

Abstract
Low-dose IL-2 represents an immunotherapy to selectively expand regulatory T cells (Tregs) to promote tolerance in patients with autoimmunity. Here we show that a fusion protein (FP) of mouse IL-2 and mouse IL-2Rα (CD25), joined by a non-cleavable linker, has greater in vivo efficacy than recombinant IL-2 at Treg expansion and control of autoimmunity. Biochemical and functional studies support a model where IL-2 interacts with CD25 in the context of this FP in trans to form inactive head-to-tail dimers that slowly dissociate into an active monomer. In vitro, IL-2/CD25 has low specific activity. However, in vivo IL-2/CD25 is long-lived to persistently and selectively stimulate Tregs. In female NOD mice, IL-2/CD25 administration increased Tregs within the pancreas and reduced the instance of spontaneous diabetes. Thus, IL-2/CD25 represents a distinct class of IL-2 FPs with the potential for clinical development for use in autoimmunity or other disorders of an overactive immune response.
Introduction
IL-2 is an extensively studied cytokine that regulates key aspects of the immune system. IL-2 was discovered as a potent T cells growth factor (1, 2) and many studies have focused on its role in promoting immune responses. In this regard, besides promoting clonal expansion of antigen-activated T cells, IL-2 drives development of CD4+ Th1 and Th2 cells, and terminally differentiated CD8+ CTLs, and also shapes T cell memory recall responses (3) while opposing development of CD4+ Th17 and T-follicular helper (Tfh) cells (4–6). Paradoxically, gene ablation of IL-2 or IL-2R subunits led to lympho-proliferation and rapid lethal systemic autoimmunity rather than to grossly impaired immune responses (7–10). This phenotype results from the loss of the essential role IL-2 plays in regulatory T cell (Treg) development and homeostasis (11–13), in part due to IL-2-dependent activation of STAT5 that directly promotes Foxp3 transcription (14).
Many clinical trials have been performed to harness the T cell activating properties of IL-2 in the context of patients with cancer and HIV/AIDS by infusion of high doses of IL-2 (typically > 500,000 units/kg, repeatedly) to boost T and NK cells. IL-2 was approved by the FDA for use in patients with melanoma and renal cell carcinoma as some (approximately 5%) exhibited complete remissions. Not only were the response rates low in these and other cancers, but this therapy was also accompanied by severe toxicity (15). IL-2 was deemed to be not effective in promoting immunity in HIV/AIDS patients. The poor efficacy of high dose IL-2 in these settings is due in part to the accompanying expansion of Tregs (15, 16). More recently, preclinical studies showed that low IL-2R signaling selectively promoted key activities of Tregs but not T effector (Teff) cells and that treatment of mice with low levels of IL-2 prevented autoimmunity (17–19). Currently, a number of patients with hyperactive pathogenic immune responses have been treated with low-dose IL-2 (0.5–2 million units). The experience thus far showed that the therapy is safe, increases Tregs in nearly all patients, and leads to clinical improvement, with no indication of reactivation of auto-aggressive T cells (20–22). However, this treatment requires daily injections and dosing is limited by the observation of increases in pro-inflammatory cytokines and increases in non-Tregs
The IL-2R exists in two functional forms. The high affinity IL-2R, which is found at high levels on Tregs but lower levels on Teff cells, assembles when IL-2 is captured by the IL-2Rα (CD25) subunit that in turn drives additional IL-2 binding and subsequent signaling by the IL-2Rβ (CD122) and γc (CD132) subunits; the intermediate affinity IL-2R occurs when IL-2 binds to directly to CD122 and CD132, where these subunits are found on many antigen-experience CD8+ T cells and most NK cells (4). IL-2 has a very short half-life in the serum (<15 min) (23, 24). Treg-directed IL-2-based therapy will likely benefit from IL-2 analogs that are longer-lasting with more selectively toward Tregs. Increased persistence of IL-2 molecules has been achieved by fusion of IL-2 to molecules such as immunoglobulins or albumin (25–27), but these molecules still have suboptimal pharmacokinetics and do not show selectivity for the high affinity IL-2R expressed by Tregs. IL-2 in complex with specific antibodies has demonstrated increased persistence and selectivity toward cells bearing the high affinity IL-2R (28). However, this represents a complicated approach that may present challenges in clinical development and material production. Alternatively, some identified mutations of IL-2 selectively interfere with the ability of IL-2 to bind to IL-2Rα or IL-2Rβ, conferring selectivity toward activating cells bearing the intermediate affinity IL-2R or high affinity IL-2R, respectively (29–33). These molecules, however, still have a short half-life, similar to recombinant IL-2 without the mutations, limiting their use in in vivo settings.
To increase IL-2 activity in the tumor microenvironment and anti-tumor immunity, minimally active IL-2/CD25 FPs have also been developed (34). In these FPs, IL-2 is linked to CD25 with a protease cleavable linker such that the IL-2 moiety is released by endogenous proteases that are over-expressed by some tumor microenvironments (34). Although this approach improved anti-tumor responses, this design is unlikely to have optimal longevity to persistently stimulate Tregs because the released IL-2 would be rapidly cleared from the circulation, comparable to free IL-2.
To persistently and effectively stimulate T cells, we constructed an IL-2/CD25 FP that contains a non-cleavable linker between IL-2 and CD25. As shown herein, the non-cleavable IL-2/CD25 FP increased the persistence of available IL-2 in the circulation due to increased molecular mass. This design was anticipated to favor CD8 and NK activation by eliminating the role of cell surface CD25 for IL-2R signaling. IL-2 was expected to bind CD25 in cis within the FP, as previously reported (34), to directly present IL-2 to cells bearing the intermediate affinity IL-2R. However, unexpectedly, the molecule showed impressive selectivity for the high affinity IL-2R of Tregs both in vivo and in vitro. Contrary to a cis interaction between IL-2 and CD25 to form monomers in the cleavable IL-2/CD25 fusion protein (34), we found that our IL-2/CD25 FP formed dimers in trans that appear to periodically dissociate, thereby allowing IL-2 to signal through cell-associated IL-2R. The data herein demonstrate that the non-cleavable IL-2/CD25 FP is highly selective to Tregs and suppresses diabetes in NOD mice.
Material and Methods
Preparation, expression and purification of IL-2/CD25
Primers for PCR amplifications are shown in Supplemental Table 1. All PCR amplification utilized Q5 hot start high-fidelity DNA polymerase (NEB). The pCIneo vector (Promega) was linearized with Sal I and PCR-amplified using primers 1/2 (Supplemental Table 1) to generate a modified vector that encoded a Xho I site and Kozak sequence at the 5’ end and a Not I site, 2 termination codons, a 6×-His affinity tag and two Gly spacer residues at the 3’end of the vector. After completion of the PCR, residual linearized unmodified vector was digested with Dpn I at 37°C for 45 min followed immediately by incubation at 80°C for 20 min to inactivate Dpn I. The modified linearized vector was gel purified for down-stream applications.
Vectors containing full length mouse IL-2 (mIL-2), mouse CD25 (mCD25) and human CD25 (hCD25) were used to generate PCR fragments necessary to prepare the FPs. PCR of mIL-2 utilized primers 3/4, 3/5, 3/6, 3/7, and 3/8 whereas PCR of mCD25 utilized primers 9/10, 9/11, 9/12, 9/13, and 9/14 to generate the fragments required to assemble FP with a 7 amino acid Gly, or 12, 16, 20, and 25 amino acid Gly/Ser linkers. PCR of hCD25 used primers 15/16 to generate the fragment required for preparation of mIL-2/hCD25 FP. All PCR fragments were gel purified. The resulting fragments were assembled by a one-step cloning reaction using the Gibson Assembly Master Mix (NEB), according to the manufacturer’s instructions, that ligated the appropriate IL-2 and CD25 PCR fragments in the desired orientation into the modified linearized pCIneo vector. After transformation of E. coli, individual clones containing FP inserts were selected and their DNA sequence was determined to verify each FP.
FPs cDNAs were cloned into pIRES-ZsGreen 1 vector (Clontech), which places the FPs upstream of the ZsGreen1 reporter to readily identify transfected cells. The modified pCIneo containing FP vectors were digested with Not I, heat inactivated, and the DNA ends were blunted using the Quick Blunting Kit (NEB, E1201). The digested DNA was purified using the QIAquick PCR Purification Kit (QIAGEN, 28104). The FP DNA fragments were released after digestion with Xho I and gel purified. The FP DNA fragments were ligated into Xho I and Sma I digested pIRES-ZsGreen 1. After transformation, the FP inserts were verified by DNA sequencing.
COS7 cells were cultured in RPMI 1640 complete medium containing 5% fetal calf serum, penicillin (100 U/ml), streptomycin (100 μg/ml), glutamine (30 μg/ml) and 2-mercaptoethanol (5 × 10−5 M). FPs were transiently expressed in COS7 cells using the FP-containing pCI-neo vector by transfection using the lipofectamine LTX reagent (Invitrogen) according to the manufactuer’s instructions. FreeStyle CHO-S cells (Invitrogen) were cultured in FreeStyle CHO Expression Medium (Invitrogen) supplemented with glutamine (8 mM), penicillin (50 U/ml) and streptomycin (50 μg/ml) and stably transfected using the FP-containing pIRES-ZsGreen 1 vector using the FreeStyle MAX Reagent (Invitrogen) according to the manufacturer’s instruction. 3–7 days after transfection, cells were selected in G418 (1 mg/ml). After selection and sufficient growth, high producing transfectants were selected by repeated cycles (5–6) of cell sorting for ZsGreen+ cells. The initial cycles isolated the 5–10% brightest cells whereas subsequent cycles isolated the 0.5–1% brightest cells.
To purify the FPs, culture medium from transfected CHO-S cells were dialyzed against PBS, pH 7.4. 1/10 volume of 500 mM sodium phosphate, 3 M NaCl, 200 mM imidazole buffer, pH7.4 was added to the culture supernatant. This material was passed over His60 Ni Superflow Resin (Clontech Laboratories) equilibrated in 50 mM sodium phosphate, 300 mM NaCl, 40 mM imidazole buffer, pH7.4 (wash buffer). The Ni column was then washed with at least 10 columns volumes of wash buffer. The FP was eluted in 1 ml fractions with 50 mM sodium phosphate, 300 mM NaCl, 300 mM imidazole buffer, pH7.4. Protein fractions were identified by O.D. 280 nM (extinction coefficient: 0.82=1 mg Vml for mIL-2/mCD25 and 0.85=1mg/ml for mIL-2/hCD25) and pooled. The FPs were then dialyzed against PBS pH 7.4, filtered sterilized, and stored in aliquots at −70°C.
IL-2 and Antibodies
Mouse IL-2 was purchased from ThermoFisher Scientic and human IL-2 (Aldesleukin/Proleukin) was manufactured by Novartis and purchased through the pharmacy. A monovalent, hetero-Fc form of mFc-mIL2 was generated by co-expressing charge-charge paired heavy chain variants of mouse IgG1 (hinge-CH2-CH3). The Fc-mIL2 was produced by co-expressing plasmids encoding for a positive charge mutant heavy chain linked at its carboxy-terminus to mIL2 (C160S) with the same region of a negative charge mutant heavy chain but lacking IL2. The Fc portion is a mouse IgG1 Fc with a D265A mutation to render it inert. Transient coexpression in HEK cells followed by protein A and size-exclusion purification steps, yielded heterodimeric protein containing a single mIL2. Identify of the monovalent Fc product was confirmed by a combination of mass spectrometry and multiple angle light scattering (MW = 72 kDa).
