

Abstract
Epithelial Na+ channel (ENaC) maturation and activation require proteolysis of both the α and γ subunits. Cleavage at multiple sites in the finger domain of each subunit liberates their autoinhibitory tracts. Synthetic peptides derived from the proteolytically released fragments inhibit the channel, likely by reconstituting key interactions removed by the proteolysis. We previously showed that a peptide derived from the α subunit's autoinhibitory sequence (α-8) binds at the α subunit's finger–thumb domain interface. Despite low sequence similarity between the α and γ subunit finger domains, we hypothesized that a peptide derived from the γ subunit's autoinhibitory sequence (γ-11) inhibits the channel through an analogous mechanism. Using Xenopus oocytes, we found here that channels lacking a γ subunit thumb domain were no longer sensitive to γ-11, but remained sensitive to α-8. We identified finger domain sites in the γ subunit that dramatically reduced γ-11 inhibition. Using cysteines and sulfhydryl reactive cross-linkers introduced into both the peptide and the subunit, we also could cross-link γ-11 to both the finger domain and the thumb domain of the γ subunit. Our results suggest that α-8 and γ-11 occupy similar binding pockets within their respective subunits, and that proteolysis of the α and γ subunits activate the channel through analogous mechanisms.
Keywords: proteolysis, allosteric regulation, cysteine-mediated cross-linking, protein conformation, epithelial sodium channel (ENaC), autoinhibition, cation channel, finger-thumb domain
Introduction
The epithelial Na+ channel (ENaC)2 is activated by double cleavage of both the α and γ subunits (1, 2). The cleavages occur in a highly variable region of the finger domains of both subunits (Fig. 1). The cleavages per se are not sufficient to activate ENaC. Rather, liberation of the intervening autoinhibitory tracts is both required and sufficient to activate the channel (2, 3). Furin, a trans-Golgi resident, pro-protein convertase, cleaves the α subunit both proximal and distal to the α-autoinhibitory tract, following RXXR motifs (4). Furin cleaves the γ subunit only proximal to the γ-autoinhibitory tract. Distal cleavage of the γ subunit is catalyzed by cell-surface resident (e.g. prostasin) or lumenal (e.g. plasmin) proteases, which are more abundant in pathophysiologic states or in response to hormonal signaling.
Figure 1.

Autoinhibitory tracts in the α and γ subunits are not conserved. A, schematic of ENaC α subunit structure based on ASIC1 structure, adapted from Ref. 40. Cylinders and arrows indicate α-helices and β-strands, respectively, and are numbered to be consistent with the structure of ASIC1 (31). Proximal and distal cleavage sites are indicated. Dashed section of schematic indicates autoinhibitory tract liberated by double cleavage; pink section within indicates key inhibitory LPHPLQRL sequence, corresponding to sequence of α-8. B, sequence alignment of mouse α (mα), mouse γ (mγ), and human γ (hγ) ENaC subunits. Predicted secondary structure of the α subunit is shown. Proximal and distal cleavage sites flank the imbedded inhibitory tracts, which are poorly conserved across subunits. Synthetic peptides corresponding to key sequences in the α and γ subunit inhibitory tracts (i.e. α-8 and γ-11) inhibit channel currents. Several proteases can cleave the γ subunit distal to the imbedded inhibitory tract in the γ subunit, including matriptase/CAP3 at a number of sites (41), and the following proteases at sites indicated by ∧: 1) TMPRSS4/channel activating protease 2 (CAP2) (42), 2) prostasin/channel activating protease 1 (CAP1) (2) and kallikrein (43), 3) plasmin (44), 4) pancreatic elastase, and 5) neutrophil elastase (45). Open triangles indicate sites mutated for experiments in Fig. 4. The closed triangle indicates the site mutated in Fig. 5.
Proteolytic ENaC activation occurs in the aldosterone-sensitive distal nephron (5), where ENaC-mediated Na+ reabsorption is coupled to K+ secretion (6). Proteases also regulate ENaC in the lung airway and alveoli (7), where ENaC regulates the airway surface liquid height (8). In these and other tissues, proteolysis of the γ subunit at the cell surface may play important physiologic roles or contribute to the pathophysiology of disease. For example, aldosterone increases prostasin expression (9), proteinuria increases lumenal plasmin concentrations (10), and inflammatory lung disease is associated with increased airway protease concentrations (11).
We previously reported that a peptide derived from the α-autoinhibitory tract (α-8, Fig. 1B) inhibits the channel by binding the finger–thumb domain interface of the α subunit (12). We also showed that α-8 and the α-autoinhibitory tract have overlapping binding sites. In addition, we identified an inhibitory peptide derived from the γ subunit (γ-11, Fig. 1B), and found that it competes with the γ-autoinhibitory tract (13).
Here we used functional measurements in Xenopus oocytes to investigate inhibition by γ-11. We show that the γ subunit thumb domain is required for γ-11 inhibition, but not for α-8 inhibition. This suggests independent binding sites for α-8 and γ-11. Using mutagenesis, we show that γ-11 inhibition depends on key residues in the γ subunit finger domain. Using a cross-linking approach, we also show that γ-11 binds to the γ subunit finger–thumb interface. Taken together, our data suggest that cleavage of the α and γ subunits activates ENaC through analogous mechanisms, i.e. by removing autoinhibitory domains from the finger–thumb interfaces of the respective subunits.
