

Abstract
Production of transforming growth factor-β (TGFβ) by T cells is key to various aspects of immune homeostasis, with defects in this process causing or aggravating immune-mediated disorders. The molecular mechanisms that lead to TGFβ generation by T cells remain largely unknown. To address this issue, we take advantage of the fact that intestinal helminths stimulate T helper 2 (Th2) cells besides triggering TGFβ generation by T lymphocytes and regulate immune-mediated disorders. We show that the Th2 cell-inducing transcription factor STAT6 is necessary and sufficient for the expression of TGFβ pro-peptide (pro-TGFβ) in T cells. STAT6 is also necessary for several helminth-triggered events, such as TGFβ-dependent suppression of alloreactive inflammation in graft-versus-host disease (GVHD), in mice. Besides STAT6, helminth-induced secretion of active TGFβ requires cleavage of pro-TGFβ by the endopeptidase furin. Thus - for the immune regulatory pathway necessary for TGFβ production by T cells - our results support a two-step model, comprised by STAT6 and furin.
Introduction
Transforming growth factor-β (TGFβ) is a pleiotropic cytokine that has strong immune regulatory activity (1–3). Defects in TGFβ circuitries lead to an increase in inflammatory cytokine production, disruption of peripheral immune tolerance and an aggravated course of immune-mediated disorders, such as inflammatory bowel disease (IBD) or graft-versus-host disease (GVHD)(1, 2, 4, 5). Mice lacking TGFβ – due to deletion of either the encoding gene (germline or conditional) or genes that encode other components of the same signaling pathway – develop spontaneous inflammation and aberrant immune reactivity(1, 2). Restoration of TGFβ pathway in patients suffering from immune-mediated disorders, such as IBD helps suppress the inflammation(5, 6). Thus, the role of TGFβ in maintaining the balance of the peripheral immune system is non-redundant as well as essential.
TGFβ is generated as an inactive pro-peptide (pro-TGFβ) and its biological effect is mediated by a C-terminal cytokine produced by proteolytic cleavage from an N terminal latency-associated peptide (LAP)(2, 3). Analyses of several animal models have suggested that although TGFβ is produced by various cell types, immune regulation depends on its expression and activation in T lymphocytes(7, 8). Polarized T helper cells, such as Th17 cells are shown to produce TGFβ (9). Besides conventional αβ or γδ T cells, TGFβ-generating T lymphocytes also include Foxp3+ regulatory T cells (Treg)(3, 7, 10, 11). Molecular mechanisms that stimulate TGFβ production by T cells are unknown. Although some proteins capable of activating the TGFβ cytokine by enzymatically cleaving the propeptide (pro-TGFβ) – such as furin - have been identified(10, 12, 13), their physiological relevance in specific contexts deserve further attention and the cellular pathways that drive the expression of pro-TGFβ remain to be established.
One set of clues to the origins of TGFβ producing T lymphocytes is that they emerge in response to either antigen stimulation in the gut(14) or colonization of the intestine by helminths(15, 16). The latter involves the stimulation of TGFβ secretion by T cells in the mesenteric lymph node (MLN) and lamina propria of the intestine(17–19). Besides stimulating TGFβ generation, helminths stimulate Th2 pathway(20).
STAT6 is a Th2-inducing transcriptional activator. Helminths modulate cytokine production by immune cells and alter intestinal mucosal function in a STAT6-dependent manner(21–23). STAT6 is activated by phosphorylation - initiated by Th2 cytokines, interleukin 4 (IL4) and IL13(24, 25). Once phosphorylated, STAT6 orchestrates the development or maturation of peripheral Th2 cells by altering the transcription of at least several hundred genes(21, 26).
In this study, we explored the role of STAT6 in helminth-induced TGFβ production and in the context of helminthic suppression of an immune-mediated disorder. In a model of graft-versus-host disease (GVHD), which is a lethal alloreactive complication of bone marrow transplantation (BMT) and amenable to helminthic immune suppression(19), we show that helminth-induced pro-TGFβ production by T cells is dependent on STAT6. STAT6 is also required for helminth-induced and TGFβ-dependent suppression of GVHD. In addition to STAT6, activation of TGFβ by cleavage, though not the expression of pro-TGFβ, requires the endopeptidase furin. Based on these results, we propose a model where TGFβ generating T cells originate from peripheral T cells with active Th2 (STAT6) signaling.
Materials and Methods
Mice and Heligmosomoides polygyrus bakeri (Hpb) administration.
Wild type (WT) C57BL/6 (H2b), WT BALB/c (H2d), STAT6–/– (H2d) and CD4 Cre (H2b) transgenic mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice with conditional (CD4 promoter-driven) deletion of endopeptidase, furin (furinfl/fl (H2b)) and mice with CD2 promoter-driven expression of a constitutively active STAT6 (STAT6 VT (H2b)) in T cells were described before(27, 28). T cell-specific deletion of furin was achieved by the expression of Cre recombinase after crossing furin fl/fl and CD4 Cre mice as described previously(12). Helminth colonization was performed by inoculating 5–6 week-old male WT BALB/c, STAT6–/–, STAT6 VT, furinfl/fl and CD4 Cre x furin fl/fl mice with 150 Heligmosomoides polygyrus bakeri (Hpb) third stage larvae (L3) by oral gavage. Infective Hpb L3 (original specimens archived at the U.S. National Helminthological Collection, no. 81930) were obtained from mouse fecal cultures of eggs by the modified Baermann method(29) and stored at 4°C until used. Mice were maintained and used in accordance with the University of Iowa Animal Care and Use Committee Guidelines.
Cell purification for GVHD induction.
Donor bone marrow (BM) cells were obtained from the femurs and tibias of uninfected, 5–8 week-old C57BL/6 mice. T-cell depleted (TCD) BM samples were prepared using mouse panT cell beads (Dynal Biotech) according the manufacturer’s instructions. Spleen cell samples from uninfected, 5–8 week-old C57BL/6 mice were magnetically enriched for untouched T cells (CD3+) using the T Cell Isolation Kit (Miltenyi Biotech).
Cell purification for in vitro cultures.
The expression of LAP and the production of TGFβ cytokine were assessed by purifying CD4+ T cells from splenic and mesenteric lymph nodes (MLN) of Hpb-infected and uninfected WT BALB/c, STAT6–/–, STAT6 VT, furinfl/fl, furinfl/fl x CD4 Cre mice using the CD4 T Cell Untouched Isolation Kit (Miltenyi Biotech). This resulted in >98% enrichment for CD4+ T cells (data not shown). To assess helminthic regulation of cytokine production by donor T cells during GVHD, donor CD3+ T cells from uninfected and Hpb-infected WT BALB/c or STAT6–/– BMT recipients were sorted from total splenocytes stained with anti-CD3 FITC and anti-H2b PE 6 days after GVHD induction, using a FACS Vantage SE DiVa cell sorter (Becton Dickinson).
Total body irradiation (TBI) and GVHD induction.
Our studies utilized an MHC I/II mismatch (H2b→H2d), acute lethal GVHD model(30). At three weeks post infection, Hbp-infected and uninfected STAT6–/– recipients (H2d) underwent total body irradiation using a Cs137 source (a total of 850 cGy in two doses given four hours apart), and were administered 10×106 T cell-depleted bone marrow (TCD-BM) cells with or without 1.5×106 splenic T lymphocytes from uninfected C57BL/6 WT donors (H2b). Mice were monitored daily for survival for up to 100 days. In parallel experiments, Hpb-infected and uninfected BALB/c wild-type and STAT6−/− mice were sacrificed 6 days after BMT for cellular and histopathological analysis.
Flow cytometry.
In experiments that did not involve BMT, spleen and MLN cells from uninfected and Hpb-infected BALB/c WT, STAT6–/–, STAT6 VT, furinfl/fl and CD4 Cre x furinfl/fl mice were isolated 3 weeks post infection. Cells were suspended at 2×107 cells/ml in PBS with 2% FCS, and Fc receptors were blocked with a 2.4G2 mAb (Clone: 145–2C11, eBiosciences). Antibodies for surface staining were: anti-CD3 PE-Cy7, anti-CD3 FITC (Clone: 145–2C11, eBiosciences), anti-CD4 PE-Cy7 (Clone: GK1.5, eBioscience), anti-LAP PE (Clone: TW7–20B9 BioLegend) vs. isotype IgG1κ PE and anti-GARP PE (Clone: F011–5, BioLegend). For intracellular Foxp3 staining, the Foxp3 staining buffer (Clone: FJK-16S, eBioscience) and anti-Foxp3 PE, Foxp3 PE-Cy7 and Foxp3 APC antibodies were used in accordance with the manufacturer’s instructions.