Donkey anti-rabbit IgG and rabbit anti-6× histidine tag were purchased from Abcam. Biotin and purified anti-mIL-2 (clone JES6–1A12), eFluorTM450 anti-mouse Foxp3 (clone FJK-16s), purified anti-mCD25 (clone PC61), and HRP-streptavidin were purchased from ThermoFisher Scientific. Antibodies to mouse Ki67 (Alexa Fluor700, clone B56) and phosphorylated mouse/human STAT5 (Alexa Fluor 488 and Alexa Fluor647, clone pY694) and hCD25 (PE and purified unconjugated antibody, clone MA251; PE, clone B56) were purchased from BD Biosciences. Antibodies to mouse CD4 (PE, clone RM4–5), mouse CD8α (PerCP/Cy5.5 and Alexa Fluor700, clone 56.6.7), mouse NK1.1 (PE, clone PK136), and mCD25 (PE, clone 3C7; PE and PE/Cy7, clone PC61) were purchased from Biolegend. Antibodies to mouse CD4 (FITC, clone GK1.5) and mIL-2 (purified antibody, clone S4B6) were prepared in house,
CD25 epitope analysis
Serial dilutions of mIL-2/mCD25 or mIL-2/hCD25, starting at 5 μg/ml, in sorter buffer were incubated for 30 min with a fixed concentration (500 ng/ml) of mAbs to mCD25 (PE-3C7, PE-7D4, or PE-PC61) or to hCD25 (PE-BC96 or PE-MA251) at a final volume of 50 μl. mCD25 transfected EL4 cells, EL4J-3.4 (35), or hCD25 transfected CHO cells (1–2 × 105), as appropriate, were added to the mixture of FP and anti-CD25 for 15 min. The cells were then washed once and then subjected to flow cytometric analysis.
IL-2 bioassay
IL-2 activity was assessed using the IL-2-dependent CTLL cell line (36) or anti-CD3 pre-activated T cell blasts. For the latter, C57BL/6 spleen cells (2 × 106/well) were cultured with RPMI 1640 complete medium containing anti-CD3 (145–2C11, 5% culture supernatant) in 24-well flat bottom tissue culture plates for 48 hr. After washing 3× with HBSS, the T cell blasts (2.5 × 104/well) were cultured in 96 well flat bottom plates with FPs samples for 24 hr, where 3H-thymidine was added during the last 4 hrs of culture. mIL-2 and hIL-2 were used in these assays as a positive control.
FACS analysis and sorting
Single cell suspensions were prepared in HBSS containing BSA (2 mg/ml) and sodium azide (1 mg/ml) and stained with mAbs (see above). Cells were subjected to FACS analysis using a BD LSR-Fortessa-HTS analyzer or cell sorting using a BD FACSAria-II. For FACS analysis, typical 100,000 events/sample were analyzed. Treg were sorted based on expression of CD4 and the Foxp3/RFP-reporter and were >97% pure. All FACS data were analyzed using BD FACS Diva Software Version 8.0.1.
Ex vivo or in vitro pSTAT5 Assay
For ex vivo pSTAT5 measurements, single cell suspensions were immediately prepared in ice cold complete RPMI 1640 media from the spleens of euthanized mice. The cell suspension (100–200 μl) was quickly transferred to a 10 × 75 mm tissue culture tube and fixed in 1.6% of paraformaldehyde at 37o for 10min. The fixed cells were permeabilized in 95% methanol for 30 min on ice or overnight in a −20o freezer. For in vitro pSTAT5 induction, spleen cells were cultured for 30 min at 37°C and then stimulated with IL-2 or FPs for 15 min at 37°C. The cells were then fixed and permeabilized as described above. The cells were then washed 2× with sorter buffer and then stained with appropriate mAbs.
Mice and in vivo treatment with IL-2/CD25
C57BL/6J, BALB/cJ and NOD-ShiLtJ, mice were purchased from Jackson Laboratories. C57BL/6-Foxp3/red fluorescent reporter (RFP) (37) and NOD-Foxp3/RFP(38) reporter mice were breed in our animal colony. All mice were 6–12 weeks of age when the experiments were initiated. All animal experiments were approved by the Animal Care and Use Committees at the University of Miami and Bristol Myers Squibb. mIL-2 (1 μg)/anti-IL-2 (JES6–1A12, 5 μg) complexes were prepared as previously described (39). These complexes, IL-2, or FPs were administered to mice by intraperitoneal injections. To assess diabetes in NOD mice, urine and blood glucose levels were monitored at least twice a week; mice were considered diabetes when blood glucose levels were >250 mg/dl. Pancreas lymphoid cells from NOD mice were prepared as previously described (40).
ELISAs for IL-2/CD25 half-life determination and anti-FP antibodies
96-well high-binding flat bottom microtiter plates (Corning 3590, VWR) were coated overnight at 4°C with of anti-mCD25 (1 μg/ml of PC61.5) or anti-hCD25 (2 μg/ml of M-A251) to measure IL-2/CD25 or with purified mIL-2/mCD25 (1 μg/ml) to measure anti-FP antibodies in 0.05M carbonate (Na2CO3)-bicarbonate (NaHCO3) coating buffer, pH9.6. Plates were washed (PBS containing 0.05% Tween-20) and then blocked at room temperature with PBS containing 1% BSA (Sigma-Aldrich) for 2 hr. Serial diluted samples in blocking buffer were added to the anti-CD25- or mIL-2/mCD25-coated plates and then they were incubated at room temperature 2 hr. To detect mIL-2/mCD25 or mIL-2/hCD25, these anti-CD25 coated plates were washed and then incubated with lab prepared biotin-conjugated anti-mIL-2 (1.0 μg/ml of JES6–1A12,) at room temperature for 1 hr. To detect antibodies to the mouse FP, the mIL-2/mCD25 coated plates were washed and then incubated with goat anti-mouse IgG (H- +L-chain reactive) at room temperature for 1 hr. All plates were washed and then incubated at room temperature with HRP-conjugated streptavidin (ThermoFisher Scientific; 1:20,000 dilution) for 1 hr. Plates were washed and developed with OptEIA TMB substrate (BD Biosciences). The reaction was terminated using 1M HCl and read immediately at 450nm (BENCHMARK PLUS, Bio-Rad). mIL-2/mCD25 and mIL-2/hCD25 concentrations were determined in comparison to a standard sample of purified FPs. Half-life of the FPs were calculated using the formulas: Kelim=ln(Cpeak)-ln(Ctrough)/tinterval and t½=0.693/Kelim where Kelim is the elimination rate constant, C is concentration and tinterval is the time interval from peak to trough. Anti-mouse FP concentrations were measured by extrapolation from the linear portion of a standard curve that was generated based on the binding of known concentrations of anti-mouse IL-2 (Jes-6.1) to the mIL-2/mCD25 coated plates.
Biochemical analysis
SDS-PAGE under reducing and non-reducing conditions and Western blot analysis under reducing condition were performed as previously described (41) using precast gel from Biorad. Proteins were visualized on SDS-PAG by staining with BlueBANBitTM stain and on Western blots by first using rabbit anti-6× His, followed by horseradish peroxidase conjugated donkey anti-rabbit IgG and then SuperSignal® West Pico Chemiluminescent Substrate (ThermoScientific). To deglycosylate mIL-2/mCD25, the purified FP (18 μg) was incubated with Deglycosylation Mix II (New England Biolabs Inc) according to the manufacturer’s instruction and then subjected to SDS-PAGE under reducing conditions.
Static and dynamic light scattering
The size and oligomeric state of the FPs were studied by size-exclusion chromatography coupled to an in-line multi-angle light scattering detector (SEC-MALS) and by dynamic light scattering (DLS). Samples were prepared by injecting 30 μg of stock sample (SEC-MALS), or by analyzing 25 μl of stock solution by DLS in a low volume 384 well plate (3540, Corning, Inc). The mAb use as a marker protein was generated at Bristol Myers Squibb and lysozyme was a 0.5% solution generated from EMD Millipore (4403–1GM). Isocratic separations were performed on a Shodex KW803 (8 mm X 300 mm) column connected to a Prominence Shimadzu UFLC system consisting of a degasser, isocratic pump, chilled sample holder with injector, UV/vis detector, and column oven in buffer containing 200 mM K2HPO4, 150 mM NaCl (pH 6.8), and 0.02% Na azide (0.1 μm filtered) running at 0.5 ml/min for 40 min per injection. Samples were injected onto the column using a Shimadzu autosampler, and data were obtained from three online detectors connected in series: a Shimadzu SPD-20 dual wavelength UV/vis spectrophotometer set for collection at 280 nm, followed by a Wyatt Technologies mini-Dawn TREOS three angle laser light scattering detector and then a Wyatt Optilab T-rEX interferometric refractometer. Data were collected and analyzed using Astra 6 (Wyatt) and LabSolutions Lite Postrun analysis (Shimadzu) software. DLS data were collected on a Wyatt DynaPro I plate reader at 25°C, using 10 acquisitions of 5 s each, and measurements were recorded in quadruplicate and averaged to give the reported values. Intensity autocorrelation functions were fitted using the “Regularization” algorithm in the Dynamics software (Wyatt Technologies).
Surface plasmon resonance
Surface plasmon resonance (SPR) studies were performed on a Biacore T100 and/or T200 instrument (GE Healthcare) at 25°C. The binding of the FP analytes were tested in 10 mM NaPO4, 130 mM NaCl, 0.05% p20 (PBS-T) (pH 7.1) on surfaces consisting of a low density (~300RU) of biot-mCD25(22–186)-BioP-TVMV-His which had been His cleaved (mCD25) and captured on a streptavidin sensor chip (GE Healthcare) or mCD122(27–241)-hFc-D/mCD132(23–263)-hFc-K heterodimeric Fc fusion (mIL-2-Rb/g) that had been captured via Protein A immobilized CM5 sensorchip surface using standard ethyl(dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide chemistry, with ethanolamine blocking (GE Healthcare). The protein analytes were injected in a titration series and regenerated back to baseline was performed by 2 X 8s injections of 10mM Glycine-HCL pH2.0 (mCD25 surface) or 2 X 15s injections of 10mM Glycine-HCL pH1.5 (Protein A surface). The data were analyzed using the Biacore T-200 Evaluation software (GE Healthcare).