Results
The γ subunit's thumb domain is required for inhibition by γ-11
We previously reported that α-8 inhibits ENaC currents by binding at the α subunit's finger–thumb domain interface (Fig. 1) (12, 14). Although there is little sequence conservation between the α and γ subunits in the regions encompassing the subunits' cleavage sites and autoinhibitory tracts (14% identity), both follow a conserved area that includes β-strand 2 and helix 1 (50% identity) and precede an area including a pair of conserved cysteines and helix 2 (53% identity). We hypothesized that γ-11 inhibits ENaC currents by binding at the γ subunit's finger–thumb interface, analogous to α-8 inhibition through α subunit binding.
We first tested whether γ-11 inhibition requires the γ subunit's thumb domain. Channels with γ subunits that lack a thumb domain are functional (15). Consistent with competition between the autoinhibitory tract in the γ subunit and γ-11, we previously demonstrated that channels that retain the γ subunit autoinhibitory tract are more poorly inhibited by γ-11 than channels that lack the γ subunit autoinhibitory tract (13). Because Xenopus oocytes expressing mouse ENaC only cleave the γ subunit once leaving the γ autoinhibitory tract in place, we deleted the thumb domain in the γ subunit using a construct lacking the autoinhibitory tract (γΔ144–186 or γΔI). This construct retains the furin site at Arg-143, so it is cleaved when expressed in oocytes. However, channels lacking both the α subunit and γ subunit autoinhibitory tracts have a high Po (16, 17), which reduces apparent γ-11 affinity because the open state is strongly favored (seemingly “locked open”) (13). To lower the Po of our baseline construct, we expressed these γ mutants with α subunits lacking furin-cleavage sites (αR205A, R231A or αF) and WT β subunits. To determine whether our constructs were locked open in our experiments, we first measured inhibition by an independent effector. We measured Na+ self-inhibition (allosteric ENaC inhibition by extracellular Na+) by rapidly increasing [Na+] in the bath while continuously recording the current at constant voltage. This maneuver rapidly increases the driving force for Na+ entry while simultaneously introducing an ENaC inhibitor to the bath, and results in a peak current that decays over several seconds (Fig. 2A). We assessed Na+ self-inhibition by measuring the magnitude of current decay relative to the peak current after rapidly increasing [Na+] (18–20). Both constructs exhibited clear Na+ self-inhibition (Fig. 2A), with control channels having 11 ± 5% inhibition (n = 18) and thumb deletion channels having 23 ± 8% inhibition (n = 30; p < 0.0001 versus control by Student's t test). These data place an upper limit on channel Po in buffers containing 110 mm Na+, suggesting neither channel is locked open. We then added γ-11 to determine the effect of removing the thumb domain on inhibition. We found that even 100 μm γ-11 poorly inhibited currents from the γ-thumb domain deletion mutant (29 ± 8%), consistent with an IC50 greater than 100 μm. In contrast, control channel currents were inhibited by 90% at 10 μm γ-11, and the measured IC50 was 1.7 ± 0.9 μm. These data suggest that the γ subunit's thumb domain plays a critical role in ENaC inhibition by γ-11.
Figure 2.
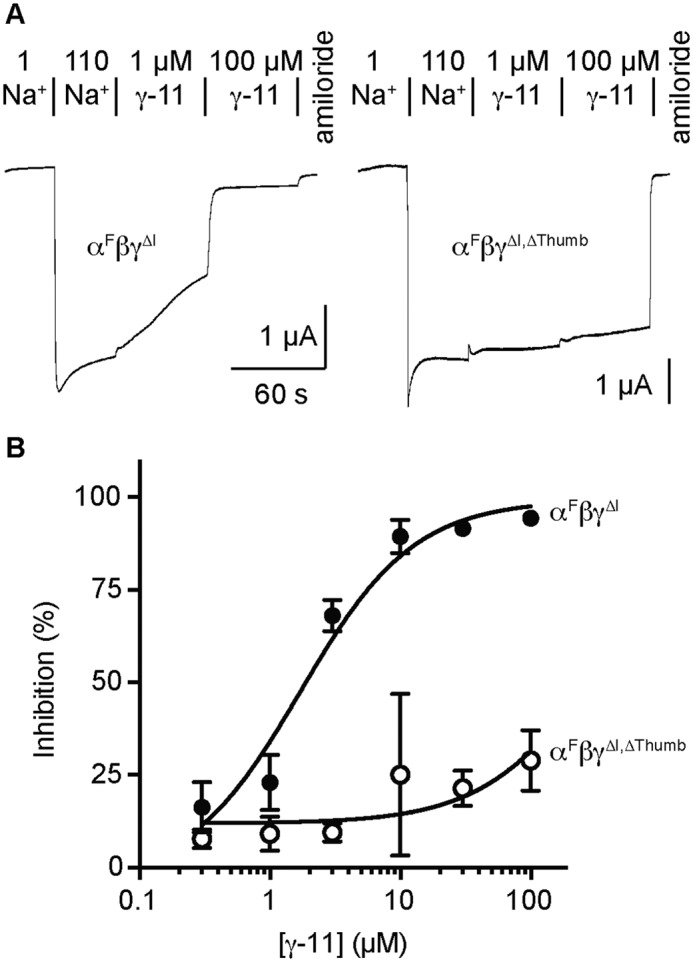
The γ subunit thumb domain is required for inhibition by γ-11. Oocytes were injected with 2 ng/subunit of cRNA encoding the ENaC subunits indicated, and whole cell recordings of currents were performed 1 day later by TEVC at −100 mV. A, representative recordings are shown; the response to two γ-11 concentrations were measured in each cell. Na+ self-inhibition was elicited by increasing bath [Na+] from 1 (1 Na+) to 110 mm (110 Na+). Indicated concentrations of γ-11 and 10 μm amiloride were diluted into 110 Na+ buffer. Current measurements were taken at the peak elicited by 110 Na+ (Ipeak), and at the end of each bath condition, giving I110Na, Iγ-11,1, Iγ-11,2, and Iamiloride. Na+ self-inhibition was determined as (1 − (Ipeak − INa110)/(Ipeak − Iamiloride)) × 100%. Peptide Inhibition was determined as (1 − (I110Na − Iγ-11,X)/(I110Na − Iamiloride)) × 100%. Baseline currents were −1.9 ± 1.2 μA for αFβγΔI and −2.1 ± 0.9 μA for αFβγΔI,ΔThumb. B, dose-response curves were plotted (mean ± S.D., n = 7–13) for αFβγΔI and αFβγΔI,ΔThumb, and fit to the Hill equation with the Hill coefficient fixed to 1. IC50 values were 1.7 ± 0.9 μm for αFβγΔI and >100 μm for αFβγΔI,ΔThumb.