In experiments involving BMT, spleen and MLN cells from uninfected and Hpb-infected BALB/c or STAT6–/– mice were isolated on day 6 post-infection and stained as detailed above. These experiments utilized, in addition, anti-H2b PE, anti-H2d PE, and anti-H2b APC antibodies (Clones: SF1–1.1, SF1–1.1.1, AF6.88.5 BD Biosciences).
In vitro cell culture and cytokine ELISA.
MLN cells from uninfected or Hpb-infected wild-type BALB/c, STAT6−/−, STAT6 VT, furinfl/fl and furinfl/fl x CD4 Cre mice without GVHD were stimulated with anti-CD3 (Clone: 145–2C11, eBioscience) and anti-CD28 (each at 1 μg/ml) (Clone: 37.51, eBioscience) for 48 hours in cell culture medium with 1% FCS and 1 mg/ml BSA(17, 18) for TGFβ ELISA and in cell culture medium with 10% FCS for interleukin 4 (IL4) ELISA (17, 18). The concentration of TGFβ cytokine in acidified and re-alkalinized supernatants was determined using antibody pairs from R&D Systems according to the manufacturer’s instructions, and the TGFβ concentration in the culture supernatants was subtracted from that in the culture medium. IFNγ and TNFα secretion was determined for sorted wild-type C57BL/6 donor splenic T cells (CD3+ and H2b+) from uninfected and Hpb-infected BALB/c WT and STAT6−/− BMT mice. Cells were stimulated with plate-bound anti-CD3 and soluble anti-CD28 (each at 1 μg/ml) for 48 hours in lymphocyte growth medium containing 10% FCS(31). Supernatants were analyzed for IFNγ, IL4 and TNFα content using antibody pairs from R&D Systems. Similar methods were employed to detect IFNγ and TNFα in sera from uninfected and Hpb-infected BALB/c WT and STAT6–/– mice 6 days after the induction of GVHD.
RNA isolation, reverse transcription and quantitative polymerase chain reaction (qPCR).
RNA from CD4-enriched T cells (CD4 T Cell Isolation Kit, Miltenyi Biotech) was isolated using TRIzol Reagent (Cat. 15596–026, Thermo Fisher Scientific). Intron sparing primers used in this study are displayed in Table 1. First-strand synthesis of cDNA from RNA was performed using SuperScript III kit (Cat. 18080–051, Thermo Fisher Scientific). Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was carried out with QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) using Power SYBR Green PCR Master Mix (Cat. 4367659, Thermo Fisher Scientific Inc, USA). As an internal control, hypoxanthine guanine phosphoribosyl transferase (HPRT) RNA levels were used to normalize gene expression. Relative transcript abundance was calculated using the 2-ᐃCT method. Data were shown as mean ± standard deviation (SD).
Table 1:
Primers used for RT-qPCR
| Gene | Sequences (5’−3’) | Fragment size (bp) |
|---|---|---|
| HPRT | F: TGAAGAGCTACTGTAATGATCAGTCAAC | 186 |
| R: GCAAGCTTGCAACCTTAACCAT | ||
| TGFβ | F: TGACGTCACTGGAGTTGTACGG | 170 |
| R: GGTTCATGTCATGGATGGTGC | ||
| Furin | F: CATGACTACTCTGCTGATGG | 148 |
| R: GAACGAGAGTGAACTTGGTC |
Histopathology.
Six days after GVHD induction, lungs and colons from uninfected or Hpb-infected mice were fixed in 4% neutral buffered formalin and processed; 6 μm sections were stained with hematoxylin and eosin. Tissues were analyzed for GVHD-related inflammation, and the severity of inflammation was scored in blinded fashion by DEE(32–36). GVHD-related colitis was graded based on both the degree of inflammation and the frequency of crypt apoptosis. Inflammation was graded as none (score: 0), mild (1), moderate (2), severe without ulcer (3) or severe with ulcer (4). Crypt apoptosis was graded based on the presence of apoptotic bodies, as follows: rare (score: 0), occasional per 10 crypts (1), 2–5 per 10 crypts (2), majority of crypts (>5) (3), majority of crypts containing more than one (4). The minimal score in this grading system for colonic disease was 0 and the maximum score was 8. GVHD-related lung inflammation was graded based on the presence of perivascular cuffing, vasculitis, peribronchiolar cuffing and alveolar hemorrhage. The minimal score in this grading system for lung inflammation was 0 and the maximum was 4.
Statistical analysis.
Differences in survival between groups were determined using the Kaplan Meier log rank test. Differences in cell number and composition, serum IFNγ and TNFα content, IFNγ and TNFα generation by splenic donor T cells, TGFβ cytokine output of in vitro stimulated cell cultures, mean fluorescence intensity (MFI) of LAP expression and histopathological GVHD scores between groups were determined using Student’s t test.
Results
Helminth-induced protection from lethal GVHD requires the host STAT6 pathway.
We previously demonstrated that helminth-induced conditioning of the host suppresses GVHD(19). Helminths stimulate the Th2 pathway in the host(20), and do so by activating genes associated with Th2 development, such as the transcription factor STAT6(23, 37). A model of acute GVHD, in which both T cell-depleted bone marrow (TCD-BM) and splenic T cells from C57BL/6 WT (H2b) donors are transplanted into Hpb-colonized or uninfected irradiated STAT6–/– (H2d) recipients, enabled us to assess whether helminth-induced immune conditioning of the host requires host cell STAT6 activity. Although Hpb infection promotes the survival of WT BMT recipient mice (Figure 1 and (19)), this was not the case for STAT6–/– hosts; in STAT6−/− animals, BMT with C57BL/6 donor cells resulted in mortality within 40 days of BMT (Figure 1). When uninfected and Hpb-infected STAT6–/– or WT BALB/c BMT mice were transplanted with only WT donor TCD-BM cells (no splenic T cells), they survived the 100-day period of the experiment (Figure 1). This suggests that STAT6 expression by host cells is critical to helminth-induced protection from lethal GVHD, and to the helminth-induced regulation of donor T cells that cause GVHD.
Figure 1. Helminthic promotion of the survival of BMT recipients requires recipient STAT6.
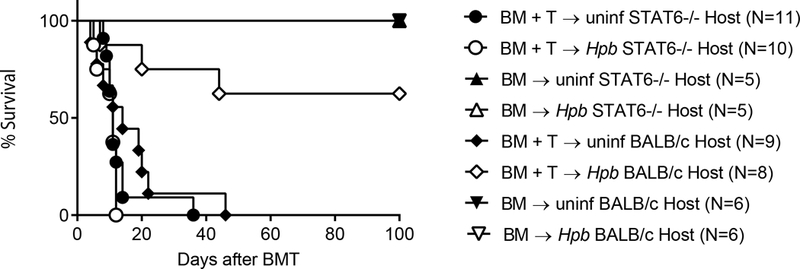
Kaplan-Meier survival curves of Heligmodomoides polygyrus bakeri (Hpb)-infected or uninfected STAT6–/– or WT BALB/c male recipients that received T cell-depleted (TCD-BM) cells (TCD BM) only or TCD-BM and total splenic T cells (TCD-BM + T) from 5–6 week-old male WT C57BL/6 mice. Cumulative data from multiple independent experiments (N: cumulative number of mice for each group) where STAT6−/− or WT BALB/c recipients received donor cells in parallel; p:NS between uninfected and Hpb-infected STAT6−/− recipients of TCD-BM + T donor cells; p<0.01 between uninfected and Hpb-infected WT BALB/c recipients of TCD-BM + T donor cells.
Helminth-induced protection from GVHD-mediated organ damage requires the host STAT6 pathway.
We demonstrated previously that helminths suppress GVHD-related end-organ damage in lung and the colon of WT BMT mice(19). Histopathological analysis of these organs in uninfected STAT6–/– BMT mice revealed severe inflammation that was not improved by helminth infection (Figure 2A-D), although in WT BALB/c BMT mice Hpb-colonization suppressed inflammation (Figure 2E-H) and as we reported previously (19). These observations suggest that helminths require the STAT6 signaling pathway of the host to suppress end-organ damage associated with GVHD.
Figure 2. Helminthic suppression of GVHD-related organ damage requires recipient STAT6.