RNAseq
RNA was extracted from sorted Tregs using TRIzol Reagent (Thermo Fisher) and column purified using miRNeasy Micro Kit (Qiagen). RNA quality was assessed using an Agilent 2100 BioAnalyzer and was quantified using Qubit RNA assay. RNA-seq libraries were prepared from 25 ng of total RNA using the KAPA’s RNA Hyperprep with RiboErase protocol and quantified using Qubit DNA and Kapa qPCR Library Quantification kit for Illumina (KapaBiosystems). Libraries were sequenced on a 75 bp paired-end run using a NextSeq 500 with a High Output Kit 150-cycle flow cell (Illumina) to generate 30 million reads per sample. Reads from RNA-seq were mapped to the Mus musculus genome GRCm38 using STAR (ver.2.5.0) aligner (42). Raw counts were generated on Ensembl gene (GENCODE M13) with featureCounts (ver.1.5.0) (43). Differential expressed genes in Tregs between the IL2/CD25 and PBS injected mice were identified using DESeq2 (44), and determined according to threshold of false discovery rate (FDR) <0.05. Pathway analysis was performed using QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN). Pathway network indicated by overlapped genes was analyzed using EnrichmentMap(45) and generated using Cytoscape (www.cytoscape.org). The RNAseq data are available from NCBI/GEO data base (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113951 ).
Statistical analysis
Data were analyzed using GraphPad Prism software. All data are expressed as mean ± SEM. The specific statistical analysis used are listed in the Figure legends.
Results
Development of IL-2/CD25
We molecularly engineered FPs consisting of the leader peptide and expressed coding sequence of mIL-2 that was linked by a variable sized sequence, i.e. G7, (G3S)3, (G3S)4, (G4S)4, and (G4S)5 to the extra-cytoplasmic region of mCD25 (Fig. 1A). A carboxyl terminal poly-histidine was added to facilitate purification of the FPs using nickel affinity chromatography. The FPs were first transiently expressed in COS7 and then tested for their ability to support proliferation of anti-CD3 pre-activated T blasts. All the FPs cells demonstrated similar bioactivity, where the highest activity was associated with 12 amino acid linker FP (Fig. 1B). This bioactivity was completely inhibited by addition of a monoclonal antibody to IL-2, confirming that IL-2 was the active component in the FP (Fig. 1C). Based on this result, we chose to characterize in more detail IL-2/CD25s with the 12 amino acid (G3S)3 linker. As mIL-2 weakly interacts to the human IL-2R (46), we also prepared a mouse/human chimeric FP (mIL-2/hCD25) (Fig. 1A), to test whether IL-2 interacts with CD25 in the context of the FP. The cDNAs encoding mIL-2/mCD25 and mIL-2/hCD25 with the (G3S)3 12 amino acid linker were stably expressed in CHO cells that were selected by sequential cell sorting 5–6 times based on expression of eGFP, which was encoded by an IRES downstream of the FPs sequences.
Figure. 1.
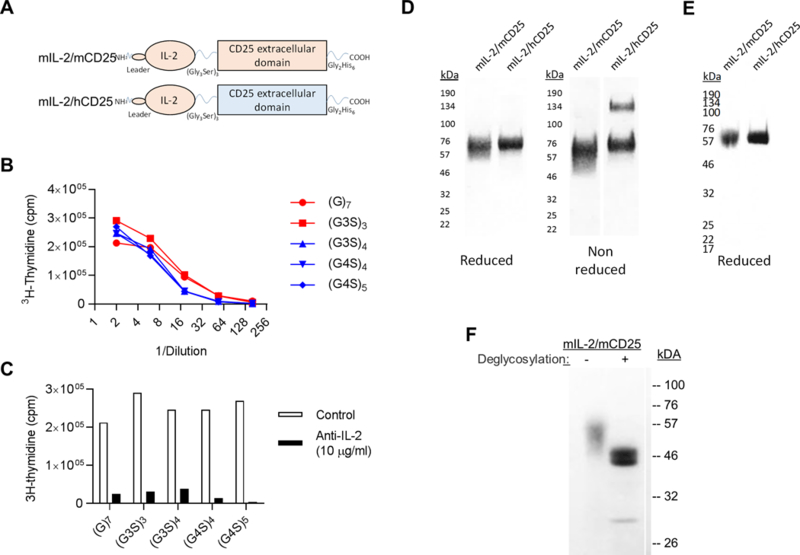
Characterization of IL-2/CD25. (A) Diagram of mIL-2/mCD25 and hybrid mIL-2/hCD25 FPs. (B, C) Bioactivity of mIL-2/mCD25 with varied linker lengths. COS7 cells were transfected with cDNAs encoding mIL-2/mCD25 with different gly/ser linkers. 3 days after transfections supernatants were tested for IL-2 activity using anti-CD3 pre-activated T cells blasts in the B) absence or C) presence of anti-IL-2 (S4B6, 10 μg/ml) (D) SDS-PAGE of purified mIL-2/mCD25 and mIL-2/hCD25 (5 μg each) under reducing and non-reducing conditions and identified by Blue BANDit™ stain (AMRESCO). (E) Western blot analysis of purified mIL-2/mCD25 and mIL-2/hCD25 under reducing conditions and detection with a primary mAb to 6×-His. (F) SDS-PAGE under reducing conditions of purified mIL-2/mCD25 after de-glycosylation using Protein Deglycosylation Mix II). IL-2/CD25s were detected using BlueBANBitTM stain.
SDS-PAGE of these two IL-2/CD25 FPs under reducing conditions revealed a somewhat heterogeneous band of ~60 kDa, consistent with IL-2 (~15 kDa) linked to a glycosylated form of the extracellular region of CD25 (~45 kDa) (Fig. 1D). SDS-PAGE under non-reducing condition revealed a similar pattern of mobility for most FPs, consistent with them as largely non-disulfide bonded proteins. One exception was some higher molecular weight material for mIL-2/hCD25 (30%), likely from an inter-chain disulfide bond through a free cysteine residue in the carboxyl region of CD25. Western blot analysis using an anti-6× His antibody to the FPs showed a single major band with the same size as the purified directly stained FPs (Fig. 1E). Treatment of mIL-2/mCD25 with a mixture of N- and O-glucosidases reduced the heterogeneity and size of this FPs to ~46 kDa, which approaches the predicted size (MW 43,579) of the protein backbone of mIL-2/mCD25 FP (Fig. 1F).
Epitope mapping and bioactivity of IL-2/CD25 FPs
Antibody blocking experiments were performed where mAbs to distinct epitopes of CD25 were first mixed with the FPs and then used to assess mAb binding to cells expressing CD25. If the CD25 epitope on the FP was accessible to a particular anti-CD25 antibody, the mAb would bind to the FP and thus inhibit the binding of the mAb to cells that expressed CD25. mIL-2/mCD25 readily bound to the 7D4 and PC61 mAbs, which define non-ligand binding regions of mCD25 (47), but not to 3C7 mAb, which binds near the IL-2-binding site of mCD25 (Fig. 2A). The mIL-2/hCD25 FP readily bound to M-A251 mAb, directed to a non-ligand binding region of hCD25, but was much less reactive to BC96, which defines an epitope near the IL-2 binding region of CD25 (Fig. 2B), though the inhibition was not as complete as seen with 3C7 and mCD25. Thus, these results suggest that in the context of these FPs, mIL-2 occupies the ligand binding site of mCD25 and hCD25, respectively, and that the mIL-2/mCD25 interaction is more stable than that of mIL-2/hCD25.
Figure 2.
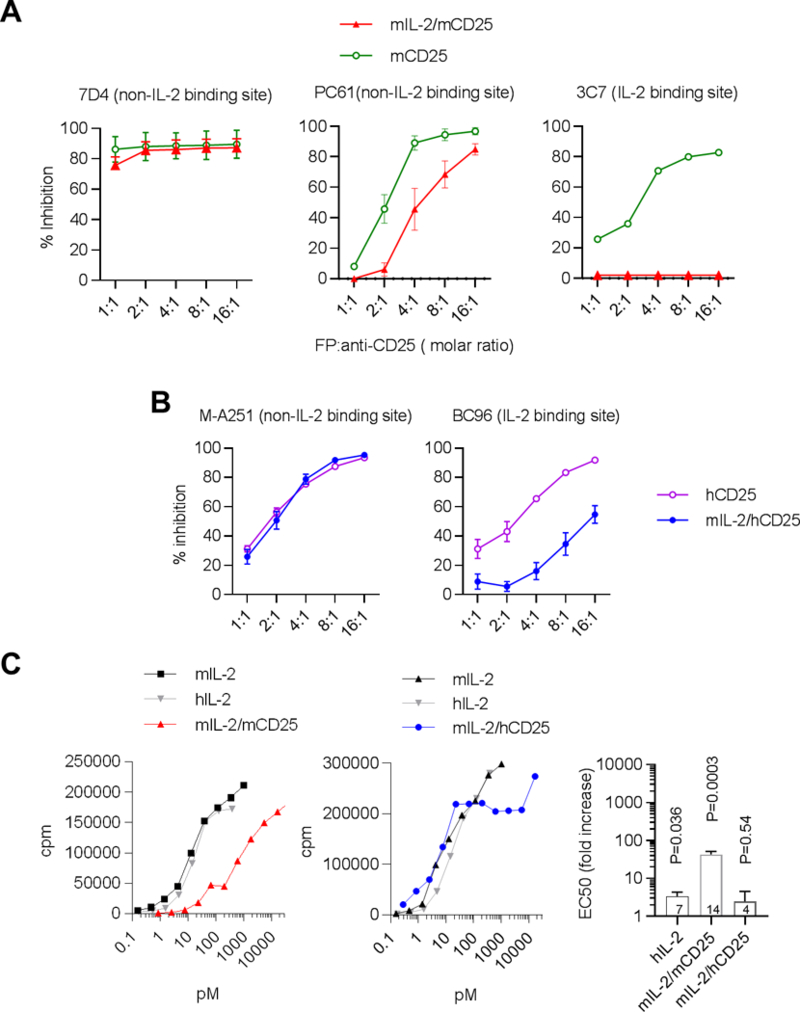
Epitope mapping and bioactivity of mIL-2/mCD25 and mIL-2/hCD25. (A,B) For epitope mapping, the indicated fluorescent-labeled mAbs were pre-incubated with various concentrations of purified mIL-2/mCD25 or soluble mCD25 (A) or mIL-2/hCD25 or soluble hCD25 (B) prior to incubation with mCD25 transfected EL4 cells (EL4J3.4) (A) or hCD25 transfected CHO cells( B) Data are represented as the percent inhibition of cellular staining with fluorescent anti-CD25 mAbs alone. Results (mean ± SEM) represent 3 independent determinations for FPs and 2 for soluble CD25. (C) Bioactivity of mIL-2/mCD25 and mIL-2/hCD25 relative to mouse and human IL-2 using the IL-2-dependent CTLL mouse cell line. The right bar graph depicts the activity of the indicated molecules to mIL-2, where the number of replicates are represents within each bar. Results represent the mean ± SEM. A two-sided one sample t-test was performed where the activity of mIL-2 was considered 1. The p value is listed above each bar. In some cases for the data in Fig. 2A and 2B, the error bars were too small to show relative to the symbols.
