenotype. Remarkably, mice could easily tolerate double the dose of cytarabine, but there was widespread mortality (70%) in mice receiving double the dose of doxorubicin. This indicated that doxorubicin is the dose-limiting agent when modeling AML induction therapy, likely due to cardio- and nephrotoxic effects. Perhaps most promising is the finding that mice that were capable of surviving the double dose of doxorubicin actually exhibit a remission like phenotype, with no symptoms of disease and virtually undetectable levels of human cells in the spleen or BM at the experimental endpoint. Further optimization of the concentration of doxorubicin is necessary to develop a treatment regimen that recapitulates clinical outcomes of AML.
Next, we examined the humanization potential of both unconditioned strains transplanted with CD34+ human umbilical cord blood cells 105 days post xenograft. Only the NRGS mice were capable of sustained, multilineage expansion of human cells, which interestingly lacked the assumed myeloid bias. Indeed, a large percent of detectable human cells were positive for the pan-myeloid mark CD33, but an almost equally large percentage of cells were CD19+ B cells, and a small population of CD3+ cells were detected selectively in NRGS mice. In contrast to previously published reports indicating that constitutive expression of human IL-3, GM-CSF, and SCF is counterproductive for the engraftment of human HSCs in human cytokine producing NOD-SCID mice[36–38], we show that unconditioned NRGS mice show durable long-term engraftment of human HSCs (CD45/CD34+) in the spleen and BM. These differences are likely attributable to intense ablative conditioning regimens (pharmacological agents or irradiation) utilized in these studies just prior to transplantation of human HSCs. Ablative conditioning may render the murine BM marrow microenvironment inhospitable to HSC seeding, or promote hematopoietic differentiation and loss of self-renewal in human HSCs. Our findings indicate that not only is myeloablative conditioning unnecessary in NRGS mice, but in fact may be detrimental to long-term durable engraftment of human HSCs.
Previously published work from members of our group has shown that NSGS mice develop a fatal macrophage activation syndrome (MAS) after transplantation of human UCB, with survival diminishing to approximately 50% 100 days post-transplant.[39] Conversely, we observed no such MAS phenotype in unconditioned NRGS mice, and all mice survived till the 105 day endpoint. Again, these differences are likely due to the ablative conditioning used in the previous study (30 mg/kg busulfan 24 hours before transplant), but may also be attributed to initial variability in transplanted human cells. This study utilized purified human CD34+ HSCs from UCB while the previous study used OKT3 treated unfractionated UCB, and this crucial difference may explain the lack of MAS in unconditioned NRGS mice. These findings indicate that only unconditioned NRGS, not NRG, mice are capable of supporting sustained, multilineage expansion of human HSCs at humanization relevant levels. In theory, a model established first with a patient’s immunity, and subsequently xenografted with the same patient’s AML, would be the most clinically relevant xenograft model of human AML. The rapid discovery of novel, efficacious AML therapeutics will require similar humanized xenograft models that not only recapitulate patient disease, but also patient immunity.
Finally, we wanted to investigate the comparative utility of both strains in studies involving a primary patient sample (not serial transplant). We obtained a fresh marrow aspirate of a patient diagnosed with acute myelomonocytic leukemia (FAB M4) from the James Graham Brown Cancer Center, which we subsequently injected into NRG and NRGS mice. Time-consuming conditioning of the mice, by either irradiation or drugs, was forgone in an effort to determine the comparative utility of each strain in high throughput studies involving experimental chemotherapeutics and primary patient derived samples. As expected all NRGS mice showed higher rates of engraftment in the BM and spleen as compared to NRG mice, and two NRG mice showed no engraftment at all. Interestingly, hepatic tumors were observed selectively in all five of the NRGS mice and this likely explaining the absence of high levels of human leukemic cells in the spleen. It remains to be determined if this phenotype is driven by the genetics/etiology of primary sample, duration of study, or intrinsic properties of the mouse liver expressing human cytokines. Regardless, these data convincingly suggest that unconditioned NRGS, but not NRG, mice are amenable to the xenotransplantation of primary patient AML, and develop patient disease in a time frame required for the investigation of novel therapeutics or SOC outcomes.
Taken together these findings highlight the great utility of both of these mouse strains in the study of human AML, specifically for modeling patient specific disease and clinical treatment outcomes. Investigators utilizing established human cell lines in their studies should selectively use NRG mice, while studies involving patient derived myeloid leukemia should only use NRGS mice for accurate recapitulation of the disease phenotype. Both strains are equally tolerant of a clinically relevant induction regimen, and treatment significantly prolongs the survival of mice receiving human leukemia xenografts. Future studies should focus on optimizing the dosing regimen to more closely match clinical conditions and endpoints, and perhaps for the study of treatment regimens for relapsed patient disease. Unconditioned NRG mice are inhospitable to human HSCs while NRGS mice show consistent multilineage expansion of human HSCs at humanization levels (>25%). Similarly, unconditioned NRGS mice show a much higher success rate of primary patient sample engraftment, as well as greatly improved engraftment efficiency. In conclusion, both NRG and NRGS mice are powerful, underused new tools for the study of both malignant and normal hematopoiesis, each with its own strengths and limitations of utility, contextually determined by the experimental design.
Highlights.
A comparison of engraftment efficiencies in unconditioned NRG and NRGS mice.
NRGS mice do not develop clinically accurate disease with cell line xenografts.
NRG mice do not efficiently engraft patient normal or leukemic cells.
NRG and NRGS mice tolerate high doses of induction therapeutics.