We postulated that the requirement for the γ subunit thumb domain was specific to inhibition by γ-11. To test specificity, we determined whether deleting the γ subunit thumb domain affected inhibition by α-8. WT ENaC subunits expressed in oocytes are cleaved by furin and do not retain the α subunit's inhibitory tract that competes with α-8 binding (16). We therefore deleted the γ subunit thumb domain in a WT ENaC background. We then determined whether the control or γ thumb domain deletion mutant was locked open by measuring Na+ self-inhibition (Fig. 3A). Extracellular Na+ inhibited αβγ by 17 ± 5% (n = 14) and αβγΔThumb by 8 ± 5% (n = 9, p < 0.01 by Student's t test). These data demonstrate that neither channel is locked open (as Na+ inhibition remains evident), but suggest a higher upper limit for the Po of αβγΔThumb as compared with WT. When we added α-8 to the bath, we found that α-8 inhibited both WT and the γ subunit thumb deletion mutant in a dose-dependent manner (Fig. 3A). Inhibition of αβγΔThumb was modestly weaker than WT at 3 μm α-8 (Fig. 3B). This may be in part due to the lower propensity for αβγΔThumb to close (13). Nonetheless, α-8 does not require the γ subunit thumb domain to inhibit ENaC, whereas γ-11 clearly does (Fig. 2B).
Figure 3.
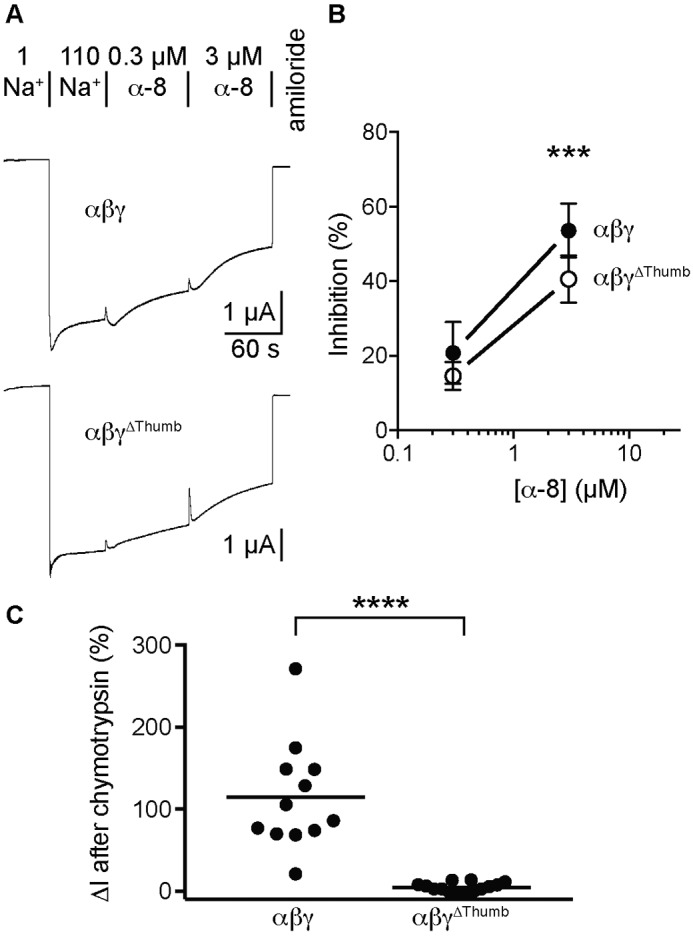
The γ subunit thumb domain is not required for inhibition by α-8. TEVC recordings of currents at −60 mV were performed 1 day after injection of 2 ng/subunit of cRNA encoding the ENaC subunits indicated. A, representative recordings of the effects of 0.3 and 3 μm α-8 are shown. Na+ self-inhibition and peptide inhibition were determined as described in the legend to Fig. 2. Baseline currents were −3.7 ± 2.3 μA for αβγ and −3.5 ± 2.0 μA for αβγΔThumb. B, dose-response curves were plotted (mean ± S.D., n = 9–14) for αβγ and αβγΔThumb, and data were analyzed by two-way ANOVA followed by Sidak's multiple comparison test. Differences were detected as the result of changing concentrations (p < 0.0001) and between groups (p < 0.0001), but not for the interaction between groups and concentrations (p = 0.11). Differences were also detected both groups at 3 μm α-8 (***, p < 0.001 versus WT). C, oocytes expressing the ENaC subunits indicated were perfused with 2 μg/ml of chymotrypsin for 2 min in 110 mm Na+ buffer to determine whether each channel could be proteolytically activated. Currents were measured by TEVC at −60 mV, and measurements were taken just prior to chymotrypsin addition, at the end of chymotrypsin addition, and at the end of 10 μm amiloride addition. The change in amiloride sensitive current in response to chymotrypsin addition is reported. ****, p < 0.0001 by Student's t test.
