

Abstract
Human T-cell leukemia virus type 1 (HTLV-1) is the first retrovirus that has conclusively been shown to cause human diseases. In HIV-1, specific interactions between the nucleocapsid (NC) domain of the Gag protein and genomic RNA (gRNA) mediate gRNA dimerization and selective packaging; however, the mechanism for gRNA packaging in HTLV-1, a deltaretrovirus, is unclear. In other deltaretroviruses, the matrix (MA) and NC domains of Gag are both involved in gRNA packaging, but MA binds nucleic acids with higher affinity and has more robust chaperone activity, suggesting that this domain may play a primary role. Here, we show that the MA domain of HTLV-1, but not the NC domain, binds short hairpin RNAs derived from the putative gRNA packaging signal. RNA probing of the HTLV-1 5′ leader and cross-linking studies revealed that the primer-binding site and a region within the putative packaging signal form stable hairpins that interact with MA. In addition to a previously identified palindromic dimerization initiation site (DIS), we identified a new DIS in HTLV-1 gRNA and found that both palindromic sequences bind specifically the NC domain. Surprisingly, a mutant partially defective in dimer formation in vitro exhibited a significant increase in RNA packaging into HTLV-1–like particles, suggesting that efficient RNA dimerization may not be strictly required for RNA packaging in HTLV-1. Moreover, the lifecycle of HTLV-1 and other deltaretroviruses may be characterized by NC and MA functions that are distinct from those of the corresponding HIV-1 proteins, but together provide the functions required for viral replication.
Keywords: RNA structure, retrovirus, RNA folding, viral replication, RNA binding protein, Gag, HTLV-1, matrix domain, nucleocapsid, RNA dimerization
Introduction
Human T-cell leukemia virus type 1 (HTLV-1),3 the first infectious and oncogenic human retrovirus discovered (1, 2), is the direct causative agent of several human pathologies, including the incurable adult T-cell leukemia/lymphoma (ATL) as well as lymphocyte-mediated HTLV-1–associated myelopathy/tropical spastic paraparesis (HAM/TSP). Approximately 5–10 million people are infected with HTLV-1 worldwide with 3–5% developing either ATL or HAM/TSP (3). Treatments for ATL are limited, leading to poor prognosis for patients, with a median survival time of 4–6 months for patients with the acute form of the disease (4). Despite clear impacts on human health, the mechanistic details of many molecular processes driving HTLV-1 replication are not well-understood.
Infectious HTLV-1 particles are assembled with two copies of single-stranded viral genomic RNA (gRNA) that are reverse transcribed into dsDNA, which is subsequently integrated into the host cell genome (5). The selective packaging of gRNA from a pool of cellular and viral mRNAs is a crucial step in the assembly of retrovirus particles (6) and is mediated by specific interactions between a gRNA packaging signal (Ψ) and the Gag polyprotein, which consists of three major domains, matrix (MA), capsid (CA), and nucleocapsid (NC). For most retroviruses, Ψ-sites are located in the 5′-untranslated region (UTR) and/or the beginning of the gag ORF (7). The highly structured 5′-UTR, which is well-conserved within individual retroviral species, regulates gRNA packaging, dimerization, initiation of reverse transcription, and translation through interactions with viral and host cell factors (8). HTLV-1 is classified as a deltaretrovirus based on virion morphology and genome sequences (9). The primary Ψ element of the closely related deltaretrovirus bovine leukemia virus (BLV) is located just downstream of the gag start codon and consists of two stem-loop (SL) elements (10, 11). Replacement of the BLV packaging signal with a similar region from either HTLV-1 or HTLV-2 led to only partial BLV replication defects, suggesting some level of conservation in the function of deltaretroviral packaging signals (11, 12).
In lentiviruses such as HIV type 1 (HIV-1), the NC domain of Gag plays a major role in selectively recruiting gRNA for packaging. This process is mediated by specific interactions between the zinc knuckles of NC and exposed single-stranded guanosines in Ψ (13, 14). HIV-1 NC also has robust nucleic acid (NA) chaperone activity, effectively annealing and refolding RNA into thermodynamically more favored structures. NC's chaperone activity involves duplex destabilization, aggregation, and rapid binding kinetics (15–17). In contrast, HTLV-1 NC shows weak NA-binding ability due to its negatively charged C-terminal domain and has very poor chaperone activity relative to other NC proteins (18–20). It has been suggested that the MA domain of Gag may perform some functions that are typically carried out by NC in other retroviruses, including NA binding and gRNA selection (20). Thus, deltaretroviruses may use a different balance of the essential roles of MA and NC domains of Gag during replication.
In addition to their established role in mediating membrane binding and localization of precursor Gag to the plasma membrane (21), retroviral MA proteins are generally capable of binding RNA with varying affinity (6, 21–26). Indeed, RNA is incorporated into NC-deficient HIV-1 particles, and either NC or an RNA-binding site in MA is required for HIV-1 assembly (27). It has been suggested that HIV-1 MA may play a supplementary role in gRNA packaging, likely by binding outside Ψ regions (28). Whereas BLV NC exhibited nonspecific RNA-binding activity, BLV MA forms a highly specific complex with the 5′ end of the gRNA dimer but not with other RNAs in vitro, suggesting that MA plays a more central role in gRNA selection than NC (29). Consistent with this observation, BLV MA binds to two SL RNAs derived from the BLV packaging signal with high affinity (10–20 nm) (30). Furthermore, mutation of conserved basic residues in BLV MA led to a significant reduction in RNA packaging efficiency without any effect on Gag membrane localization (31). Similarly, individual mutations of HTLV-1 MA basic residues abolished viral infectivity and greatly reduced viral particle production but did not affect intracellular targeting of Gag (32). Moreover, HTLV-2 MA bound to SL2 of its putative Ψ with higher affinity and specificity than NC and facilitated HIV-1 trans-activation response element (TAR) RNA/DNA annealing more efficiently than NC (33), demonstrating its specificity for packaging signal elements and potential chaperone activity. HTLV-2 MA contains 11 basic residues throughout the primary sequence, including a cluster in α-helix II at the surface of the protein that is important for NA binding (33). HTLV-1 MA shares 58% sequence identity with HTLV-2 MA and retains 8 of the 11 basic residues (32) present in HTLV-2. The high homology between deltaretroviral MA domains suggests conserved functions in gRNA packaging. However, the role of HTLV-1 MA in gRNA interaction and selection remains unknown.
To begin to characterize the interactions between HTLV-1 Gag and gRNA, in vitro binding assays and RNA probing/cross-linking experiments were performed using the two RNA-interacting Gag domains, MA and NC. An experimentally derived secondary structure of the entire HTLV-1 5′ leader was determined using high-throughput selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE). SHAPE and cross-linking studies performed in the presence of MA or NC showed a clear preference for NC binding to two putative dimerization initiation sites (DISs) located in the U5 sequence of the 5′-UTR. The importance of the DISs for dimerization and RNA packaging was probed using in vitro and cell culture–based assays, respectively. In contrast to NC binding, MA binding was more promiscuous with binding sites identified throughout the 5′-UTR, including the putative packaging signal. These results support divergent roles for MA and NC proteins in the HTLV-1 viral lifecycle with potentially important implications for gRNA packaging and dimerization.
Results
HTLV-1 MA, but not NC, binds to putative packaging signal stem-loop RNAs
Purification of recombinant full-length HTLV-1 Gag has not yet been reported; however, investigations using isolated deltaretroviral Gag domains suggest key differences from the more well-characterized HIV-1 system. Here, the RNA-binding specificities of HTLV-1 Gag MA and NC domains were studied. In HTLV-2, MA interacts with the putative HTLV-2 packaging signal with higher affinity and specificity than NC (33). To examine whether HTLV-1 MA and NC play similar roles as the analogous proteins in HTLV-2, fluorescence anisotropy (FA) binding assays were performed using fluorescently labeled RNAs derived from the previously proposed HTLV-1 Ψ element (SL(450–470), SL(474–508), and SL(447–512) in Fig. 1A) (11, 12). HTLV-1 SL(450–470) and SL(474–508) RNA sequences derived from a provirus cloned from the lymphocytic cell line CS-1 were previously used to replace BLV SL1 and SL2 individually and simultaneously. The chimeric BLV viruses with either SL1 or SL2 replaced with the corresponding HTLV-1 sequence replicated at a reduced level compared with WT BLV virus but with higher efficiency than mutants where either BLV SL1 or SL2 was mutated, leaving only one functional BLV SL. These data suggest that HTLV-1 SL elements can function as packaging elements in the context of BLV gRNA and may act as packaging signals for HTLV-1 (12). In the HTLV-1CH isolate studied here, nucleotide (nt) 491 is a G instead of A, resulting in a slightly different Mfold-predicted structure for SL(474–508) alone and in the context of SL(447–512) (11, 12), but stable SL structures are still predicted. Binding to a nonspecific 20-nt single-stranded (ss) DNA derived from the HIV-1 genome (ssDNA20) was also investigated. HTLV-1 MA bound ssDNA20, SL(474–508), and SL(447–512) with similar apparent Kd values (∼1–2 μm) but more weakly to SL(450–470) (Kd ∼ 8 μm) (Fig. 2A and Table 1). Surprisingly, although HTLV-1 NC displayed ∼2-fold stronger binding to ssDNA compared with MA, it failed to bind HTLV-1 SL(450–470) or SL(474–508).
Figure 1.
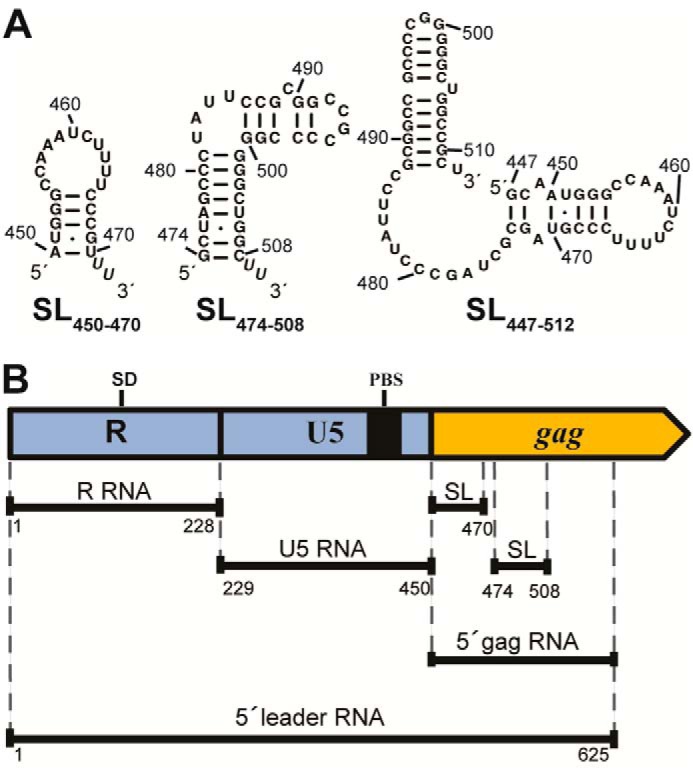
HTLV-1 gRNA-derived 5′ leader constructs (HTLV-1CH isolate) used in this study. A, Mfold-predicted secondary structures (101) of short gRNA constructs SL(450–470), SL(474–508), and SL(447–512) derived from the putative HTLV-1 packaging signal within the gag coding region. B, schematic representation of HTLV-1 5′ leader and three constructs encompassing the 5′-UTR (R and U5) and the 5′ end of the gag coding region (5′ gag). The locations of SL(450–470) and SL(474–508) as well as the SD and PBS are indicated.