Even though mIL-2 is positioned near the binding sites for both FPs, they exhibited markedly different bioactivity in vitro using the IL-2-dependent CTLL cell line. The bioactivity of purified mIL-2/mCD25 (Fig. 2C, left) was 42-fold lower than an equivalent amount of recombinant mIL-2. In contrast, mIL-2/hCD25 chimeric FP (Fig. 2C, right) exhibited bioactivity only 2.5-fold lower than for mIL-2. This observation, combined with the differential efficacy in the antibody block assay, supports the conclusion that mIL-2 interacts less effectively with hCD25 in the context of the chimeric FP. Thus, the relatively low bioactivity of mIL-2/mCD25 is likely accounted for by an effective interaction of mIL-2 with the IL-2 binding site in mCD25 in the fully mouse FP, which limits the ability of IL-2 to interact with and stimulate the IL-2R associated with CTLL cells.
IL-2/CD25 forms stable dimers
Size exclusion high performance liquid chromatography of these FPs under native conditions using Sephacryl S-200 revealed that the bioactivity and detection (280 nm) of most FPs were resolved at a molecular weight greater than the marker protein aldolase (Mr 158,000) (Fig. 3A). These data raise the possibility that the interaction of IL-2 with CD25 in the context of the FP is more complex than a simple cis interactions as a monomer. The apparent molecular masses of the native FPs complexes were determined by SEC-MALS. This analysis showed that the WT and the chimeric FP were dimers, with no evidence for an IL-2/CD25 monomer (Fig. 3B). This analysis predicts a size of 111 kDa (76 kDa protein; 35 kDa carbohydrate) for mIL-2/mCD25 or mIL-2/hCD25, which is twice the expected mass of approximately 55.5 kDa for the monomer, and is consistent with the formation of a dimer. DLS revealed that the hydrodynamic radius of these FPs were 5.6–5.8 nm (Fig. 3C). This size is much larger than expected for a 44 kDa non-glycosylated globular monomer protein (3.0 nm), but is consistent to what would be expected for a non-spherical glycosylated dimer. The hydrodynamic radius for the FPs was also shown to be comparable to that of a standard mAb and substantially larger than mIL-2 or mCD25 alone. Thus, we conclude that IL-2 interacts with CD25 in trans to predominately produce non-covalent dimers and that such dimers are also formed by the chimeric mIL-2/hCD25 FP, despite the weaker interaction observed between mIL-2 and hCD25. A trans interaction could exist in two main forms. One is an inverted head-to-tail dimer (Fig. 3D). The other is a linear dimer. The inverted head-to-tail dimer form will increase the avidity of the IL-2 interaction with CD25 and would favor production of stable dimer, which we observed in the biochemical analysis. Linear dimers are expected to be unstable, due to the fast-on fast-off rates associated with IL-2 binding to CD25, and should have produced a higher levels of FP monomers, if this was the operative model for IL-2/CD25.
Figure 3.
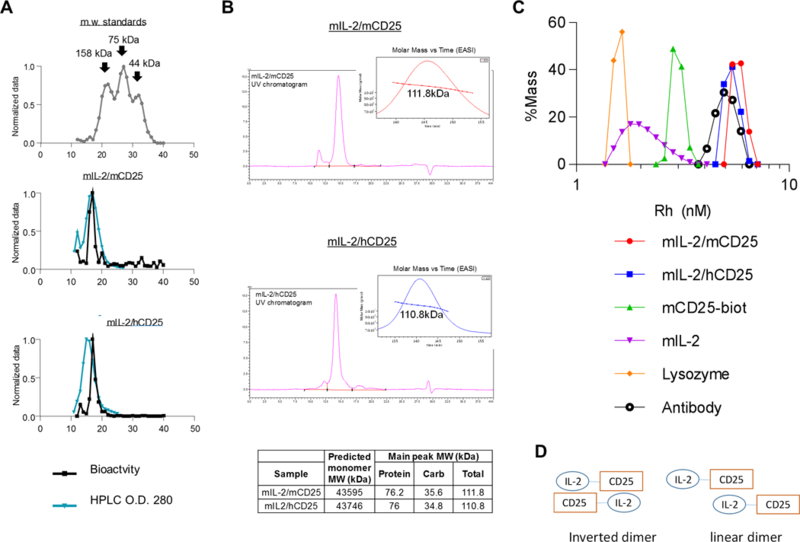
Molecular characteristics of IL-2/CD25. (A) Size determination of IL-2/CD25 by HPLC. The indicated purified proteins were subjected to HPLC using Sephacryl S-200 HR. The elution profile (black line) by O.D. (280 nm) and bioactivity (blue lines) using CTLL cells are shown. The highest values for the O.D. or cpm for 3H-thymidine proliferation were normalized to 1. (B) UV tracing of elution profiles of SEC-MALS of mIL-2/mCD25 and mIL-2/hCD25. The inset plots the molar mass vs. time light scattering signal of the main peak. The predicted MW of the protein backbone and observed MW of the FPs (right), where Carb reflect the amount of glycosylation. (C) Dynamic light scattering histograms of mIL-2/mCD25 and mIL-2/hCD25, with recombinant mIL-2 and mCD25, lysosozyme, and antibody, serving as reference proteins. Rh refers to the hydrodynamic radius. D. Models of FP dimers.
Binding properties of IL-2/CD25 to purified IL-2R subunits
SPR was performed to measure the binding of the FPs to stably captured CD25 (IL-2Rα) and CD122/CD132 (IL-2Rβ/γc) heterodimers (Fig. 4A). As expected, mIL-2 readily bound to mCD25 (Kd 9.8 nM) and mIL-2Rβ/γc (Kd 0.4 nM) but less efficiently bound to hCD25 (Kd ~234-fold weaker). The IL-2/CD25 FPs also exhibited similar differential response dependent on the target species, but with much lower affinities than IL-2 alone. Notably, the binding of mIL-2/mCD25 to mCD25 was not saturable at the top concertation of 4000 nM (Fig. 4A) and had an estimated apparent Kd 1000-fold weaker than mIL-2 to mCD25. Likewise, mIL-2/hCD25 bound to mCD25 more weakly than did free mIL-2, but with 10-fold stronger affinity than the fully mouse FP. A direct comparison of the binding characteristics of these 3 proteins at similar concentration to mCD25 is shown in Fig. 4B. This apparent affinity difference between the FPs and mIL-2 is primarily due to their slower association to CD25. Neither of these FPs showed appreciable binding to hCD25.
Figure 4.
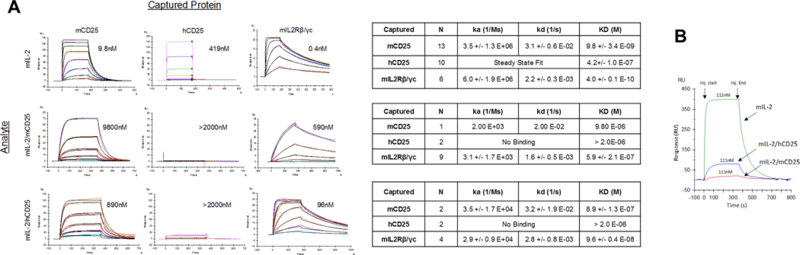
Binding properties of IL-2/CD25 FPs to captured IL-2R by Surface Plasmon Resonance (SPR). (A) Binding of mIL-2, mIL-2/mCD25, and mIL-2/hCD25 to captured mouse and human CD25 or mouse IL-2Rβ/γc heterodimers by SPR. The left shows representative binding sensograms and the right indicates the association and dissociation constants and the Kd for the binding of each protein to CD25 or IL-2Rβ/γc. The top concentrations for IL-2 was 1000 nM for binding to captured CD25 or IL-2Rβ/γc, except for mIL-2 to mIL-2Rβ/γc, which was 111 nM and 4000nM for the FPs. (B) Direct comparison of the binding levels of the indicated proteins to captured mCD25, where the ligand concentrations are show in the graph.
The binding of mIL-2/mCD25 to mIL-2Rβ/γc was also not saturable up to the highest concentration tested, yielding an apparent Kd that was ~680-fold weaker than the binding of mIL-2 to mIL-2Rβ/γc (Fig. 4A). In comparison to mIL-2/mCD25, mIL-2/hCD25 bound with a stronger affinity to mIL-2Rβ/γc, but this binding was still 45-fold weaker than that observed for mIL-2. However, similar to IL-2, mIL-2/mCD25 and mIL-2/hCD25 bound more strongly to the immobilized intermediate affinity IL-2Rβ/γc than the low affinity IL-2Rα (CD25). Thus, the capacity of the FPs to form dimers due to a trans interaction with CD25 constrains the IL-2 moiety of the FP and hinders binding to the purified IL-2R subunits. This effect is more striking for mIL-2/mCD25, where mIL-2 binds more tightly to mCD25 than hCD25. The low specific activity of mIL-2/mCD25 in vitro (Fig. 2C) is likely the result of slow dissociation of mIL-2 from mCD25, creating a low equilibrium concentration of available monomeric FP to provide IL-2 to the high affinity IL-2R of the CTLL cells. This dissociation of the dimer to the monomers is expected to occur more readily for mIL-2/hCD25, with a corresponding increase in the bioactivity of this FP, as observed.
IL-2/CD25 selectively activates the high affinity IL-2R
To test the reactivity of IL-2/CD25 to cells bearing the high and intermediate affinity IL-2R, we compared the capacity of mIL-2/mCD25, mIL-2/hCD25 FPs and mIL-2 to stimulate tyrosine phosphorylation of STAT5 (pSTAT5). When mouse spleen cells were cultured with mIL-2 or mIL-2/mCD25 for 15 min, Tregs, which constitutively express the high affinity IL-2R, were the main population that induced pSTAT5 (Fig. 5A). At a high dose of mIL-2, but not mIL-2/mCD25 or mIL-2/hCD25, pSTAT5 was induced in some CD8+ T and NK cells (Fig. 5B). Representative flow plots of these dose-response studies for C57BL/6 and NOD CD4+ Foxp3+ and CD4+Foxp3- T cells are shown in Supplemental Fig. 1. Thus, IL-2/CD25 does not substantially bind and activate STAT5 in cells such as NK and CD8 T cells, which primarily express the intermediate affinity IL-2R, consisting of IL-2Rβ and γc. The inability to activate these cells at concentrations below 1 μM further indicates that the dimer of the FPs blocks not only IL-2 binding to CD25, but also reduced its binding to the intermediate affinity IL-2R. Quantifying the dose response data showed that Tregs were 555- and 76-fold less responsive to mIL-2/mCD25 and mIL-2/hCD25, respectively when compared to mIL-2 (Fig. 5C), consistent with the trends for the bioassay and SPR results. Similar results were seen for NOD Tregs (Supplemental Fig. 1). Overall, these data in conjunction with the biochemical, CTLL bioassay, and SPR binding data indicate that in the context of the FP, IL-2 interacts with CD25 to form IL-2/CD25 tight dimers, lowering its capacity to activate IL-2-dependent signaling in vitro while favoring selective responsiveness of Tregs to low-dose IL-2/CD25.
Figure 5.
