

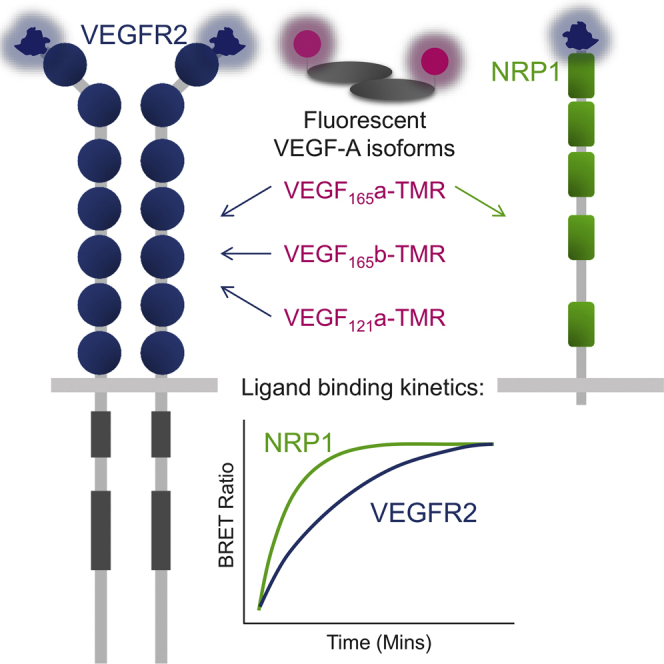
Summary
Fluorescent VEGF-A isoforms have been evaluated for their ability to discriminate between VEGFR2 and NRP1 in real-time ligand binding studies in live cells using BRET. To enable this, we synthesized single-site (N-terminal cysteine) labeled versions of VEGF165a, VEGF165b, and VEGF121a. These were used in combination with N-terminal NanoLuc-tagged VEGFR2 or NRP1 to evaluate the selectivity of VEGF isoforms for these two membrane proteins. All fluorescent VEGF-A isoforms displayed high affinity for VEGFR2. Only VEGF165a-TMR bound to NanoLuc-NRP1 with a similar high affinity (4.4 nM). Competition NRP1 binding experiments yielded a rank order of potency of VEGF165a > VEGF189a > VEGF145a. VEGF165b, VEGF-Ax, VEGF121a, and VEGF111a were unable to bind to NRP1. There were marked differences in the kinetic binding profiles of VEGF165a-TMR for NRP1 and VEGFR2. These data emphasize the importance of the kinetic aspects of ligand binding to VEGFR2 and its co-receptors in the dynamics of VEGF signaling.
Key words: VEGFR2, neuropilin-1, NanoBRET, ligand binding kinetics, VEGF isoforms, receptor mechanisms
Graphical Abstract
Highlights
-
•
VEGF165a, VEGF121a, and VEGF165b were single-site labeled with tetramethylrhodamine
-
•
NanoBRET quantified that VEGF-A isoforms have similar binding properties at VEGFR2
-
•
NRP1 expressed in live cells does not bind VEGF165b, VEGF121a, VEGF-Ax, or VEGF111a
-
•
VEGFR2 and NRP1 have markedly distinct kinetic profiles binding VEGF165a-TMR
Peach et al. have used fluorescent VEGF-A isoforms to demonstrate that they can discriminate between VEGFR2 and its co-receptor NRP1 in real-time ligand binding studies in live cells. This precision chemical biology approach showed that fluorescent VEGF165a binds more rapidly to NRP1 than VEGFR2.
Introduction
Angiogenesis, the growth of new blood vessels from pre-existing vasculature, is critical in both physiology and pathology for maintaining an adequate supply of oxygen and nutrients (Chung and Ferrara, 2011). Vascular endothelial growth factor A (VEGF-A) is an essential mediator of both angiogenesis and vascular permeability that signals via its cognate receptor VEGF receptor 2 (VEGFR2) (Koch et al., 2011, Shibuya, 2011). VEGF binds to VEGFR2 at the extracellular immunoglobulin (Ig)-like domains 2 and 3 (D2/D3) of the receptor (Ruch et al., 2007). VEGF binding stimulates receptor dimerization and initiates conformational changes across the VEGFR2 dimer interface that result in auto- and transphosphorylation of intracellular tyrosine residues (Cunningham et al., 1997). Subsequent recruitment of adaptor proteins and activation of downstream signaling cascades leads to cell proliferation, migration, and survival (Koch et al., 2011). VEGFR2 is overexpressed in many solid tumors and leads to activation of pro-angiogenic signaling, which promotes tumorigenesis. As a consequence, a number of anti-angiogenic therapeutics have been targeted at the VEGF/VEGFR2 axis (Ferrara and Adamis, 2016).
VEGFR2 signaling is selectively enhanced by its co-receptor neuropilin-1 (NRP1), a transmembrane glycoprotein that lacks kinase activity and whose upregulation in malignant tumors is correlated to aggressive cancer phenotypes (Jubb et al., 2012, Goel and Mercurio, 2013, Lee et al., 2014). NRP1 is a multifaceted co-receptor that can also bind structurally and functionally unrelated class 3 semaphorins (Djordjevic and Driscoll, 2013, Guo and Vander Kooi, 2015). However, its functional role in vessel development is evident from the severe cardiovascular abnormalities exhibited in Nrp1 knockout mice (Kitsukawa et al., 1997, Kawasaki et al., 1999, Gu et al., 2003). NRP1 selectively potentiates VEGFR2-mediated endothelial cell motility and vascular permeability without promoting proliferation, driving arterial vessel development in vivo (Chittenden et al., 2006, Fantin et al., 2011, Lanahan et al., 2013). While it lacks kinase activity, NRP1 has a short cytoplasmic tail containing a serine-glutamate-alanine motif that interacts with PDZ domain-containing synectin (Cai and Reed, 1999, Wang et al., 2006, Prahst et al., 2008), through which NRP1 may modulate VEGFR2 trafficking or expression (Ballmer-Hofer et al., 2011). VEGF interacts with NRP1 via a C-terminal arginine residue, whereas N-terminal residues on VEGF are responsible for VEGFR2 binding (Djordjevic and Driscoll, 2013, Guo and Vander Kooi, 2015).
VEGF is an anti-parallel disulfide-linked homodimer with multiple endogenous isoforms resulting from alternative mRNA splicing or encoded by separate genes that each elicit different signaling outcomes (Woolard et al., 2009). Alternative splicing of the VEGF-A gene (Vegfa) results in isoforms of varying lengths that include the prototypical pro-angiogenic isoform VEGF165a and a freely diffusible VEGF121a isoform lacking interactions with heparin (Harper and Bates, 2008). Isoforms with a carboxy terminus substituting CDKPRR for SLTRKD, including VEGF165b and the more recently identified VEGF-Ax, have reported anti-angiogenic activity in vivo (Woolard et al., 2004, Cébe Suarez et al., 2006, Eswarappa et al., 2014). Distinct signaling outcomes downstream of VEGFR2 have been suggested to result from different abilities of distinct VEGF isoforms to bind to NRP1 (Simons et al., 2016, Peach et al., 2018). Despite existing anti-cancer therapeutics targeting VEGF and its known modulation by NRP1, there is limited quantitative information on the binding characteristics of specific isoforms at full-length VEGFR2 and NRP1 in living cells.
Significant advances in our understanding of ligand binding to G-protein-coupled receptors (GPCRs), and more recently RTKs, have resulted from the development of fluorescent ligand technologies that use bioluminescence resonance energy transfer (BRET) (Stoddart et al., 2015, Stoddart et al., 2018). NanoBRET is a proximity-based assay that can quantify interactions between a fluorescent ligand and a receptor fused at its N terminus to a small, bright nanoluciferase (NanoLuc) (Machleidt et al., 2015, Stoddart et al., 2015, Kilpatrick et al., 2017). Having developed a technique to stoichiometrically label VEGF165a with the red-shifted fluorophore tetramethylrhodamine (TMR) (Kilpatrick et al., 2017), we synthesized fluorescent variants of “anti-angiogenic” VEGF165b and freely diffusible VEGF121a to probe their pharmacology at full-length VEGFR2 and its co-receptor NRP1 in living cells at 37°C. We report here the binding affinities and real-time binding kinetics of VEGF-A isoforms to NanoLuc-tagged VEGFR2 and NRP1. We also demonstrate that fluorescent analogs of VEGF165b and VEGF121a can be used to selectively bind to VEGFR2 but not NRP1 in living cells.
Results
Generation and Characterization of Stoichiometrically Labeled VEGF165b-TMR and VEGF121a-TMR
Synthesis and purification of fluorescent VEGF-A isoforms VEGF165b and VEGF121a (Figure 1A) labeled at a single N-terminal cysteine residue with 6-TMR-PEG-CBT were prepared as described by Kilpatrick et al. (2017). In brief, VEGF isoforms were expressed as secreted N-terminal HaloTag fusions. The linker connecting HaloTag and the VEGF isoforms contained a modified tobacco etch virus recognition site (EDLYFQC), which upon proteolytic cleavage released a VEGF isoform with an N-terminal cysteine residue that can be specifically labeled via 2-cyanobenzothiazole (CBT) condensation.
Figure 1.

Functional Characterization of VEGF165b-TMR and VEGF121a-TMR Activities
(A) Schematic illustrating exons present in different VEGF-A isoforms following alternative mRNA splicing, including the region from post-translational readthrough (PTR) in VEGF-Ax.
(B and C) NFAT production in HEK293T cells stably expressing wild-type VEGFR2 in response to 5 hr stimulation with VEGF165b-TMR or VEGF165b prepared identically to the fluorescent analog (B), or VEGF121a-TMR or unlabeled equivalent VEGF121a (C). Data are mean ± SEM (5 independent experiments, duplicate wells) expressed as a percentage of the response to 10 nM VEGF165a measured in the same experiment.