Figure 2.
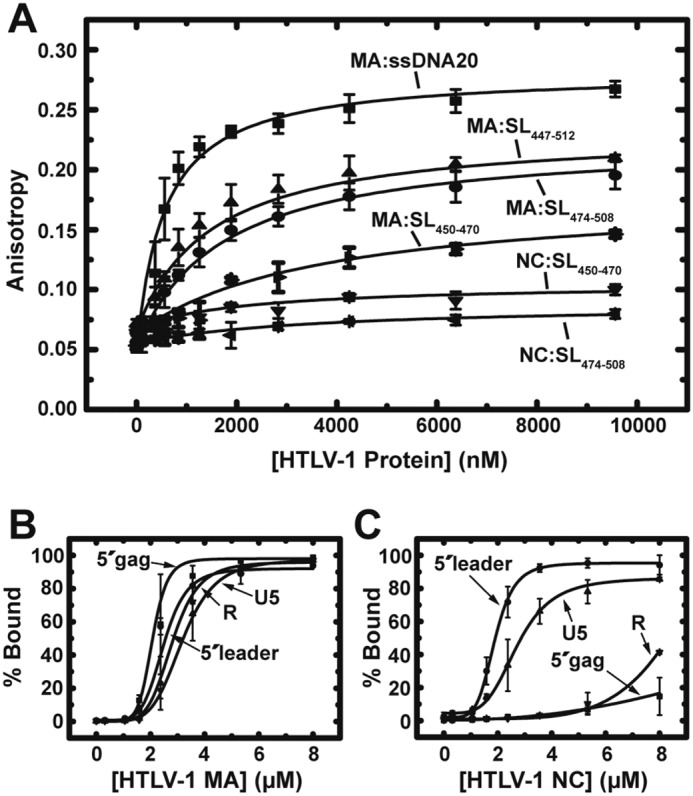
Binding assays with HTLV-1 MA and NC. A, results of FA binding assays wherein 20 nm fluorescently labeled ssDNA20 or RNA was titrated with increasing concentrations of HTLV-1 MA or NC proteins. The averages of three experiments with standard deviations are shown. B and C, results of EMSA binding studies investigating HTLV-1 MA (B) and NC (C) binding interactions with the 5′ leader. The percentage of bound RNA is plotted as a function of protein concentration with curves fit to the Hill equation (96). Error bars represent standard deviations from two independent experiments.
Table 1.
Binding parameters of HTLV-1 proteins to HTLV-1 gRNA-derived constructs
| RNA |
Kd (μm) |
|
|---|---|---|
| MA | NC | |
| ssDNA20a | 1.2 ± 0.3 | 0.6 ± 0.1 |
| SL(450–470)a | 8.1 ± 2.6 | NB |
| SL(474–508)a | 1.9 ± 0.2 | NB |
| SL(447–512)a | 1.6 ± 0.2 | — |
| Rb | 2.8 ± 0.3 | Weak |
| U5b | 3.2 ± 0.3 | 2.7 ± 0.4 |
| 5′ gagb | 2.1 ± 1.0 | Weak |
| 5′ leaderb | 2.5 ± 0.3 | 1.9 ± 0.2 |
a Apparent equilibrium dissociation constants (Kd) determined from FA assays performed in 20 mm Hepes, pH 7.5, 40 mm NaCl, 10 mm KCl, and 1 mm MgCl2. Values are the average of at least three measurements with standard deviations indicated. NB indicates that no binding was detected with up to ∼10 μm protein. A dash indicates that the value was not determined.
b Apparent Kd values determined from EMSAs performed in 20 mm Hepes, pH 7.5, 60 mm NaCl, 10 mm KCl, 1 mm MgCl2, and 5% (v/v) glycerol. Values are the average of at least two measurements with standard deviations indicated. “Weak” indicates that binding affinity could not be determined.
HTLV-1 MA and NC display different specificities for 5′ leader gRNA constructs
Electrophoretic mobility shift assays (EMSAs) were used to investigate the interactions of HTLV-1 MA and NC with longer RNAs derived from the HTLV-1 gRNA. In addition to the full-length 625-nt 5′ leader, which encompasses the HTLV-1 5′-UTR plus 176 nt of the gag coding region, binding to three different truncation constructs was examined: R (nt 1–228), U5 (nt 229–450) and 5′ gag (nt 450–625) (Fig. 1B). HTLV-1 MA bound to all four gRNA constructs with moderate affinity (∼2–3 μm) (Fig. S1A, Fig. 2B, and Table 1). The binding of MA to 5′ gag and the full 5′ leader, which both contain the putative Ψ sequence, was ∼1.5-fold stronger than the binding to R and U5. HTLV-1 NC displayed much stronger binding to U5 and 5′ leader RNAs (Kd ∼ 2–3 μm) relative to 5′ gag and R where binding was too weak to determine a Kd (Fig. S1B, Fig. 2C, and Table 1). These data suggest that HTLV-1 NC specifically binds to the U5 region in the 5′ leader. The binding constants obtained via EMSA for the longer RNAs cannot be directly compared with those measured for the shorter RNAs by FA (Table 1) due to the different experimental methods used.
Structural analysis and secondary structure modeling of HTLV-1 5′ leader
Conserved structural features in the 5′ leader generally serve as recognition elements for specific retroviral Gag binding (7, 8, 34–36). To determine the secondary structure of the full-length HTLV-1 5′ leader, we used SHAPE, a high-throughput RNA structure probing technique (37). SHAPE reagents react with the ribose 2′-hydroxyl group of all 4 nt, displaying high reactivity with less constrained, unpaired residues and low reactivity with base-paired or structurally constrained regions (37–39). To increase resolution at the beginning of the leader, a 15-nt linker was appended to the 5′ end of the 625-nt 5′ leader (38). This RNA folded into a homogeneous monomer under our folding conditions (Fig. S2A) and was probed using N-methylisatoic anhydride (NMIA). Three sets of primers complementary to different regions of the 5′ leader ∼150 nt apart were used in independent primer extension reactions, allowing optimal resolution over the entire RNA. The NMIA reactivity profile for the full-length 5′ leader RNA is shown in Fig. S2B. The regions with overlapping data from different primers correlated closely and accounted for ∼26% of the nt positions probed (Fig. S2C). Seventy-two percent of nt that showed high or intermediate SHAPE reactivity are predicted to be located in single-stranded regions, loops, and bulges in the lowest-energy structure (Fig. 3). A small number of base-paired residues located proximal to the end of a helix or bulge also showed intermediate to high SHAPE reactivity.
Figure 3.

SHAPE analysis of HTLV-1 5′ leader RNA. The secondary structure model was predicted by incorporating the averaged NMIA reactivities for each nt from three to seven independent experiments as pseudo free-energy constraints in RNAstructure (99, 100). Nucleotides are colored according to SHAPE reactivity as indicated in the key. The PBS region, a previously identified DIS (DIS-2) (40, 47), a newly identified DIS (DIS-1), SL(450–470), SL1, and the U5:AUG interaction are labeled and indicated by black boxes or lines. The AUG start codon is highlighted using a red box. Gray nt indicate regions where SHAPE data could not be accurately analyzed due to primer binding and signal distortion near the RNA ends.
The secondary structure model revealed several notable elements, including a highly structured R region, a partially palindromic loop-containing hairpin that was previously identified as a putative DIS (named DIS-2) (40, 41), a surprisingly highly structured primer-binding site (PBS) domain, and an SL (named SL1) in the previously predicted HTLV-1 Ψ region (11, 12) (Fig. 3). A previous secondary structure model for nt 1–192, proposed based on enzymatic probing experiments (42), differs somewhat from our results in the region between nt 119 and 175. Our SHAPE-predicted structure has three small SLs in this region (Fig. 3) instead of a long base-paired SL with several small internal bulges. In good agreement, nt 124–129, 149–152, 159, and 167–169, which are predicted to be in ss regions in our structure, were also predicted to be unpaired in the previous structure (42). DIS-2 (nt 377–389) adopts an SL structure in our model, which is consistent with the computer modeling and Mfold predictions reported in previous studies (40, 43), but slightly different from the structure probed by enzymatic and chemical methods (41). In contrast to the HIV-1 PBS, the HTLV-1 PBS sequence is located in a remarkably stable, highly base-paired region in the 5′ leader RNA. Seven consecutive nt in the PBS sequence showed no NMIA reactivity and were located on one side of the base-paired stem (Fig. 3).
The nt immediately preceding SL1 (nt 469–475) are also predicted to be base-paired even though they showed intermediate NMIA reactivity. It is possible that this short helix is dynamic with “breathing” between open and closed states. Finally, base pairing was observed between nt in the U5 region and those in and around the AUG start codon (U5:AUG) in the HTLV-1 5′ leader (Fig. 3). A similar interaction has been proposed to occur in the HIV-1 5′ leader RNA and is predicted to be conserved in HIV-1 and other distantly related retroviruses (44, 45). The residues on one side of the U5:AUG helix showed intermediate SHAPE reactivity, whereas those on the other side displayed little or no reactivity, supporting a possible structural switch in this region as previously proposed in HIV-1 (46).
Identification of a new DIS in the HTLV-1 5′ leader
Previous studies wherein a 37-nt region encompassing DIS-2 was deleted only led to a 20–25% decrease in HTLV-1 infectivity (47), suggesting that additional features of the HTLV-1 5′ leader may play a role in facilitating genome dimerization. Our SHAPE-predicted secondary structure revealed two additional SL structures that contain palindrome-like sequences and are therefore good DIS candidates: nt 343–355 (DIS-1; Fig. 3, boxed; see also Fig. 4) and nt 494–504 in SL1 (Figs. 3 and 4). Both regions have residues in the loop with high to medium NMIA reactivity. In addition, DIS-1 and DIS-2 are not perfect palindromes due to the presence of a single purine in the middle of the sequence. One side of the predicted stem region in each case also showed medium reactivity, suggesting a dynamic structure in this region.
Figure 4.

Dimerization study of the HTLV-1 5′ leader. A, mutations introduced to disrupt the palindromic loops at nt positions 348–350 (M348), 382–386 (M382), and 498–500 (M498) of the full-length 5′ leader RNA. In each case, stable GAGA tetraloops were substituted for WT sequences individually or simultaneously (3M) (highlighted in red). B, schematic showing tetraloop mutations introduced into the loop regions of the full 5′ leader RNA. The predicted intermolecular contacts formed by each palindromic sequence are shown next to each putative DIS. C, native agarose gel showing the results of dimerization assays of WT and mutated HTLV-1 5′ leader. RNAs (0.5 μm) were either folded in low-salt buffer (50 mm Hepes, pH 7.5, and 1 mm MgCl2) (lanes 2, 4, 6, 8, and 10) or folded in low-salt buffer followed by incubation in dimerization buffer (50 mm Hepes, pH 7.5, 150 mm NaCl, and 1 mm MgCl2) at 37 °C for 1 h (lanes 3, 5, 7, 9, and 11). Lane 1 is a 100-bp DNA ladder. Markers for monomeric (M) and dimeric (D) RNAs are shown. The identity of the slowest migrating band is unknown but may represent a trimer. The native gel is representative of three independent experiments.