We then tested the ability of chymotrypsin to activate both αβγ and αβγΔThumb channels. Because Xenopus oocytes endogenously express furin, activation of WT ENaC by exogenous proteases occurs though cleavage of the γ subunit distal to the autoinhibitory tract. Based on our hypothesis of γ subunit autoinhibitory tract function, we predicted that channels lacking the γ subunit thumb domain would be poorly activated by proteolytic removal of the γ subunit autoinhibitory tract. Indeed, we found that chymotrypsin robustly activated WT channels, but not channels lacking the γ subunit thumb domain (Fig. 3C). These data suggest that the γ subunit thumb domain is required for proteolytic activation resulting from removal of the γ subunit autoinhibitory tract. These data are consistent with the notion that the γ autoinhibitory tract and γ-11 both require the γ thumb domain to affect inhibition.
Sites in the γ subunit finger domain affect γ-11 inhibition
Tryptophan mutagenesis of specific sites in the α subunit's finger domain weakened α-8 inhibition (12). Several of these sites are conserved in the α and γ subunits (Fig. 1B, open triangles). We tested our hypothesis of γ-11 binding and inhibition by mutating these sites to tryptophan and determining the effect on γ-11 inhibition (Fig. 4). We introduced mutations into an αFβγΔI background as described above for experiments in Fig. 2. Two of the mutants, L212W and A231W, had time-dependent current rundown that was 4–5-fold greater than control channels, confounding measurements of peptide inhibition (data not shown). As a result, we excluded these mutants from our analyses. We first assessed Na+ self-inhibition to determine the effect of mutation on inhibition by an independent effector. Two mutants showed Na+ self-inhibition similar to control, and one, T224W, had minimal Na+ self-inhibition (3 ± 2%, n = 7), suggesting it may be locked open (Fig. 4, A and B). Examining the effect of the γ subunit finger domain mutation on γ-11 inhibition, we found that G209W, T224W, and N242W greatly reduced γ-11 inhibition (Fig. 4, A and C). Because T224W was also poorly inhibited by an independent effector, the reduction in inhibition by γ-11 may not be due to direct interference with the mechanism of γ-11 inhibition. In contrast G209W and N242W had Na+ self-inhibition similar to control channels, suggesting that mutation at these sites in the γ subunit finger domain specifically interfere with inhibition by γ-11.
Figure 4.

Mutations at specific sites in the γ subunit finger domain weaken inhibition by γ-11. Oocytes were injected with 1–4 ng/subunit of cRNA encoding the ENaC subunits indicated. Whole cell recordings of currents were performed 1 day later by TEVC at −60 mV. A, representative recordings of the effects of 0.5 and 1.5 μm γ-11 are shown. Na+ self-inhibition and peptide inhibition were determined as described in the legend to Fig. 2. Baseline currents (all on a αFβγΔI background) were −5.3 ± 2.5 μA for control, −1.3 ± 1.5 μA for G209W, −0.81 ± 0.51 μA for γT224W, and −1.2 ± 0.7 μA for N242W. B, Na+ self-inhibition values are plotted for individual experiments with the mean indicated by a bar. Data were log transformed prior to statistical analysis. C, values for inhibition by 1.5 μm γ-11 are plotted for individual experiments with the mean indicated by a bar. Data in B and C were analyzed by one-way ANOVA followed by Tukey's multiple comparison test. ***, p < 0.001; ****, p < 0.0001 versus control.
γ-11 derivatives cross-link to sites in the γ subunit finger and thumb domains
Our data suggest a role for the γ subunit finger and thumb domains for γ-11 binding and inhibition. To determine whether γ-11 binds at the γ subunit finger–thumb interface, we attempted to cross-link the peptide to the channel by using sulfhydryl reactive cross-linkers and cysteines introduced at specific sites in both the peptide and the channel. In experiments with mouse ENaC and cysteine derivatives of mouse γ-11, we found that peptide inhibition was poorly reversible making cross-link detection difficult. When we tested human ENaC with cysteine derivatives of human γ-11, we found that inhibition was largely reversible. Human γ-11 inhibited αβγΔI with an IC50 of 10.1 ± 2.4 μm (n = 7), which is ∼2.9-fold weaker than the analogous mouse combination (13). We speculate that the increased reversibility of human γ-11 may be due to its decreased hydrophobicity (mγ-11: RFLNLIPLLVF versus hγ-11: RFSHRIPLLIF). We found that human γ-11 derivatives with cysteine added to the beginning (CRFSHRIPLLIF) or end (RFSHRIPLLIFC) of the sequence, or with cysteine substitution at the third position (RFCHRIPLLIF), inhibited the channel (Fig. 5A).
Figure 5.

γ-11 cross-links to sites in the γ subunit finger and thumb domains. Oocytes were injected with 1 ng/subunit of cRNA encoding human α lacking furin cleavage sites (αF), WT β, and γ subunits lacking the autoinhibitory tract (γΔI; control) with cysteine mutations as indicated. Sites tested were γS220C in the finger domain (A and B) and γQ426C in the thumb domain (C and D). A and C, currents were measured by two-electrode voltage clamp at −60 mV. Channels were labeled by perfusing oocytes with 10 μm MTS-4-MTS for 15 s, followed by a 30-s wash (−). Channels were then inhibited by the γ-11 cysteine derivative indicated for 60 s, followed by a wash step (−) to assess reversibility of peptide inhibition. Baseline currents were −1.0 ± 3.3 μA for γS220C, and −3.6 ± 0.6 μA for γQ426C. B and D, reversibility of peptide inhibition was measured by comparing the currents at the end of the final wash step to the currents just before peptide addition and the currents at the end of peptide addition (see schematic in A and C). Data were analyzed by one-way ANOVA followed by Tukey's multiple comparison test. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.