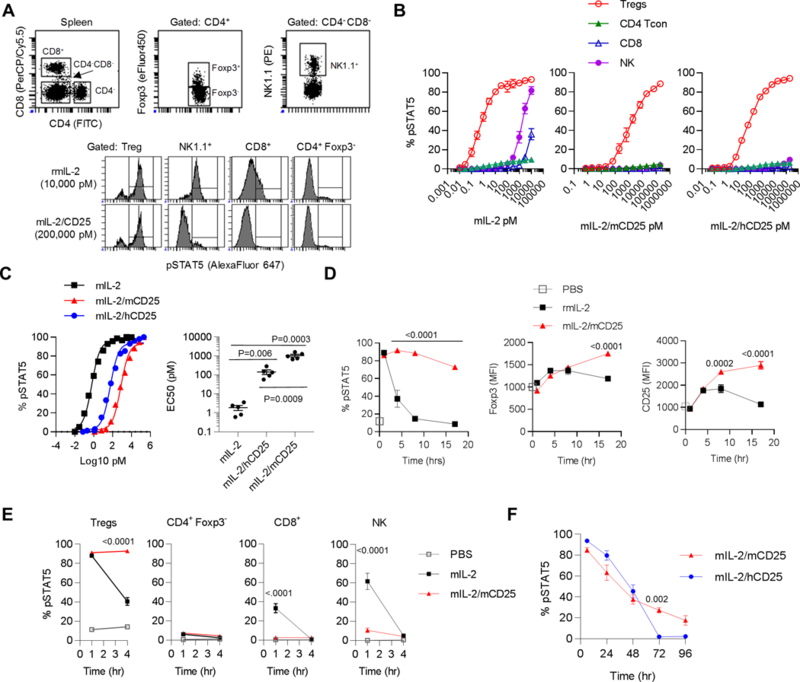
Activation of STAT5 by IL-2/CD25. (A) Representative FACS analysis of induction of pSTAT5 after incubation of spleen cells from female C57BL/6 mice with mIL-2 or mIL-2/mCD25 for 15 min. (B) Dose-response curves of in vitro STAT5 activation (mean ± SEM) of the indicated cell populations in the spleen after incubation with IL-2 (n=3), mIL-2/mCD25 (n=2), or mIL-2/hCD25 (n=2) for 15 min. (C) Representative normalized dose-responses curves (left) for CD4+ Foxp3+ Tregs; the EC50s (right) of each dose-response (n=5) were determined by non-linear regression analysis. Shown is the mean ± SEM, where the number in the graph represents the p values of an unpaired two-sided t-test. (D-F) Time-course of in vivo responsiveness of the indicated cells after injection of female C57BL/6 mice with IL-2 (5 μg), mIL-2/mCD25 (20 μg), or mIL-2/hCD25 (20 μg). At the indicated time, spleen cells were harvested and immediately processed for ex vivo pSTAT5 staining or (D) FACS analysis for Foxp3 and CD25. Shown is the mean ± SEM. The number in each graph represents the p value for the analysis of mIL-2/mCD25 vs mIL-2 (D, E) or mIL-2/mCD25 vs.mIL-2/hCD25 (F) by two-way ANOVA using Tukey”s multiple comparison test. (D) Responses by Tregs to mIL-2 and mIL-2/mCD25. (E) Responsiveness by various cells populations to mIL-2 and mIL-2/mCD25. (F) Comparison of the responses by Tregs to mIL-2/mCD25 or mIL-2/hCD25. In some cases for the data in Fig. 4, the error bars were too small to show relative to the symbols.
IL-2/CD25 is much more active than IL-2 in vivo
Initially, C57BL/6 mice received an equivalent number of mIL-2 and mIL-2/mCD25 molecules and pSTAT5 of Tregs was assessed directly ex vivo. Thirty minutes post-treatment, mIL-2 and mIL-2/mCD25 induced pSTAT5 in most Tregs. However, this response to mIL-2 quickly waned while that induced by mIL-2/mCD25 was sustained for at least 16 hr (Fig. 5D). mIL-2/mCD25 also more persistently upregulated Foxp3 and CD25 in Tregs than mIL-2. Furthermore, mIL-2/mCD25 showed limited ability to activate CD4+ Foxp3-, CD8+, and NK cells (Fig. 5E). These cells initially showed only a minimal response that was substantially muted, particularly for the latter two cell populations. Therefore, in marked contrast to the in vitro activity of IL-2/CD25, these data indicate that mIL-2/mCD25 is much more active than IL-2 in selectively stimulating Tregs in vivo.
We also compared mIL-2/mCD25 to mIL-2/hCD25 to activate pSTAT5 in vivo. These results showed that mIL-2/hCD25 was slightly more active than mIL-2/mCD25 initially, but the response of the former was less persistent (Fig. 5F). Thus, the tighter dimer formed by mIL-2/mCD25, which renders it less active in vitro, promotes sustained Treg activation in vivo.
IL-2 has a serum half-life of <15 min (23), whereas, mIL-2/mCD25 showed much more robust pharmacokinetics with a half-life of approximately 16.2 hrs in C57BL/6 mice (Fig. 6A). The shorter half-life of the chimeric mIL2/hCD25 FP (10h) relative to mIL-2/mCD25 coupled with its lower overall exposure in the serum likely accounts for the less persistent activation of Tregs (Fig. 5F). Thus, the stronger associated of IL-2 to CD25 in dimeric mIL-2/mCD25 promotes the availability of IL-2 for a more extended period of time by slower dissociation of the dimer into a biological active monomer.
Figure 6.
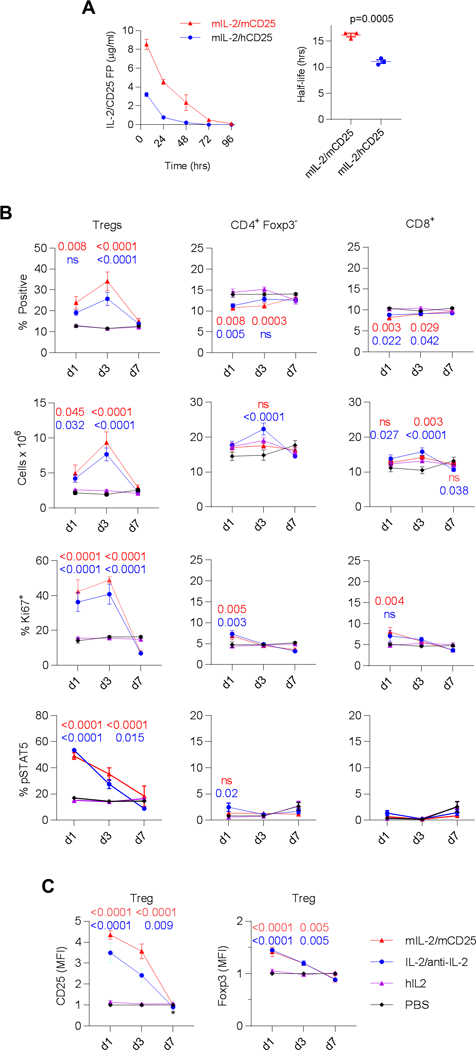
Pharmacokinetics and pharmacodynamics of IL-2/CD25. (A) Female C57/B6 mice were injected i.p. with 20 μg of mIL-2/mCD25 or mIL-2/hCD25. Serum was collected at the indicated time points and the levels of the FPs were determined by ELISA developed to specifically detect these molecules. Shown are the mean ± SEM where an unpaired two-sided t-test was used to compare the half-life of the two FPs. (B, C) Male C57BL/6 mice received mIL-2/mCD25 (8 μg) and mIL-2 (1.0 μg)/anti-IL-2 (5 μg of JES6–12A1) (days 1, 3 and 5) and hIL-2 (25,000 U) or PBS (daily for 5 days); the indicated cell populations from the spleen was assessed 1, 3 and 7 days after the last injection. Shown is the mean ± SEM. The number in each graph represents the p value for the analysis of mIL-2/mCD25 (upper value) or mIL-2/anti-mIL-2 complex (lower values) to hIL-2 by two-way ANOVA using Tukey’s multiple comparison test. (B) Effect on the indicated parameter for Tregs and conventional CD4+ and CD8+ T cells. (C) The levels of CD25 and Foxp3 on Tregs. In some cases for the data in Fig. 5, the error bars were too small to show relative to the symbols.
Additional in vivo studies focused on the homologous mIL-2/mCD25 FP. A time course study showed a substantial and maximal increase in the fraction and number of Tregs, which increased 4.8-fold, from the spleen of C57BL/6 mice 3 days post-treatment with mIL-2/mCD25 (Fig. 6B) and these cells are proliferating based on expression of Ki67. A similar effect was noted for mice treated with IL-2/anti-IL-2 agonist complexes, although the increased in numbers was slightly less (3.9-fold). pSTAT5 (Fig. 6B), which peaked 24 hr post-treatment, CD25, and Foxp3 (Fig. 6C) were also upregulated in Tregs by these two agents. IL-2 as administered in this regimen had no effect on all these parameters. Importantly, CD4+ Foxp3- and CD8+ T cells were minimally affected when considering their proportions and numbers in the spleen (Fig. 6B), consistent with a selective effect on Tregs by mIL-2/mCD25 and IL-2/anti-IL-2 complexes.
When BALB/c mice received similar low doses of mIL-2/mCD25, Treg numbers increased approximately 5–6-fold for mIL-2/mCD25, but only approximately 4-fold for mFc-mIL-2 (Supplemental Fig. 2A). Again, at these doses of the FP, changes in cell numbers were minimal for CD4+ and CD8+ conventional T cells and lower than observed for mice treated with mFc-mIL-2. At higher and more frequent dosing of mIL-2/mCD25 or mIL-2, Tregs increased much more robustly with mIL-2/mCD25 than mIL-2 such that Tregs proportions became substantially over-represented in relationship to conventional CD4+ and CD8+ T cells only for the mice treated with the FP (Supplemental Fig. 2B). However, the numbers of CD4+ and CD8+ T cells more markedly increased in these mice. Overall, these data illustrate that mIL-2/mCD25 is a long-lasting molecule in vivo with a greater capacity to selectively boost Tregs, when compared to mIL-2, mIL-2/anti-IL2 complexes or mFc-mIL-2.
IL-2/CD25 induced substantial genes activation in Tregs
To understand the molecular consequences after mIL-2/mCD25 stimulates Tregs, RNAseq was performed for Tregs that were purified from the spleens of C57BL/6 mice that received the mIL-2/mCD25 3 days previously, when Treg increases were maximal. A total of 20,240 genes were detected and 5,032 of these were differentially expressed (DE) by 1.5-fold with false discovery rate q value (q) less than 0.05 (2,631, up-regulated; 2,400 down-regulated). Top 2,692 DE genes were selected with more stringent criteria (genes with more than 1.5-fold change, p<10−5 and q<0.001) for further analysis with Ingenuity Pathway Analysis (IPA) to reveal their biological functional significance. IPA predicts activation status of a pathway (or up-stream regulator) using an activation z-score (z), with z>0 and q<0.05 indicating that the mIL-2/mCD25 stimulation results an increasing activity of the pathway (or up-stream regulator). Bio-function analysis of the top DE genes showed activation of biological processes associated with cell proliferation (z=5.4, q<10−21); cell survival (z=5.2, q<10−28) and cell viability (z=5.1, q<10−25). Canonical Pathway analysis (Fig. 7A) showed high concordance with the bio-function analysis revealing many inter-related pathways that control cell growth and signaling. Some pathways highly enriched and up-regulated include: estrogen-mediated S-phase entry (z=2.5; ratio: 65%; q<10−8), mitotic roles of Polo-like kinase (z=2.3, ratio: 44 %; q<10−8), cell cycle regulation by BTG family proteins (z=2.1; ratio: 40%; q<10−4); RhoA signaling (z=3.3; ratio: 23%; q=0.001), regulation of actin-based motility by Rho (z=3.1; ratio: 23%; q=0.007), and actin cytoskeleton signaling (z=1.1; ratio: 21%; q=0.004). Regarding up-stream regulators of gene expression driving these pathways, Erbb2 (z=6.69; q<10−34), Myc (z=6.63; q<10−28) and the E2f family of transcription factors (z=5.6; q<10−22), were among the highest activated. These molecular data are consistent with the large increased in Treg numbers, which expressed the proliferative marker Ki67 (Fig. 6B) and indicate that IL-2/CD25 acts as a Treg growth factor.