(D and E) VEGFR2 phosphorylation in HEK293T cells stably expressing NanoLuc-VEGFR2 in response to 20 min stimulation with 30 nM unlabeled VEGF165b (D) or VEGF121a (E). Data are presented for VEGF165b or VEGF121a obtained from a commercial source (R&D Systems) or prepared identically to the TMR analogs (Analogue), or for the fluorescent TMR-labeled variants of each VEGF-A isoform. As a negative control, cells were pre-incubated with 1 μM cediranib for 30 min and stimulated in its presence. Cells were fixed (3% paraformaldehyde [PFA]/PBS), permeabilized (0.025% Triton-X-PBS), blocked for non-specific binding, incubated with an antibody specific for phosphorylated tyrosine 1212, and nuclei stained with H33342. Cells were imaged using an IX Micro widefield platereader (20× objective) and quantified using a granularity algorithm (MetaXpress, Molecular Devices). Data were baseline corrected for non-specific binding (secondary antibody only) and expressed as a percentage normalized to cediranib-treated wells (0%) and response to 30 nM VEGF165a (100%) from 5 independent experiments. Statistical analysis performed using a one-way ANOVA and Sidak's multiple comparisons showed no significance.
(F and G) Comparison of the extent of HUVEC proliferation in response to stimulation with VEGF165b or VEGF165b-TMR (F) and VEGF121a or VEGF121a-TMR (G) isoforms. Following serum deprivation, HUVECs were stimulated in duplicate wells for 48 hr with 0.3, 3, or 30 nM ligand (37°C/5% CO2), then fixed using 3% PFA/PBS and nuclei stained with H33342. Cells were imaged using an IX Micro widefield platereader (4× objective) with nuclei counted using a granularity algorithm (MetaXpress, Molecular Devices). Data are expressed as a percentage of the response to 3 nM VEGF165a and represent mean ± SEM from 6 independent experiments. Statistical analyses were performed using a one-way ANOVA and Sidak's multiple comparisons: *p < 0.05.
See also Figures S1 and S2; Tables S1 and S2.
Labeling specificity of VEGF165b-TMR (Figure S1) and VEGF121a-TMR (Figure S2) were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of labeled and unlabeled VEGF isoforms that were digested with multiple proteases as described previously for VEGF165a-TMR (Kilpatrick et al., 2017). This analysis indicated highly efficient and selective labeling of the N-terminal cysteine residue of each VEGF isoform (Figures S1 and S2). 6-TMR-PEG-CBT chemical modification (817 Da) was identified exclusively on the N-terminal cysteine residue of each VEGF isoform at 97% (VEGF165b-TMR) and 94%–99% (VEGF121a-TMR) labeling efficiency (Tables S1 and S2). We did not observe non-specific labeling of any of the other cysteine residues present in either VEGF165b-TMR or VEGF121a-TMR. Fluorescence SDS-PAGE analysis of the purified VEGF165b-TMR and VEGF121a-TMR isoforms in the presence or absence of 100 mM DTT confirmed that, in non-reducing conditions, both VEGF isoforms were largely present as homodimers (Figures S1 and S2). Deglycosylation by PNGase provided evidence that the purified VEGF165b-TMR was glycosylated (Figure S1). However, for VEGF121a-TMR, treatment with PNGase strongly suggested that it was present in both glycosylated and non-glycosylated forms under normal conditions (Figure S2). To confirm the concentrations of VEGF165b-TMR and VEGF121a-TMR (and their dimeric nature), we also undertook fluorescence correlation spectroscopy studies in the presence and absence of 10 mM DTT as described by Kilpatrick et al. (2017) (Figures S1 and S2).
Agonist Activity of Fluorescent VEGF Isoforms in HEK293 Cells and HUVECs
To determine whether the N-terminal TMR labeling of VEGF165b and VEGF121a influenced their VEGFR2 agonist activity, we used a calcium-based nuclear factor of activated T cells (NFAT) reporter gene assay (Carter et al., 2015) to measure signaling downstream of wild-type VEGFR2 expressed in HEK293T cells lacking VEGFR1 or NRP1 (Figure S3). Figure 1 shows the agonist activity of VEGF165b-TMR (Figure 1B) and VEGF121a-TMR (Figure 1C) compared with the agonist actions of equivalent unlabeled VEGF isoforms prepared in a manner identical to that of the fluorescent variant. Each ligand evoked a submaximal response compared with the response obtained with 10 nM VEGF165a (Figures 1B and 1C), consistent with previous work with unlabeled VEGF165b and VEGF121a (Carter et al., 2015, Kilpatrick et al., 2017). However, a comparison of the EC50 values of VEGF165b-TMR and VEGF121a-TMR indicated that the fluorescent ligands had EC50 values that were an order of magnitude higher than their unlabeled counterparts (VEGF165b-TMR pEC50 = 8.16 ± 0.11 versus VEGF165b pEC50 = 9.16 ± 0.09; VEGF121a-TMR pEC50 = 8.57 ± 0.07 versus VEGF121a pEC50 = 9.51 ± 0.09; n = 5 in each case). However, in each case the TMR-labeled VEGF isoform produced a maximum response similar to that obtained with the unlabeled VEGF165b or VEGF121a (Figures 1B and 1C). Although untransfected HEK293T cells did show some low-level expression of endogenous VEGFR2 (Figure S3), neither untransfected nor NanoLuc-NRP1-expressing cells produced a measurable NFAT signal in response to VEGF165a (data not shown).
The agonist effect of the two fluorescent ligands was also evaluated for pY1212 phosphorylation of VEGFR2 using a phosphospecific antibody (Figures 1C and 1D). At 30 nM, both ligands were able to stimulate pY1212 phosphorylation to the same extent as the equivalent unlabeled versions of VEGF165b and VEGF121a (Figures 1C and 1D).
Finally, we also investigated agonist activity of these VEGF-A isoforms in human umbilical vein endothelial cells (HUVECs) that endogenously express both VEGFR2 and NRP1 (Figures S3, 1C, and 1D). Immunolabeling of HUVECs showed a minimal presence of endogenous VEGFR1 (Figure S3). Both unlabeled isoforms stimulated a concentration-dependent increase in HUVEC cell proliferation (Figures 1E and 1F). VEGF165b produced a maximum response that was only circa 60% of that obtained with 3 nM VEGF165a (Figure 1E). In contrast, VEGF121a produced a response similar to that obtained with VEGF165a (Figure 1F). Both fluorescent ligands, however, evoked much lower maximal responses (30% for VEGF165b-TMR; 40% for VEGF121a-TMR) than those obtained with their unlabeled counterparts (Figures 1E and 1F), indicative of partial agonist activity. In keeping with this, the EC50 values of the fluorescent isoforms for HUVEC cell proliferation were, however, very similar to the unlabeled VEGF165b and VEGF121a (Figures 1E and 1F). This contrasted markedly with the full agonist response determined previously with VEGF165a-TMR in HUVECs (Kilpatrick et al., 2017).
Binding of VEGF165a-TMR, VEGF165b-TMR, and VEGF121a-TMR to VEGFR2
Initial imaging studies were undertaken to monitor the spatial aspects of VEGF isoform binding to HaloTag-labeled VEGFR2 expressed in HEK293T cells (labeled with membrane-impermeant HaloTag-AlexaFluor488 substrate; Figure 2). Under basal conditions, VEGFR2 was located on both the cell membrane and within intracellular sites (indicative of constitutive internalization; Kilpatrick et al., 2017; Figure 2). Following 60-min stimulation with 10 nM VEGF165a-TMR (Kilpatrick et al., 2017), VEGF165b-TMR, or VEGF121a-TMR, there was a clear co-localization with HaloTag-VEGFR2 at both the cell membrane and increased internalized receptor (Figure 2).
Figure 2.

Co-localization of Fluorescent VEGF-A Isoform Binding and HaloTag-VEGFR2
Confocal images of HEK293T cells stably expressing HaloTag-VEGFR2 (green) stimulated with vehicle or 10 nM VEGF165a-TMR, VEGF165b-TMR, or VEGF121a-TMR (red) for 1 hr at 37°C. Cells were imaged live using a Zeiss LSM710 and are representative images of 3 independent experiments. Scale bar, 10 μm. See also Figures S3 and S4.
NanoBRET was also used to quantify the real-time binding of the three fluorescent VEGF-A isoforms to NanoLuc-tagged VEGFR2 expressed in living HEK293T cells at 37°C. The assay is based on the close proximity (<10 nm) required for bioluminescence energy transfer between the fluorophore of a receptor-bound fluorescent ligand (BRET acceptor) and the N-terminal NanoLuc (BRET donor) of the receptor. Saturable binding of VEGF165a-TMR, VEGF165b-TMR, and VEGF121a-TMR to NanoLuc-VEGFR2 was clearly demonstrated, and this was largely prevented in the presence of 100 nM unlabeled competitor (Figures 3A–3C). Derived equilibrium binding constants revealed that each isoform bound with nanomolar affinity with a rank order VEGF165a-TMR > VEGF121a-TMR > VEGF165b-TMR (Table 1). Real-time binding kinetics measured every 30 s at 37°C showed VEGFR2 binding peaked within 20 min for each VEGF-TMR isoform (Figures 3D–3F). Kinetic binding experiments were conducted with five separate concentrations of VEGF-TMR isoform, which enabled a global fit of the data to provide estimates for kon and koff for each fluorescent ligand. These data showed that VEGF165a-TMR had a faster kon than VEGF121a-TMR and VEGF165b-TMR, but each isoform had similar koff rates (Table 1). The ratio of koff/kon also provided an estimate of the kinetically derived KD values, which were very similar to those obtained from equilibrium measurements (Table 1).
Figure 3.

Binding Characteristics of Fluorescent VEGF Isoforms to NanoLuc-VEGFR2 Expressed in HEK293 Cells
(A–C) HEK293T cells expressing N-terminal NanoLuc-VEGFR2 were incubated with increasing concentrations of (A) VEGF165a-TMR, (B) VEGF165b-TMR, or (C) VEGF121a-TMR, in the presence and absence of 100 nM unlabeled VEGF, added simultaneously to define non-specific binding (60 min; 37°C). BRET ratios are expressed as mean ± SEM from 5 independent experiments with duplicate wells. Where not shown, error bars are within the size of the symbol.