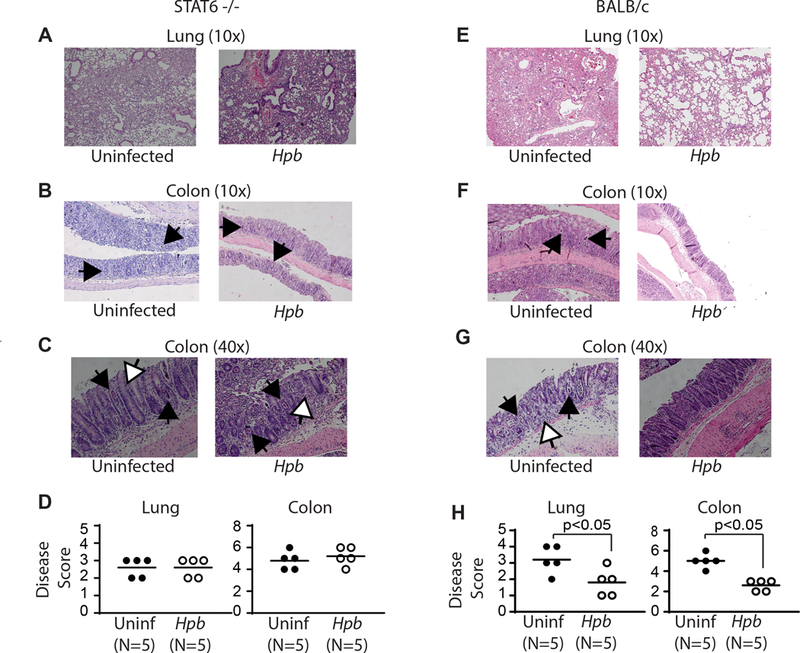
Histopathology of lung (A) and colonic (B-C) tissues from uninfected and Hpb-infected STAT6–/– or WT BALB/c BMT mice six days after BMT. Histopathological analysis was performed in 6-μm-thick, hematoxylin-and-eosin-stained samples. (D) Severity of inflammation in lung and colon, with scoring as described in Materials and Methods. GVHD-related colitis was characterized by mononuclear cell infiltrates, apoptotic cells filling crypts (black arrows) and apoptotic bodies (white arrows). Dot plot distribution with mean of cumulative data from multiple samples. Each dot represents a single sample and the bars the mean values (N=number of samples; p: NS between uninfected (Uninf; black filled dots) and Hpb-infected (white empty dots) STAT6−/− recipients; p<0.05 between uninfected (Uninf; black filled dots) and Hpb-infected (white empty dots) WT BALB/c recipients).
Next we investigated helminthic modulation of the generation of inflammatory cytokines by C57BL/6 WT donor T cells. CD3+ and H2b+ splenocytes from uninfected and Hpb-infected STAT6–/– or BALB/c WT BMT mice were sorted by FACS, 6 days after BMT. Our analysis revealed that helminths suppressed the secretion of inflammatory cytokines (IFNγ and TNFα) by C57BL/6 WT donor T cells, as well as serum inflammatory cytokine content, in WT BALB/c but not in STAT6–/– BMT recipients (Figure 3). These results imply that the STAT6 pathway of the host – associated with helminth-induced Th2 polarization – is critical for helminthic regulation of GVHD.
Figure 3. Helminthic suppression of inflammatory cytokine generation in GVHD requires host STAT6.
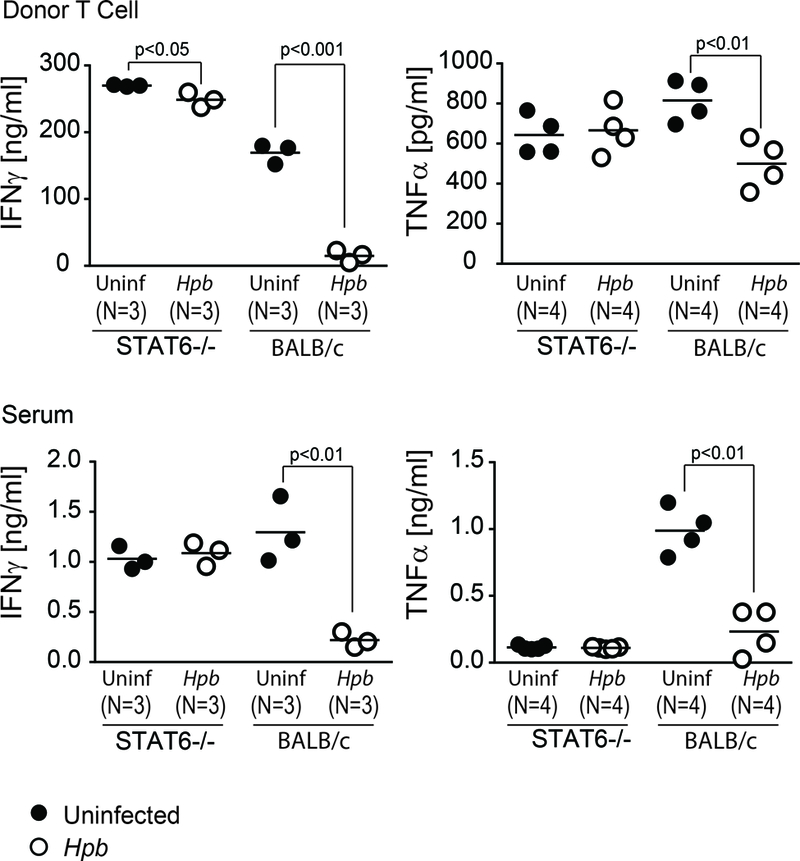
IFNγ and TNFα concentrations in donor T cells (upper panels) and serum (lower panels) of uninfected control (black filled dots) and Hpb-infected (white empty dots) STAT6–/– or WT (BALB/c) BMT recipients (N=the number of mice) six days after BMT. Cytokine concentrations were analyzed by ELISA, and results are displayed as mean with dot plot distribution. Dot plot distribution with mean of multiple samples (N) with each dot representing the mean for a single sample from multiple determinations (≥3) and each bar representing the mean from multiple samples; p values are provided in each panel.
Helminthic induction of T cell-stimulated TGFβ secretion is dependent on STAT6.
Given that helminthic regulation of GVHD is dependent on TGFβ(19), we investigated whether T cell-stimulated secretion of TGFβ following colonization by Hpb is dependent on STAT6. To this end we stimulated MLN cells from Hpb-infected or uninfected STAT6–/– and WT BALB/c mice with anti-CD3 and anti-CD28 in vitro. Unlike the MLN cells from colonized WT mice, those from colonized STAT6–/– mice did not secrete TGFβ in response T-cell stimulation (Figure 4).
Figure 4. Helminth-induced generation of active TGFβ requires STAT6.
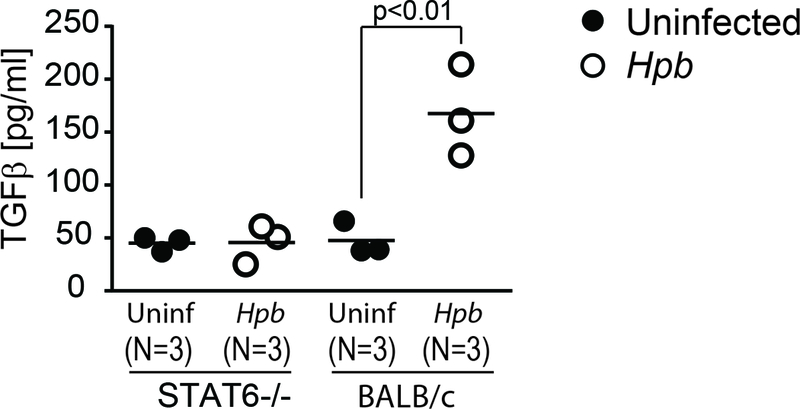
Mesenteric lymph node (MLN) T cells from Hpb-infected and uninfected 8–9 week-old male STAT6–/– or WT (BALB/c) mice were cultured in vitro with plate-bound anti-CD3 and soluble anti-CD28 for 48 hours. Supernatants were analyzed for TGFβ cytokine content by ELISA. Dot plot distribution with mean of multiple samples (N) with each dot representing the mean for a single sample from multiple determinations (≥3) and each bar representing the mean from multiple samples; p values are provided in each panel.
TGFβ is first generated as a propeptide (pro-TGFβ). The expression of pro-TGFβ can be determined by flow cytometry and by the quantification of mean fluorescence intensity (MFI) of the N-terminal portion of pro-TGFβ, the latency associated peptide (LAP)(38). We took advantage of this to determine whether helminthic induction of pro-TGFβ expression is dependent on STAT6. Pro-TGFβ content in the spleen and the MLN cell population was analyzed by flow cytometry in WT BALB/c mice and in STAT6−/− counterparts three weeks after the animals were administered 150 L3 Hpb larvae by gastric gavage. Although helminth infection led to an increase in LAP expression on Foxp3– CD4 T cells and Foxp3+ Tregs in the WT BALB/c mice, the levels of LAP on both T-cell subsets remained low in the STAT6–/– mice (Figure 5A and B). To determine whether helminth-induced and STAT6-dependent increase in TGFβ expression is regulated transcriptionally, we analyzed TGFβ mRNA synthesis by quantitative PCR (qPCR) in purified splenic or MLN CD4 T cells from uninfected, Hpb-colonized STAT6−/− or WT BALB/c mice. We did not observe an increase in TGFβ mRNA expression after helminth infection in samples from WT mice and we also did not observe an increase in samples from WT mice compared to samples from STAT6−/− counterparts (Figure 5C).