To establish whether any of the three candidate DIS sequences plays a role in gRNA dimerization, we substituted 3–5 nt in each loop with a stable GAGA tetraloop sequence individually (M348, M382, and M498 constructs) and simultaneously (3M construct) in the context of the full-length 5′ leader RNA (Fig. 4, A and B). Under low-salt conditions, the major conformation for all folded RNAs is monomeric (Fig. 4C, lanes 2, 4, 6, 8, and 10). A small population of dimer can be detected for WT and two of the mutant RNA constructs. After 1-h incubation in dimerization buffer at 37 °C, all RNAs except the M348 and 3M RNAs showed an increase in the dimer population (Fig. 4C, lanes 3, 5, 7, 9, and 11). A small amount of monomer band smearing was observed in the M348 and 3M RNAs after incubation in dimerization buffer, but no evidence of stable dimer formation was detected (Fig. 4C, lanes 5 and 11), indicating that mutation of loop nt 348–350 abolished the dimerization capability of the HTLV-1 5′ leader. These results support the conclusion that DIS-1 plays a role in dimerization of the 5′ leader in vitro.
Identification of HTLV-1 MA-binding sites and MA-induced RNA structural changes in the 5′ leader
Cross-linking with SHAPE (XL-SHAPE) combines two complementary techniques, SHAPE and UV cross-linking, to allow simultaneous identification of protein-binding sites as well as any concurrent RNA conformational changes associated with the interaction (48). SHAPE allows for identification of secondary structure changes or sites of increased or decreased flexibility upon protein binding, whereas UV cross-linking identifies specific nt that are in very close proximity to a bound protein. We applied XL-SHAPE to study the interactions between HTLV-1 Gag domains and the 5′ leader RNA. In good agreement with the EMSA binding studies (Fig. 2B), HTLV-1 MA cross-linked with a variety of sites throughout the 5′ leader, including clusters of 3 or more residues located at nt 121–127 downstream of the splice donor (SD; nt 118–119), nt 401–405 just upstream of the PBS sequence, nt 513–517 in the SL1 bulge, and nt 564–566 downstream of SL1 (Fig. S3A and Fig. 5A). MA binding to SL1 resulted in significantly increased SHAPE reactivity in this region (nt 513–525) (Fig. S3B and Fig. 5A). In contrast, MA binding to the sequence proximal to the PBS results in decreased SHAPE reactivity in several nearby sites (nt 374, 383, and 399). Importantly, cross-linking and SHAPE-reactivity changes in the sequences adjacent to the SD, upstream of the PBS, and in SL1 are MA dose-dependent (Fig. S4), consistent with specific binding to these regions. The majority (95%) of MA cross-links were to regions predicted to be ss with a preference for A (40%) and C (34%) over G (6%) and U (20%). In comparison, the nt distribution of ss residues in the 5′ leader is A (21%), C (36%), G (11%), and U (32%), indicating that MA may prefer to bind ss adenosines.
Figure 5.

Results of XL-SHAPE analysis on HTLV-1 MA (A) and NC (B) binding to the HTLV-1 5′ leader RNA. Arrows indicate sites with decreased (red) and increased (blue) NMIA reactivity upon protein binding, and violet stars indicate protein cross-linking sites. All identified sites correspond to positions with calculated reactivity changes of ≥0.3 and p < 0.05 based on an unpaired two-tail Student's t test. Experiments were performed in triplicate. Potential intermolecular dimerization of DIS-1 and DIS-2 sequences is shown in B (upper left, boxed) with SHAPE reactivity changes upon NC binding indicated (nt color scheme within the boxes is the same as in Fig. 3).
Identification of HTLV-1 NC-binding sites and NC-induced RNA structural changes in the HTLV-1 5′ leader
In contrast to the results obtained with MA, EMSA binding assays with HTLV-1 NC showed specificity for binding to the U5 region (Fig. 2C), which contains DIS-1, DIS-2, and the PBS sequence. XL-SHAPE was used to map the direct interactions between HTLV-1 NC and the 5′ leader as well as RNA conformational changes induced by protein binding. NC primarily cross-linked to three sites in the U5 region and one site in the R region, whereas no cross-linking was observed to 5′ gag (Fig. S3C and Fig. 5B). Two cross-linking sites (DIS-1 and DIS-2) overlapped with two clusters of decreased SHAPE reactivity, indicating that the binding of NC leads to decreased backbone flexibility in this region (Fig. S3D and Fig. 5B). The cross-linking and SHAPE reactivity in these two regions showed a dose-dependent change upon titration of HTLV-1 NC (Fig. S5), indicating specific binding. It is possible that the decreased backbone flexibility is caused by the dimerization (i.e. intermolecular base pairing) of these loop regions as indicated by the decreased SHAPE reactivity in DIS-1 and DIS-2 upon NC binding (Fig. 5, upper left, boxed insets). Similar to MA, NC also cross-linked to a site just downstream of the SD in the highly structured R domain (nt 123). NC did not cross-link to sufficient sites to determine a clear nt preference, but cross-linking was observed to A, C, and G residues. Binding of both proteins also resulted in structural changes in the predicted ss region between nt 192 and 247 (Fig. 5, A and B).
Impact of DIS mutants on RNA packaging into HTLV-1–like particles
To help assess the role of DIS-1 and DIS-2 in HTLV-1 replication, the same tetraloop mutations tested in vitro, M348 and M382, and a double mutation of these sites (2M construct) were tested for their impact on RNA packaging in a cell culture–based assay where virus-like particles (VLPs) are produced by expression of the HTLV-1 Gag protein. The three mutant RNA sequences were introduced into a mammalian gene expression construct, pHT-UTR-MA, which generates an 810-nt-long RNA encompassing the entire 625-nt HTLV-1 5′ leader. This plasmid was cotransfected into mammalian cells with a HTLV-1 Gag expression construct that has been previously validated to produce HTLV-1–like particles (49–51). The amount of Gag was detected and quantified from VLPs as well as from particle-producing cells (Fig. 6, A and B). Total viral RNAs from VLPs were quantified in a two-step, RT-qPCR analysis (Fig. 6C). The RNA packaging efficiencies of the mutants were analyzed and compared with that of the HT-UTR-MA parent. This analysis allowed for the relative -fold change in RNA packaging efficiencies between the mutants and the parental viral RNA. Intriguingly, we observed that the M348 mutant led to a significant increase in RNA packaging efficiency, whereas the M382 and 2M mutants had RNA packaging efficiencies comparable with that of the HT-UTR-MA parental RNA (Fig. 6D).
Figure 6.

HTLV-1 RNA packaging efficiency of the WT and mutant HT-UTR-MA RNA. A, serial dilution of the HTLV-1–like particles for protein quantification. Top, immunoblot displaying a serial dilution (1–20 μl of lysate per loading) of HTLV-1–like particles. Bottom, protein band intensities (in arbitrary units) (y axis) were plotted against the volume of protein loaded (x axis) to evaluate the linearity and accuracy of protein quantification. The linear equation and an R2 value are indicated. B, immunoblots of both VLPs and cell lysates were probed with anti-HA antibodies, and cell lysate blots were normalized for expression based upon glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The amount of protein was determined based on the linear standard shown in A. Lane 1, WT HT-UTR-MA; lane 2, M348 mutant; lane 3, M382 mutant; lane 4, 2M mutant. C, relative expression level of WT or mutant HT-UTR-MA RNAs in VLPs as detected by a two-step quantitative RT-PCR assay. The expression level of the M348, M382, and 2M mutants was normalized to that from WT HT-UTR-MA. The results of four independent experiments are shown with the mean value indicated by a horizontal bar. D, relative -fold change of RNA packaging efficiency. The RNA packaging efficiency (y axis) was calculated as indicated under “Experimental procedures.” The RNA packaging efficiency of the M348, M382, and 2M mutants was normalized to that from WT HT-UTR-MA. The results of four independent experiments are shown with the mean value indicated by a horizontal bar. n.s., no significant difference.
Discussion
Many critical functional elements are present in the retroviral 5′ leader sequence. These include two overlapping (or near-overlapping) cis-elements important for viral RNA dimerization and packaging. The latter ensures genome recognition by the Gag protein during virus assembly (52, 53), whereas the DIS ensures that two copies of gRNA are packaged into particles (54, 55). Among retroviruses, packaging of two gRNAs into virus particles is believed to be required for ensuring particle viability and infectivity (56). However, the timing of the critical molecular interactions that ensure that one gRNA dimer is packaged per particle remains an open question in the field. This is particularly true for HTLV-1 where many of the details regarding the virus assembly process have lagged behind due to the lack of tractable model systems and difficulties in studying the virus in cell culture (5).
In HIV-1, the primary DIS is in the SL1 hairpin within Ψ, which has a 6-nt palindromic loop sequence. This motif allows the formation of a “kissing loop” interaction with a second gRNA molecule (55). Although deletion or base substitution in the DIS loop abolishes in vitro dimerization of a 615-nt fragment of HIV-1 gRNA (57), DIS alone is not sufficient for dimerization in cells. The DIS stem-loop is dispensable for HIV-1 replication in peripheral blood mononuclear cells, although gRNA packaging is reduced by 50% for the DIS mutant compared with the WT (58). Furthermore, viral RNAs extracted from DIS-deletion mutants are still dimeric (55, 58–60). Therefore, although the SL1 DIS plays a role in enhancing dimerization and gRNA packaging in HIV-1, other elements may also be involved in RNA dimerization (55).
The SHAPE-derived secondary structure of the HTLV-1 5′ leader RNA determined here has potentially important functional implications. The DIS-2 loop is predicted to dimerize with an internal A:A mismatch (Fig. 5B, boxed). Rather than a two-step mechanism involving a kissing loop initiation proposed for other retroviruses (57, 61–63), HTLV-1 DIS-2 was previously identified and proposed to dimerize in a single step with immediate duplex formation (41) and to form a trinucleotide loop containing a central A and an unusual C:synG bp closing the loop (41). In the present study, an additional DIS, DIS-1 (nt 343–355), was identified (Fig. 3). DIS-1 contains a palindromic sequence with a predicted G:G mismatch upon dimerization (Fig. 5B, boxed). Mutating the loop of DIS-1 completely abolished dimerization of the HTLV-1 5′ leader RNA in vitro, whereas mutating the DIS-2 loop region had no detectable effect (Fig. 4C). However, SHAPE reactivity changes in the presence of NC implicate both DIS-1 and DIS-2 as potential contributors to dimerization (see below). A previous study showed that mutating each half of the DIS-2 stem region decreased, but did not abolish, the dimerization capability (43). Although we cannot rule out the possibility that DIS-2 uses a different dimerization mechanism that is insensitive to the mutation we tested, we propose that DIS-1 likely acts as the primary site of dimer initiation. As expected for a primary DIS, DIS-1 is highly conserved across HTLV-1 isolates with nt 341–357 showing strict conservation (present in 245 of 320 (77%) sequences from the HTLV-1 molecular epidemiology database) and another 53 sequences (16.6%) containing compensatory mutations to maintain dimer base pairing (64). Use of two DISs has precedent in retroviruses as Moloney murine leukemia virus contains two DISs that independently contribute to gRNA dimerization in vitro (63). Despite significant efforts to increase the yield of RNA dimer observed by native gel electrophoresis by varying salt concentration, RNA concentration, temperature, and incubation time (data not shown), the overall yield of dimerized HTLV-1 5′ leader RNA remained relatively low (<50%). This is in sharp contrast to the robust dimerization that can be achieved in the case of HIV-1 5′-UTR sequences (23, 65–67).