Based on our model of α-8 inhibition through binding the α subunit (14), we predicted that the C terminus of bound γ-11 lies near two conserved Ser residues just before the predicted helix α2. We therefore generated γS220C in a human γΔI background, and expressed hαFβγΔIS220C (S220C) or hαFβγΔI (control) channels in oocytes (Fig. 5). We first perfused oocytes with 10 μm 1,4-butanediyl bismethanethiosulfonate (MTS-4-MTS) for 15 s (Fig. 5A). We and others have reported that longer exposures to bifunctional methanethiosulfonate (MTS) compounds irreversibly inhibit ENaC (21–24). This step labels accessible cysteines on the channel with MTS-4-MTS, potentially leaving an unreacted MTS group tethered to the mutated site. After a wash step to remove MTS-4-MTS from the bath, we added γ-11 cysteine derivatives. If the bound inhibitory peptide presents a cysteine in close proximity to an MTS group tethered to the channel, the bound peptide may react and become tethered to the channel. To detect this possibility, we removed the inhibitory peptide from the bath and measured reversibility of peptide inhibition (Fig. 5A). We reasoned that reduced reversibility with specific combinations of γ subunit mutant and γ-11 derivative suggests cross-linking. We observed reduced reversibility with the γS220C/RFSHRIPLLIFC combination as compared with control channels with the same γ-11 derivative, or with the same mutant but a different γ-11 derivative (RFCHRIPLLIF), or with the same mutant and peptide but omitting MTS-4-MTS (Fig. 5, A and B). These data suggest that the C terminus of γ-11 cross-links to the γ subunit finger domain.
We also predicted that the N terminus of bound γ-11 lies at the finger–thumb domain interface. To test our hypothesis, we generated γQ426C in a human γΔI background, and expressed hαFβγΔIQ426C (Q426C) or hαFβγΔI (control) channels in oocytes (Fig. 5, C and D). In agreement with our hypothesis, we observed reduced reversibility of peptide inhibition with the γQ426C/CRFSHRIPLLIF combination than we did with control channels and the same γ-11 derivative, or with γQ426C and a different γ-11 derivative (RFCHRIPLLIF), or with γQ426C/CRFSHRIPLLIF but omitting MTS-4-MTS. These data are consistent with a model of γ-11 inhibition where γ-11 inhibits the channel by binding the γ subunit finger–thumb interface.
Discussion
The ENaC α and γ subunits share no sequence homology in the region that includes the cleavage sites and autoinhibitory tracts (Fig. 1B). Despite this, our data suggest that the inhibitory mechanisms of peptides derived from these autoinhibitory tracts (α-8 and γ-11) are analogous. Each peptide inhibits the channel by binding to the finger–thumb domain interface of the subunit from which it is derived, and each peptide acts allosterically (Fig. 6).
Figure 6.

Model of ENaC proteolytic activation and autoinhibitory tract-derived peptide inhibition. ENaC α and γ subunits are synthesized with autoinhibitory tracts flanked by cleavage sites (upper left). The autoinhibitory tracts facilitate interactions between the finger (β2–α1 loop, helix α2 and preceding β-strands) and thumb domains (α4–α5 loop) that favor a closed conformation of the channel pore. Upon double cleavage of either subunit, the respective autoinhibitory tract is removed, releasing interactions between the top of the thumb domain and the finger domain (upper right). The new arrangement at the finger–thumb domain interface may alter the conformation of the β9–α4 and α5–β10 loops, which interact with the pore forming transmembrane helices (TM2 and TM2). The new structural arrangement favors the open conformation of the channel pore. Addition of either α-8 or γ-11 recapitulates key interactions at the respective finger–thumb domain interface that inhibit the channel, reversing the functional effect of double cleavage.
Our previous work suggested distinct binding sites for each peptide. We showed that the α subunit autoinhibitory tract and α-8 likely have overlapping binding sites (14). We also showed that the γ subunit autoinhibitory tract competes with γ-11 inhibition (13). Accordingly, here we have shown that the γ subunit thumb domain is essential for γ-11 inhibition (Fig. 2), but not for α-8 inhibition (Fig. 3). Yet our data also suggest analogous binding sites for α-8 and γ-11. We previously showed that mutations at specific sites in the finger domain of the α subunit diminished the efficacy of α-8 inhibition (12). Mutations at two of the equivalent sites in the γ subunit reduced inhibition by γ-11 (Fig. 4). We have also shown that each peptide can be cross-linked at their N terminus to the respective thumb domain, and at their C terminus to the respective finger domain (Fig. 5) (21).
An allosteric mechanism for peptide inhibition is supported by several observations. Despite being positively charged, inhibition by an α subunit-derived peptide was not voltage dependent and did not compete with amiloride, a bona fide pore blocker (3). This likely rules out a pore-blocking mechanism analogous to amiloride. The sites we have identified for α-8 and γ-11 are far from the putative channel gate in the transmembrane helices (see Fig. 1A). Furthermore, we have shown that the finger–thumb domain interfaces of both the α and γ subunits can be manipulated independently of peptide addition to modulate channel function (21, 22). These data suggest that inhibitory peptide binding occurs at dynamic domain interfaces whose conformations are functionally coupled to the conformation of the channel pore. Additionally, the finger and thumb domains of proteins in the ENaC protein family have been implicated in channel regulation by other factors. For ENaC, these include Na+ (19, 25, 26), protons (27), other cations (28), Cl− (29), and laminar shear stress (30). For ASIC1, these include protons (31, 32), coral snake MitTx (33), and tarantula psalmotoxin (34).