Figure 5. Helminth-induced expression of pro-TGFβ requires STAT6.
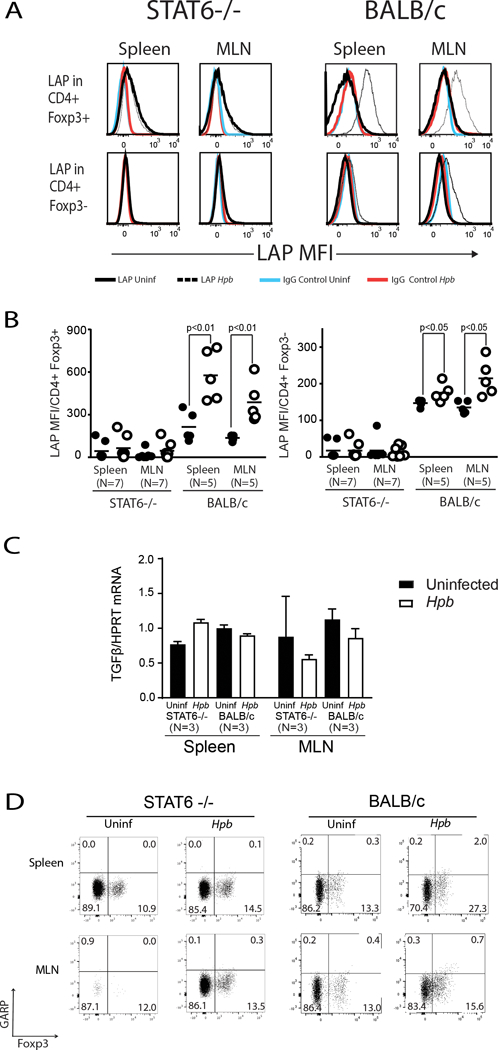
Freshly isolated splenic and MLN cells from Hpb-infected and uninfected 8–9 week-old male STAT6–/– or WT (BALB/c) mice were stained for CD3, CD4, Foxp3 and latency-associated peptide (LAP) or CD3, CD4, Foxp3 and control IgG. (A) Histograms showing mean fluorescence intensity (MFI) of LAP or control IgG staining on Foxp3+ CD4 or Foxp3- CD4 T cells from Hpb-infected (white empty dots) or uninfected (black filled dots) mice. (B) Dot plots representing cumulative data from experiments as in A, showing mean±SD for LAP staining from several independent determinations (N: the number of independent determinations; p values as indicated on the figure). (C) TGFβ mRNA content in purified splenic or MLN CD4 T cells from uninfected (Black rectangles) or helminth-infected (white rectangles) STAT6−/− or WT BALB/c mice were displayed as the ratio of TGFβ mRNA to HPRT mRNA. (N: number of independent measurements; p: not significant between uninfected and helminth-infected groups). (D) Spleen and MLN cells from uninfected and helminth-infected STAT6−/− or WT BALB/c mice were stained for CD3, CD4, Foxp3 and GARP. Cells were gated on CD3+ CD4+ T cells. Numbers represent the percentage of events in each quadrant. Data are representative example from at least two samples for each set.
Cell surface expression of LAP is also dependent on cell surface protein, glycoprotein A repetitions predominant protein (GARP), also called thrombospondin. GARP or thrombospondin serves as an anchor for cell surface LAP and is involved in regulating the bioavailability and activation of TGFβ (39). The expression of GARP in resting splenic or MLN Foxp3- CD4 T cell or Foxp3+ Tregs from WT mice did not increase after helminth infection (Figure 5D) in parallel to an increase in LAP expression (Figure 5A and B), although the expression of GARP increased significantly after in vitro stimulation with anti-CD3/28 (Supplemental Figure 1), as previously reported (40). Helminth infection was associated with a subtle increase in GARP expression in Foxp3+ but not Foxp3- CD4 T cells and same pattern of increase was evident in samples from WT BALB/c as well as STAT6−/− mice (Figure 5D). Hence the increase in LAP expression in T cells after helminth infection is not due to an increase in expression of the surface anchor protein, GARP. Together, these results suggest that STAT6 is critical to cellular expression of the TGFβ pro-peptide.
STAT6-dependent induction of TGFβ in the recipient is critical for the expansion of GVHD-regulating donor Tregs.
The STAT6-dependent generation of TGFβ that follows helminth infection is critical for the previously observed increase in Treg expansion after BMT(19). In the spleen and MLN of WT BALB/c BMT mice, the population of WT donor Foxp3+ Tregs increased, in terms of both total number and percentage of the CD3+ population, following helminth infection (Figure 6 and 7), as we had shown previously(19). However, in STAT6–/– BMT recipients, both numbers remained low. The number of recipient Tregs – which regulate GVHD during the early days after BMT(41) – likewise remained low in Hpb-infected STAT6–/– recipients (Figure 6 and 7). Add-back of TGFβ to STAT6–/– T cell cultures restored the generation of Tregs (Supplemental Figure 2), as described previously(42). These findings suggest that STAT6-dependent induction of TGFβ generation is critical to expansion of Foxp3+ Treg populations that suppress GVHD.
Figure 6. Helminth-induced increase in percentage of Foxp3+ Tregs requires host cell STAT6.
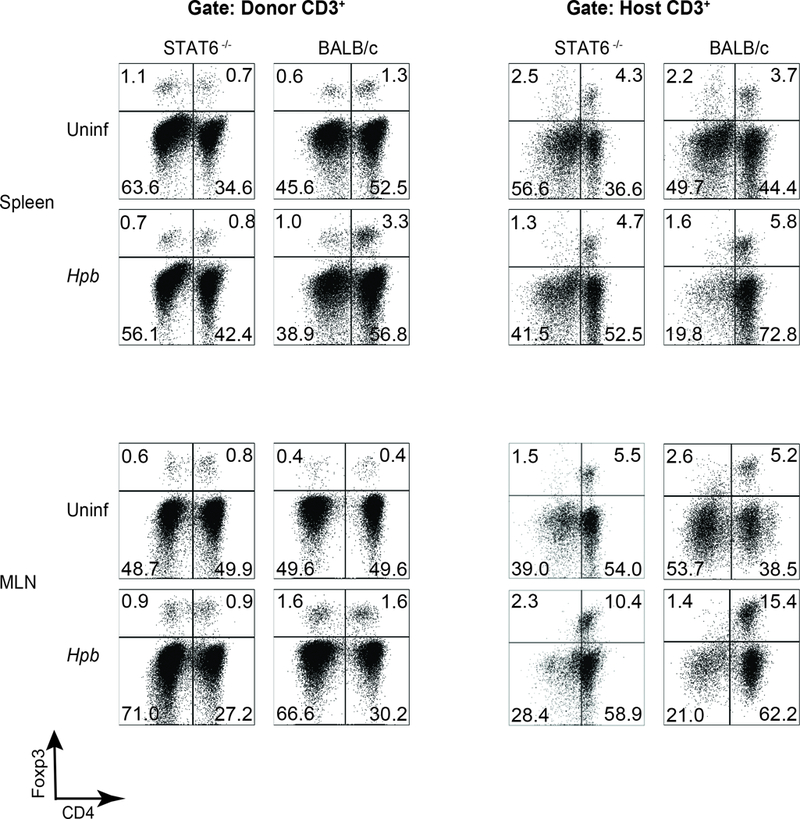
Representative dot plot FACS analysis from spleen (upper rows) and MLN (lower rows) cells isolated from uninfected (Uninf) and Hpb-infected (Hpb) STAT6–/– or WT (BALB/c) mice 6 days after BMT. Spleen and MLN cells were stained for CD3, CD4, H2b, H2d and Foxp3. Cells were gated on donor or host CD3+ T cells. Numbers represent the percentage of events in each quadrant.
Figure 7. Helminth-induced increase in Foxp3+ Treg percentage and number requires host cell STAT6 signaling.