(D–F) Time course of (D) VEGF165a-TMR, (E) VEGF165b-TMR, or (F) VEGF121a-TMR ligand binding kinetics at NanoLuc-VEGFR2. Cells treated with furimazine were left to equilibrate for 5 min before addition of 1–20 nM fluorescent VEGF ligand or vehicle, and measurements were taken every 30 s for 20 min (37°C). Baseline BRET ratios are corrected to vehicle at time zero. Data represent mean ± SEM from 5 independent experiments, and individual curves were fitted with a simple exponential association model.
(G–I) Displacement of (G) VEGF165a-TMR, (H) VEGF165b-TMR, or (I) VEGF121a-TMR binding by unlabeled VEGF-Ax. Increasing concentrations of VEGF-Ax were added in duplicate wells simultaneously with 5 separate fixed concentrations (0.25–3 nM) of VEGF165a-TMR, VEGF165b-TMR, or VEGF121a-TMR (60 min, 37°C). Raw BRET ratios from 5 independent experiments are shown as mean ± SEM with bars illustrating vehicle (white bars) or fluorescent VEGF-TMR alone.
Table 1.
Binding Characteristics of Fluorescent Ligands Binding to VEGFR2 or NRP1
| Fluorescent Ligand | Receptor | Saturation KD (nM) | Kinetic KD (nM) | kon (min−1 M−1) | koff (min−1) |
|---|---|---|---|---|---|
| VEGF165a-TMR | NanoLuc-VEGFR2 | 2.03 ± 0.51 | 6.64 ± 4.37 | 1.54 × 107 ± 0.38 × 107 | 0.06 ± 0.02 |
| VEGF165b-TMR | NanoLuc-VEGFR2 | 9.53 ± 1.36 | 11.3 ± 3.54 | 7.29 × 106 ± 1.84 × 106 | 0.06 ± 0.01 |
| VEGF121a-TMR | NanoLuc-VEGFR2 | 5.54 ± 1.34 | 5.75 ± 0.46 | 8.51 × 106 ± 0.81 × 106 | 0.05 ± 0.00 |
| VEGF165a-TMR | NanoLuc-NRP1 | 4.41 ± 1.34 | 4.95 ± 1.25 | 7.11 × 107 ± 2.33 × 107 | 0.26 ± 0.05 |
Equilibrium binding parameters for fluorescent VEGF isoforms derived from saturation and kinetic NanoBRET experiments, showing equilibrium dissociation (KD), association rate (kon), and dissociation rate (koff) constants at NanoLuc-VEGFR2 and NanoLuc-NRP1. Data are expressed as mean ± SEM determined from 5 independent experiments.
To gain some insight into whether NanoLuc-VEGFR2 or HaloTag-VEGFR2 were markedly overexpressed in our HEK293T cells, we compared their relative expression levels with those of native untransfected HEK293T and HUVECs using quantitative immunohistochemistry with a selective VEGFR2 antibody (Figure S4). These data showed that the expression levels of the tagged VEGFR2 variants were low and below the native expression level of VEGFR2 in HUVECs (Figure S4).
Using VEGF165a-TMR, VEGF165b-TMR, and VEGF121a-TMR as three distinct fluorescent probes, increasing concentrations of unlabeled VEGF-Ax were used to inhibit the specific binding of each concentration of fluorescent ligand to NanoLuc-VEGFR2 (0.25–3 nM) (Figures 3G–3I). These data were used to derive pKi values for VEGF-Ax assuming mass action interactions (Table S3). Binding affinities were also derived from similar experiments with a comprehensive panel of unlabeled VEGF-A isoforms at NanoLuc-VEGFR2 (Table S3). pKi values obtained for each competing ligand were not significantly different between the fluorescent VEGF probes used (one-way ANOVA).
Real-Time Binding of Fluorescent VEGF165a to NRP1
We were also able to apply the NanoBRET technology to the type I single transmembrane co-receptor NRP1. NanoLuc was fused to the extracellular N terminus of NRP1 and expressed in HEK293T cells to isolate binding of the different fluorescent VEGF-A isoforms to full-length NRP1. Specific binding of VEGF165a-TMR to NanoLuc-NRP1 was clearly observed with minimal non-specific binding following incubation for 60 min (KD = 4.41 ± 1.34 nM, n = 5; Figure 4A). Kinetic binding measurements also revealed that specific binding of VEGF165a-TMR to NanoLuc-NRP1 was reached within 4 min and exhibited faster kon (7.11 ± 2.33 × 107 min−1 M−1) and koff (0.26 ± 0.05 min−1) rate constants than were achieved with this ligand at NanoLuc-VEGFR2 (Figure 4B and Table 1). However, the equilibrium dissociation constants were very similar for VEGF165a-TMR between NRP1 and VEGFR2 (Table 1). Displacing each concentration of VEGF165a-TMR (0.5–5 nM) by increasing concentrations of unlabeled VEGF165a showed competitive inhibition, yielding a pKi of 9.54 ± 0.21 (Figure 4C, n = 5; Table S3). A linear relationship was observed between the IC50 and VEGF165a-TMR concentration at NanoLuc-NRP1 (R2 = 0.95, p < 0.005; Figure 4D).
Figure 4.

Binding Characteristics of VEGF165a Binding to NanoLuc-NRP1
(A) Increasing concentrations of VEGF165a-TMR were added to HEK293T cells stably expressing N-terminal NanoLuc-NRP1 in the presence and absence of 100 nM unlabeled VEGF165a to determine non-specific binding, and cells were incubated for 60 min at 37°C. Raw BRET ratios are expressed as mean ± SEM from 5 independent experiments.
(B) Time course of VEGF165a-TMR binding to NanoLuc-NRP1. BRET ratios were baseline corrected to vehicle, curves were fitted to a simple exponential association model, and data are shown as mean ± SEM from 5 independent experiments.
(C) Inhibition of the binding of VEGF165a-TMR (0.5, 1, 2, 3, and 5 nM) to NanoLuc-NRP1 by increasing concentrations of unlabeled VEGF165a added simultaneously and incubated for 60 min at 37°C. Raw BRET ratios from 5 independent displacement experiments using duplicate wells are shown as mean ± SEM with bars representing vehicle (white) or VEGF165a-TMR only.
(D) Linear regression analysis (R2 = 0.95; p < 0.005) of the relationship between IC50 values determined in (C) and VEGF165a-TMR concentration. The y intercept provides an estimate for the Ki of competing VEGF165a (0.10 nM), while the slope (0.09) represents the ratio Ki/KD thus yielding an estimated KD = 1.11 nM for VEGF165a-TMR at NanoLuc-NRP1.
NRP1 Expressed in Living Cells Does Not Bind VEGF165b, VEGF121a, VEGF-Ax, or VEGF111a
To investigate how the three distinct fluorescent VEGF isoforms interacted with NRP1, we used VEGF165a-TMR alongside VEGF165b-TMR and VEGF121a-TMR to image fluorescent ligand binding to HaloTag-NRP1 expressed in HEK293T cells and labeled with membrane-impermeant Alexa Fluor 488. Upon both vehicle and fluorescent ligand application, HaloTag-NRP1 remained at the cell surface (Figure 5A). While 10 nM VEGF165a-TMR co-localized with HaloTag-NRP1 when imaged after 60 min, no binding of VEGF165b-TMR and VEGF121a-TMR to HaloTag-NRP1 was detected (Figure 5A). This latter observation was confirmed using NanoBRET, whereby no saturable binding was detected between VEGF165b-TMR or VEGF121a-TMR and NanoLuc-NRP1 (Figure 5B). Using 3 nM VEGF165a-TMR as a fluorescent probe, only unlabeled VEGF165a, VEGF145a, and VEGF189a displaced binding from NRP1 (Figure 5C). Full competition ligand binding experiments allowed pKi values at NanoLuc-NRP1 to be determined for these latter VEGF-A isoforms (Table S3). Quantitative immunohistochemistry analysis confirmed that NanoLuc-NRP1 and HaloTag-NRP1 were expressed at low levels in HEK293T cells (Figure S4).
Figure 5.

Selective Binding of VEGF Isoforms at NRP1
(A) Confocal live cell imaging of fluorescently labeled VEGF-TMR isoforms binding to N-terminal HaloTag-NRP1 stably expressed in HEK293T cells. HaloTag-NRP1 was tagged with the membrane-impermeant HaloTag-AF488 dye (green) and then incubated with 10 nM VEGF165a-TMR, VEGF165b-TMR, or VEGF121a-TMR (red) for 60 min at 37°C. Cells were imaged using an LSM710 confocal microscope and images are representative of those obtained in 3 independent experiments. Scale bar, 10 μm.
(B) NanoLuc-NRP1 HEK293T cells were incubated with increasing concentrations of VEGF165a-TMR, VEGF165b-TMR, or VEGF121a-TMR and incubated for 60 min at 37°C. Raw BRET ratios are expressed as mean ± SEM from 3–4 independent experiments.
(C) Inhibition of VEGF165a-TMR (3 nM) by competing unlabeled VEGF isoforms (30 nM), added simultaneously and incubated for 60 min at 37°C. Data are normalized to 3 nM VEGF165a-TMR (100%, black bar) and represent mean ± SEM pooled from 5 independent experiments. Statistical analyses were performed using Welch's t test: ***p ≤ 0.001; ****p ≤ 0.0001.
NanoBRET was also used to investigate ligand binding at a previously identified VEGF binding-dead mutant NRP1 Y297A, lacking a key residue in the b1 domain responsible for VEGF binding (Fantin et al., 2014). Having also confirmed membrane expression of HaloTag-NRP1 Y297A using live cell imaging, co-localization was absent for all three fluorescent VEGF isoforms (Figure 6A). Analogous BRET experiments showed that VEGF165a-TMR did not interact with NanoLuc-NRP1 Y297A, yielding BRET ratios that did not differ from vehicle (Figure 6B). This confirmed NRP1 Y297A as a mutant deficient for VEGF binding.