By extension, our data suggest a mechanism for the proteolytic activation of ENaC (Fig. 6). The cleavage sites in the α and γ subunits are necessarily exposed to allow for proteolytic enzyme access. This may have led to a labile sequence that is highly variable. Prior to proteolysis, key residues between the cleavage sites coordinate residues across the finger–thumb domain interface. These include the β2–α1 loop, helix α2, and preceding β-strands in the finger domain, and the α4–α5 loop in the thumb domain (Figs. 4 and 5) (12, 14). This arrangement at the finger–thumb domain interface favors the closed state at the channel gate. Upon proteolysis at both the proximal and distal sites in either the α or γ subunits, the respective autoinhibitory tract is liberated, releasing interactions that had constrained the finger–thumb domain interface. We propose that autoinhibitory tract release-associated changes to the finger–thumb domain interface are coupled to conformational changes of the loops connecting the thumb domain to the palm domain. It remains unclear whether new interactions are facilitated by removal of the autoinhibitory tract. This new arrangement at the finger–thumb domain interface favors an open channel gate. The magnitude of the shift to favor the open state is greater in the case of double cleavage of the γ subunit than it is for the α subunit (17).
How and why the ENaC α and γ subunits evolved to include autoinhibitory domains that can be proteolytically liberated remains unknown. Activation from a pro-form is a common theme in biology that is often mediated by proteases, but is unusual for channels or transporters. Delaying channel activation until arrival at the plasma membrane is mediated by phosphatidylinositol 4,5-bisphosphate (PIP2) signaling (35, 36), as for many other channels (37). Proteolytic activation may have evolved to allow for a pool of inactive channels that can be rapidly recruited in response to a stimulus. Or, because uncleaved channels transport Na+ only at low [Na+] and cleaved channels transport Na+ at both low and high [Na+] (16), having a mixed population of channels may allow for fine tuning of Na+ reabsorption. ENaC subunits are expressed widely, including in Na+ transporting epithelia, in Na+ sensing epithelia, and tissues where ENaC may play a mechanosensing role. Proteolytic activation may have allowed for ENaC functional tuning, facilitating a variety of physiologic roles. Which functional roles drove the development of proteolytic ENaC regulation remains an open question.
Experimental procedures
Materials
All peptides were synthesized and HPLC-purified by GenScript Corp. (Piscataway, NJ), and were modified by N-terminal acetylation and C-terminal amidation. MTS-4-MTS was purchased from Toronto Research Chemicals (North York, ON).
Plasmids and site-directed mutagenesis
cDNA encoding ENaC subunits in pBluescript SK(−) (Stratagene, San Diego, CA) were previously described (38). Plasmid containing γΔThumb was a generous gift from Shaohu Sheng. Site-directed mutagenesis was performed with the QuikChange II XL Site-directed Mutagenesis Kit (Agilent, Santa Clara, CA). Primers were designed using QuikChange Primer Design (Agilent) and were obtained from Integrated DNA Technologies (Coralville, IA) to modify mouse or human ENaC subunits. Direct sequencing was used to confirm constructs.
ENaC functional expression in Xenopus oocytes
Plasmids were linearized with restriction endonucleases (New England Biolabs, Ipswich, MA) at 37 °C overnight and purified using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). Linearized plasmids were transcribed using mMessage mMachine T3 Transcription Kit (Invitrogen) and purified using the RNeasy MiniElute Cleanup Kit (Qiagen). Oocytes from Xenopus laevis were harvested and defolliculated using type II collagenase (Sigma). 1–4 ng of cRNA per ENaC subunit was injected into stage V or VI Xenopus oocytes using a Nanoject II (Drummond, Broomall, PA). Xenopus oocytes were stored at 18 °C in modified Barth's saline (88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 15 mm HEPES, 0.3 mm Ca(NO3)2, 0.41 mm CaCl2, and 0.82 mm MgSO4, pH 7.4) supplemented with 10 μg/ml of sodium penicillin, 10 μg/ml of streptomycin sulfate, and 100 μg/ml of gentamicin sulfate. The protocol for harvesting oocytes from X. laevis was approved by the University of Pittsburgh's Institutional Animal Care and Use Committee.
Measurement of ENaC currents
ENaC current was measured using the two-electrode voltage clamp (TEVC) technique using an Axoclamp 900A voltage clamp amplifier (Molecular Devices, Sunnyvale, CA) and pClamp 10.5 software (Molecular Devices). A 20-μl recording chamber (AutoMate Scientific, Berkeley, CA) was used with perfusion (3–5 ml/min) controlled by an eight-channel pinch valve system (AutoMate Scientific). Measurement of ENaC currents were made in Na-110 buffer (110 mm NaCl, 2 mm KCl, 2 mm CaCl2, and 10 mm HEPES (pH 7.4)). At the end of each experiment, currents were measured in 10 μm amiloride (stock solution of 100 mm amiloride in dimethyl sulfoxide (DMSO) stored at room temperature diluted into Na-110 buffer) to determine the amiloride-sensitive component of the current.
Na+ self-inhibition measurements
Na+ self-inhibition was measured as previously described (39). Briefly, under constant voltage conditions, [Na+] was rapidly raised from 1 mm Na+ (with 109 mm N-methyl-d-glucamine chloride replacing 109 mm NaCl in Na-110 buffer) to 110 mm Na+. Because ENaC Na+ self-inhibition is slow relative to bath exchange, acutely raising [Na+] from a low concentration (where the channel open probability (Po) is high) results in a peak inward current that subsequently declines. Current reduction from the peak reflects Na+ self-inhibition.