The introduction of RNA sequences encoding DIS and Ψ into nonviral RNAs or in the construction of minimal retroviral vectors is a validated approach for establishing the identity of DIS and Ψ cis-acting elements (56, 68, 69). In this study, we tested the ability of a small RNA containing the DIS and Ψ sequences from HTLV-1 to be packaged into HTLV-1–like particles. We surprisingly observed that a mutant that is less capable of allowing dimer formation in vitro leads to a significant increase in RNA packaging into HTLV-1–like particles in cell culture–based assays (Fig. 6). Moreover, a double mutant that also disrupts in vitro dimerization is packaged as efficiently as WT. These data imply that efficient dimerization may not be a strict requirement for RNA packaging in HTLV-1. Importantly, the data suggest that particular mutations can have differential effects on RNA dimerization in vitro and RNA packaging in cells and indicate that efficient RNA packaging with HTLV-1 may occur in the absence of efficient RNA dimerization. Alternatively, these mutants may retain dimerization ability in vivo despite the observed in vitro properties. Previous studies showed that deletion of a 32-nt region encompassing DIS-2 did not affect viral production and resulted in only a 20–25% decrease in infectivity (47). Ongoing studies seek to determine whether HTLV-1 particles containing DIS-1 mutant RNAs possess infectivity.
Another surprising finding was the presence of a highly structured PBS sequence. This differs from the relatively unstructured PBS sequences in other retroviruses such as HIV-1 (37, 70, 71). Moreover, the G-C–rich stem is in contrast to the results of nt-bias studies showing the HTLV-1 gRNA to be C-rich but also very G-poor (72, 73). This highly stable PBS hairpin would require destabilization of both tRNAPro and the PBS for efficient primer annealing, suggesting the need for a strong chaperone protein. However, HTLV-1 NC is a very poor NA chaperone compared with other retroviral NC proteins (18). A recent study showed that both primer tRNAPro and a 3′ 18-nt fragment of tRNAPro are packaged into viral particles and are capable of functioning as primers for HTLV-1 reverse transcription in vitro (74). If HTLV-1 uses the 18-nt fragment, it would serve to lower the energy barrier for primer annealing. Alternatively, HTLV-1 Gag or cellular factors such as RNA helicase A may serve as robust chaperones to facilitate the tRNA annealing reaction (75). Similar to the primer activation signal identified in HIV-1 (76, 77), the stabilized PBS stem may serve as a structural regulator of reverse transcription initiation.
We predict the presence of a U5:AUG interaction in the HTLV-1 5′ leader (Fig. 3). This interaction is conserved in HIV-1 and other retroviruses (44, 45) and plays a role in HIV-1 gRNA dimerization (67). The HIV-1 U5:AUG interaction is supported by a recent NMR study that proposed it as part of a structural switch that can expose the DIS and NC-binding sites to facilitate gRNA dimerization and packaging (46). Here, we show that this interaction likely also exists in deltaretroviral 5′ leaders. Such high conservation among different retroviral genera highlights its functional importance, but its precise role in HTLV-1 replication requires further investigation.
The 5′ leader secondary structure is also predicted to contain a long (56-nt) single-stranded stretch immediately downstream of the 5′ Rex response element (nt 54–191) (Fig. 3). It is notable that these residues display mostly medium to low reactivity, suggesting the potential for transient stacking or base-pairing interactions. This stretch may also serve as a flexible linker between RNA domains with different functional roles, thus allowing each to perform their function without interference from the other. This is reminiscent of retroviral polyproteins such as Gag in which the MA, CA, and NC domains are separated by primarily unstructured linkers (78). Furthermore, in the absence of a robust NA chaperone protein, the unstructured segment could facilitate reverse transcription and minus-strand transfer (if also present in the 3′ R region).
The previously predicted SL(450–470) (predicted using FOLDRNA and Mfold computer programs; see Fig. 1) of the putative Ψ element located upstream of SL1 is notably absent in the SHAPE-derived secondary structure (11, 12) (Fig. 3, nt 450–470, dashed line). Instead, this region is primarily ss with intermediate SHAPE reactivity. This finding is consistent with lack of NC binding and only weak binding of MA to SL(450–470) (Table 1), suggesting that this sequence may not be functionally important in MA binding or RNA packaging. In contrast, the Mfold-predicted hairpin spanning nt 488–511 in the context of SL(447–512) (Fig. 1) is consistent with our binding and SHAPE data. In particular, we predict formation of SL1 (nt 477–523) with high SHAPE reactivity observed in the loop region (nt 497–501), intermediate reactivity in a large internal loop, and lower reactivity in the predicted stem regions (Fig. 3).
XL-SHAPE probing upon MA binding to the 5′ leader provided information on binding sites, revealing a preference for ss A residues, and conformational changes that resulted from these interactions (Fig. 5A). A cluster of nt in SL1 cross-linked to MA compared with only one nt in the 450–470 region (Fig. 5A), consistent with our FA data showing that MA has a binding preference for SL(474–508) compared with SL(450–470) (Table 1 and Fig. 2A). SHAPE reactivity increases were observed within and proximal to SL1 upon MA binding as well as in the 30–240-nt region (Fig. 5A). This is similar to the destabilization observed throughout the HIV-1 5′ leader upon HIV-1 Gag binding as elucidated by XL-SHAPE (48). BLV MA–gRNA interaction sites have previously been mapped to regions spanning the PBS to the 5′-terminal 60 residues in the gag coding region (29, 79), which are very similar to the probing results for HTLV-1 MA reported here.
Interestingly, MA binding induced large SHAPE reactivity increases for nt 408 and 409 within the PBS stem. Although not significant by our statistical analysis due to large error bars, the increased SHAPE reactivity is reasonable given the binding of MA to the adjacent nt just upstream of the PBS (Fig. 5A and Fig. S3B). The structural changes induced by MA binding are potentially important in facilitating tRNA annealing to the PBS. Indeed, in HTLV-2, MA has been reported to be a better NA chaperone than NC (33). Similarly, HIV-2 MA displayed NA chaperone activity and facilitated tRNALys,3 annealing (80). We propose that deltaretroviral Gag may facilitate primer annealing via destabilization of the PBS hairpin primarily through interactions with the MA domain.
Retroviral NC domains are generally responsible for specific gRNA selection through interactions with Ψ elements as well as serving as NA chaperones for primer annealing and reverse transcription (15, 81, 82). Although BLV NC does not bind to the proposed primary Ψ signal (SL1–SL2), it binds to a region just upstream of the gag start codon, which also contains two SL structures, with high affinity (83). The latter region was also previously implicated in gRNA packaging (84). In this study, we report that HTLV-1 NC does not bind to the putative HTLV-1 Ψ element (Table 1 and Fig. 2A) but specifically recognizes the U5 region as well as one site in R (Figs. 2C and 5B). Because HTLV-1 Ψ regions have not been fully characterized, it is possible that HTLV-1 Ψ, like BLV Ψ, is discontinuous (10, 83) and includes elements that bind to NC in the U5 region. Retroviral packaging signals and dimerization sites usually overlap, and studies have suggested that gRNA dimerization and packaging are coupled (46, 85, 86). Here, we show that HTLV-1 NC specifically binds to both of the experimentally identified DISs in the HTLV-1 5′ leader, indicating a potential role for NC in promoting gRNA dimerization. Interestingly, NC cross-linking was observed exclusively at purine residues (3 Gs and 1 A), similar to HIV-1 NC's well-established preference for ss G residues (13, 87, 88). Two clusters of decreased SHAPE reactivity in the palindromic sequences of DIS-1 and DIS-2, proximal to NC cross-linking sites, were observed upon titration with NC (Fig. 5B and Fig. S3B), suggesting that these residues may become base-paired as a result of NC-induced dimerization of the 5′ leader. We did not observe an NC-dependent increase in dimer formation by native gel analysis (data not shown); however, this may be the result of a requirement for full-length Gag. To our knowledge, full-length HTLV-1 Gag has not been successfully purified. The inability to use Gag is an important caveat of this study because gRNA packaging and dimerization occur in the context of the immature Gag polyprotein (7, 89). Our current study using mature HTLV-1 MA and NC proteins provides the basis for future studies using full-length Gag.
In summary, retroviral genomes are limited in size, requiring them to act both as genetic material and as functional components in the lifecycle. Indeed, the multistep process of selective gRNA packaging and assembly is primarily orchestrated through multiple functional components of the Gag polyprotein and the gRNA 5′ leader. Here, RNA probing was used to derive the secondary structure of the full-length HTLV-1 5′ leader, revealing features that are potentially important for replication. We experimentally identified a component of the predicted Ψ element that is specifically recognized by MA. Additionally, a new primary DIS is also reported that interacts with NC. Although retroviruses may have evolved to package RNA dimers to maintain infectivity, our data suggest that efficient dimerization may not be required for packaging by HTLV-1. Taken together, our observations imply that packaging of viral RNA dimers represents a finely tuned balance between packaging and dimerization, which has likely evolved to be optimal for sustaining viral infectivity.
Experimental procedures
Preparation of proteins and RNAs
HTLV-1 MA and NC proteins were expressed and purified from Escherichia coli. A codon-optimized gene encoding HTLV-1 MA (CH isolate) was designed using a codon optimization tool (Integrated DNA Technologies) and purchased from Integrated DNA Technologies. The optimized MA gene, which includes an N-terminal GST tag followed by a His tag and a tobacco etch virus (TEV) protease cleavage site (ENLYFQG), was cloned into BamHI and XhoI sites of pET42b (Novagen). Hereafter, this plasmid will be referred to as pET42b-HtlvMA. The TEV site allows for removal of the GST- and His-affinity tags. Briefly, pET42b-HtlvMA was transformed into BL21-CodonPlus(DE3)-RP competent cells (Stratagene). Protein expression was induced using 1 mm isopropyl β-d-1-thiogalactopyranoside at 37 °C for 4 h. Cells were harvested and resuspended in binding buffer (20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 5 mm imidazole, and one tablet of protease inhibitor (Roche Applied Science)), sonicated, and loaded onto a His-Select nickel column (Sigma-Aldrich). Protein elution was performed in binding buffer containing 100 mm imidazole. Fractions containing HTLV-1 MA fusion protein were dialyzed into storage buffer (50 mm Tris-HCl, pH 8.0, 200 mm NaCl, 5 mm β-mercaptoethanol, and 1 mm DTT). The HTLV-1 MA fusion protein was cleaved with 1 mg of TEV protease/20 mg of protein to remove GST and His tags during dialysis. The expression vector for TEV protease was a gift from Robert J. Gorelick (AIDS and Cancer Virus Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research). TEV protease was purified as described previously (90). After dialysis, the cleaved HTLV-1 MA protein was further purified on a Superdex 75 (GE Healthcare) gel filtration column using storage buffer as the running buffer. Fractions containing HTLV-1 MA protein were combined and concentrated using 10,000 molecular-weight-cutoff centrifugal filter units (Amicon) by centrifuging at 3,200 × g at 4 °C. The concentration was determined using a Bradford assay (Bio-Rad), and protein, which was revealed to be >95% pure by SDS-PAGE and Coomassie Blue staining (Fig. S6), was stored at −20 °C (91).