Figure 6.

The NRP1 Mutant Y297A Is Unable to Bind Any VEGF Isoforms
(A) Live confocal imaging of HEK293T cells stably expressing mutant HaloTag-NRP1 Y297A (green) labeled with membrane-impermeant HaloTag-AF488 dye (green). Cells were stimulated with 10 nM VEGF165a-TMR, VEGF165b-TMR, or VEGF121a-TMR for 60 min at 37°C. Cells were imaged using an LSM710 confocal microscope, and images are representative images of 3 independent experiments. Scale bar, 10 μm.
(B) NanoBRET measurements of the effect of unlabeled VEGF isoforms (30 nM) on the binding of 3 nM VEGF165a-TMR to wild-type NanoLuc-NRP1 or NanoLuc-NRP1 Y297A stably expressing HEK293T for 60 min (37°C). Raw BRET ratios are expressed as mean ± SEM pooled from 4 independent experiments.
Discussion
In the present study we have evaluated the ability of three fluorescent analogs of VEGF (VEGF165a, VEGF165b, and VEGF121a) to discriminate between VEGFR2 and NRP1 in living cells in real time. To enable this, we prepared single-site (N-terminal cysteine) labeled versions of VEGF165b and VEGF121a essentially as described previously for VEGF165a (Kilpatrick et al., 2017). These fluorescent ligands were used in combination with HEK293T cells stably expressing N-terminal NanoLuc-tagged VEGFR2 or NRP1 to evaluate the selectivity of VEGF isoforms for these two membrane proteins. The close proximity requirements (<10 nm) of the interaction between fluorescent ligand and receptor protein in order for bioluminescence transfer to occur (for NanoBRET measurement) ensured a high specificity of interaction, regardless of the extent of endogenous receptor expression. This was important since, although HEK293T did not express endogenous NRP1 (Figure S3), endogenous VEGFR2 were detected in a subpopulation of untransfected HEK293T cells. Furthermore, following expression of HaloTag-labeled NRP1, the endogenous expression of VEGFR2 appeared to increase (Figure S3). The expression level of VEGFR1 was, however, minimal in both untransfected HEK293T cells and those transfected with tagged variants of either VEGFR2 or NRP1.
VEGF165a-TMR, VEGF165b-TMR, and VEGF121a-TMR each exhibited saturable binding to NanoLuc-VEGFR2 expressed in HEK293T cells with nanomolar affinity. Furthermore, there were minimal levels of non-specific binding detected with each fluorescent ligand. Analysis of the real-time binding characteristics of each fluorescent ligand indicated that all three fluorescent VEGF variants had very similar kon and koff rate constants, and indeed their off rates were very slow (koff = 0.05–0.06 min−1). pKi values were obtained for a panel of seven unlabeled VEGF-A isoforms, including the recently described VEGF-Ax (Eswarappa et al., 2014), from competition experiments using all three of the fluorescent probes. All seven ligands had comparable nanomolar binding affinities for VEGFR2 ranging between 0.2 and 1.4 nM, in agreement with previous studies (Peach et al., 2018), suggesting that potential differences in signaling responses of these isoforms is not due to binding alone (Whitaker et al., 2001, Cébe Suarez et al., 2006, Eswarappa et al., 2014, Kilpatrick et al., 2017). There was no evidence of probe dependence in the measurement of these equilibrium constants, suggesting that the interactions could be described by simple mass action interactions.
VEGF165a-TMR bound to NanoLuc-NRP1 in living cells with a high affinity (4.41 nM) similar to that observed at NanoLuc-VEGFR2 (2.03 nM). However, in marked contrast VEGF165b-TMR and VEGF121a-TMR did not bind to NanoLuc-NRP1 (measured via NanoBRET) at concentrations up to 20 nM. This observation was corroborated by live cell confocal imaging, which showed that VEGF165b-TMR (10 nM) and VEGF121a-TMR (10 nM) bound to HaloTag-VEGFR2 but not to HaloTag-NRP1. The importance of residue Y297 (Fantin et al., 2014) of NRP1 for the binding of VEGF165a was confirmed in HEK293T cells expressing a Y297A mutant of NRP1. Competition binding experiments at NanoLuc-NRP1 yielded a rank order of pKi values of VEGF165a > VEGF189a > VEGF145a. In contrast, VEGF165b, VEGF-Ax, VEGF121a, and VEGF111a were unable to displace 3 nM VEGF165a-TMR at concentrations up to 30 nM. These observations support previous reports that these isoforms may be unable to bind NRP1 (Woolard et al., 2009). There have, however, been conflicting reports regarding VEGF121a binding to NRP1 (reviewed in Sarabipour and Mac Gabhann, 2017). Thus, although radioligand binding and solid-phase biotinylation assays have shown no interaction between VEGF121a and NRP1 (Cébe Suarez et al., 2006, Kawamura et al., 2008, Xin et al., 2016), low-affinity binding was detected using immobilized monomeric NRP1 and surface plasmon resonance (SPR) or isolated NRP1 b1/b2 domains (Pan et al., 2007, Parker et al., 2012, Delcombel et al., 2013).
A key feature of the present study is the ability to study the binding of VEGF-A isoforms to full-length VEGFR2 and NRP1 in living cells and in real time. This ensures that the interactions studied are of physiological relevance (Djordjevic and Driscoll, 2013). The lack of binding of VEGF165b, VEGF-Ax, VEGF121a, and VEGF111a to NRP1 is seen at concentrations up to 20 nM, which are far in excess of the predicted physiological levels of these ligands (<1 nM; Clegg and Mac Gabhann, 2017). These data suggest that VEGF165b-TMR and VEGF121a-TMR can be used as selective fluorescent probes for VEGFR2, even in cells that also express endogenous NRP1.
Real-time analysis of the binding of VEGF165a-TMR to NanoLuc-NRP1 expressed in HEK293T cells enabled the kinetics of ligand binding to be monitored to these membrane proteins for the first time. Despite comparable equilibrium dissociation constants determined by saturation and kinetic binding experiments, VEGF165a-TMR had faster binding kinetics at NRP1 compared with VEGFR2. Maximum specific binding to NanoLuc-NRP1 could be achieved within 5 min largely as a consequence of its very fast koff (0.26 min−1). These data suggest that in cells expressing both VEGFR2 and NRP1, VEGF165a will bind more quickly to NRP1 than to VEGFR2, particularly at low agonist concentrations. This may have important implications for the dynamics of VEGF signaling, and emphasize the need to understand the kinetic aspects of ligand binding to VEGFR2 and its co-receptors as well as the temporal aspects of intracellular signaling. Thus, since
for 1 nM VEGF165a-TMR using the parameters provided in Table 1, the t1/2 for association to VEGFR2 will be 9.2 min while that for NRP1 will be 2.1 min. For 10 nM VEGF165a-TMR the t1/2 values are 3.2 min and 0.7 min for VEGFR2 and NRP1, respectively.
Imaging ligand/receptor interactions using a membrane-impermeant HaloTag label also highlighted distinct differences in the subcellular distributions of VEGFR2 and NRP1, and the consequences of incubation with VEGF165a. HaloTag-VEGFR2 was constitutively internalized in the absence of ligand stimulation. This agrees with previous antibody-based imaging in HUVECs and human microvascular endothelial cells (Gampel et al., 2006, Basagiannis and Christoforidis, 2016, Basagiannis et al., 2016), and our own studies using VEGFR2 stably expressed in HEK293T cells (Kilpatrick et al., 2017). Furthermore, VEGF165a-TMR, VEGF165b, and VEGF121a were able to stimulate VEGFR2 internalization. In contrast, HaloTag-NRP1, labeled with a cell-impermeant HaloTag dye, was largely expressed on the cell membrane of HEK293T cells and remained at the cell surface despite 60 min of stimulation with a high concentration of VEGF165a-TMR. Furthermore, VEGF165a-TMR only labeled membrane-expressed NRP1. Other groups have shown an intracellular NRP1 distribution using permeabilized fluorescent antibody labeling (Narazaki and Tosato, 2006, Ballmer-Hofer et al., 2011). However, it is clear from the present work that cell membrane NRP1 is the primary target for VEGF165a and that this VEGF-A isoform does not stimulate internalization of NRP1.
It has been previously noted that fluorescent ligands can have pharmacological properties very different from their unlabeled counterparts and that they should be evaluated as new chemical entities (Stoddart et al., 2015, Stoddart et al., 2016). We have previously shown that VEGF165a-TMR behaves very similarly to VEGF165a in its ability to (1) stimulate NFAT reporter gene responses in HEK293T cells expressing wild-type VEGFR2 and (2) enable proliferation of HUVECs (Kilpatrick et al., 2017). However, both VEGF165b-TMR and VEGF121a-TMR behave differently in functional assays to VEGF165b and VEGF121a prepared in an identical way to the fluorescent probes. Thus, in NFAT assays the EC50 values obtained with both VEGF165b-TMR (pEC50 = 8.28) and VEGF121a-TMR (pEC50 = 8.57) were an order of magnitude higher (less potent) than the non-fluorescent versions. However, these EC50 values were very similar to the pKD values obtained from saturation binding studies (7.9–8.1 for VEGF165b-TMR and 8.2–8.4 for VEGF121a-TMR) and from competition binding studies (9.29–9.30 for VEGF165b and 9.16–9.59 for VEGF121a). This suggests that the differences were predominantly affinity based and that there was little signal amplification in the NFAT assay. Comparison of the agonist effects of fluorescent VEGF165b and VEGF121a on pY1212 phosphorylation, however, indicated that they produced the same maximal response as their unlabeled counterparts. In the HUVEC proliferation assay both VEGF165b-TMR and VEGF121a-TMR appeared to be of lower efficacy than the non-fluorescent ligands but still showed partial agonism in stimulating HUVEC proliferation. Taken together, these data suggest that VEGF165b-TMR and VEGF121a-TMR, unlike VEGF165a-TMR, are lower-affinity and lower-efficacy agonists at VEGFR2 than their unlabeled analogs. Furthermore, the extent of agonist activity appears to depend on the signaling pathway being monitored. This may indicate an ability for these fluorescent analogs to exhibit some signaling bias in a similar way to that seen with G-protein-coupled receptors (Smith et al., 2018).