Cross-linking ENaC and inhibitory peptides
Stock solutions of 10 mm MTS-4-MTS were prepared in DMSO, and stored under desiccant at −20 °C for up to 2 months. Stock solutions of γ-11 derivatives were made in DMSO at 10 mm, and stored at −20 °C. Working solutions of MTS-4-MTS were prepared immediately before each experiment by diluting stock solutions into Na-110 buffer. Working solutions of amiloride and γ-11 derivatives were prepared on the day of the experiment by diluting stocks into Na-110 buffer. Currents from ENaC expressing oocytes were continuously recorded while clamped at −60 mV. The bath was perfused with Na-110 at a rate of 3 ml/min throughout the experiment. After currents became stable (45 s), 10 μm MTS-4-MTS was added for 15 s, followed by a 45-s wash with Na-110 buffer. Then 3 μm γ-11 derivative in Na-110 was added for 1 min, followed by a 45–60-s wash with Na-110 buffer. At the end of each experiment, 10 μm amiloride was added for 10 s. Current measurements were taken just prior to peptide addition (baseline), at the end of peptide addition (inhibited), after the final wash (washout), and after amiloride addition. Amiloride-insensitive currents were subtracted from all measured currents. Reversibility was calculated by: 100% × (washout − inhibited)/(baseline − inhibited).
Statistical analyses
Group comparisons were performed by Student's t test or by ANOVA followed by a Tukey multiple comparison test, as appropriate. Prism 7 (GraphPad, Inc., La Jolla, CA) was used to perform statistical calculations and curve fitting. Adjusted values of p < 0.05 were considered significant.
Author contributions
D. M. B., R. N. T., and O. B. K. formal analysis; D. M. B., R. N. T., and O. B. K. investigation; D. M. B., R. N. T., and O. B. K. writing-review and editing; O. B. K. conceptualization; O. B. K. funding acquisition; O. B. K. methodology; O. B. K. writing-original draft; O. B. K. project administration.
This work was supported by NIDDK, National Institutes of Health, Grant R01 DK098204 (to O. B. K.) and the Pittsburgh Center for Kidney Research was supported by NIDDK Grant P30 DK079307, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ENaC
- epithelial Na+ channel
- MTS
- methanethiosulfonate
- MTS-4-MTS
- 1,4-butanediyl bismethanethiosulfonate
- TEVC
- two-electrode voltage clamp
- Po
- open probability
- DMSO
- dimethyl sulfoxide
- ANOVA
- analysis of variance.
References
- 1. Hughey R. P., Bruns J. B., Kinlough C. L., Harkleroad K. L., Tong Q., Carattino M. D., Johnson J. P., Stockand J. D., and Kleyman T. R. (2004) Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 279, 18111–18114 10.1074/jbc.C400080200 [DOI] [PubMed] [Google Scholar]
- 2. Bruns J. B., Carattino M. D., Sheng S., Maarouf A. B., Weisz O. A., Pilewski J. M., Hughey R. P., and Kleyman T. R. (2007) Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the g-subunit. J. Biol. Chem. 282, 6153–6160 10.1074/jbc.M610636200 [DOI] [PubMed] [Google Scholar]
- 3. Carattino M. D., Sheng S., Bruns J. B., Pilewski J. M., Hughey R. P., and Kleyman T. R. (2006) The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its a subunit. J. Biol. Chem. 281, 18901–18907 10.1074/jbc.M604109200 [DOI] [PubMed] [Google Scholar]
- 4. Kleyman T. R., Carattino M. D., and Hughey R. P. (2009) ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J. Biol. Chem. 284, 20447–20451 10.1074/jbc.R800083200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frindt G., Gravotta D., and Palmer L. G. (2016) Regulation of ENaC trafficking in rat kidney. J. Gen. Physiol. 147, 217–227 10.1085/jgp.201511533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pearce D., Soundararajan R., Trimpert C., Kashlan O. B., Deen P. M., and Kohan D. E. (2015) Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 10, 135–146 10.2215/CJN.05760513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Donaldson S. H., Hirsh A., Li D. C., Holloway G., Chao J., Boucher R. C., and Gabriel S. E. (2002) Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 277, 8338–8345 10.1074/jbc.M105044200 [DOI] [PubMed] [Google Scholar]
- 8. Jiang C., Finkbeiner W. E., Widdicombe J. H., McCray P. B. Jr., and Miller S. S. (1993) Altered fluid transport across airway epithelium in cystic fibrosis. Science 262, 424–427 10.1126/science.8211164 [DOI] [PubMed] [Google Scholar]
- 9. Narikiyo T., Kitamura K., Adachi M., Miyoshi T., Iwashita K., Shiraishi N., Nonoguchi H., Chen L. M., Chai K. X., Chao J., and Tomita K. (2002) Regulation of prostasin by aldosterone in the kidney. J. Clin. Invest. 109, 401–408 10.1172/JCI0213229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Svenningsen P., Bistrup C., Friis U. G., Bertog M., Haerteis S., Krueger B., Stubbe J., Jensen O. N., Thiesson H. C., Uhrenholt T. R., Jespersen B., Jensen B. L., Korbmacher C., and Skøtt O. (2009) Plasmin in nephrotic urine activates the epithelial sodium channel. J. Am. Soc. Nephrol. 