A pET32a-based plasmid, a gift from Robert J. Gorelick, expresses HTLV-1 NC containing an N-terminal His tag and thioredoxin fusion with a TEV protease cleavage site to allow removal of the tags (20). The fusion protein was purified under native conditions. Briefly, the expression plasmid was transformed into BL21(DE3)pLysS (Stratagene). After induction with 0.1 mm isopropyl β-d-1-thiogalactopyranoside overnight at room temperature, cells were harvested and resuspended in buffer A (25 mm Hepes, pH 7.5, 500 mm NaCl, 5 mm imidazole, 0.1 mm ZnCl2, and one tablet of protease inhibitor). After lysis by sonication, polyethylenimine was added to a final concentration of 0.6% to precipitate NAs. The protein was precipitated by adding ammonium sulfate to 60% saturation. After centrifuging, the pellet containing the precipitated protein was resuspended in buffer A and purified on a His-Select nickel column by eluting with a linear 5–200 mm imidazole gradient. The fractions containing fusion protein were combined, concentrated, and dialyzed into buffer B (25 mm Hepes, pH 7.5, 200 mm NaCl, 0.1 mm ZnCl2, and 0.5 mm DTT). The TEV cleavage was performed during overnight dialysis as described above. After dialysis, the protein was further purified on a Superdex 75 gel filtration column run in buffer B. Fractions containing HTLV-1 NC protein were combined and concentrated using 3,500 molecular-weight-cutoff centrifugal filter units (Amicon) by centrifuging at 3,200 × g at 4 °C. The protein concentration was determined by measuring the absorbance at 280 nm (A280) and using an extinction coefficient of 11,740 m−1 cm−1 (20). The purified protein was stored at −20 °C.
All RNAs used in this study were kept in buffers prepared with diethylpyrocarbonate-treated water. HTLV-1 SL(450–470) (5′-AUG GGC CAA AUC UUU UCC CGU UU-3′) and SL(474–508) (5′-GCU AGC CCU AUU CCG CGG CCG CCC CGG GGG CUG GCU U-3′) were purchased from Dharmacon (GE healthcare). The two Us in italics are not encoded by HTLV-1 and were added to facilitate fluorescent labeling and ensure free rotation of the dye. All other HTLV-1 gRNA-derived constructs were in vitro transcribed from digested plasmids using T7 RNA polymerase and previously established methods (92). The following HTLV-1 gRNA constructs were derived from the proviral plasmid ACHneo (93) (a gift from Patrick L. Green, Department of Veterinary Biosciences, The Ohio State University) and cloned into KpnI and PstI sites of pUC19 (New England Biolabs) in front of a T7 RNA polymerase promoter: HTLV-1 SL(447–512) derived from nt 447–512, R derived from nt 1–228, U5 derived from nt 229–450, 5′ gag derived from 450–625, and 5′ leader derived from nt 1–625. A 15-nt 5′-extension (GGC CTT CGG GCC AAG) was added to these gRNA constructs to facilitate SHAPE analysis (38). To probe potential dimerization sites in the 5′ leader, three palindromic loop regions, nt 348–350, nt 382–386, and nt 498–500, were mutated to GAGA tetraloops individually and simultaneously using site-directed, ligase-independent mutagenesis (SLIM) (94). The individually mutated RNAs were named as M348, M382, and M498, respectively, and the triple mutation was designated as 3M (Fig. 4, A and B). The RNA concentrations were determined using the following extinction coefficients: SL(474–508), 22.0 × 104 m−1 cm−1; SL(474–508), 32.5 × 104 m−1 cm−1; SL(447–512), 57.1 × 104 m−1 cm−1; R, 22.2 × 105 m−1 cm−1; U5, 21.7 × 105 m−1 cm−1; 5′ gag, 17.4 × 105 m−1 cm−1; 5′ leader, 59.4 × 105 m−1 cm−1; M348, M382, M498, and 3M, 58.0 × 105 m−1 cm−1.
All RNAs were folded before use as follows. HTLV-1 R, U5, 5′ gag, and 5′ leader were folded by heating to 80 °C for 2 min in 50 mm Hepes, pH 7.5, followed by 60 °C for 2 min. MgCl2 was then added to a final concentration of 1 mm followed by incubation at 37 °C for 30 min and cooling on ice for at least 30 min. HTLV-1 SL1, SL2, and SL1–SL2 were folded similarly except 10 mm Mg2+ was added following the 60 °C incubation.
FA binding assays
Fluorescently labeled HTLV-1 SL(450–470), SL(474–508), and SL(447–512) RNAs were prepared by labeling with fluorescein-5-thiosemicarbazide (Thermo Fisher Scientific) at the 3′ end as described (95). A 20-nt DNA oligonucleotide (ssDNA20; 5′-FAM-CTT CTT TGG GAG TGA ATT AG-3′) was purchased from Integrated DNA Technologies. FA was used to determine the NA-binding affinity of HTLV-1 MA or NC as described previously (18). Briefly, fluorescently labeled RNAs (20 nm) were folded as described above and incubated in the dark at room temperature for 30 min with various concentrations of HTLV-1 MA or NC (0–9.6 μm) in 20 mm Hepes, pH 7.5, 40 mm NaCl, 10 mm KCl, and 1 mm MgCl2. FA measurements were carried out on a SpectraMax M5 plate reader (Molecular Devices). Binding affinities were determined by fitting FA data to a 1:1 binding model as described previously (18).
EMSAs
HTLV-1 R, U5, 5′ gag, and 5′ leader RNAs were internally labeled with 32P, including a mixture of 2 mm [α-32P]GTP (PerkinElmer Life Sciences) and 2 mm GTP in an otherwise standard 50-μl in vitro transcription reaction. Internally labeled RNAs (100 pm) were folded as described above and incubated with varying concentrations of HTLV-1 MA or NC (0–8 μm) in 20 mm Hepes, pH 7.5, 60 mm NaCl, 10 mm KCl, 1 mm MgCl2, and 5% (v/v) glycerol) in a total volume of 10 μl at room temperature for 30 min. Samples were mixed with 2 μl of 6× native gel loading dye (50% glycerol, xylene cyanol, and bromphenol blue) and resolved on 6 (R, U5, and 5′ gag) or 4.5% (5′ leader) native polyacrylamide gels containing 1 mm MgCl2 and run at 4 °C. Gels were dried, exposed overnight, and visualized on a Typhoon FLA 9500 (GE Healthcare). Bands were quantified using ImageJ software (National Institutes of Health). The fraction of bound RNA was determined by dividing the band intensity in the bound population by the sum of the bound plus free band intensities. Kd values were determined by fitting data to a modified Hill equation as follows,
| (Eq. 1) |
where f is the fraction bound; [Pt] is total protein concentration; m and b are normalization factors that represent the fraction of bound RNA at the start and end points, respectively; and n is the Hill coefficient (2, 96).
SHAPE probing
Purified in vitro transcribed HTLV-1 5′ leader RNA was probed by SHAPE using NMIA (Sigma-Aldrich) as described with minor variations (37, 39). Briefly, the RNA (3 μm) was folded in buffer containing 50 mm Hepes, pH 7.5, and 1 mm MgCl2 as described above and then diluted to 0.5 μm in 20 mm Hepes, pH 7.5, 60 mm NaCl, 10 mm KCl, and 1 mm MgCl2. Each reaction (9 μl) was initiated with 1 μl of NMIA (80 mm stock in DMSO) or neat DMSO for the (+)- and (−)-reactions, respectively, and incubated at 37 °C for 22 min. RNA was recovered by ethanol precipitation. SHAPE experiments to probe HTLV-1 MA– and NC–5′ leader interactions were carried out as follows. Folded RNA (0.5 μm) was incubated with or without HTLV-1 MA or NC in a total volume of 9 μl in a buffer containing 20 mm Hepes, pH 7.5, 60 mm NaCl, 10 mm KCl, and 1 mm MgCl2. For the RNA-only sample, protein storage buffer was added to keep the final buffer conditions the same for each reaction. Three different MA or NC concentrations (1.5, 3, and 5 μm) were added to the RNA and incubated at room temperature for 30 min. NMIA (1 μl of an 80 mm stock) was added to initiate each reaction, and SHAPE experiments were performed as described above. The following five reactions were included in each experiment: a control reaction of RNA with DMSO, RNA with NMIA, and RNA with NMIA in the presence of three different concentrations of HTLV-1 MA or NC. After incubating at 37 °C for 22 min, RNAs were recovered by ethanol precipitation. All samples were subsequently processed by reverse transcription primer extension reactions and capillary electrophoresis as described below.
Cross-linking experiments
The HTLV-1 5′ leader RNA was folded and incubated with three concentrations of HTLV-1 MA or NC as described above. Samples (9 μl) were cross-linked on ice in a Stratalinker 2400 UV cross-linker (Stratagene) using a total energy of 400 mJ/cm2. Non-cross-linked samples were incubated on ice for the same amount of time as the cross-linked samples. The following five reactions were included in each experiment: no protein/no UV background, RNA only with cross-linking, and RNA incubated with three different concentrations of HTLV-1 MA or NC with cross-linking. Following the cross-linking reactions, 1 μl of 5% SDS was added (0.5% final) followed by addition of 1 μl of Proteinase K (800 units/ml; New England Biolabs) and incubation at 55 °C for 60 min. RNA was recovered by phenol-chloroform extraction and ethanol precipitation. All samples were subsequently processed by reverse transcription primer extension reactions and capillary electrophoresis as described below.
Reverse transcription and sequencing
RNA samples from either SHAPE probing or cross-linking experiments were resuspended in 9 μl of diethylpyrocarbonate-treated water. A mixture of dNTP (1 μl of 10 mm each) and 2 μl of 5 μm 5′-NEDTM–labeled primer (Applied Biosystems, Life Technologies) were added to each sample followed by incubation at 85 °C for 1 min, 60 °C for 5 min, 35 °C for 5 min, and 50 °C for 10 min. Reverse transcription mixture (8 μl) containing 1 μl of SuperScript III (200 units/μl), 4 μl of 5× first-strand buffer, 2 μl of 0.1 m DTT, and 1 μl of RNaseOUT (40 units/μl) was added to each tube. Primer extension reactions were performed at 55 °C for 1 h and inactivated at 70 °C for 15 min following the manufacturer's protocol (Invitrogen). To hydrolyze the RNA, 1 μl of 4 m NaOH was added to each tube, and reactions were heated at 95 °C for 3 min, cooled on ice, and neutralized with 2 μl of 2 m HCl. For SHAPE studies of RNA alone, three sets of 5′-NED–labeled primers were used: 5′-CGG CAG TCA GTC GTG AAT GAA AG-3′, 5′-GGA ATA AAG GGG CGC TCG TAT CC-3′, and 5′-GGA CAG ATC CTG AGC GGT GTT TC-3′, which anneal to HTLV-1 5′ leader nt positions 302–324, 435–457, and 627–640, respectively. For SHAPE and cross-linking experiments probing protein–5′ leader interactions, only two primers were used (302–324 and 627–640).
Dideoxy sequencing reactions were conducted on the same DNA template used for in vitro transcription of the HTLV-1 5′ leader using the Thermo Sequenase Cycle Sequencing kit (Affymetrix) and the same NED-labeled primers described above. Optimized sequencing reactions containing 200 ng of DNA template and 1 pmol of NED-labeled primer were heated to 95 °C for 1 min and then subjected to 60 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min in a T100 thermal cycler (Bio-Rad). Sequencing reactions were performed independently with each of the three different NED-labeled primers.
Primer extension products from the SHAPE experiments, cross-linking experiments, and sequencing reactions were analyzed by capillary electrophoresis. cDNA pellets were resuspended in formamide and mixed with GeneScanTM 600 LIZ® Size Standard (Applied Biosystems) for intercapillary alignment. Samples were resolved on an Applied Biosystems 3730 DNA Analyzer (Plant-Microbe Genomics Facility, Ohio State University).
Data analysis and RNA structure modeling
Raw electropherograms were converted to normalized SHAPE reactivity using the recently described RNA capillary-electrophoresis analysis tool (RiboCAT) (97). Briefly, RiboCAT uses internal size standards to align signals from different sample-containing capillaries as well as a peak-sharpening algorithm to enhance peak identification. For SHAPE probing experiments of RNA alone, (−)-reaction peak areas were scaled to the (+)-reaction based on the average of the lowest 20% of peak areas in each trace prior to normalization as described (97). At least three independent experiments were performed for each of the three primers used. Data from each independent trial were analyzed individually and averaged to generate mean ± S.D. SHAPE reactivities.