In summary, fluorescent VEGF isoforms were used to probe the pharmacology of VEGFR2 and its co-receptor NRP1 in living cells in real time at 37°C. Despite approved therapeutics targeting VEGF/VEGFR2 (Ferrara and Adamis, 2016), this is the first comprehensive ligand binding study of the interactions of a range of VEGF isoforms with both full-length VEGFR2 and NRP in living cells. The real-time sensitivity of NanoBRET revealed clear differences in the kinetic binding profiles of VEGF165a-TMR for NRP1 and VEGFR2, despite this ligand having a very similar equilibrium dissociation binding constant for each membrane protein. All VEGF isoforms studied had a similar high affinity for VEGFR2 but not all isoforms interacted with NRP1. In particular, VEGF165b-TMR and VEGF121a-TMR were not able to bind to NRP1 at physiologically relevant concentrations. These two partial agonist ligands should therefore be important and selective probes for the study of VEGFR2 in cells also expressing NRP1. Furthermore, our study also emphasizes the importance of the kinetic aspects of ligand binding to VEGFR2 and its co-receptors in the overall dynamics of VEGF signaling.
Significance
VEGF-A is an essential mediator of angiogenesis that signals via VEGFR2. We have synthesized fluorescent VEGF-A isoforms and demonstrate that they can discriminate between VEGFR2 and its co-receptor NRP1 in real-time ligand binding studies. We have used a precision chemical biology approach in live cells to accurately define the binding characteristics of specific VEGF-A isoforms and to determine which isoforms can bind to NRP1 at concentrations required to occupy VEGFR2. Only VEGF165a, VEGF145a, and VEGF189a are able to also bind to NRP1. Furthermore, we have shown that while VEGF165a-TMR has a similar equilibrium binding affinity for VEGFR2 and NRP1, it binds more rapidly to NRP1 than to VEGFR2. We have also shown that VEGF165a-TMR has a shorter residence time (1/koff) at NRP1 (3.8 min) than VEGFR2 (16.6 min). These fluorescent ligands should therefore serve as valuable probes to interrogate the roles of VEGFR2 and NRP1 in angiogenesis and signaling.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-VEGFR1 | Sigma Aldrich | Cat# V4762 RRID:AB_477622 |
| Mouse monoclonal anti-VEGFR2 | Sigma Aldrich | Cat# V9134 RRID:AB_477630 |
| Goat polyclonal anti-Neuropilin-1 | Santa Cruz | Cat# SC7239 RRID:AB_2150835 |
| Rabbit monoclonal anti-VEGFR2 phosphoY1212 | Cell Signalling Technology | Cat# 2477S RRID:AB_331374 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| VEGF165a | R&D Systems (Abingdon, UK) | Cat# 293-VE |
| VEGF165b | R&D Systems (Abingdon, UK) | Cat# 3045-VE |
| VEGF121a | R&D Systems (Abingdon, UK) | Cat# 4644-VS |
| VEGF145a | R&D Systems (Abingdon, UK) | Cat# 7626-VE |
| VEGF189a | R&D Systems (Abingdon, UK) | Cat# 8147-VE |
| VEGF111a | R&D Systems (Abingdon, UK) | Cat# 5336-VE |
| VEGF-Ax | R&D Systems (Abingdon, UK) | Cat# 9018-VE |
| HaloTag AlexaFluor 488 membrane impermeant substrate | Promega Corporation (Wisconsin, USA) | Cat# G1002 |
| bisBenzimide H 33342 trihydrochloride | Sigma Aldrich | Cat# B2261 |
| Formaldehyde solution 4% | Sigma Aldrich | Cat# F8775 |
| Cediranib | Sequoia Research Products | Cat# SRP01883c |
| Chromasolv | Sigma Aldrich | Cat# 34877 |
| Rhodamine 6G | Sigma Aldrich | Cat# R4127 |
| Triton-X-100 (laboratory grade) | Sigma Aldrich | Cat# X100 |
| DTT 1,4-Dithiothreitol | Sigma Aldrich | Cat# DTT-RO |
| PNGase F | Promega Corporation (Wisconsin, USA) | Cat# V4831 |
| Protease-free bovine serum albumin | Milpore | Cat# 126609 |
| Protease-free bovine serum albumin | Sigma Aldrich | Cat# 03117332001 |
| Secondary chick anti-mouse | Invitrogen | Cat# A21463 |
| Secondary donkey anti-goat | Invitrogen | Cat# A11056 |
| Secondary chick anti-rabbit AlexaFluor-488 | ThermoFisher Scientific, USA | Cat# A-21441 |
| Chicken serum | Sigma Aldrich | Cat# C5405 |
| Donkey serum | Sigma Aldrich | Cat# D9663 |
| ProLong Gold antifade reagent | ThermoFisher Scientific, USA | Cat# P10144 |
| Dulbecco’s Modified Eagle’s Medium | Sigma Aldrich | Cat# D6429 |
| Fetal Bovine Serum | Sigma Aldrich | Cat# F2442 |
| Medium 200 (Gibco) | ThermoFisher Scientific, USA | Cat# M-200-500 |
| Large Vessel Endothelial Supplement (LVES 50x) (Gibco) | ThermoFisher Scientific, USA | Cat# A1460801 |
| Poly-D-Lysine hydrobromide | Sigma Aldrich | Cat# P6407 |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Sigma Aldrich | Cat# D8537 |
| Trypsin-EDTA solution x10 | Sigma Aldrich | Cat# T4174 |
| Critical Commercial Assays | ||
| HaloTag Mammalian Protein Detection and Purification System | Promega Corporation (Wisconsin, USA) | Cat# G6795 |
| ONE-Glo™ Luciferase | Promega Corporation (Wisconsin, USA) | Cat# E6120 |
| Nano-Glo luciferase assay system (Furimazine) | Promega Corporation (Wisconsin, USA) | Cat# N1130 |
| Experimental Models: Cell Lines | ||
| Human: GloResponse™ NFAT-RE-luc2P HEK293 cell line (female) | Promega Corporation (Wisconsin, USA) | Cat# E8510 |
| Human: HUVEC cells (newborn male, single donor) | ThermoFisher Scientific | Cat# C0035C. Lot number: 1606186. |
| Human: HEK293T cells (female) | ATCC (Virginia, USA) | Cat# CRL-3216 |
| Recombinant DNA | ||
| NanoLuc-VEGFR2 | Promega Corporation (Wisconsin, USA) | Custom synthesis |
| NanoLuc-NRP1 | Promega Corporation (Wisconsin, USA) | Custom synthesis |
| NanoLuc-NRP1 Y297A | Promega Corporation (Wisconsin, USA) | Custom synthesis |
| HaloTag-VEGFR2 | Promega Corporation (Wisconsin, USA) | Custom synthesis |
| HaloTag-NRP1 | Promega Corporation (Wisconsin, USA) | Custom synthesis |
| HaloTag-NRP1 Y297A | Promega Corporation (Wisconsin, USA) | Custom synthesis |
| VEGF165a | Gene Dynamics LLC (Oregon, USA) | Custom synthesis |
| VEGF165b | Gene Dynamics LLC (Oregon, USA) | Custom synthesis |
| VEGF121a | Gene Dynamics LLC (Oregon, USA) | Custom synthesis |
| pFN21 HaloTag CMV Flexi Vector (modified to contain a IL-6 secretion sequence and a EPTTEDLYFQCDN linker sequence) | Promega Corporation (Wisconsin, USA) | Cat# G2821 |
| Software and Algorithms | ||
| GraphPad Prism 7.02 | GraphPad Software, La Jolla California USA |
www.graphpad.com |
| Zen 2010 | Zeiss, Germany | www.zeiss.com |
| MetaXpress | Molecular Devices, USA | www.moleculardevices.com |
| Other | ||
| Black 96-well plates | Greiner Bio-One | Cat# 655090 |
| White 96-well plates | Greiner Bio-One | Cat# 655098 |
| 8-well plates | Nunc Lab-Tek, Thermo Fisher Scientific | Cat# 155411 |
| Coverslips (18x18mm; 1.5H) | Zeiss, Germany | Cat# 474030-9000-000 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen J. Hill (stephen.hill@nottingham.ac.uk).
Experimental Model and Subject Details
HUVECs (obtained from a single newborn male donor) and HEK293T (female) cells were transfected and cultured as described in Method Details.
Method Details
Cell Culture
HEK293T cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich, USA) supplemented with 10% Fetal Calf Serum (FCS; Sigma-Aldrich, USA) at 37°C/5% CO2. Cells were passaged at 70-80% confluency using Phosphate Buffered Saline (PBS; Lonza, Switzerland) and trypsin (0.25% w/v in versene; Lonza). Stable and transient transfections were performed using FuGENE HD (Promega Corporation, USA) at a reagent to cDNA ratio of 3:1. Human umbilical vein endothelial cells (HUVECs; C0035C, Thermo Fisher Scientific, USA) were grown at 37°C/5% CO2 in Medium 200 containing 10% Large Vessel Endothelial Supplement (LVES, 50X; Thermo Fisher Scientific, USA) and passaged at 80-90% confluency between passages 4 to 9.