Acknowledgements
We would like to acknowledge all members of the Beverly and Siskind Labs for their advice and technical support. This work was supported by the James Graham Brown Cancer Center, University of Louisville School of Medicine and Kosair Pediatric Cancer Program (to LJB), NIH/NCI RO1 (R01CA193220, to LJB), NIH/NCI R50 (R50CA211404 to MW) and the Pediatric Avatar Program at CCHMC.
Footnotes
Authorship and Conflict of Interest
All experiments were performed by A. Barve, all animal husbandry and H&E immunohistochemical preparation and staining was done by L. Casson, M. Krem helped with sample acquisition and experimental design, L. J. Beverly assisted in all experimental design and data interpretation, material, technical, and editing support were aided by J. Mulloy and M. Wunderlich. The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sykes SM and Scadden DT, Modeling human hematopoietic stem cell biology in the mouse. Semin Hematol, 2013. 50(2): p. 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manz MG and Di Santo JP, Renaissance for mouse models of human hematopoiesis and immunobiology. Nat Immunol, 2009. 10(10): p. 1039–42. [DOI] [PubMed] [Google Scholar]
- 3.Cook GJ and Pardee TS, Animal models of leukemia: any closer to the real thing? Cancer Metastasis Rev, 2013. 32(1–2): p. 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito R, et al. , Current advances in humanized mouse models. Cell Mol Immunol, 2012. 9(3): p. 208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuber J, et al. , Mouse models of human AML accurately predict chemotherapy response. Genes Dev, 2009. 23(7): p. 877–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Francia G, et al. , Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer, 2011. 11(2): p. 135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lapidot T, et al. , A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature, 1994. 367(6464): p. 645–8. [DOI] [PubMed] [Google Scholar]
- 8.Bonnet D and Dick JE, Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med, 1997. 3(7): p. 730–7. [DOI] [PubMed] [Google Scholar]
- 9.Rashidi A and Uy GL, Targeting the microenvironment in acute myeloid leukemia. Curr Hematol Malig Rep, 2015. 10(2): p. 126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meads MB, Hazlehurst LA, and Dalton WS, The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res, 2008. 14(9): p. 2519–26. [DOI] [PubMed] [Google Scholar]
- 11.Nervi B, et al. , Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood, 2009. 113(24): p. 6206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majeti R, et al. , CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell, 2009. 138(2): p. 286–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colmone A, et al. , Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science, 2008. 322(5909): p. 1861–5. [DOI] [PubMed] [Google Scholar]
- 14.Flanagan SP, ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res, 1966. 8(3): p. 295–309. [DOI] [PubMed] [Google Scholar]
- 15.Bosma GC, Custer RP, and Bosma MJ, A severe combined immunodeficiency mutation in the mouse. Nature, 1983. 301(5900): p. 527–30. [DOI] [PubMed] [Google Scholar]
- 16.Dvir A, et al. , Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc Natl Acad Sci U S A, 1992. 89(24): p. 11920–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodwin JF and Knudsen KE, Beyond DNA repair: DNA-PK function in cancer. Cancer Discov, 2014. 4(10): p. 1126–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shultz LD, et al. , Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol, 1995. 154(1): p. 180–91. [PubMed] [Google Scholar]
- 19.Fortier JM and Graubert TA, Murine models of human acute myeloid leukemia. Cancer Treat Res, 2010. 145: p. 183–96. [DOI] [PubMed] [Google Scholar]
- 20.Ito M, et al. , NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood, 2002. 100(9): p. 3175–82. [DOI] [PubMed] [Google Scholar]
- 21.Shultz LD, et al. , Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol, 2005. 174(10): p. 6477–89. [DOI] [PubMed] [Google Scholar]
- 22.Sugamura K, et al. , The interleukin-2 receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu Rev Immunol, 1996. 14: p. 179–205. [DOI] [PubMed] [Google Scholar]
- 23.Wunderlich M, et al. , AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood, 2013. 121(12): p. e90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishikawa F, et al. , Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood, 2005. 106(5): p. 1565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feuring-Buske M, et al. , Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia, 2003. 17(4): p. 760–3. [DOI] [PubMed] [Google Scholar]
- 26.Wunderlich M, et al. , AML xenograft efficiency is significantly improved in NOD/SCIDIL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia, 2010. 24(10): p. 1785–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yahata T, et al. , Functional human T lymphocyte development from cord blood CD34+ cells in nonobese diabetic/Shi-scid, IL-2 receptor gamma null mice. J Immunol, 2002. 169(1): p. 204–9. [DOI] [PubMed] [Google Scholar]
- 28.Shultz LD, et al. , NOD/LtSz-Rag1nullPfpnull mice: a new model system with increased levels of human peripheral leukocyte and hematopoietic stem-cell engraftment. Transplantation, 2003. 76(7): p. 1036–42. [DOI] [PubMed] [Google Scholar]
- 29.Wunderlich M, et al. , OKT3 prevents xenogeneic GVHD and allows reliable xenograft initiation from unfractionated human hematopoietic tissues. Blood, 2014. 123(24): p. e134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saurabh K, et al. , Dissecting the in vivo leukemogenic potency of BCLxl. J Leuk (Los Angel), 2014. 2(5): p. 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Othus M, et al. , Fate of patients with newly diagnosed acute myeloid leukemia who fail primary induction therapy. Biol Blood Marrow Transplant, 2015. 21(3): p. 559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li S, Mason CE, and Melnick A, Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr Opin Genet Dev, 2016. 36: p. 100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Kouchkovsky I and Abdul-Hay M, ‘Acute myeloid leukemia: a comprehensive review and 2016 update’. Blood Cancer J, 2016. 6(7): p. e441. [DOI] [PMC free article] [PubMed] [Google Scholar]