For SHAPE experiments to probe protein–RNA interactions, RiboCAT was used to calculate the peak areas for each of the five reactions. These reactions were then scaled to the RNA-only background reaction using the same scaling method described above. The normalized SHAPE reactivity values were determined by subtracting the RNA-only background and dividing the resulting values by the average of the top 10% of NMIA-treated, free-RNA values. Three independent trials were performed. The differences between the SHAPE reactivity of the free RNA and that of the reaction in the presence of the highest protein concentration were analyzed by an unpaired two-tail Student's t test. Absolute differences in SHAPE reactivity ≥0.3 arbitrary units and a p value <0.05 were considered statistically significant (98).
Cross-linking data from three independent trials were analyzed and scaled similarly. The normalization differed in that the background-subtracted peak areas were divided by the average of the top 10% of values from the cross-linking reaction carried out with the highest protein concentration. The differences between the normalized UV-exposed free-RNA reactivity (background) values and those of the cross-linking reactions with the highest protein concentration were subjected to the same criteria for significance as the SHAPE data.
RNA structure modeling of the HTLV-1 5′ leader was carried out using RNAstructure (99, 100) by incorporating SHAPE reactivity values as pseudo energy constraints for secondary structure calculations. Structures were drawn using XRNA (rna.ucsc.edu/rnacenter/xrna/xrna.html).4
RNA dimerization assays
WT and mutant 5′ leader RNAs (0.6 μm) were folded in a low-salt buffer containing 50 mm Hepes, pH 7.5, and 1 mm MgCl2 as described above. Each folded RNA was diluted to 0.5 μm by adding 5× dimerization buffer to achieve a final concentration of 50 mm Hepes, pH 7.5, 150 mm NaCl, and 1 mm MgCl2 to initiate dimerization. Samples were incubated at 37 °C for 1 h and then on ice for 5 min. A control reaction was also performed with the samples kept in the low-salt buffer during the 37 °C incubation. After incubation, samples were mixed with 2 μl of 6× native gel loading dye, loaded onto an ethidium bromide–prestained 2% native agarose gel containing 1 mm MgCl2, and run at 4 °C in TBM buffer (45 mm Tris borate buffer with 0.2 mm MgCl2) at 12 V/cm. Gels were imaged on an AlphaImagerTM (AlphaInnotech).
Plasmid construction for cell culture–based assays
A 625-nt region containing the 5′-UTR and the 5′ end of the HTLV-1 gag gene (i.e. encoding the N-terminal end of MA) was cloned into the HindIII and XbaI restriction sites of a pEGFP-N3 (Clontech) derivative where the EGFP gene sequence was removed. The gag gene start codon was mutated from AUG to GUG to prevent protein translation of the truncated ORF. The resulting plasmid, pHT-UTR-MA, produces an RNA transcript from the transcription start site in the cytomegalovirus promoter region to the poly(A) signal sequence that is 810 nt in length.
Determination of RNA packaging efficiency
Cotransfection of 3 × 106 HEK293T/17 cells with a codon-optimized HTLV-1 Gag expression construct derivative with a C-terminal HA epitope tag (pN3-HTLV-1-Gag-HA) (49–51) and pHT-UTR-MA was carried out using GenJet (SignaGen Laboratories). Cells and cell culture supernatants were harvested at 48 h posttransfection. Total RNA from cells was harvested by first lysing cells using a QIAShredder (Qiagen, Valencia, CA), and RNA was purified using an RNeasy kit (Qiagen). VLPs were harvested from cell culture supernatants by filtering through a 0.22-μm syringe filter prior to ultracentrifugation in a Beckman 50.2 Ti rotor at 35,000 rpm for 2.5 h at 4 °C. VLP pellets were washed twice with phosphate-buffered saline, and viral RNA was extracted using a High Pure Viral RNA kit (Roche Applied Science) followed by phenol-chloroform extraction and ethanol precipitation overnight. RNAs obtained from VLPs and cells were treated with DNase to eliminate carryover of DNAs prior to RT-qPCR analyses. RNA quantification was carried out using a two-step RT-qPCR assay with cellular and VLP RNAs as a template with the iScript cDNA synthesis kit to synthesize cDNA and the SYBR Green qPCR kit (Bio-Rad) for the qPCR amplifications. The primers used to amplify the RNA transcribed from pHT-UTR-MA were 5′-CTCAAGATCGCCCTGGAGAC and 5′-ATAACCCTTGGGCAGCAGAC. Amplifications were performed using the CFX96 Real Time Detection System (Bio-Rad). To analyze VLP RNA levels, a serial dilution of pHT-UTR-MA (0.5 fg to 1 pg) was used to standardize RT-qPCR analyses.
For quantification of Gag protein, VLPs from transfected cells were harvested by filtering the supernatant though a 0.22-μm syringe filter before ultracentrifugation in a Beckman 50.2 Ti rotor at 35,000 rpm for 2.5 h at 4 °C. VLP pellets were washed twice with phosphate-buffered saline and resuspended in 20 μl of phosphate-buffered saline containing protease inhibitor mixture. Cells were then washed with phosphate-buffered saline, trypsinized, and washed twice with phosphate-buffered saline. Cells were suspended in 500 μl of phosphate-buffered saline, 0.1% Triton X-100, and protease inhibitor mixture and incubated on ice for 20 min. The lysate was then collected by centrifugation. The VLP and cellular lysate samples were electrophoresed on 4–20% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. HTLV-1 Gag was detected with a primary mouse anti-HA antibody (16B12; BioLegend) at a 1:1,000 dilution followed by a horseradish peroxidase–conjugated goat anti-mouse IgG at a 1:5,000 dilution. Band intensities were quantified with the ChemiDoc MP Imaging system (Bio-Rad). RNA packaging efficiencies were determined by the ratio of the amount of RNA detected from VLPs to the amount of RNA detected from cells divided by the ratio of the amount of Gag present in VLPs to the amount of Gag present in cells (31, 33).
Author contributions
W. W., J. H., W. A. C., R. J. B., H. M. H., L. M. M., and K. M.-F. conceptualization; W. W. data curation; W. W., J. H., and Y.-C. S. formal analysis; W. W. validation; W. W., J. H., Y.-C. S., W. A. C., R. J. B., H. M. H., and K. M.-F. investigation; W. W. and L. M. M. visualization; W. W., J. H., Y.-C. S., W. A. C., R. J. B., H. M. H., L. M. M., and K. M.-F. methodology; W. W., W. A. C., and K. M.-F. writing-original draft; W. W., J. H., Y.-C. S., W. A. C., R. J. B., H. M. H., L. M. M., and K. M.-F. writing-review and editing; W. A. C., L. M. M., and K. M.-F. supervision; W. A. C., L. M. M., and K. M.-F. funding acquisition; K. M.-F. resources; K. M.-F. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Robert J. Gorelick and Patrick L. Green for providing materials.
This work was supported by National Institutes of Health Grants R01 GM065056 and P50 GM103368 (HIV Interactions and Viral Evolution Center, which supported development of the software used for XL-SHAPE data analysis) (to K. M.-F.) and R01 GM098550 (to L. M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- HTLV-1
- human T-cell leukemia virus type 1
- ATL
- adult T-cell leukemia/lymphoma
- HAM/TSP
- HTLV-1–associated myelopathy/tropical spastic paraparesis
- gRNA
- genomic RNA
- MA
- matrix
- CA
- capsid
- NC
- nucleocapsid
- BLV
- bovine leukemia virus
- SL
- stem-loop
- NA
- nucleic acid
- SHAPE
- selective 2′-hydroxyl acylation analyzed by primer extension
- DIS
- dimerization initiation site
- FA
- fluorescence anisotropy
- ss
- single-stranded
- NMIA
- N-methylisatoic anhydride
- PBS
- primer-binding site
- XL-SHAPE
- cross-linking with SHAPE
- SD
- splice donor
- TEV
- tobacco etch virus
- RiboCAT
- RNA capillary-electrophoresis analysis tool
- HIV-1
- HIV type 1
- nt
- nucleotide(s)
- EMSA
- electrophoretic mobility shift assay
- VLP
- virus-like particle
- qPCR
- quantitative PCR
- GST
- glutathione S-transferase
- EGFP
- enhanced GFP
- HA
- hemagglutinin.