DNA Constructs
For N terminal NanoLuc tagged wildtype VEGFR2 (NM_002253; Genscript, New Jersey, USA) or NRP1 constructs (NM_003873.5; Kazusa DNA Research Institute (Japan) the appropriate cDNA was cloned into a pF-sNnK CMV/neo vector (Promega Corporation; N1321) encoding the secretory signal peptide sequence of IL-6 fused onto the N terminus of NanoLuc. This resulted in open reading frames which encoded a secreted NanoLuc fused via a Gly-Ser-Ser-Gly (AIA) linker to the N terminus of wildtype VEGFR2 or NRP1 (termed NanoLuc VEGFR2 or NRP1 respectively). For N terminal HaloTag constructs, wildtype VEGFR2 or NRP1 cDNA was cloned into a pFN21A CMV/neo flexi vector (Promega Corporation; G2821) encoding a fusion of the secretory signal peptide sequence of IL-6 onto the N terminus of HaloTag. The resultant ORFs encoded a secreted HaloTag fused via a EPTTEDLYFQSDN(AIA) linker to the N terminus of NRP1 (HaloTag VEGFR2 or NRP1).
Fluorescent Ligand Synthesis
VEGF-A isoforms VEGF165a, VEGF165b and VEGF121a labelled at a single N-terminal cysteine residue with 6- tetramethylrhodamine (TMR)-PEG-CBT were synthesised and purified using the HaloTag mammalian protein detection and purification system (G6795; Promega Corporation, USA) alongside unlabelled analogues prepared identically (as described in Kilpatrick et al., 2017). To generate labelled isoforms, the HaloTEV proteolytic release was done in the presence of 100μM TCEP and 4x molar excess of 6-TMR-PEG-CBT. This step generated VEGF isoforms with an N-terminal cysteine that served as single point of conjugation with 6-TMR-PEG-CBT. The purified labelled isoforms were dialyzed for 24 hours (50mM HEPES, 150mM NaCl) to remove the unconjugated 6-TMR-PEG-CBT and TCEP and stored in 2.5mg/ml protease-free bovine serum albumin (BSA; Millipore, USA) at -80°C. Labelling specificity and efficiency was determined using liquid chromatography-tandem mass spectrometry (LC-MS). SDS-PAGE assays in the presence and absence of 100mM dithiothreitol (DTT; Sigma-Aldrich, UK) or PNGase (Promega Corporation, USA) were used to measure dimerisation and glycosylation status respectively (detailed in Kilpatrick et al., 2017). Ligands were stored in 2.5mg/ml protease-free bovine serum albumin (BSA; Millipore, USA). Labelling specificity and efficiency was determined using liquid chromatography-tandem mass spectrometry (LC-MS). SDS-PAGE assays in the presence and absence of dithiothreitol (DTT; Sigma-Aldrich, UK) or PNGase (Promega Corporation, USA) were used to measure dimerisation and glycosylation status respectively (detailed in Kilpatrick et al., 2017).
NFAT Luciferase Reporter Gene Assay
HEK293T cells stably expressing both wild type VEGFR2 and the Firefly luciferase reporter gene ReLuc2P (Promega Corporation, USA) inserted downstream of the NFAT promoter were used to monitor NFAT-induced gene transcription following VEGFR2 activation (Carter et al., 2015). On the day of experimentation, cells grown to 95-100% confluency were plated in white-sided 96 well plates (Greiner Bio-One, 655089) at 44,000 cells/well, and incubated for 1 hour in 100μl/well serum free DMEM/0.1% BSA (37°C/5% CO2). Cells were stimulated in duplicate wells with increasing concentrations of VEGF121a-TMR, VEGF165b-TMR or equivalent unlabelled VEGF isoforms (synthesised in an identical manner to the fluorescent variant), then incubated for 5 hours at 37°C/5% CO2. ONE-Glo Luciferase reagent (Promega Corporation, USA) was then added at 100μl/well and luminescence was measured using a TopCount platereader (Perkin Elmer, UK) following a 5 minute delay allowing reagent to react with luciferase and background luminescence to subside.
VEGFR2 Phosphorylation Assay
HEK293T cells stably expressing NanoLuc-VEGFR2 were seeded at 15,000 cells/well in black flat-bottomed 96-well plates (Greiner Bio-One, 655090) pre-coated with poly-D-lysine (0.01mg/ml in PBS). Following 24 hours, cells were serum starved and grown for another 24 hours (37°C/5% CO2), with additional 1 hour serum starving step prior to experimentation. For negative control wells, cells were pre-incubated for 30 minutes with 1μM cediranib (Sequoia Research Products, UK). Cells were then stimulated for 20 minutes with 30nM VEGF165b-TMR or VEGF121a-TMR, commercially available VEGF165a, VEGF165b or VEGF121a (R&D Systems) and VEGF165b or VEGF121a prepared identically to the fluorescent analogues, in the presence of absence of negative control 1μM cediranib. Cells were washed with 100μl/well PBS, fixed with 3% paraformaldehyde (PFA)/PBS for 20min at room temperature (RT), washed (3x5min PBS), permeabilised with 0.025% Triton-X-100 in PBS, washed (3x5min PBS) and incubated with 3% BSA/1% glycine/PBS to reduce non-specific binding (30mins, RT). After washing (3x5min PBS), cells were blocked with 10% chick serum in PBS (30min, RT) and incubated at 4°C overnight with rabbit monoclonal anti-VEGFR2 phosphoY1212 (Cell Signalling, 2477) diluted 1:200 in 10% chick serum/PBS. Cells were washed (3x5min PBS) and incubated in the dark with secondary antibody chick anti-rabbit AlexaFluor488 (Thermo Fisher, A21441). Nuclei were stained with 2mg/ml H33342 (15min, RT), washed and stored at 4°C in PBS. Cells were imaged using an ImageXpress Micro widefield platereader (Molecular Devices, USA) with a 20x objective at 4 sites per well using FITC and DAPI filters (exposure 1500ms and 25ms respectively).
HUVEC Proliferation Assay
HUVECs (passage 4-9) were seeded at 5,000 cells/well in black flat-bottomed 96-well plates (Greiner Bio-One, 655090) in 10% LVES/Medium 200. Following 24 hours of cell growth at 37°C/5% CO2, plating medium was replaced with Medium 200 containing 0.1% serum for 24 hours. Cells were then stimulated with commercially available VEGF121a or VEGF165b (R&D Systems), VEGF121a-TMR or VEGF165b-TMR (Promega Corporation, USA) at 0.3nM, 3nM or 30nM (in 0.1% serum/medium), or positive control 3nM VEGF165a (R&D Systems). Following 48 hour stimulation at 37°C/5% CO2, cells were washed with 100μl/well PBS, fixed with 3% PFA/PBS (20 minutes, room temperature ) and nuclei stained with 2mg/ml H33342 (15 minutes, RT). Nuclei were imaged using an ImageXpress Micro widefield platereader (Molecular Devices, USA) with a 4x objective using a DAPI filter (4 sites per well, 25ms exposure time).
Measuring Ligand Binding Using NanoBRET
HEKT293 cells stably expressing full-length wild-type VEGFR2, NRP1 or NRP1 Y297A, tagged on the N-terminus with the 19kDa luciferase NanoLuc, were seeded 24 hours prior to experimentation at 35,000 cells/well on white 96-well clear bottomed plates (Greiner Bio-One, 655089) pre-coated with poly-D-lysine (0.01mg/ml in PBS), and incubated at 37°C/5% CO2. Having identified a natural polymorphism (V297I) in the NanoLuc-VEGFR2 construct used previously (Kilpatrick et al., 2017), experiments performed with VEGF165a-TMR verified no distinction from wild type VEGFR2 (Figures 1 and 3). Medium was replaced with Hank’s buffered saline solution (HBSS) containing 0.1% BSA. For full displacement experiments, cells were co-incubated with increasing concentrations of unlabelled ligand (R&D Systems) or vehicle (HBSS/0.1% BSA), as well as fixed concentrations of fluorescently labelled VEGF165a-TMR, VEGF165b-TMR or VEGF121a-TMR in duplicate wells (0.25nM, 0.5nM, 1nM, 2nM, 3nM). Additional displacement experiments incubated NanoLuc-VEGFR2 or NanoLuc-NRP1 cells with 3nM VEGF-TMR in the presence and absence of 30nM competing unlabelled VEGF. For saturation experiments, increasing concentrations of VEGF165a–TMR, VEGF165b-TMR or VEGF121a-TMR were added in the presence or absence of a high concentration of corresponding unlabelled ligand (100nM, ∼100-fold greater than the estimated KD value). Following 60min stimulation in the dark at 37°C, the NanoLuc substrate furimazine (final concentration 10μM) was added to each well and equilibrated for 5 minutes to enable NanoLuc-mediated furimazine oxidation and resulting bioluminescence emission. Emissions were recorded using the PHERAstar FS platereader (BMG Labtech) using filters measuring NanoLuc emissions at 450nm (30nm bandpass), then TMR emissions using a longpass filter at 550nm for NanoLuc-VEGFR2 cells or 610nm for cells expressing wild type or mutant NanoLuc-NRP1. BRET ratios were calculated as fluorescence over luminescence emissions. NanoBRET kinetic experiments were performed at 37°C throughout and required furimazine pre-treatment 5 minutes prior to addition of VEGF165a-TMR, VEGF165b-TMR or VEGF121a-TMR (1nM to 20nM). BRET ratios were then calculated every 30 seconds for up to 120 minutes.
Live Cell Confocal Imaging
HEKT293 cells stably expressing HaloTag-VEGFR2, HaloTag-NRP1 or HaloTag-NRP1 Y297A were seeded 48 hours prior to imaging at 20,000 cells/well in 8-well plates (Nunc Lab-Tek, Thermo Fisher Scientific) pre-coated with poly-D-lysine (0.01mg/ml in PBS), then replaced with serum free DMEM following 24 hours. Cells were treated with 0.5μM membrane impermeant HaloTag Alexa Fluor 488 substrate (Promega Corporation, USA) in HBSS/0.1% BSA for 30 min (37°C). Cells were then washed twice and replaced with HBSS/0.1% BSA prior to incubation with 10nM VEGF165a-TMR, VEGF165b-TMR or VEGF121a-TMR in the dark at 37°C. Cells were imaged live using an LSM710 confocal microscope fitted with a 63x Pan Apochromat oil objective (1.4NA) using Argon488 and Argon 546 laser excitation (3% power), a long pass 540 filter and a pin hole diameter of 1 Airy unit. All images were taken at 1024x1024 pixels per frame with 8 averages.