Figure 7.
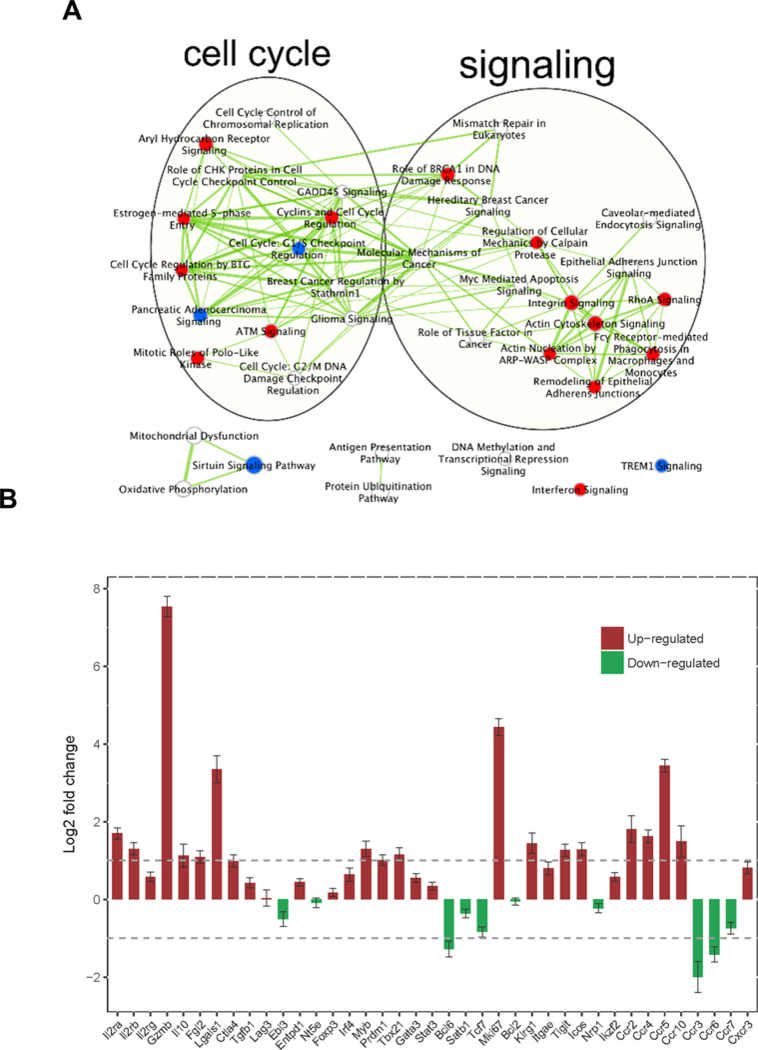
Molecular consequences of mIL-2/mCD25 on Tregs. Male C57BL/6-Foxp3/RFP mice received a single injection (i.p.) of mIL-2/mCD25 (20 μg) or PBS. 3 days later, Tregs were sorted from the spleens and RNA was isolated for RNAseq. (A) Enriched canonical pathways grouped according to overlapped top differentially expressed (DE) genes. The canonical pathways were selected with a p value <0.05 and adjusted p value < 0.25. Red indicates up-regulated versus down-regulated (blue), and undetermined (white). Thickness of edges (line) indicates the number of DE genes overlapped between the pathways. (B) IL-2/CD25-dependent expression of selected immune related genes in Tregs. Error bars represent standard errors. Reference lines indicate fold-change of 0.5 and 2, respectively.
Genes involved in immune system processes were not enriched in mIL-2/mCD25-stimulated Tregs. However, several notable changes were associated with genes that are expected to enhance the Treg function (Fig. 7B). Notably, RNAs encoding molecules involved in Treg suppressive function (Gzmb, Il10, Fgl2, Ctla4) increased. Transcriptional regulators (Irf4, Myb, Prdm1) and surface molecule (Klrg1, Itgae, Tigit, Icos), associated with highly activated effector Tregs, were also upregulated by mIL-2/mCD25. Furthermore, molecules promoting migration to lymph nodes (CD62L, CCR7) decreased while chemokine receptors (Ccr2, Ccr4, Ccr5, Ccr10, Cxcr3) that favor trafficking of cells to inflamed tissues sites increased. As expected, Ki67 mRNA was highly increased, consistent with high proliferation of mIL-2/mCD25-stimuated Tregs. Thus, mIL-2/mCD25 is not just a Treg growth factor but also a molecule that appears to promote more activated and suppressive Tregs with an increased propensity to migrate into tissue-sites, favorable attributes to counteract an overactive immune response, especially one associated with inflamed tissues.
IL-2/CD25 promotes immune tolerance by increasing Tregs in autoimmune target tissue
A number of pre-clinical mouse studies have shown that low-dose IL-2 suppresses the development of autoimmunity, including diabetes in NOD mice (18, 19). To assess whether mIL-2/mCD25 might support tolerance, young 6 week old NOD mice were treated for 5 weeks with two doses of mIL-2/mCD25. Diabetes development was significantly delayed only in the mice that received 4 μg of mIL-2/mCD25 administered every 3.5 days (Fig. 8A). When another cohort of mice received these doses of mIL-2/mCD25 for 2 weeks, Treg increases were noted in the spleen, pancreatic lymph node (PLN) and the pancreas stimulated with 4 μg, but not 1 μg, of mIL-2/mCD25 (Fig. 8B). The effect on the pancreas was particularly striking, supporting the idea that mIL-2/mCD25 promotes increases in Tregs in non-lymphoid tissue sites. These data establish that a 4 μg dose is the minimal required to boost Tregs and prevent autoimmunity.
Figure 8.
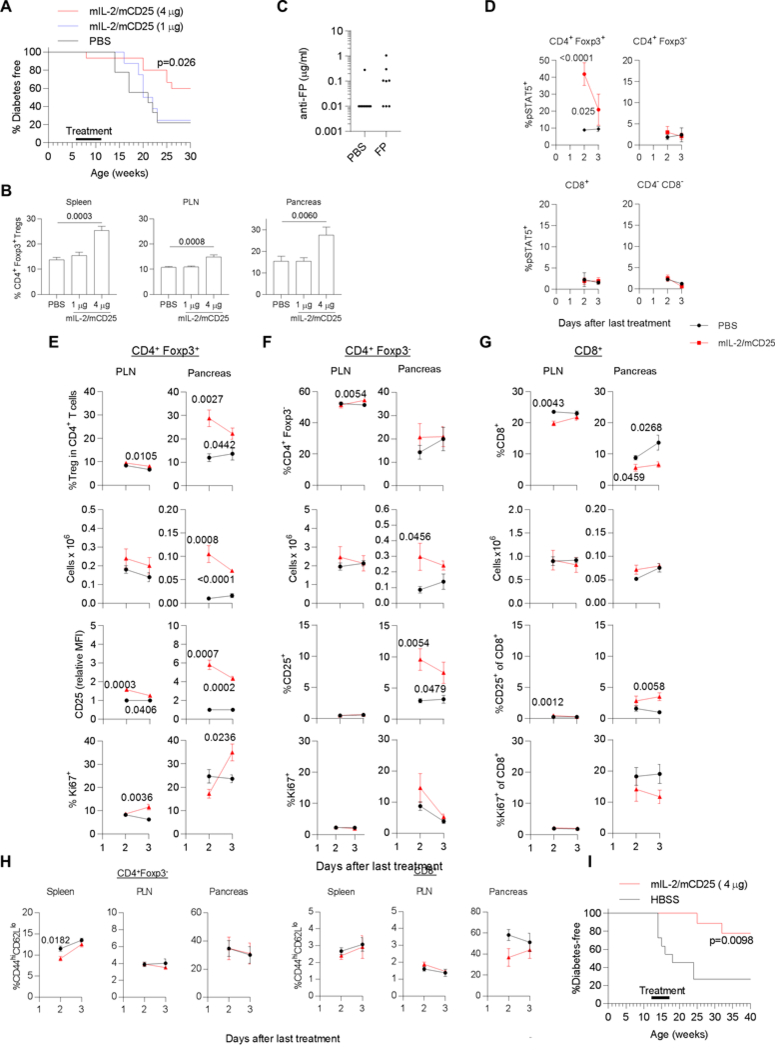
The effect of IL-2/CD25 on diabetes in female NOD mice. (A) 6–7 week old female NOD mice received the indicate dose of mIL-2/mCD25 i.p. or PBS and diabetes instances were assessed until 30 weeks of age. Data were analyzed by Mantal-Cox log rank test. (B) Female NOD-Foxp3/RFP mice were injected (i.p.) twice per week 3.5 days apart with mIL-2/mCD25 or PBS for 2 weeks. 3 days after the last injection the proportion of Tregs within the CD4+ T cell compartment was determined for the indicated tissues, where PLN refers to the pancreatic lymph node. Shown is the mean ± SEM, where the number of replicates is shown within the bars of each graph. Data were analyzed by Kruskal-Wallis one-way ANOVA. (C-H) A distinct cohort of 6–7 week old female NOD mice were treated twice per week 3.5 days apart with PBS or mIL-2/mCD25 (4 μg) for 5 weeks. Two to 3 days after the last treatment, serum was collected, the mice were euthanized, the spleen and PLN were collected and lymphoid cells were isolated from the pancreas. (C) Levels of antibodies reactive to the FP (n=8). (D) In vivo pSTAT5 levels were assessed directly ex vivo for the indicated cell subsets by immediate fixation of the spleen cell suspension followed by staining and FACS analysis. (E-G) The cell number and phenotype of CD4+ Foxp3+ Tregs (E) and CD4+ Foxp3- (F) and CD8+ (G) T conventional cells were assessed for the indicated tissues. (H) The proportion of CD44hi CD62Llo activated T cells were determined for CD4+ Foxp3- and CD8+ conventional T cells, as indicated. Each time point (D-H) consisted of 5 mice/group. All data were assessed by a two-sided non-paired t-test and any p value <0.05 is shown in the graph when comparing FP vs the PBS control. (I) 12 week old female NOD mice were treated with PBS (n=11) or an initial 8 ug dose of mIL-2/mCD25 (n=9). Subsequently, the mice received PBS or mIL-2/mCD25 (4 μg) every 3.5 days for 5 weeks. Diabetes instances were assessed until 40 weeks of age. Data were analyzed by Mantel-Cox log rank test.