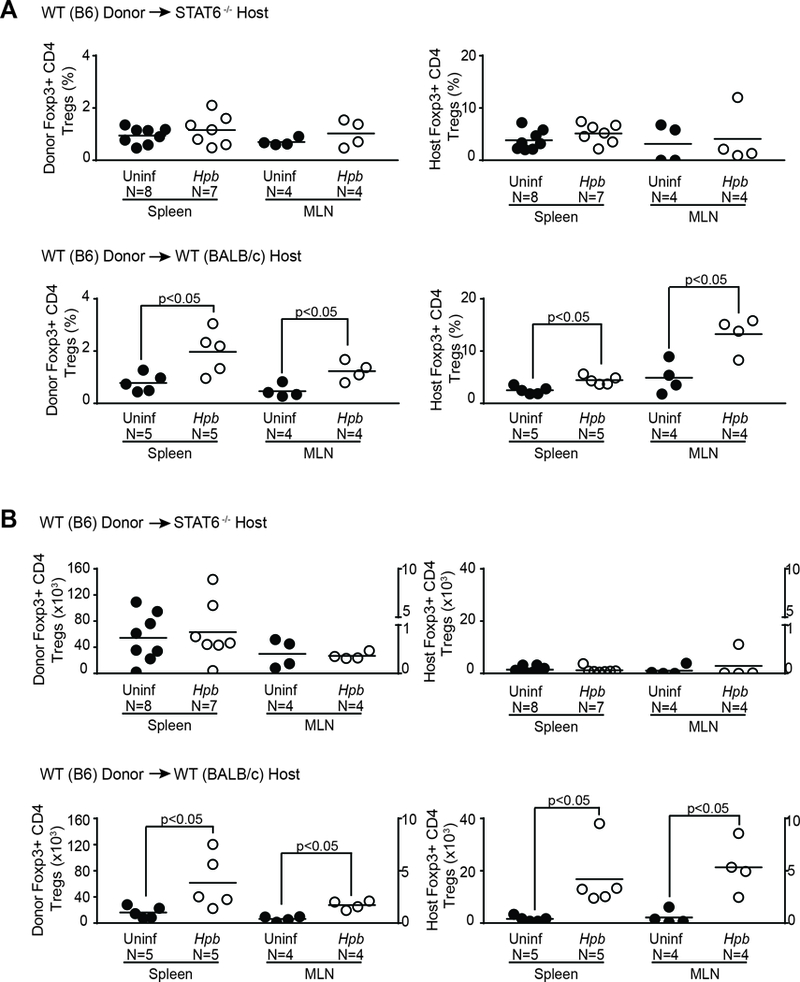
Data from multiple samples (N) analyzed as detailed in Figure 6, displayed as dot-plot distribution with means (bar), showing percentage (A) or number (B) of Foxp3+ CD4 Tregs. (A) Each dot represents the percentage of donor Foxp3+ donor CD4 Tregs among total donor T cells or the percentage of host (recipient) Foxp3+ CD4 Tregs among total host (recipient) T cells. P values as shown in each graph between Hpb-infected (Hpb; white empty dots) vs. uninfected (Uninf; black filled dots) groups. (B) The total number of splenic Foxp3+ CD4 Tregs (donor or host (recipient)-derived) was calculated from the percentage of donor or host Tregs and the number of cells isolated from a single spleen. The total number of MLN Foxp3+ CD4 Tregs (donor or host (recipient)-derived) was calculated from the percentage of donor or host Tregs and the number of cells isolated from MLN of a single mouse or pooled MLN from multiple mice. Each dot represents one experiment with bars representing mean values from multiple experiments. Left side Y-axis shows data for splenic cell analysis, whereas right-sided Y axis displays the results from MLN cell analysis.
STAT6 is necessary and sufficient to induce expression of the pro-TGFβ.
To determine whether STAT6 is sufficient to stimulate the generation of pro-TGFβ, we used a transgenic mouse in which T cells express a constitutively active form of STAT6 (STAT6 VT), under the control of a CD2 promoter(28). LAP expression was higher in Foxp3– CD4 T cells and Foxp3+ Tregs isolated from the spleen and MLN of STAT6VT+ (VT(+)) mice than in their VT- and WT counterparts (Figure 8A and B). Nonetheless, the secretion of active TGFβ cytokine by the VT+ cells was not higher than that by their VT- (VT(−)) and WT counterparts (Figure 9A). TGFβ mRNA content did not increase significantly in VT+ samples compared to their VT- counterparts, although TGFβ mRNA content was slightly higher in CD4 T cells from MLN samples (Figure 8C). This difference between spleen and MLN samples was not statistically significant (Figure 8C). We also analyzed LAP content in culture supernatants by ELISA. Nonetheless, the sensitivity of the ELISA was too low (detecting LAP concentrations >150–300 pg/ml) to permit analysis (data not shown). We also stained cells for T cell markers and GARP to determine whether the increase in LAP expression on VT+ T cells is caused by an increase in GARP expression. GARP expression was slightly increased in Foxp3+ CD4 Tregs – and not in Foxp3- CD4 T cells – in samples from VT+ mice (Figure 8D). The magnitude increase of GARP expression in Foxp3+ Tregs from VT+ mice was less than the magnitude increase of LAP expression in the same T cell subset (Figure 8A, B and D). Although expression of a constitutively active STAT6 protein (STAT6 VT) increases LAP expression in Foxp3- CD4 T cells, STAT6 VT does not increase the expression of GARP protein in Foxp3- CD4 T cells (Figure 8A, B and D). Hence, the increase in LAP (pro-TGFβ) expression in T cells from STAT6 VT mice is not due to enhanced expression of GARP. Our results also made us draw the conclusion that STAT6-mediated regulation of pro-TGFβ protein expression is posttranscriptionally regulated. Furthermore, STAT6 is necessary and sufficient to promote expression of the pro-TGFβ protein, but that other pathways are required for its proteolytic cleavage and secretion.
Figure 8. Continuous expression of active STAT6 in T cells is sufficient to stimulate the expression of pro-TGFβ.
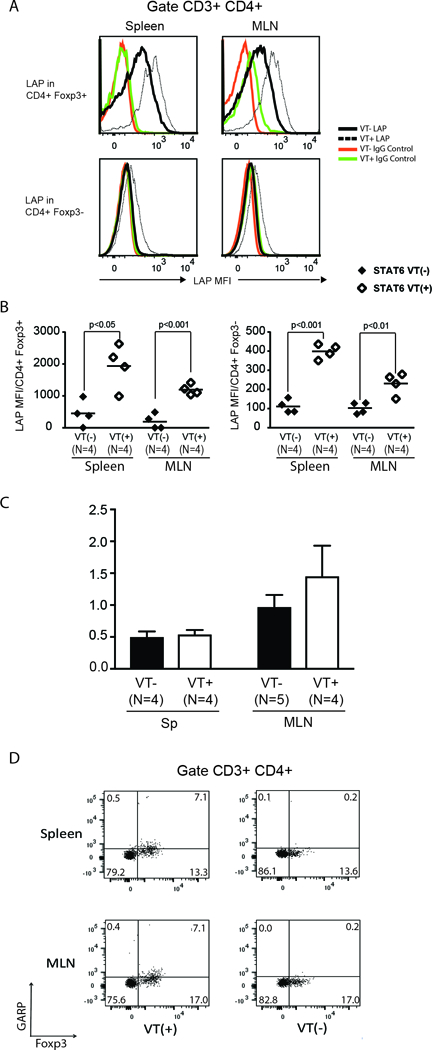
(A) Mean fluorescence intensity of LAP staining on Foxp3+ CD4+ and Foxp3- CD4+ T cells from spleen and MLN of STAT6 VT (STAT6 VT+; white histograms) mice and WT (STAT6 VT-) littermates (gray histograms). (B) Cumulative data showing mean±SD from several experiments (n: the number of independent experiment. Each dot represents a single experiment with bars representing mean values from multiple experiments; p value as indicated in each panel). (C) TGFβ mRNA content in purified splenic or MLN CD4 T cells from STAT6 VT- (Black rectangles) or STAT6 VT+ (white rectangles) mice were displayed as the ratio of TGFβ mRNA to HPRT mRNA. (N: number of independent measurements; p: not significant between STAT6 VT- and STAT6 VT+ groups). (D) Spleen and MLN cells from uninfected and helminth-infected STAT6 VT+ (VT(+)) or STAT6 VT- (VT(−)) mice were stained for CD3, CD4, Foxp3 and GARP. Cells were gated on CD3+ CD4+ T cells. Numbers represent the percentage of events in each quadrant. Data are representative example from three samples for each set.
Figure 9. Furin expression by T cells is necessary for the generation of active TGFβ but not for pro-TGFβ.