Fluorescence Correlation Spectroscopy (FCS)
Solution based FCS measurements were performed in Nunc LabTek 8-well chambered coverglasses (Thermo-Fisher Scientific, UK) using a LSM510 NLO Confocor3 microscope equipped with a c-Apochromat 40/1.2NA water immersion objective (Zeiss, Germany). The confocal volume was placed 200μm in solution above the surface of the coverglass. Calibration of beam paths was performed using 20nM Rhodamine 6G (Diffusion coefficient (D) = 2.8 10-10 m2/s; Sigma Aldrich, UK) in high performance liquid chromatography grade water (Chromasolv; Sigma Aldrich) with 488nm and 561nm laser lines using 10x10sec reads. A range of VEGF165b-TMR or VEGF121a-TMR (2-10nM) solutions were prepared in HBSS/0.1% BSA in the presence or absence of 10mM DTT. DTT containing ligand solutions were preincubated for 30min. FCS recordings were collected with 2 sets of 10x10secs reads using 561nm laser excitation (20% power; AOTF set to 10; equivalent to 0.39kW/cm2) with fluorescence emissions collected using a long pass 580 (LP580) filter.
Immunofluorescence Labelling
For confocal imaging (Figure S3), HUVECs, wild type HEKT293T cells or HEK293T cells expressing NanoLuc-VEGFR2 or NanoLuc-NRP1 were seeded onto poly-D-lysine coated high resolution coverslips (Zeiss, Germany; 18mmx18mm, 1.5H) at 300,000 cells/well and grown in 6 well culture plates 24 hours prior to experimentation. On the day of the assay, coverslips were transferred to humidified wells lined with parafilm and PBS to avoid dryness and washed 3x5min with PBS. Cells were fixed with 3% paraformaldehyde (PFA)/PBS for 20min at room temperature (RT), washed (3x5min PBS) and incubated with 3% BSA/1% glycine/PBS to reduce non-specific binding (30mins, RT). After washing, cells were blocked with 4% chick serum or donkey serum for VEGFR2 and NRP1 staining respectively (PBS, 30min, RT). This was then replaced with primary antibody diluted 1:200 in 4% serum/PBS and incubated overnight at 4°C (anti-VEGFR1 mAb produced in mice, Sigma V4762; anti-VEGFR2 mAb (mouse), Sigma V9134; anti-NRP1 goat pAb Santa Cruz 7239). The following day, cells were washed and incubated in the dark with secondary antibody diluted 1:500 in 4% serum/PBS for 1 hour at room temperature (VEGFR1 and VEGFR2 chick anti-mouse AlexaFluor488, Invitrogen A21463; NRP1 donkey anti-goat AlexaFluor546, Invitrogen A11056). Coverslips were washed, mounted onto slides using ProLong Diamond (Thermo Fisher Scientific), sealed and stored at 4°C. Coverslips were imaged using a Confocal Zeiss LSM880 fitted with a 63x Pan Apochromat oil objective (1.4NA) using Argon488 or DPSS561 laser excitation at 2% laser power with a pinhole diameter of 1 Airy unit.
To quantify relative receptor expression (Figure S4), HUVECs, wild type HEK293T cells or HEK293T cells expressing NanoLuc- and HaloTag- labelled VEGFR2 or NRP1 were seeded at 25,000 cells/well in black 96-well plates pre-coated with poly-D-lysine (0.01mg/ml in PBS) and grown for 24 hours (37°C/5% CO2). Cells were fixed with 3% PFA/PBS then followed an identical immunofluorescence staining protocol as above in 96-well plates with VEGFR2 mouse mAb (Sigma V9134) and NRP1 goat pAb (Santa Cruz 7239). Having labelled with respective secondary antibodies, cells were washed with PBS, nuclei were stained with 2mg/ml H33342 (15 minutes, RT), washed and stored in PBS at 4°C. Cells were imaged using an ImageXpress Micro widefield platereader with a 20x objective at 4 sites per well, with a FITC or TRITC filter for VEGFR2 or NRP1 respectively (500ms exposure time) and a DAPI filter imaging nuclei (25ms exposure time).
Quantification and Statistical Analysis
Data Analysis
All data are presented as mean ± S.E.M. and were analysed using GraphPad Prism 7.02 (San Diego, CA, USA). Equilibrium binding and functional assays were analysed as described in Kilpatrick et al. (2017). A power calculation was performed to confirm sample number for statistical comparisons of pKi values obtained with different fluorescent ligands. This was done on the basis of 5 separate experiments with the anticipated standard deviation obtained in similar experiments and a calculation of the statistical power to detect a significant change of pKi of 0.3 log units. This yielded a power of 0.99, i.e. there was a 99% chance of detecting a significant change in pKi value of 0.3 log units. Statistical analyses using one-way ANOVA are described in the corresponding figure legends or within the text. Significance was defined as p<0.05.
High Content Imaging
Images obtained with the ImageXpress Micro widefield platereader at 4 sites per well were quantified using MetaXpress 2.0 (Molecular Devices, USA). Nuclei were quantified with diameter 5-25μm and 100 graylevel intensity above background. VEGFR2 phosphorylation was quantified (Figures 1D and 1E) using a granularity algorithm, granules were defined as 6-12μm diameter with a graylevel intensity of 50 above background. Granularity was quantified per cell, baseline-corrected to non-specific binding (secondary antibody only) and normalised to cediranib-treated wells (0%) and response to 30nM VEGF165a (100%). Quantifying relative receptor expression (Figure S4) using a multiwavelength cell scoring algorithm, regions were defined as 2-15μm in size. Due to distinctions in secondary antibodies, VEGFR2 (FITC) was defined as intensity over 200 graylevels and NRP1 (TRITC) over 50 graylevels. Fluorescence was quantified as integrated intensity per cell and baseline-corrected per experiment to non-specific fluorescence (secondary antibody only).
FCS Autocorrelation Analysis
Autocorrelation analysis was performed using Zen 2010 software (Zeiss, Germany) with all traces fit using a single one component, free 3D Brownian diffusion model, with a pre-exponential included to account for the triplet state of the fluorophore.
Data and Software Availability
GraphPad Prism 7.02 (San Diego, CA, USA) was used to analyse the quantified data and produce the graphs. Zen 2010 (Zeiss; Germany) was used to perform autocorrelation analysis for FCS. MetaXpress 2.0 (Molecular Devices, USA) was used to quantify VEGFR2 phosphorylation and receptor expression labelled with immunofluorescence following high content imaging on the widefield platereader.
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council (grant number BB/L019418/1) and Promega Corporation. C.P. was funded by an A.J. Clark studentship from the British Pharmacological Society (BPS). We thank Dave Good and Sergy Levin for the synthesis of 6-TMR-PEG-CBT and Mike Rosenblatt for the LC-MS/MS analysis. We also thank the School of Life Sciences Imaging (SLIM) team for maintenance and support for high content and confocal imaging facilities.
Author Contributions
Conceptualization, S.J.H., J.W., and L.E.K.; Methodology, S.J.H., L.E.K., R.F.-O., and K.Z.; Formal analysis, C.J.P., L.E.K., S.J.H. and R.F.-O.; Investigation, C.J.P., L.E.K., R.F.-O., M.B.R., S.J.H., and J.W.; Writing – Original Draft, C.J.P., L.E.K., and S.J.H.; Writing – Review and Editing, C.J.P., L.E.K., K.V.W., M.B.R., R.F.-O, J.W., and S.J.H; Supervision, S.J.H., J.W., and L.E.K.
Declaration of Interests
R.F.-O., M.B.R., K.Z., and K.V.W. are employees of Promega Corporation, which has proprietary rights over the NanoBRET assay, HaloTag technology, and CBT labeling technology.
Published: July 26, 2018
Footnotes
Supplemental Information includes four figures and three tables and can be found with this article online at https://doi.org/10.1016/j.chembiol.2018.06.012.
Contributor Information
Jeanette Woolard, Email: jeanette.woolard@nottingham.ac.uk.
Stephen J. Hill, Email: stephen.hill@nottingham.ac.uk.