Another cohort of NOD mice were similarly treated with an initial 4 μg dose of IL-2/CD25 followed by twice weekly injections of the FP for 5 weeks, analogous to the therapeutic experiment. Serum from these mice was collected after this time to test whether repeated dosing of the FP elicited anti-FP antibodies. (Fig. 8C). The levels of such antibodies was at or lower than 1 μg/ml.
To study the selectivity of the responses by Treg to the FP, ex vivo analysis of Treg and T conventional cells was evaluated. FACS plots to illustrate the gating for CD25 and Ki67 are shown in Supplemental Fig. 3A. When these cells were directly examined ex vivo 2 and 3 days after the last administration of the FP, STAT5 was substantially activated in only Tregs (Fig. 8D). Correspondingly, the proportion of Tregs within total CD4+ T cells and Treg numbers increased that was particularly striking for the pancreas (Fig. 8E); these effects were accompanied by upregulation of CD25 and increases in the proliferative marker Ki67 (Fig. 8E). Interestingly, maximal pSTAT5 and CD25 upregulation preceded the increase in Ki67 (Fig 8D, E), consistent with IL-2R signaling and gene activation promoting cell cycle progression of the Tregs.
With respect to CD4+ Foxp3- and CD8+ T cells, the proportion of CD4+ Foxp3- did not increase (Fig. 8F) and CD8+ T cells decreased (Fig. 8G). This lower fraction of CD8+ T cells in the pancreas was accompanied by fewer Ki67+ CD8+ T cells (Fig. 8G). The total number of CD4+ Foxp3- in the pancreas variably increased (Fig. 8F). In contrast to Tregs, this increased cellularity was not accompanied by activation of STAT5 (Fig. 8D) or more Ki67+ cell, and only a small fraction of CD4+ CD25- and CD8+ T cells in the pancreas showed increased expression of CD25 (Fig. 8F, G). Thus, the CD4+ CD25- and CD8+ T cell compartments did not show a profile of substantial response to IL-2 in the pancreas, the key site of inflammation.
We also assessed whether the small increase in absolute numbers of CD4+ Foxp3- T cells might be due to broad systemic activation of these cells. However, the proportion of activated CD44hi CD62Llo conventional CD4+ and CD8+ T cells did not increase after administering the FP for 5 weeks and a trend for a lower proportion of these activated CD8+ T cells was found for the pancreas (Fig. 8H; Supplemental Fig. 3B). Thus, these data, coupled with the lower proportion of CD8+ T cells with decreased Ki67 expression in the pancreas, raise the possibility that enhancing Tregs in NOD mice might act to directly suppress CD8+ T cells.
In a second therapeutic experiment, older 12 week old mice with more established disease receive IL-2/CD25. In this experiment the treatment period coincided with the onset of diabetes. Under these conditions, development of diabetes was also significantly prevented, an effect which persisted even when animals were no longer exposed to drug (Fig. 8I). Collectively, these data show that IL-2/CD25 selectively enhances Tregs, especially in the pancreas, the target tissue of autoimmunity, and delays the development of diabetes in NOD mice, including such mice with more established disease, without the generation of high anti-FP antibodies.
Discussion
In the present report we describe a new analog of IL-2, a FP of IL-2 linked to CD25 with a non-cleavable Gly/Ser linker. Although homologous mIL-2/mCD25 showed poor capacity to stimulate IL-2R-bearing T cells in vitro, it was highly active in vivo, especially to increase the frequency of Tregs. Molecular studies are consistent with a model where IL-2/CD25 exists predominately as a stable dimer that slowly dissociates to IL-2/CD25 monomers, which is the biologically active form of this FP. This mechanism of action is distinctive from a previous report where IL-2 was linked to CD25 using a protease cleavable linker (34), where the biologically active form is free IL-2 liberated from the FP protein by a protease. Moreover, this biologic is distinctive from other approaches to promote IL-2 selectivity and longevity in that we achieve these properties without the introduction of mutations in either IL-2 or CD25 or fusion to immunoglobulin. Thus, our approach minimizes immunogenicity, which is critical since low-dose IL-2 is likely to be a chronic therapy where even small changes in the cytokine or CDR3 of Ig might result elicit an immune response to lower or abrogate its activity.
Based on past work (34), we expected that IL-2 would interact in cis with CD25 to constrain the availability of IL-2 to cells expressing the IL-2R. One possibility was that IL-2 in the context of CD25 would present IL-2 to cells that express the intermediate affinity IL-2R, found on NK and CD8 memory cells, perhaps in a manner analogous to IL-15/IL-15Rα or IL-2 bound to certain anti-IL-2 mAbs (28, 48). Alternatively, IL-2 might transiently dissociate from CD25 to make IL-2 available to stimulate cells bearing the IL-2R, in a manner similar to free IL-2. In this scenario, IL-2 might stimulate cells expressing the high and intermediate affinity IL-2R, depending upon the levels of available IL-2. Indeed, this second possibility appears operative but not due to a cis interaction between IL-2 and CD25, but a trans interactions leading to IL-2/CD25 dimers as shown by sizing through SEC-MALS, DLS and HPLC chromatography. SPR studies are consistent with an equilibrium that favors the dimer with a small percentage of the monomer, which is the active form that binds to the IL-2R (Fig. 9A). Correspondingly, to achieve equilivent bioactivity and direct binding in vitro to purified IL-2R subunits by SPR, much higher concentrations of IL-2/CD25 were required when compared to recombinant IL-2. We cannot discriminate, however, whether IL-2/CD25 fully dissociates into monomers or whether one IL-2 moiety becomes transiently free from CD25 in the dimer to permit binding to cellular IL-2Rs (Fig. 9B). We believe this latter possibility is not the dominant mode of action as the other IL-2 moiety of the FP would still be bound the to CD25 in trans and reformation of the dimer complex would likely be favored over binding to cell surface IL-2R due to the close proximity of the FP-CD25 to transiently released IL-2.
Figure. 9.

Models to generate biologically active IL-2 from dimers of IL-2/CD25. A) Dissociation of IL-2/CD25 dimers to monomer. B) Release of one IL-2 moiety from IL-2/CD25 dimers. The percentage of dimers (99%) at any one point in time is estimated from the reduced (approximately 40-fold) bioactivity of the FP and its decreased binding (approximately 1000-fold) to immobilized CD25 or IL-2Rβ/γc when compared to recombinant IL-2.
We have two lines of evidence that IL-2 interacts with the ligand binding site of CD25. First, antibody blocking studies indicated that the 3C7 and BC96 mAbs, directed at the ligand binding sites of mouse and hCD25, respectively, could not bind to mIL-2/mCD25 or mIL-2/hCD25. This finding is consistent with IL-2 binding to CD25 in the context of the FP, but this result might also simply reflect steric hindrance of antibody binding. However, second and more direct evidence is that mIL-2/hCD25 stimulated pSTAT5 activation and proliferation in vitro with greater potency than mIL-2/mCD25. mIL-2 interacts more weakly with hCD25 than with mCD25 (46). The higher activity of mIL-2/hCD25 is readily explained by enhanced dissociation of mIL-2 from hCD25 that increases the concentration of free monomers, facilitating stimulation of IL-2R bearing cells. We noted that mIL-2/hCD25 was also detected as a dimer. Thus, even though the affinity of mIL-2 for hCD25 is relatively low, when mIL-2 is placed in tight proximity to hCD25, trans interactions between these two proteins readily occurs. In addition, the amino acid identity between the extracytoplasmic domains of mouse and hCD25 is 58% over 220 amino acids. Thus, the capacity of the mIL-2/hCD25 chimeric FP to produce functional dimers and the similar activity in vitro of mIL-2/mCD25 with different linker lengths makes plain that considerable variation in the amino acid sequence in CD25 and the linker is permissive to produce an IL-2/CD25 dimer with an extend half-life in vivo. This observation suggests that CD25 could be modified in homologous IL-2/CD25 FPs to yield variants that form dimers of distinctive stability and activity.
Our in vivo and in vitro studies clearly demonstrate that IL-2/CD25 is selective for Tregs. This is particularly striking when one considers the consistent increased proportion of Tregs relative to CD4+ and CD8+ T conventional cells by IL-2/CD25, which was more evident when compared to IL-2, Fc-IL-2, or IL-2/anti-IL-2 complexes. After treatment of mice with mIL-2/mCD25, only Tregs showed substantial activation of STAT5, upregulation of IL-2-responsive CD25, and induction of proliferation based on Ki67 expression that was accompanied by a gene profile consistent with cycling T cells. At low doses, IL-2/CD25 led to a marked increased proportion and number of Tregs, but not CD4+ Foxp3- or CD8 T cells, in the spleen of several mouse strains and, similarly, the proportion and absolute numbers of Tregs were increased in the pancreas of NOD mice. In contrast to the robust and consistent Treg induction, IL2/CD25 induced only a modest increase in the total number, but not the proportion of, CD4+ Foxp3- T cells in the pancreas of NOD mice. Further, the proportion of CD8+ T cells decreased in the pancreas of FP-treated mice. These data are consistent with the FP to favorably amplify Tregs. This effect likely controlled diabetes in NOD mice, in part based on a greater capacity to suppress autoreactive T cells within the inflamed pancreas, and in part by the increased Tregs to more favorably compete for IL-2 that may particularly lower responses of CD8+ Teff cells, as noted by others (49). The selectivity of the fusion protein toward Tregs is accounted for by a mechanism where the FP exists primarily as a persistent inactive dimer leading to low levels of active monomer (Fig. 9). Accordingly, the levels of available IL-2 from the fusion protein are adequate to readily stimulate Tregs due to their high levels of the high affinity IL-2R but are not sufficient to substantially stimulate Teff cells with lower levels of the high affinity IL-2R or CD8+ T memory and NK cells that express the intermediate affinity IL-2R. Thus, homologous IL-2/CD25 is endowed with a feature associated with several mutants of IL-2 (31, 32), i.e. selectivity toward the high affinity IL-2R and Tregs, that is absent for native recombinant IL-2.
Much interest has emerged in using low-dose IL-2 in the setting of autoimmunity, as it represents a direct approach to boost Tregs in many patients (22). Low-dose IL-2 has the potential to regulate the immune system to re-establish immune tolerance in a natural manner, in contrast to the typical current approaches that non-specifically block the immune system with immunosuppressive drugs that often exhibit toxic side-effects and leave patients vulnerable to infections. However, recombinant IL-2 has a short half-life in vivo which limits its practical use as a drug availability to stimulate Tregs. Indeed, to boost Tregs in this type of immunotherapy, IL-2 must be injected frequently to overcome its short half-life and the margin between effective stimulation of Tregs and that of undesired immune activation is low and may limit efficacy (20, 21). We found that the biological half-life of mIL-2/mCD25 was 17–20 hrs. This increased pharmacokinetics of IL-2/CD25 is due to the increased size of approximately 110 kDa of the IL-2/CD25 dimer and the slow-release of active IL-2/CD25 monomer. Our in vivo studies showed increases in Tregs on a per molecule basis that was far superior to that achieved by recombinant IL-2 and comparable to agonist mIL-2/anti-mIL-2 complexes. Moreover, mIL-2/mCD25 prevented the development of diabetes in NOD mice, illustrating that the Tregs induced by the FP are functional and capable of controlling autoimmunity. Although much more needs to be learned about the in vivo mechanism of action of IL-2/CD25 and its potency to mediate immune tolerance, these results demonstrate that IL-2/CD25 FP offers potential advantages over low-dose recombinant IL-2 to selectively and persistently boost Tregs. The in vivo activity of IL-2/CD25 FPs may surpass IL-2-immunoglobulin FPs and engineered IL-2 muteins that target the high affinity IL-2R due to the longer half-life and unique mode of action.