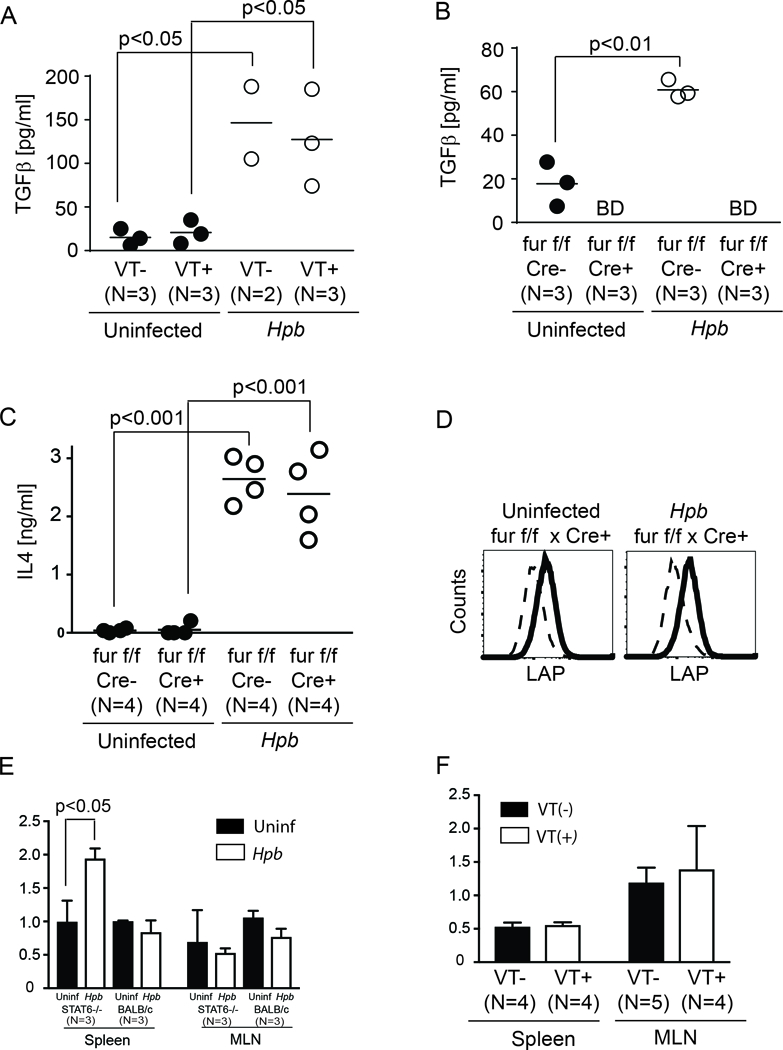
(A) Dot plots showing TGFβ concentration in MLN T cells isolated from uninfected (black filled dots) or Hpb-infected (white empty dots) STAT6 VT+ and VT- mice, as assessed by TGFβ ELISA using supernatants from 48-hr anti-CD28 stimulated cultures. Cumulative data from several independent experiments. Dot plot distribution with mean of multiple samples (N) with each dot representing the mean for a single sample from multiple determinations (≥3) and each bar representing the mean from multiple samples; p values are provided in each panel. (B) Dot plot distribution of TGFβ concentration in MLN T cells isolated from uninfected (black filled dots) or Hpb-infected (white empty dots) furin-expressing (Cre-) or furin-deficient (Cre+) mice, as assessed by TGFβ ELISA as described in A. Dot plot distribution with mean of multiple samples (N) with each dot representing the mean for a single sample from multiple determinations (≥3) and each bar representing the mean from multiple samples; p values as shown; BD: below detection. (C) Dot plot distribution of IL4 concentration - as measured by ELISA - in MLN T cells isolated and cultured as in Figure 9B. Dot plot distribution with mean of multiple samples (N) with each dot representing the mean for a single sample from multiple determinations (≥3) and each bar representing the mean from multiple samples; p values as shown; BD: below detection. (D) LAP expression (bold) compared to isotype control antibody staining (dashed) in furin deficient (Cre+) uninfected or Hpb-colonized MLN T cells (representative example from 3 independent experiments). (E) Furin mRNA content in purified splenic or MLN CD4 T cells from uninfected (Black rectangles) or helminth-infected (white rectangles) STAT6−/− or WT BALB/c mice were displayed as the ratio of furin mRNA to HPRT mRNA. (N: number of independent measurements; p: not significant between uninfected and helminth-infected groups, unless indicated otherwise on the figure). (F) Furin mRNA content in purified splenic or MLN CD4 T cells from STAT6 VT- (Black rectangles) or STAT6 VT+ (white rectangles) mice were displayed as the ratio of TGFβ mRNA to HPRT mRNA. (N: number of independent measurements; p: not significant between STAT6 VT- and STAT6 VT+ groups).
Generation of active TGFβ from pro-TGFβ requires furin.
To determine whether helminths induce an increase in TGFβ cytokine secretion in MLN cells from STAT6 VT+ mice, we stimulated CD3+ MLN T cells from Hpb-infected and uninfected STAT6 VT mice in vitro for 48 hours. As shown by ELISA, TGFβ secretion by STAT6 VT+ T cells increased significantly in the presence of helminths (Figure 9A). Helminth-induced increase in TGFβ secretion by VT+ T cells was similar to helminth-induced increase in TGFβ secretion by VT- T cells.
Helminths can condition TGFβ immunity utilizing host pathways or their own products(43), and a previous study had demonstrated that T-cell-specific activation of TGFβ requires the endopeptidase furin(12). To explore the possibility that helminth-induced generation of active TGFβ requires furin, we analyzed the cellular expression of pro-TGFβ as well as the secretion of active TGFβ in uninfected and Hpb-infected T cells from CD4 Cre x furinfl/fl mice, whose T cells have a genetic deficiency for furin due to the T cell-specific activity of Cre recombinase. The secretion of active TGFβ remained undetectable in furin−/− CD4 T cell samples (Figure 9B), although helminth infection stimulated Th2 pathway - as shown by IL4 production - and LAP expression in MLN cells from the furinfl/fl x CD4 Cre mice (Figure 9C and D). The expression of furin mRNA did not increase after helminth infection in WT BALB/c samples, although furin mRNA content increased in splenic CD4 T cells from STAT6−/− mice after colonization with Hpb (Figure 9E). Furin mRNA content did not change significantly in splenic or MLN CD4 T cells between STAT6 VT+ and VT- samples (Figure 9F). Hence the expression of furin is not associated with Th2 polarization associated with helminth infection or the expression of a constitutively active STAT6 protein. These results lead us to propose a model for T cell TGFβ production in the periphery, where STAT6 is necessary and sufficient for the expression of pro-TGFβ and where an endopeptidase such as furin is required for production of the active cytokine and its secretion by T cells. Other cellular proteins such as GARP (thrombospondin) also play a role in surface expression of TGFβ.
Discussion
In this study, we took the advantage of a helminth infection model, in which T cells are stimulated to generate inactive pro-TGFβ, secrete active TGFβ, and undergo Th2 maturation. Our strategy enabled us to shed light on the molecular mechanisms that lead to TGFβ production by T cells. First, we demonstrated that the Th2-specific transcription factor STAT6 is necessary and sufficient for the generation of pro-TGFβ. Second, we showed that Th2-inducing transcription factor STAT6 is required for helminth-mediated suppression of GVHD. Third, our findings revealed that the endopeptidase furin is required for secretion of the cleaved and active C-terminal cytokine after helminth infection. Together, these results constitute evidence for the existence of a “two-step” cellular program that is critical for T cell TGFβ generation. The combination of furin-mediated and STAT6-dependent TGFβ generation by T cells hints at a mechanism that might account for persistent activity of cellular pathways, which leads to Th2 differentiation and cellular TGFβ generation after helminth infection. According to this “two-step” model, T cells with active Th2 signaling (STAT6) develop into TGFβ generating T cells in the context of endopeptidase activity.
In conditions without helminth infection, Th2 cytokines and TGFβ can counter-regulate each other, with only one of these pathways dominating the immune response. TGFβ is a potent inhibitor of the Th2 pathway(1) where T cells deficient for TGFβ signaling due to T cell-specific over-expression of a dominant-negative TGFβ receptor II (TGFβ RII DN) spontaneously differentiate into Th2 cells(44, 45). After helminth infection, TGFβ - generated in response to Th2 stimulation - can still exert some level of inhibition on an induced Th2 pathway, as evident from the fact that, when T cells from TGFβ RII DN mice are exposed to helminths, the already robust Th2 cytokine production is further enhanced(46).
Likewise, elements of Th2 pathway can inhibit TGFβ-dependent cellular events. For example, Th2 cytokine IL4 prevents the generation of Foxp3+ Tregs from naïve CD4 T cells(47). IL4 activates STAT6 - and similarly - STAT6 inhibits TGFβ-dependent generation of Tregs(42). Nonetheless, the percentage of Tregs is reduced in STAT6–/– mice and increased in mice whose T cells express a constitutively active form of STAT6(48). Based on these studies, it has been proposed that STAT6 not only inhibits TGFβ-mediated activation of Tregs but also constitutes a nonredundant signal that stimulates expansion and the maintenance of peripheral Tregs(48). We propose that the effects of active STAT6 on Tregs depends on a second (cleavage) step: in the absence of furin or another endopeptidase that cleaves pro-TGFβ, active TGFβ will not be generated and STAT6 will inhibit the TGFβ-dependent activation of Tregs; and in the presence of furin, TGFβ will be generated to stimulate the expansion of functional Tregs.