Supplemental Information
References
- Ballmer-Hofer K., Andersson A.E., Ratcliffe L.E., Berger P. Neuropilin-1 promotes VEGFR-2 trafficking through Rab11 vesicles thereby specifying signal output. Blood. 2011;118:816–826. doi: 10.1182/blood-2011-01-328773. [DOI] [PubMed] [Google Scholar]
- Basagiannis D., Christoforidis S. Constitutive endocytosis of VEGFR2 protects the receptor against shedding. J. Biol. Chem. 2016;291:16892–16903. doi: 10.1074/jbc.M116.730309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basagiannis D., Zografou S., Murphy C., Fotsis T., Morbidelli L., Ziche M., Bleck C., Mercer J., Christoforidis S. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation via macropinocytosis. J. Cell Sci. 2016;129:4091–4104. doi: 10.1242/jcs.188219. [DOI] [PubMed] [Google Scholar]
- Cai H., Reed R.R. Cloning and characterization of neuropilin-1-interacting protein: a PSD- 95/Dlg/ZO-1 domain-containing protein that interacts with the cytoplasmic domain of neuropilin-1. J. Neurosci. 1999;19:6519–6527. doi: 10.1523/JNEUROSCI.19-15-06519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter J., Wheal A., Hill S., Woolard J. Effects of receptor tyrosine kinase inhibitors on VEGF165a- and VEGF165b-stimulated gene transcription in HEK-293 cells expressing human VEGFR2. Br. J. Pharmacol. 2015;172:3141–3150. doi: 10.1111/bph.13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cébe Suarez S., Pieren M., Cariolato L., Arn S., Hoffman U., Bogucki A., Manlius C., Wood J., Ballmer-Hofer K. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell. Mol. Life Sci. 2006;63:2067–2077. doi: 10.1007/s00018-006-6254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittenden T.W., Claes F., Lanahan A.A., Autiero M., Palac R.T., Tkachenko E.V., Elfenbein A., Ruiz de Almodovar C., Dedkov E., Tomanek R. Selective regulation of arterial branching morphogenesis by synectin. Dev. Cell. 2006;10:783–795. doi: 10.1016/j.devcel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Chung A.S., Ferrara N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- Clegg L.E., Mac Gabhann F. A computational analysis of in vivo VEGFR activation by multiple co-expressed ligands. PLoS Comput. Biol. 2017;13:e1005445. doi: 10.1371/journal.pcbi.1005445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham S.A., Arrate M.P., Brock T.A., Waxham M.N. Interactions of FLT-1 and KDR with phospholipase Cγ: identification of the phosphotyrosine binding sites. Biochem. Biophys. Res. Commun. 1997;240:635–639. doi: 10.1006/bbrc.1997.7719. [DOI] [PubMed] [Google Scholar]
- Delcombel R., Janssen L., Vassy R., Gammons M., Haddad O., Richard B., Letourneur D., Bates D., Hendricks C., Waltenberger J. New prospects in the roles of the C-terminal domains of VEGF-A and their cooperation for ligand binding, cellular signaling and vessels formation. Angiogenesis. 2013;16:353–371. doi: 10.1007/s10456-012-9320-y. [DOI] [PubMed] [Google Scholar]
- Djordjevic S., Driscoll P.C. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov. Today. 2013;18:447–455. doi: 10.1016/j.drudis.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Eswarappa S.M., Potdar A.A., Koch W.J., Fan Y., Vasu K., Lindner D., Willard B., Graham L.M., Dicorleto P.E., Fox P.L. Programmed translational readthrough generates antiangiogenic VEGF-Ax. Cell. 2014;157:1605–1618. doi: 10.1016/j.cell.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A., Schwarz Q., Davidson K., Normando E.M., Denti L., Ruhrberg C. The cytoplasmic domain of neuropilin 1 is dispensable for angiogenesis, but promotes the spatial separation of retinal arteries and veins. Development. 2011;138:4185–4191. doi: 10.1242/dev.070037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A., Herzog B., Mahmoud M., Yamaji M., Plein A., Denti L., Ruhrberg C., Zachary I. Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF-A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development. 2014;141:556–562. doi: 10.1242/dev.103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N., Adamis A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016;15:385–403. doi: 10.1038/nrd.2015.17. [DOI] [PubMed] [Google Scholar]
- Gampel A., Moss L., Jones M.C., Brunton V., Norman J.C., Mellor H. VEGF regulates the mobilization of VEGFR2/KDR from an intracellular endothelial storage compartment. Blood. 2006;108:2624–2631. doi: 10.1182/blood-2005-12-007484. [DOI] [PubMed] [Google Scholar]
- Goel H.L., Mercurio A.M. VEGF targets the tumour cell. Nat. Rev. Cancer. 2013;13:871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C., Rodriguez E.R., Reimert D.V., Shu T., Fritzsch B., Richards L.J., Kolodkin A.L., Ginty D.D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell. 2003;5:45–57. doi: 10.1016/s1534-5807(03)00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.F., Vander Kooi C.W. Neuropilin functions as an essential cell surface receptor. J. Biol. Chem. 2015;290:29120–29126. doi: 10.1074/jbc.R115.687327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper S.J., Bates D. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat. Rev. Cancer. 2008;8:880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubb A.M., Strickland L.A., Liu S.D., Mak J., Schmidt M., Koeppen H. Neuropilin-1 expression in cancer and development. J. Pathol. 2012;226:50–60. doi: 10.1002/path.2989. [DOI] [PubMed] [Google Scholar]
- Kawamura H., Li X., Harper S., Bates D., Claesson-Welsh L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008;68:4683–4692. doi: 10.1158/0008-5472.CAN-07-6577. [DOI] [PubMed] [Google Scholar]
- Kawasaki T., Kitsukawa T., Bekku Y., Matsuda Y., Sanbo M., Yagi T., Fujisawa H. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–4902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- Kilpatrick L.E., Friedman-Ohana R., Alcobia D.C., Riching K., Peach C.J., Wheal A.J., Briddon S.J., Robers M.B., Zimmerman K., Machleidt T. Real-time analysis of the binding of fluorescent VEGF165a to VEGFR2 in living cells: effect of receptor tyrosine kinase inhibitors and fate of internalized agonist-receptor complexes. Biochem. Pharmacol. 2017;136:62–75. doi: 10.1016/j.bcp.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsukawa T., Shimizu M., Sanbo M., Hirata T., Taniguchi M., Bekku Y., Yagi T., Fujisawa H. Neuropilin–semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron. 1997;19:995–1005. doi: 10.1016/s0896-6273(00)80392-x. [DOI] [PubMed] [Google Scholar]
- Koch S., Tugues S., Li X., Gualandi L., Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011;437:169–183. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- Lanahan A., Zhang X., Fantin A., Zhuang Z., Rivera-Molina F., Speichinger K., Prahst C., Zhang J., Wang Y., Davis G. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell. 2013;25:156–168. doi: 10.1016/j.devcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.W., Lee J.E., Yoo C.Y., Ko M.S., Park C.S., Yang S.H. NRP-1 expression is strongly associated with the progression of pituitary adenomas. Oncol. Rep. 2014;32:1537–1542. doi: 10.3892/or.2014.3392. [DOI] [PubMed] [Google Scholar]
- Machleidt T., Woodroofe C.C., Schwinn M.K., Méndez J., Robers M., Zimmerman K., Otto P., Daniels D.L., Kirkland T.A., Wood K.B. NanoBRET—a novel BRET platform for the analysis of protein-protein interactions. ACS Chem. Biol. 2015;10:1797–1804. doi: 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- Narazaki M., Tosato G. Ligand-induced internalization selects use of common receptor neuropilin-1 by VEGF165 and semaphorin3A. Blood. 2006;107:3892–3901. doi: 10.1182/blood-2005-10-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q., Chathery Y., Wu Y., Rathore N., Tong R., Peale F., Bagri A., Tessier-Lavigne M., Koch A.W., Watts R.J. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J. Biol. Chem. 2007;282:24049–24056. doi: 10.1074/jbc.M703554200. [DOI] [PubMed] [Google Scholar]
- Parker M.W., Xu P., Guo H.F., Vander Kooi C.W. Mechanism of selective VEGF-a binding by neuropilin-1 reveals a basis for specific ligand inhibition. PLoS One. 2012;7:e49177. doi: 10.1371/journal.pone.0049177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peach C.J., Mignone V.W., Augusta Arruda M., Alcobia D.C., Hill S.J., Kilpatrick L.E., Woolard J. Molecular pharmacology of VEGF-A isoforms: binding and signalling at VEGFR2. Int. J. Mol. Sci. 2018;19:1264. doi: 10.3390/ijms19041264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahst C., Héroult M., Lanahan A.A., Uziel N., Kessler O., Shraga-Heled N., Simons M., Neufeld G., Augustin H.G. Neuropilin-1-VEGFR-2 complexing requires the PDZ-binding domain of neuropilin-1. J. Biol. Chem. 2008;283:25110–25114. doi: 10.1074/jbc.C800137200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruch C., Skiniotis G., Steinmetz M.O., Walz T., Ballmer-Hofer K. Structure of a VEGF–VEGF receptor complex determined by electron microscopy. Nat. Struct. Mol. Biol. 2007;14:249–250. doi: 10.1038/nsmb1202. [DOI] [PubMed] [Google Scholar]
- Sarabipour S., Mac Gabhann F. VEGF-A121a binding to neuropilins—a concept revisited. Cell Adh. Migr. 2017 doi: 10.1080/19336918.2017.1372878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer. 2011;2:1097–1105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M., Gordon E., Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016;17:611–625. doi: 10.1038/nrm.2016.87. [DOI] [PubMed] [Google Scholar]
- Smith J.S., Lefkowitz R.J., Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018;17:243–269. doi: 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart L.A., Johnstone E.K.M., Wheal A.J., Goulding J., Robers M.B., Machleidt T., Wood K.V., Hill S.J., Pfleger K.D.G. Application of BRET to monitor ligand binding to GPCRs. Nat. Methods. 2015;12:661–663. doi: 10.1038/nmeth.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart L.A., White C.W., Nguyen K., Hill S.J., Pfleger K.D. Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding. Br. J. Pharmacol. 2016;173:3028–3037. doi: 10.1111/bph.13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart L.A., Kilpatrick L.E., Hill S.J. NanoBRET approaches to study ligand binding to GPCRs and RTKs. Trends Pharmacol. Sci. 2018;39:136–147. doi: 10.1016/j.tips.2017.10.006. [DOI] [PubMed] [Google Scholar]
- Wang L., Mukhopadhyay D., Xu X. C terminus of RGS-GAIP-interacting protein conveys neuropilin-1-mediated signaling during angiogenesis. FASEB J. 2006;20:1513–1515. doi: 10.1096/fj.05-5504fje. [DOI] [PubMed] [Google Scholar]
- Whitaker G.B., Limberg B.J., Rosenbaum J.S. Vascular endothelial growth factor receptor-2 and neuropilin-1 form a receptor complex that is responsible for the differential signaling potency of VEGF165 and VEGF121. J. Biol. Chem. 2001;276:25520–25531. doi: 10.1074/jbc.M102315200. [DOI] [PubMed] [Google Scholar]
- Woolard J., Wang W., Bevan H.S., Qiu Y., Morbidelli L., Pritchard-Jones R.O., Cui T., Sugiono M., Waine E., Perrin R. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64:7822–7835. doi: 10.1158/0008-5472.CAN-04-0934. [DOI] [PubMed] [Google Scholar]
- Woolard J., Bevan H.S., Harper S.J., Bates D. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation. 2009;16:572–592. doi: 10.1080/10739680902997333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H., Zhong C., Nudleman E., Ferrara N. Evidence for pro-angiogenic functions of VEGF-Ax. Cell. 2016;167:275–284. doi: 10.1016/j.cell.2016.08.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.