Supplementary Material
Acknowledgments
We thank Michael Dee (University of Miami) for technical assistance, Priyanka A. Madia, Anjaneya Chimalakonda, Surendran Rajendran, Adam Jardel and Karlene Melero (Bristol Myers Squib) for help with pharmacokinetic analysis and preparation of mFC-mIL-2, the FACS cores at the Diabetes Research Institute and Sylvester Comprehensive Cancer Center (SCCC) at the University of Miami and the Oncogenomics Core at SCCC.
This work was supported by the NIH (R01 DK093866), Wallace H. Coulter Center, the Diabetes Research Institute, American Diabetes Association, and Bristol Myers Squibb
Abbreviations:
- DE
differentially expressed
- DLS
dynamic light scattering
- FP
fusion protein
- hCD25
human CD25
- IPA
Ingenuity pathway analysis
- mCD25
mouse CD25
- mIL-2
mouse IL-2
- RFP
red fluorescent protein
- PLN
pancreatic lymph node
- pSTAT5
phosphotyrosine STAT5
- SEC-MALS
size-exclusion chromatography coupled to an in-line multi-angle light scattering detector
- SPR
surface plasmon resonance; red fluorescent protein
- Teff
T effector cell
- Treg
regulatory T cell
- Tfh
T-follicular helper cell
Footnotes
Disclosure
The University of Miami and Thomas Malek have a patent pending on IL-2/CD25 fusion proteins and this technology has been licensed to Bristol Myers Squibb by the University of Miami for commercial development.
References
- 1.Morgan DA, Ruscetti FW, and Gallo R 1976. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 193: 1007–1008. [DOI] [PubMed] [Google Scholar]
- 2.Gillis S, and Smith KA 1977. Long term culture of tumour-specific cytotoxic T cells. Nature 268: 154–156. [DOI] [PubMed] [Google Scholar]
- 3.Williams MA, Tyznik AJ, and Bevan MJ 2006. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 441: 890–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malek TR 2008. The biology of interleukin-2. Annu Rev Immunol 26: 453–479. [DOI] [PubMed] [Google Scholar]
- 5.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, and O’Shea J J 2007. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26: 371–381. [DOI] [PubMed] [Google Scholar]
- 6.Johnston RJ, Choi YS, Diamond JA, Yang JA, and Crotty S 2012. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med 209: 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, and Horak I 1993. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 75: 253–261. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, Schmits R, Simard JJL, Ohashi PS, Griesser H, Taniguchi T, Paige CJ, and Mak TW 1995. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science 268: 1472–1476. [DOI] [PubMed] [Google Scholar]
- 9.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, and Alt FW 1995. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3: 521–530. [DOI] [PubMed] [Google Scholar]
- 10.Kundig TM, Schorle H, Bachmann MF, Hengartner H, Zinkernagel RM, and Horak I 1993. Immune responses in interleukin-2-deficient mice. Science 262: 1059–1061. [DOI] [PubMed] [Google Scholar]
- 11.Malek TR, Yu A, Vincek V, Scibelli P, and Kong L 2002. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity 17: 167–178. [DOI] [PubMed] [Google Scholar]
- 12.Fontenot JD, Rasmussen JP, Gavin MA, and Rudensky AY 2005. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 6: 1142–1151. [DOI] [PubMed] [Google Scholar]
- 13.Cheng G, Yu A, Dee MJ, and Malek TR 2013. IL-2R signaling is essential for functional maturation of regulatory T cells during thymic development. J Immunol 190: 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, Laurence A, Robinson GW, Shevach EM, Moriggl R, Hennighausen L, Wu C, and O’Shea JJ 2007. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood 109: 4368–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atkins MB 2002. Interleukin-2: clinical applications. Semin Oncol 29: 12–17. [DOI] [PubMed] [Google Scholar]
- 16.Ahmadzadeh M, and Rosenberg SA 2006. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood 107: 2409–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu A, Zhu L, Altman NH, and Malek TR 2009. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity 30: 204–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, and Bluestone JA 2008. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28: 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, Cagnard N, Carpentier W, Tang Q, Bluestone J, Chatenoud L, Klatzmann D, Salomon BL, and Piaggio E 2010. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 207: 1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP 3rd, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, and Soiffer RJ 2011. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med 365: 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, Sene D, Cacoub P, and Klatzmann D 2011. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med 365: 2067–2077. [DOI] [PubMed] [Google Scholar]
- 22.Klatzmann D, and Abbas AK 2015. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 15: 283–294. [DOI] [PubMed] [Google Scholar]
- 23.Lotze MT, Frana LW, Sharrow SO, Robb RJ, and Rosenberg SA 1985. In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J Immunol 134: 157–166. [PubMed] [Google Scholar]
- 24.Konrad MW, Hemstreet G, Hersh EM, Mansell PW, Mertelsmann R, Kolitz JE, and Bradley EC 1990. Pharmacokinetics of recombinant interleukin 2 in humans. Cancer Res 50: 2009–2017. [PubMed] [Google Scholar]
- 25.Yao Z, Dai W, Perry J, Brechbiel MW, and Sung C 2004. Effect of albumin fusion on the biodistribution of interleukin-2. Cancer Immunol Immunother 53: 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell CJ, Sun Y, Nowak UM, Clark J, Howlett S, Pekalski ML, Yang X, Ast O, Waldhauer I, Freimoser-Grundschober A, Moessner E, Umana P, Klein C, Hosse RJ, Wicker LS, and Peterson LB 2015. Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J Autoimmun 56: 66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng XX, Steele AW, Hancock WW, Kawamoto K, Li XC, Nickerson PW, Li Y, Tian Y, and Strom TB 1999. IL-2 receptor-targeted cytolytic IL-2/Fc fusion protein treatment blocks diabetogenic autoimmunity in nonobese diabetic mice. J Immunol 163: 4041–4048. [PubMed] [Google Scholar]
- 28.Boyman O, Kovar M, Rubinstein MP, Surh CD, and Sprent J 2006. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311: 1924–1927. [DOI] [PubMed] [Google Scholar]
- 29.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, Pande VS, Fathman CG, Boyman O, and Garcia KC 2012. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 484: 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmenate T, Pacios A, Enamorado M, Moreno E, Garcia-Martinez K, Fuente D, and Leon K 2013. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. J Immunol 190: 6230–6238. [DOI] [PubMed] [Google Scholar]
- 31.Rao BM, Driver I, Lauffenburger DA, and Wittrup KD 2004. Interleukin 2 (IL-2) variants engineered for increased IL-2 receptor alpha-subunit affinity exhibit increased potency arising from a cell surface ligand reservoir effect. Mol Pharmacol 66: 864–869. [DOI] [PubMed] [Google Scholar]
- 32.Shanafelt AB, Lin Y, Shanafelt MC, Forte CP, Dubois-Stringfellow N, Carter C, Gibbons JA, Cheng SL, Delaria KA, Fleischer R, Greve JM, Gundel R, Harris K, Kelly R, Koh B, Li Y, Lantz L, Mak P, Neyer L, Plym MJ, Roczniak S, Serban D, Thrift J, Tsuchiyama L, Wetzel M, Wong M, and Zolotorev A 2000. A T-cell-selective interleukin 2 mutein exhibits potent antitumor activity and is well tolerated in vivo. Nat Biotechnol 18: 1197–1202. [DOI] [PubMed] [Google Scholar]
- 33.Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W, Fischer S, Oh J, Spolski R, Weiskopf K, Kohrt H, Foley JE, Rajagopalan S, Long EO, Fowler DH, Waldmann TA, Garcia KC, and Leonard WJ 2015. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity 42: 826–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puskas J, Skrombolas D, Sedlacek A, Lord E, Sullivan M, and Frelinger J 2011. Development of an attenuated interleukin-2 fusion protein that can be activated by tumour-expressed proteases. Immunology 133: 206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saragovi H, and Malek T 1987. The murine interleukin 2 receptor. Irreversioble cross-linking of radiolabeled interleukin 2 to high affinity interleukin 2 receptors reveals a noncovalently associated subunit. J. Immunol. 139: 1918–1926. [PubMed] [Google Scholar]
- 36.Malek TR, Robb RJ, and Shevach EM 1983. Identification and initial characterization of a rat monoclonal antibody reactive with the murine interleukin 2 receptor-ligand complex. Proc. Natl. Acad. Sci. USA 80: 5694–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wan YY, and Flavell RA 2007. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 445: 766–770. [DOI] [PubMed] [Google Scholar]
- 38.Miska J, Abdulreda MH, Devarajan P, Lui JB, Suzuki J, Pileggi A, Berggren PO, and Chen Z 2014. Real-time immune cell interactions in target tissue during autoimmune-induced damage and graft tolerance. J Exp Med 211: 441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng G, Yuan X, Tsai MS, Podack ER, Yu A, and Malek TR 2012. IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J Immunol 189: 1780–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dwyer CJ, Bayer AL, Fotino C, Yu L, Cabello-Kindelan C, Ward NC, Toomer KH, Chen Z, and Malek TR 2017. Altered homeostasis and development of regulatory T cell subsets represent an IL-2R-dependent risk for diabetes in NOD mice. Sci Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malek TR, and Korty PE 1986. The murine interleukin 2 receptor. IV. Biochemical characterization. J Immunol 136: 4092–4098. [PubMed] [Google Scholar]
- 42.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao Y, Smyth GK, and Shi W 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- 44.Love MI, Huber W, and Anders S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merico D, Isserlin R, Stueker O, Emili A, and Bader GD 2010. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5: e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu KD, Greene WC, and Goldsmith MA 1996. The alpha chain of the IL-2 receptor determines the species specificity of high-affinity IL-2 binding. Cytokine 8: 613–621. [DOI] [PubMed] [Google Scholar]
- 47.Moreau JL, Nabholz M, Diamantstein T, Malek T, Shevach E, and Theze J 1987. Monoclonal antibodies identify three epitope clusters on the mouse p55 subunit of the interleukin 2 receptor: relationship to the interleukin 2-binding site. Eur J Immunol 17: 929–935. [DOI] [PubMed] [Google Scholar]
- 48.Rubinstein MP, Kovar M, Purton JF, Cho JH, Boyman O, Surh CD, and Sprent J 2006. Converting IL-15 to a superagonist by binding to soluble IL-15R{alpha}. Proc Natl Acad Sci U S A 103: 9166–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, and Rudensky AY 2016. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. 17:1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.