References
- 1. Poiesz B. J., Ruscetti F. W., Gazdar A. F., Bunn P. A., Minna J. D., and Gallo R. C. (1980) Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 77, 7415–7419 10.1073/pnas.77.12.7415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoshida M., Miyoshi I., and Hinuma Y. (1982) Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. U.S.A. 79, 2031–2035 10.1073/pnas.79.6.2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gessain A., and Cassar O. (2012) Epidemiological aspects and world distribution of HTLV-1 infection. Front. Microbiol. 3, 388 10.3389/fmicb.2012.00388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsukasaki K., Hermine O., Bazarbachi A., Ratner L., Ramos J. C., Harrington W. Jr., O'Mahony D., Janik J. E., Bittencourt A. L., Taylor G. P., Yamaguchi K., Utsunomiya A., Tobinai K., and Watanabe T. (2009) Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J. Clin. Oncol. 27, 453–459 10.1200/JCO.2008.18.2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martin J. L., Maldonado J. O., Mueller J. D., Zhang W., and Mansky L. M. (2016) Molecular studies of HTLV-1 replication: an update. Viruses 8, E31 10.3390/v8020031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jewell N. A., and Mansky L. M. (2000) In the beginning: genome recognition, RNA encapsidation and the initiation of complex retrovirus assembly. J. Gen. Virol. 81, 1889–1899 10.1099/0022-1317-81-8-1889 [DOI] [PubMed] [Google Scholar]
- 7. D'Souza V., and Summers M. F. (2005) How retroviruses select their genomes. Nat. Rev. Microbiol. 3, 643–655 10.1038/nrmicro1210 [DOI] [PubMed] [Google Scholar]
- 8. Lu K., Heng X., and Summers M. F. (2011) Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 410, 609–633 10.1016/j.jmb.2011.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weiss R. A. (1996) Retrovirus classification and cell interactions. J. Antimicrob. Chemother. 37, Suppl. B, 1–11 10.1093/jac/37.suppl_B.1 [DOI] [PubMed] [Google Scholar]
- 10. Mansky L. M., Krueger A. E., and Temin H. M. (1995) The bovine leukemia virus encapsidation signal is discontinuous and extends into the 5′ end of the gag gene. J. Virol. 69, 3282–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mansky L. M., and Wisniewski R. M. (1998) The bovine leukemia virus encapsidation signal is composed of RNA secondary structures. J. Virol. 72, 3196–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mansky L. M., and Gajary L. C. (2002) The primary nucleotide sequence of the bovine leukemia virus RNA packaging signal can influence efficient RNA packaging and virus replication. Virology 301, 272–280 10.1006/viro.2002.1578 [DOI] [PubMed] [Google Scholar]
- 13. De Guzman R. N., Wu Z. R., Stalling C. C., Pappalardo L., Borer P. N., and Summers M. F. (1998) Structure of the HIV-1 nucleocapsid protein bound to the SL3 ψ-RNA recognition element. Science 279, 384–388 10.1126/science.279.5349.384 [DOI] [PubMed] [Google Scholar]
- 14. Amarasinghe G. K., De Guzman R. N., Turner R. B., Chancellor K. J., Wu Z. R., and Summers M. F. (2000) NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 of the ψ-RNA packaging signal. Implications for genome recognition. J. Mol. Biol. 301, 491–511 10.1006/jmbi.2000.3979 [DOI] [PubMed] [Google Scholar]
- 15. Levin J. G., Guo J., Rouzina I., and Musier-Forsyth K. (2005) Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid. Res. Mol. Biol. 80, 217–286 10.1016/S0079-6603(05)80006-6 [DOI] [PubMed] [Google Scholar]
- 16. Cruceanu M., Gorelick R. J., Musier-Forsyth K., Rouzina I., and Williams M. C. (2006) Rapid kinetics of protein-nucleic acid interaction is a major component of HIV-1 nucleocapsid protein's nucleic acid chaperone function. J. Mol. Biol. 363, 867–877 10.1016/j.jmb.2006.08.070 [DOI] [PubMed] [Google Scholar]
- 17. Narayanan N., Gorelick R. J., and DeStefano J. J. (2006) Structure/function mapping of amino acids in the N-terminal zinc finger of the human immunodeficiency virus type 1 nucleocapsid protein: residues responsible for nucleic acid helix destabilizing activity. Biochemistry 45, 12617–12628 10.1021/bi060925c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stewart-Maynard K. M., Cruceanu M., Wang F., Vo M. N., Gorelick R. J., Williams M. C., Rouzina I., and Musier-Forsyth K. (2008) Retroviral nucleocapsid proteins display nonequivalent levels of nucleic acid chaperone activity. J. Virol. 82, 10129–10142 10.1128/JVI.01169-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Darugar Q., Kim H., Gorelick R. J., and Landes C. (2008) Human T-cell lymphotropic virus type 1 nucleocapsid protein-induced structural changes in transactivation response DNA hairpin measured by single-molecule fluorescence resonance energy transfer. J. Virol. 82, 12164–12171 10.1128/JVI.01158-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qualley D. F., Stewart-Maynard K. M., Wang F., Mitra M., Gorelick R. J., Rouzina I., Williams M. C., and Musier-Forsyth K. (2010) C-terminal domain modulates the nucleic acid chaperone activity of human T-cell leukemia virus type 1 nucleocapsid protein via an electrostatic mechanism. J. Biol. Chem. 285, 295–307 10.1074/jbc.M109.051334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parent L. J., and Gudleski N. (2011) Beyond plasma membrane targeting: role of the MA domain of Gag in retroviral genome encapsidation. J. Mol. Biol. 410, 553–564 10.1016/j.jmb.2011.04.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alfadhli A., McNett H., Tsagli S., Bächinger H. P., Peyton D. H., and Barklis E. (2011) HIV-1 matrix protein binding to RNA. J. Mol. Biol. 410, 653–666 10.1016/j.jmb.2011.04.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rye-McCurdy T., Olson E. D., Liu S., Binkley C., Reyes J. P., Thompson B. R., Flanagan J. M., Parent L. J., and Musier-Forsyth K. (2016) Functional equivalence of retroviral MA domains in facilitating ψ RNA binding specificity by Gag. Viruses 8, E256 10.3390/v8090256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kutluay S. B., Zang T., Blanco-Melo D., Powell C., Jannain D., Errando M., and Bieniasz P. D. (2014) Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 159, 1096–1109 10.1016/j.cell.2014.09.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Todd G. C., Duchon A., Inlora J., Olson E. D., Musier-Forsyth K., and Ono A. (2017) Inhibition of HIV-1 Gag-membrane interactions by specific RNAs. RNA 23, 395–405 10.1261/rna.058453.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones C. P., Datta S. A., Rein A., Rouzina I., and Musier-Forsyth K. (2011) Matrix domain modulates HIV-1 Gag's nucleic acid chaperone activity via inositol phosphate binding. J. Virol. 85, 1594–1603 10.1128/JVI.01809-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ott D. E., Coren L. V., and Gagliardi T. D. (2005) Redundant roles for nucleocapsid and matrix RNA-binding sequences in human immunodeficiency virus type 1 assembly. J. Virol. 79, 13839–13847 10.1128/JVI.79.22.13839-13847.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Purohit P., Dupont S., Stevenson M., and Green M. R. (2001) Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 7, 576–584 10.1017/S1355838201002023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katoh I., Kyushiki H., Sakamoto Y., Ikawa Y., and Yoshinaka Y. (1991) Bovine leukemia virus matrix-associated protein MA(p15): further processing and formation of a specific complex with the dimer of the 5′-terminal genomic RNA fragment. J. Virol. 65, 6845–6855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qualley D. F., Lackey C. M., and Paterson J. P. (2013) Inositol phosphates compete with nucleic acids for binding to bovine leukemia virus matrix protein: implications for deltaretroviral assembly. Proteins 81, 1377–1385 10.1002/prot.24281 [DOI] [PubMed] [Google Scholar]
- 31. Wang H., Norris K. M., and Mansky L. M. (2003) Involvement of the matrix and nucleocapsid domains of the bovine leukemia virus Gag polyprotein precursor in viral RNA packaging. J. Virol. 77, 9431–9438 10.1128/JVI.77.17.9431-9438.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Le Blanc I., Rosenberg A. R., and Dokhélar M. C. (1999) Multiple functions for the basic amino acids of the human T-cell leukemia virus type 1 matrix protein in viral transmission. J. Virol. 73, 1860–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun M., Grigsby I. F., Gorelick R. J., Mansky L. M., and Musier-Forsyth K. (2014) Retrovirus-specific differences in matrix and nucleocapsid protein-nucleic Acid interactions: implications for genomic RNA packaging. J. Virol. 88, 1271–1280 10.1128/JVI.02151-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heng X., Kharytonchyk S., Garcia E. L., Lu K., Divakaruni S. S., LaCotti C., Edme K., Telesnitsky A., and Summers M. F. (2012) Identification of a minimal region of the HIV-1 5′-leader required for RNA dimerization, NC binding, and packaging. J. Mol. Biol. 417, 224–239 10.1016/j.jmb.2012.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Banks J. D., and Linial M. L. (2000) Secondary structure analysis of a minimal avian leukosis-sarcoma virus packaging signal. J. Virol. 74, 456–464 10.1128/JVI.74.1.456-464.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gherghe C., Lombo T., Leonard C. W., Datta S. A., Bess J. W. Jr, Gorelick R. J., Rein A., and Weeks K. M. (2010) Definition of a high-affinity Gag recognition structure mediating packaging of a retroviral RNA genome. Proc. Natl. Acad. Sci. U.S.A. 107, 19248–19253 10.1073/pnas.1006897107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilkinson K. A., Gorelick R. J., Vasa S. M., Guex N., Rein A., Mathews D. H., Giddings M. C., and Weeks K. M. (2008) High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 6, e96 10.1371/journal.pbio.0060096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Merino E. J., Wilkinson K. A., Coughlan J. L., and Weeks K. M. (2005) RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 127, 4223–4231 10.1021/ja043822v [DOI] [PubMed] [Google Scholar]
- 39. Watts J. M., Dang K. K., Gorelick R. J., Leonard C. W., Bess J. W. Jr, Swanstrom R., Burch C. L., and Weeks K. M. (2009) Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460, 711–716 10.1038/nature08237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Monie T., Greatorex J., and Lever A. M. (2001) Oligonucleotide mapping of the core genomic RNA dimer linkage in human T-cell leukaemia virus type-1. Virus Res. 78, 45–56 10.1016/S0168-1702(01)00283-0 [DOI] [PubMed] [Google Scholar]
- 41. Monie T. P., Greatorex J. S., Zacharias M., and Lever A. M. (2004) The human T-cell lymphotropic virus type-I dimerization initiation site forms a hairpin loop, unlike previously characterized retroviral dimerization motifs. Biochemistry 43, 6085–6090 10.1021/bi030237i [DOI] [PubMed] [Google Scholar]
- 42. Askjaer P., and Kjems J. (1998) Mapping of multiple RNA binding sites of human T-cell lymphotropic virus type I rex protein within 5′- and 3′-Rex response elements. J. Biol. Chem. 273, 11463–11471 10.1074/jbc.273.19.11463 [DOI] [PubMed] [Google Scholar]
- 43. Greatorex J. S., Laisse V., Dockhelar M. C., and Lever A. M. (1996) Sequences involved in the dimerisation of human T cell leukaemia virus type-1 RNA. Nucleic Acids Res. 24, 2919–2923 10.1093/nar/24.15.2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abbink T. E., and Berkhout B. (2003) A novel long distance base-pairing interaction in human immunodeficiency virus type 1 RNA occludes the Gag start codon. J. Biol. Chem. 278, 11601–11611 10.1074/jbc.M210291200 [DOI] [PubMed] [Google Scholar]
- 45. Damgaard C. K., Andersen E. S., Knudsen B., Gorodkin J., and Kjems J. (2004) RNA interactions in the 5′ region of the HIV-1 genome. J. Mol. Biol. 336, 369–379 10.1016/j.jmb.2003.12.010 [DOI] [PubMed] [Google Scholar]
- 46. Lu K., Heng X., Garyu L., Monti S., Garcia E. L., Kharytonchyk S., Dorjsuren B., Kulandaivel G., Jones S., Hiremath A., Divakaruni S. S., LaCotti C., Barton S., Tummillo D., Hosic A., et al. (2011) NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 334, 242–245 10.1126/science.1210460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Le Blanc I., Greatorex J., Dokhélar M. C., and Lever A. M. (2000) A 37 base sequence in the leader region of human T-cell leukaemia virus type I is a high affinity dimerization site but is not essential for virus replication. J. Gen. Virol. 81, 105–108 10.1099/0022-1317-81-1-105 [DOI] [PubMed] [Google Scholar]