STAT6 is activated by the Th2 cytokines, IL4 and IL13, and it consecutively stimulates expression of the master regulator of the Th2 pathway, GATA3 (49). Activation of STAT6 or GATA3 can inhibit Tregs(42, 50). Nonetheless, in Hpb-infected and Th2-polarized mice, Foxp3+ Tregs express abundant GATA3(51) and this expression is critical to the maintenance of suppressive function. We showed previously that helminth-induced expansion of Foxp3+ Tregs and the suppression of inflammation in a BMT-based GVHD model is dependent on TGFβ(19). Here we build on these findings by showing that helminth-induced generation of TGFβ is dependent on STAT6, with constitutive STAT6 signaling in T cells being sufficient to induce the expression of pro-TGFβ and helminth infection activating pro-TGFβ in a furin-dependent manner. The role of GATA3 in this phenomenon and in T cell TGFβ generation is unexplored.
TGFβ production by T cells was previously characterized in the context of oral tolerance, where the presentation of antigen through the gut promotes the generation of T cell clones that produce not only TGFβ but also IL4(8). A similar outcome was observed after helminth infection, with T cell being stimulated to produce TGFβ and IL4(31, 46). Further studies of oral tolerance have shown that the delivery of antigen through the gut results in the generation of Tregs that express pro-TGFβ(52). These pro-TGFβ+ T cells execute T cell tolerance in a TGFβ-dependent manner. As pro-TGFβ is an inactive protein, it is unknown how Tregs that express pro-TGFβ regulate immunity in a TGFβ-dependent manner. Cytokine activation by endopeptidase-mediated proteolytic cleavage of pro-TGFβ provides a possible explanation for how pro-TGFβ expressing Tregs suppress immune responses through TGFβ-dependent pathways.
In our study we focused on the effects of GARP (thrombospondin) for the attachment of pro-TGFβ on T cell surface and on the effects of furin as an endopeptidase for cleavage of pro-TGFβ. However, other cellular proteins have also been shown to effect this cleavage and TGFβ activation. Besides GARP (thrombospondin), these cellular proteins include matrix metalloproteases and integrins (10, 13, 53, 54), such as αVβ8 integrin. Our results do not attest to an important role of GARP in STAT6-dependent increase of GARP expression on T cells for the following reasons: First, the magnitude increase of GARP expression does not parallel the STAT6-dependent fold-increase of LAP expression in Foxp3+ CD4 Tregs (Figures 5 and 8). Second, helminth-induced increase in GARP expression was of similar extent in Foxp3+ CD4 Tregs from STAT6−/− and BALB/c WT samples (Figure 5). Last, in Foxp3- CD4 T cells, LAP expression increased in a STAT6-dependent manner but GARP expression did not increase (Figures 5 and 8). The increase in GARP expression on Foxp3+ CD4 Tregs can be a result of Treg activation, because helminth infection activates the immune system in vivo and expression of STAT6 VT protein in T cells gives them an activated phenotype (28). Nonetheless, in vitro activation of T cells with a saturating dose of anti-CD3 and 28 results in a robust increase in GARP expression in both, Foxp3+ CD4 Tregs and Foxp3- CD4 T cells (Supplemental Figure 1). This discrepancy can be related to a difference between T cell activation in vivo and in vitro. It is also possible that the increase of GARP expression in T cell subsets, is related to other, less defined factors.
Next we focused on furin for the cleavage of TGFβ. Previous reports indicated that furin and integrins can have nonredundant roles in activating TGFβ in different cellular contexts(10, 12) and our results suggest that furin is critical to helminth-induced TGFβ production by T cells. The involvement of multiple mechanisms might ensure that TGFβ is activated under various inflammatory or homeostatic conditions to prevent immune pathologies(55). This idea is supported by studies in mice deficient for matrix metalloproteases where cleavage of TGFβ and activation of the cytokine appears to be intact (56).
Interestingly, transient expression of active TGFβ in T cells via a transgene results in immune regulation(38), whereas its continuous expression in T lymphocytes augments T-cell apoptosis, inhibits T-cell proliferation and leads to thymic atrophy(57). Thus a check-point for TGFβ activation besides protein expression may be essential for regulation of the immune system. Expression of the activating enzyme by cells other than T lymphocytes(58) – which are the source of the majority of pro-TGFβ – could potentially also provide such a check-point and further contribute to the complexity of TGFβ-mediated immune regulation that includes additional intra- and extracellular proteins to modulate TGFβ circuitries.
Collectively, the findings presented here identify a novel pathway of TGFβ production that opens new avenues for research. Follow-up studies on the mechanisms underlying STAT6-mediated expression of pro-TGFβ, the activity of furin, and the contributions of other proteins that are capable of cleaving pro-TGFβ in various contexts involving polarized immune responses will lead to a deeper understanding of this multistep pathway of TGFβ secretion. A more concrete understanding of the key players in TGFβ regulation will have important implications for our understanding of immune regulation in the gut. Further in detail characterization of the TGFβ generation by T cells is expected to lead to the development of treatments, which might be effective for diseases such as lethal and devastating GVHD.
Supplementary Material
Acknowledgments
This study was supported by research funds from the National Institute of Health R56 AI 116715 (MNI); R01 HL56067, AI34495, HL11879 (BRB); R01 AI095282 (MHK), Department of Veterans Affairs BX002906 (MNI), BX002715 (DEE), FWO Vlaanderen G.0738.15N, Belgium (JWC), the Sigrid Juselius Foundation and the Academy of Finland (MP).
Footnotes
Conflict of Interest Disclosure
The authors declare no conflict of financial interest
References
- 1.Li MO, and Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134(3):392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li MO, Wan YY, Sanjabi S, Robertson AK, and Flavell RA. Transforming growth factor-beta regulation of immune responses. AnnuRevImmunol. 2006;24(99–146. [DOI] [PubMed] [Google Scholar]
- 3.Travis MA, and Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol. 2014;32(51–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banovic T, Macdonald KP, Morris ES, Rowe V, Kuns R, Don A, Kelly J, Ledbetter S, Clouston AD, and Hill GR. TGF-beta in allogeneic stem cell transplantation: friend or foe? Blood. 2005;106(6):2206–14. [DOI] [PubMed] [Google Scholar]
- 5.Sedda S, Marafini I, Dinallo V, Di Fusco D, and Monteleone G. The TGF-beta/Smad System in IBD Pathogenesis. Inflamm Bowel Dis. 2015;21(12):2921–5. [DOI] [PubMed] [Google Scholar]
- 6.Monteleone G, and Pallone F. Mongersen, an Oral SMAD7 Antisense Oligonucleotide, and Crohn’s Disease. N Engl J Med. 2015;372(25):2461. [DOI] [PubMed] [Google Scholar]
- 7.Li MO, Wan YY, and Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26(5):579–91. [DOI] [PubMed] [Google Scholar]
- 8.Weiner HL, da Cunha AP, Quintana F, and Wu H. Oral tolerance. Immunol Rev. 2011;241(1):241–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, and Li MO. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34(3):396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Worthington JJ, Kelly A, Smedley C, Bauche D, Campbell S, Marie JC, and Travis MA. Integrin alphavbeta8-Mediated TGF-beta Activation by Effector Regulatory T Cells Is Essential for Suppression of T-Cell-Mediated Inflammation. Immunity. 2015;42(5):903–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rezende RM, da Cunha AP, Kuhn C, Rubino S, M’Hamdi H, Gabriely G, Vandeventer T, Liu S, Cialic R, Pinheiro-Rosa N, et al. Identification and characterization of latency-associated peptide-expressing gammadelta T cells. Nat Commun. 2015;6(8726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, Andersson J, Shevach EM, Quezado M, Bouladoux N, et al. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature. 2008;455(7210):246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oida T, Zhang X, Goto M, Hachimura S, Totsuka M, Kaminogawa S, and Weiner HL. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. Journal of immunology. 2003;170(5):2516–22. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Kuchroo VK, Inobe J, Hafler DA, and Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265(5176):1237–40. [DOI] [PubMed] [Google Scholar]
- 15.Grencis RK. Immunity to helminths: resistance, regulation, and susceptibility to gastrointestinal nematodes. Annu Rev Immunol. 2015;33(201–25. [DOI] [PubMed] [Google Scholar]
- 16.Weinstock JV, and Elliott DE. Helminth Infections Decrease Host Susceptibility to Immune-Mediated Diseases. J Immunol. 2014;193(7):3239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ince MN, Elliott DE, Setiawan T, Metwali A, Blum A, Chen HL, Urban JF, Flavell RA, and Weinstock JV. Role of T cell TGF-beta signaling in intestinal cytokine responses and helminthic immune modulation. EurJImmunol. 2009;39(7):1870–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ince MN, Elliott DE, Setiawan T, Blum A, Metwali A, Wang Y, Urban JF Jr, and Weinstock JV. Heligmosomoides polygyrus induces TLR4 on murine mucosal T cells that produce TGFbeta after lipopolysaccharide stimulation. JImmunol. 2006;176(2):726–9. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Chen HL, Bannick N, Henry M, Holm AN, Metwali A, Urban JF Jr., Rothman PB, Weiner GJ, Blazar BR, et al. Intestinal helminths regulate lethal acute graft-versus-host disease and preserve the graft-versus-tumor effect in mice. J Immunol. 2015;194(3):1011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gause WC, and Maizels RM. Macrobiota - helminths as active participants and partners of the microbiota in host intestinal homeostasis. Curr Opin Microbiol. 2016;32(14–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elo LL, Jarvenpaa H, Tuomela S, Raghav S, Ahlfors H, Laurila K, Gupta B, Lund RJ, Tahvanainen J, Hawkins RD, et al. Genome-wide profiling of interleukin-4 and STAT6 transcription factor regulation of human Th2 cell programming. Immunity. 2010;32(6):852–62. [DOI] [PubMed] [Google Scholar]
- 22.Metwali A, Blum A, Elliott DE, and Weinstock JV. Interleukin-4 receptor alpha chain and STAT6 signaling inhibit gamma interferon but not Th2 cytokine expression within schistosome granulomas. Infect Immun. 2002;70(10):5651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madden KB, Yeung KA, Zhao A, Gause WC, Finkelman FD, Katona IM, Urban JF Jr., and Shea-Donohue T. Enteric nematodes induce stereotypic STAT6-dependent alterations in intestinal epithelial cell function. Journal of immunology. 2004;172(9):5616–21. [DOI] [PubMed] [Google Scholar]
- 24.Paul WE. History of interleukin-4. Cytokine. 2015;75(1):3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goenka S, and Kaplan MH. Transcriptional regulation by STAT6. Immunol Res. 2011;50(1):87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei L, Vahedi G, Sun HW, Watford WT, Takatori H, Ramos HL, Takahashi H, Liang J, Gutierrez-Cruz G, Zang C, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32(6):840–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roebroek AJ, Taylor NA, Louagie E, Pauli I, Smeijers L, Snellinx A, Lauwers A, Van de Ven WJ, Hartmann D, and Creemers JW. Limited redundancy of the proprotein convertase furin in mouse liver. J Biol Chem. 2004;279(51):53442–50. [DOI] [PubMed] [Google Scholar]
- 28.Bruns HA, Schindler U, and Kaplan MH. Expression of a constitutively active Stat6 in vivo alters lymphocyte homeostasis with distinct effects in T and B cells. J Immunol. 2003;170(7):3478–87. [DOI] [PubMed] [Google Scholar]
- 29.Persson L A modified baermann apparatus for the recovery of infective nematode larvae from herbage and manure. Zentralbl Veterinarmed B. 1974;21(7):483–8. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann P, Ermann J, Edinger M, Fathman CG, and Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. JExpMed. 2002;196(3):389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Setiawan T, Metwali A, Blum AM, Ince MN, Urban JF Jr, Elliott DE, and Weinstock JV. Heligmosomoides polygyrus promotes regulatory T-cell cytokine production in the murine normal distal intestine. InfectImmun. 2007;75(9):4655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pillai AB, George TI, Dutt S, and Strober S. Host natural killer T cells induce an interleukin-4-dependent expansion of donor CD4+CD25+Foxp3+ T regulatory cells that protects against graft-versus-host disease. Blood. 2009;113(18):4458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pillai AB, George TI, Dutt S, Teo P, and Strober S. Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation. JImmunol. 2007;178(10):6242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr., Crawford JM, and Ferrara JL. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88(8):3230–9. [PubMed] [Google Scholar]
- 35.Kaplan DH, Anderson BE, McNiff JM, Jain D, Shlomchik MJ, and Shlomchik WD. Target antigens determine graft-versus-host disease phenotype. JImmunol. 2004;173(9):5467–75. [DOI] [PubMed] [Google Scholar]
- 36.Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, and Serody JS. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood. 2009;113(6):1365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shea-Donohue T, Sun R, Bohl JA, McLean LP, and Zhao A. Enteric nematodes and the path to up-regulation of type 2 cytokines IL-4 and IL-13. Cytokine. 2015;75(1):62–7. [DOI] [PubMed] [Google Scholar]
- 38.Carrier Y, Yuan J, Kuchroo VK, and Weiner HL. Th3 cells in peripheral tolerance. I. Induction of Foxp3-positive regulatory T cells by Th3 cells derived from TGF-beta T cell-transgenic mice. Journal of immunology. 2007;178(1):179–85. [DOI] [PubMed] [Google Scholar]
- 39.Metelli A, Salem M, Wallace CH, Wu BX, Li A, Li X, and Li Z. Immunoregulatory functions and the therapeutic implications of GARP-TGF-beta in inflammation and cancer. J Hematol Oncol. 2018;11(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oida T, and Weiner HL. TGF-beta induces surface LAP expression on murine CD4 T cells independent of Foxp3 induction. PLoS One. 2010;5(11):e15523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inoue T, Ikegame K, Kaida K, Okada M, Yoshihara S, Tamaki H, Fujimori Y, Soma T, and Ogawa H. Host Foxp3+CD4+ Regulatory T Cells Act as a Negative Regulator of Dendritic Cells in the Peritransplantation Period. J Immunol. 2016;196(1):469–83. [DOI] [PubMed] [Google Scholar]
- 42.Takaki H, Ichiyama K, Koga K, Chinen T, Takaesu G, Sugiyama Y, Kato S, Yoshimura A, and Kobayashi T. STAT6 Inhibits TGF-beta1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem. 2008;283(22):14955–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grainger JR, Smith KA, Hewitson JP, McSorley HJ, Harcus Y, Filbey KJ, Finney CA, Greenwood EJ, Knox DP, Wilson MS, et al. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. JExpMed. 2010;207(11):2331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorelik L, Fields PE, and Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. JImmunol. 2000;165(9):4773–7. [DOI] [PubMed] [Google Scholar]
- 45.Gorelik L, and Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12(2):171–81. [DOI] [PubMed] [Google Scholar]
- 46.Ince MN, Elliott DE, Setiawan T, Metwali A, Blum A, Chen HL, Urban JF, Flavell RA, and Weinstock JV. Role of T cell TGF-beta signaling in intestinal cytokine responses and helminthic immune modulation. Eur J Immunol. 2009;39(7):1870–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, and Qin FX. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2007;104(46):18169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanchez-Guajardo V, Tanchot C, O’Malley JT, Kaplan MH, Garcia S, and Freitas AA. Agonist-driven development of CD4+CD25+Foxp3+ regulatory T cells requires a second signal mediated by Stat6. J Immunol. 2007;178(12):7550–6. [DOI] [PubMed] [Google Scholar]
- 49.Paul WE. What determines Th2 differentiation, in vitro and in vivo? Immunol Cell Biol. 2010;88(3):236–9. [DOI] [PubMed] [Google Scholar]
- 50.Mantel PY, Kuipers H, Boyman O, Rhyner C, Ouaked N, Ruckert B, Karagiannidis C, Lambrecht BN, Hendriks RW, Crameri R, et al. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol. 2007;5(12):e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, Hall JA, Yagi R, Naik S, Bhairavabhotla R, et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest. 2011;121(11):4503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen ML, Yan BS, Bando Y, Kuchroo VK, and Weiner HL. Latency-associated peptide identifies a novel CD4+CD25+ regulatory T cell subset with TGFbeta-mediated function and enhanced suppression of experimental autoimmune encephalomyelitis. Journal of immunology. 2008;180(11):7327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Q, and Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163–76. [PMC free article] [PubMed] [Google Scholar]
- 54.Worthington JJ, Fenton TM, Czajkowska BI, Klementowicz JE, and Travis MA. Regulation of TGFbeta in the immune system: an emerging role for integrins and dendritic cells. Immunobiology. 2012;217(12):1259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fenton TM, Kelly A, Shuttleworth EE, Smedley C, Atakilit A, Powrie F, Campbell S, Nishimura SL, Sheppard D, Levison S, et al. Inflammatory cues enhance TGFbeta activation by distinct subsets of human intestinal dendritic cells via integrin alphavbeta8. Mucosal Immunol. 2017;10(3):624–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Annes JP, Munger JS, and Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116(Pt 2):217–24. [DOI] [PubMed] [Google Scholar]
- 57.Schramm C, Huber S, Protschka M, Czochra P, Burg J, Schmitt E, Lohse AW, Galle PR, and Blessing M. TGFbeta regulates the CD4+CD25+ T-cell pool and the expression of Foxp3 in vivo. Int Immunol. 2004;16(9):1241–9. [DOI] [PubMed] [Google Scholar]
- 58.Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, Wang Y, Bernstein X, Huang X, Reichardt LF, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449(7160):361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.