- 48. Kenyon J. C., Prestwood L. J., and Lever A. M. (2015) A novel combined RNA-protein interaction analysis distinguishes HIV-1 Gag protein binding sites from structural change in the viral RNA leader. Sci. Rep. 5, 14369 10.1038/srep14369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maldonado J. O., Cao S., Zhang W., and Mansky L. M. (2016) Distinct morphology of human T-cell leukemia virus type 1-like particles. Viruses 8, E132 10.3390/v8050132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin J. L., Cao S., Maldonado J. O., Zhang W., and Mansky L. M. (2016) Distinct particle morphologies revealed through comparative parallel analyses of retrovirus-like particles. J. Virol. 90, 8074–8084 10.1128/JVI.00666-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin J. L., Mendonça L. M., Angert I., Mueller J. D., Zhang W., and Mansky L. M. (2017) Disparate contributions of human retrovirus capsid subdomains to Gag-Gag oligomerization, virus morphology, and particle biogenesis. J. Virol. 91, e00298–17 10.1128/JVI.00298-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bieniasz P., and Telesnitsky A. (2018) Multiple, switchable protein:RNA interactions regulate human immunodeficiency virus type 1 assembly. Annu. Rev. Virol. 5, 165–183 10.1146/annurev-virology-092917-043448 [DOI] [PubMed] [Google Scholar]
- 53. Kuzembayeva M., Dilley K., Sardo L., and Hu W. S. (2014) Life of ψ: how full-length HIV-1 RNAs become packaged genomes in the viral particles. Virology 454–455, 362–370 10.1016/j.virol.2014.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dubois N., Marquet R., Paillart J. C., and Bernacchi S. (2018) Retroviral RNA dimerization: from structure to functions. Front. Microbiol. 9, 527 10.3389/fmicb.2018.00527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moore M. D., and Hu W. S. (2009) HIV-1 RNA dimerization: it takes two to tango. AIDS Rev. 11, 91–102 [PMC free article] [PubMed] [Google Scholar]
- 56. Torrent C., Gabus C., and Darlix J. L. (1994) A small and efficient dimerization/packaging signal of rat VL30 RNA and its use in murine leukemia virus-VL30-derived vectors for gene transfer. J. Virol. 68, 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Skripkin E., Paillart J. C., Marquet R., Ehresmann B., and Ehresmann C. (1994) Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc. Natl. Acad. Sci. U.S.A. 91, 4945–4949 10.1073/pnas.91.11.4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hill M. K., Shehu-Xhilaga M., Campbell S. M., Poumbourios P., Crowe S. M., and Mak J. (2003) The dimer initiation sequence stem-loop of human immunodeficiency virus type 1 is dispensable for viral replication in peripheral blood mononuclear cells. J. Virol. 77, 8329–8335 10.1128/JVI.77.15.8329-8335.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Berkhout B., and van Wamel J. L. (1996) Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J. Virol. 70, 6723–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Song R., Kafaie J., Yang L., and Laughrea M. (2007) HIV-1 viral RNA is selected in the form of monomers that dimerize in a three-step protease-dependent process; the DIS of stem-loop 1 initiates viral RNA dimerization. J. Mol. Biol. 371, 1084–1098 10.1016/j.jmb.2007.06.010 [DOI] [PubMed] [Google Scholar]
- 61. Paillart J. C., Skripkin E., Ehresmann B., Ehresmann C., and Marquet R. (1996) A loop-loop “kissing” complex is the essential part of the dimer linkage of genomic HIV-1 RNA. Proc. Natl. Acad. Sci. U.S.A. 93, 5572–5577 10.1073/pnas.93.11.5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Girard P. M., Bonnet-Mathonière B., Muriaux D., and Paoletti J. (1995) A short autocomplementary sequence in the 5′ leader region is responsible for dimerization of MoMuLV genomic RNA. Biochemistry 34, 9785–9794 10.1021/bi00030a016 [DOI] [PubMed] [Google Scholar]
- 63. Ly H., and Parslow T. G. (2002) Bipartite signal for genomic RNA dimerization in Moloney murine leukemia virus. J. Virol. 76, 3135–3144 10.1128/JVI.76.7.3135-3144.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Araujo T. H., Souza-Brito L. I., Libin P., Deforche K., Edwards D., de Albuquerque-Junior A. E., Vandamme A. M., Galvao-Castro B., and Alcantara L. C. (2012) A public HTLV-1 molecular epidemiology database for sequence management and data mining. PLoS One 7, e42123 10.1371/journal.pone.0042123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Webb J. A., Jones C. P., Parent L. J., Rouzina I., and Musier-Forsyth K. (2013) Distinct binding interactions of HIV-1 Gag to ψ and non-ψ RNAs: implications for viral genomic RNA packaging. RNA 19, 1078–1088 10.1261/rna.038869.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tran T., Liu Y., Marchant J., Monti S., Seu M., Zaki J., Yang A. L., Bohn J., Ramakrishnan V., Singh R., Hernandez M., Vega A., and Summers M. F. (2015) Conserved determinants of lentiviral genome dimerization. Retrovirology 12, 83 10.1186/s12977-015-0209-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Song R., Kafaie J., and Laughrea M. (2008) Role of the 5′ TAR stem–loop and the U5-AUG duplex in dimerization of HIV-1 genomic RNA. Biochemistry 47, 3283–3293 10.1021/bi7023173 [DOI] [PubMed] [Google Scholar]
- 68. Liu Y., Nikolaitchik O. A., Rahman S. A., Chen J., Pathak V. K., and Hu W. S. (2017) HIV-1 sequence necessary and sufficient to package non-viral RNAs into HIV-1 particles. J. Mol. Biol. 429, 2542–2555 10.1016/j.jmb.2017.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jewell N. A., and Mansky L. M. (2005) Packaging of heterologous RNAs by a minimal bovine leukemia virus RNA packaging signal into virus particles. Arch. Virol. 150, 1161–1173 10.1007/s00705-004-0476-7 [DOI] [PubMed] [Google Scholar]
- 70. Damgaard C. K., Dyhr-Mikkelsen H., and Kjems J. (1998) Mapping the RNA binding sites for human immunodeficiency virus type-1 gag and NC proteins within the complete HIV-1 and -2 untranslated leader regions. Nucleic Acids Res. 26, 3667–3676 10.1093/nar/26.16.3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Berkhout B. (1997) The primer binding site on the RNA genome of human and simian immunodeficiency viruses is flanked by an upstream hairpin structure. Nucleic Acids Res. 25, 4013–4017 10.1093/nar/25.20.4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van Hemert F., van der Kuyl A. C., and Berkhout B. (2014) On the nucleotide composition and structure of retroviral RNA genomes. Virus Res. 193, 16–23 10.1016/j.virusres.2014.03.019 [DOI] [PubMed] [Google Scholar]
- 73. van Hemert F., van der Kuyl A. C., and Berkhout B. (2016) Impact of the biased nucleotide composition of viral RNA genomes on RNA structure and codon usage. J. Gen. Virol. 97, 2608–2619 10.1099/jgv.0.000579 [DOI] [PubMed] [Google Scholar]
- 74. Ruggero K., Guffanti A., Corradin A., Sharma V. K., De Bellis G., Corti G., Grassi A., Zanovello P., Bronte V., Ciminale V., and D'Agostino D. M. (2014) Small noncoding RNAs in cells transformed by human T-cell leukemia virus type 1: a role for a tRNA fragment as a primer for reverse transcriptase. J. Virol. 88, 3612–3622 10.1128/JVI.02823-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xing L., Liang C., and Kleiman L. (2011) Coordinate roles of Gag and RNA helicase A in promoting the annealing of formula to HIV-1 RNA. J. Virol. 85, 1847–1860 10.1128/JVI.02010-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Beerens N., Groot F., and Berkhout B. (2001) Initiation of HIV-1 reverse transcription is regulated by a primer activation signal. J. Biol. Chem. 276, 31247–31256 10.1074/jbc.M102441200 [DOI] [PubMed] [Google Scholar]
- 77. Ooms M., Cupac D., Abbink T. E., Huthoff H., and Berkhout B. (2007) The availability of the primer activation signal (PAS) affects the efficiency of HIV-1 reverse transcription initiation. Nucleic Acids Res. 35, 1649–1659 10.1093/nar/gkm046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bell N. M., and Lever A. M. (2013) HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol. 21, 136–144 10.1016/j.tim.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 79. Katoh I., Yasunaga T., and Yoshinaka Y. (1993) Bovine leukemia virus RNA sequences involved in dimerization and specific gag protein binding: close relation to the packaging sites of avian, murine, and human retroviruses. J. Virol. 67, 1830–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pachulska-Wieczorek K., Błaszczyk L., Biesiada M., Adamiak R. W., and Purzycka K. J. (2016) The matrix domain contributes to the nucleic acid chaperone activity of HIV-2 Gag. Retrovirology 13, 18 10.1186/s12977-016-0245-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rein A., Henderson L. E., and Levin J. G. (1998) Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: significance for viral replication. Trends Biochem. Sci. 23, 297–301 10.1016/S0968-0004(98)01256-0 [DOI] [PubMed] [Google Scholar]
- 82. Darlix J. L., Lapadat-Tapolsky M., de Rocquigny H., and Roques B. P. (1995) First glimpses at structure-function relationships of the nucleocapsid protein of retroviruses. J. Mol. Biol. 254, 523–537 10.1006/jmbi.1995.0635 [DOI] [PubMed] [Google Scholar]
- 83. Yildiz F. Z., Babalola K., and Summers M. F. (2013) Identification of a high affinity nucleocapsid protein binding element from the bovine leukemia virus genome. Virus Res. 171, 278–286 10.1016/j.virusres.2012.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kurg A., Sommer G., and Metspalu A. (1995) An RNA stem-loop structure involved in the packaging of bovine leukemia virus genomic RNA in vivo. Virology 211, 434–442 10.1006/viro.1995.1425 [DOI] [PubMed] [Google Scholar]
- 85. Laughrea M., Jetté L., Mak J., Kleiman L., Liang C., and Wainberg M. A. (1997) Mutations in the kissing-loop hairpin of human immunodeficiency virus type 1 reduce viral infectivity as well as genomic RNA packaging and dimerization. J. Virol. 71, 3397–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. D'Souza V., and Summers M. F. (2004) Structural basis for packaging the dimeric genome of Moloney murine leukaemia virus. Nature 431, 586–590 10.1038/nature02944 [DOI] [PubMed] [Google Scholar]
- 87. Keane S. C., Heng X., Lu K., Kharytonchyk S., Ramakrishnan V., Carter G., Barton S., Hosic A., Florwick A., Santos J., Bolden N. C., McCowin S., Case D. A., Johnson B. A., Salemi M., et al. (2015) Structure of the HIV-1 RNA packaging signal. Science 348, 917–921 10.1126/science.aaa9266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fisher R. J., Fivash M. J., Stephen A. G., Hagan N. A., Shenoy S. R., Medaglia M. V., Smith L. R., Worthy K. M., Simpson J. T., Shoemaker R., McNitt K. L., Johnson D. G., Hixson C. V., Gorelick R. J., Fabris D., et al. (2006) Complex interactions of HIV-1 nucleocapsid protein with oligonucleotides. Nucleic Acids Res. 34, 472–484 10.1093/nar/gkj442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rein A. (2010) Nucleic acid chaperone activity of retroviral Gag proteins. RNA Biol. 7, 700–705 10.4161/rna.7.6.13685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tropea J. E., Cherry S., and Waugh D. S. (2009) Expression and purification of soluble His6-tagged TEV protease. Methods Mol. Biol. 498, 297–307 10.1007/978-1-59745-196-3_19 [DOI] [PubMed] [Google Scholar]
- 91. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 92. Milligan J. F., Groebe D. R., Witherell G. W., and Uhlenbeck O. C. (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 15, 8783–8798 10.1093/nar/15.21.8783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Anderson M. D., Ye J., Xie L., and Green P. L. (2004) Transformation studies with a human T-cell leukemia virus type 1 molecular clone. J. Virol. Methods 116, 195–202 10.1016/j.jviromet.2003.11.016 [DOI] [PubMed] [Google Scholar]
- 94. Chiu J., March P. E., Lee R., and Tillett D. (2004) Site-directed, Ligase-Independent Mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 32, e174 10.1093/nar/gnh172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jones C. P., Saadatmand J., Kleiman L., and Musier-Forsyth K. (2013) Molecular mimicry of human tRNALys anti-codon domain by HIV-1 RNA genome facilitates tRNA primer annealing. RNA 19, 219–229 10.1261/rna.036681.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ryder S. P., Recht M. I., and Williamson J. R. (2008) Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol. Biol. 488, 99–115 10.1007/978-1-60327-475-3_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cantara W. A., Hatterschide J., Wu W., and Musier-Forsyth K. (2017) RiboCAT: a new capillary electrophoresis data analysis tool for nucleic acid probing. RNA 23, 240–249 10.1261/rna.058404.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Steen K. A., Rice G. M., and Weeks K. M. (2012) Fingerprinting noncanonical and tertiary RNA structures by differential SHAPE reactivity. J. Am. Chem. Soc. 134, 13160–13163 10.1021/ja304027m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Reuter J. S., and Mathews D. H. (2010) RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129 10.1186/1471-2105-11-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mathews D. H. (2014) RNA secondary structure analysis using RNAstructure. Curr. Protoc. Bioinformatics 46, 12.6.1–25 10.1002/0471250953.bi1206s46 [DOI] [PubMed] [Google Scholar]
- 101. Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.