20, 299–310 10.1681/ASN.2008040364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Butterworth M. B., Zhang L., Heidrich E. M., Myerburg M. M., and Thibodeau P. H. (2012) Activation of the epithelial sodium channel (ENaC) by the alkaline protease from Pseudomonas aeruginosa. J. Biol. Chem. 287, 32556–32565 10.1074/jbc.M112.369520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kashlan O. B., Boyd C. R., Argyropoulos C., Okumura S., Hughey R. P., Grabe M., and Kleyman T. R. (2010) Allosteric inhibition of the epithelial Na+ channel (ENaC) through peptide binding at peripheral finger and thumb domains. J. Biol. Chem. 285, 35216–35223 10.1074/jbc.M110.167064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Passero C. J., Carattino M. D., Kashlan O. B., Myerburg M. M., Hughey R. P., and Kleyman T. R. (2010) Defining an inhibitory domain in the γ subunit of the epithelial sodium channel. Am. J. Physiol. Renal Physiol. 299, F854–F861 10.1152/ajprenal.00316.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kashlan O. B., Adelman J. L., Okumura S., Blobner B. M., Zuzek Z., Hughey R. P., Kleyman T. R., and Grabe M. (2011) Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel a subunit reveals a mechanism of channel activation by proteases. J. Biol. Chem. 286, 649–660 10.1074/jbc.M110.167098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sheng S., Chen J., Mukherjee A., Yates M. E., Buck T. M., Brodsky J. L., Tolino M. A., Hughey R. P., and Kleyman T. R. (September 18, 2018) Thumb domains of the three epithelial Na+ channel subunits have distinct functions. J. Biol. Chem. 10.1074/jbc.RA118.003618 10.1074/jbc.RA118.003618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheng S., Carattino M. D., Bruns J. B., Hughey R. P., and Kleyman T. R. (2006) Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am. J. Physiol. Renal Physiol. 290, F1488–F1496 10.1152/ajprenal.00439.2005 [DOI] [PubMed] [Google Scholar]
- 17. Carattino M. D., Hughey R. P., and Kleyman T. R. (2008) Proteolytic processing of the epithelial sodium channel γ subunit has a dominant role in channel activation. J. Biol. Chem. 283, 25290–25295 10.1074/jbc.M803931200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bize V., and Horisberger J. D. (2007) Sodium self-inhibition of human epithelial sodium channel: selectivity and affinity of the extracellular sodium sensing site. Am. J. Physiol. Renal Physiol. 293, F1137–F1146 10.1152/ajprenal.00100.2007 [DOI] [PubMed] [Google Scholar]
- 19. Kashlan O. B., Blobner B. M., Zuzek Z., Tolino M., and Kleyman T. R. (2015) Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J. Biol. Chem. 290, 568–576 10.1074/jbc.M114.606152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sheng S., Perry C. J., and Kleyman T. R. (2004) Extracellular Zn2+ activates epithelial Na+ channels by eliminating Na+ self-inhibition. J. Biol. Chem. 279, 31687–31696 10.1074/jbc.M405224200 [DOI] [PubMed] [Google Scholar]
- 21. Kashlan O. B., Blobner B. M., Zuzek Z., Carattino M. D., and Kleyman T. R. (2012) Inhibitory tract traps the epithelial Na+ channel in a low activity conformation. J. Biol. Chem. 287, 20720–20726 10.1074/jbc.M112.358218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blobner B. M., Wang X. P., and Kashlan O. B. (2018) Conserved cysteines in the finger domain of the epithelial Na+ channel α and γ subunits are proximal to the dynamic finger–thumb domain interface. J. Biol. Chem. 293, 4928–4939 10.1074/jbc.M117.819367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collier D. M., Tomkovicz V. R., Peterson Z. J., Benson C. J., and Snyder P. M. (2014) Intersubunit conformational changes mediate epithelial sodium channel gating. J. Gen. Physiol. 144, 337–348 10.1085/jgp.201411208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Snyder P. M., Olson D. R., and Bucher D. B. (1999) A pore segment in DEG/ENaC Na+ channels. J. Biol. Chem. 274, 28484–28490 10.1074/jbc.274.40.28484 [DOI] [PubMed] [Google Scholar]
- 25. Winarski K. L., Sheng N., Chen J., Kleyman T. R., and Sheng S. (2010) Extracellular allosteric regulatory subdomain within the γ subunit of the epithelial Na+ channel. J. Biol. Chem. 285, 26088–26096 10.1074/jbc.M110.149963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maarouf A. B., Sheng N., Chen J., Winarski K. L., Okumura S., Carattino M. D., Boyd C. R., Kleyman T. R., and Sheng S. (2009) Novel determinants of epithelial sodium channel gating within extracellular thumb domains. J. Biol. Chem. 284, 7756–7765 10.1074/jbc.M807060200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collier D. M., Peterson Z. J., Blokhin I. O., Benson C. J., and Snyder P. M. (2012) Identification of extracellular domain residues required for epithelial Na+ channel activation by acidic pH. J. Biol. Chem. 287, 40907–40914 10.1074/jbc.M112.417519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheng S., Perry C. J., and Kleyman T. R. (2002) External nickel inhibits epithelial sodium channel by binding to histidine residues within the extracellular domains of α and γ subunits and reducing channel open probability. J. Biol. Chem. 277, 50098–50111 10.1074/jbc.M209975200 [DOI] [PubMed] [Google Scholar]
- 29. Collier D. M., and Snyder P. M. (2011) Identification of epithelial Na+ channel (ENaC) intersubunit Cl− inhibitory residues suggests a trimeric agb channel architecture. J. Biol. Chem. 286, 6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi S., Blobner B. M., Kashlan O. B., and Kleyman T. R. (2012) Extracellular finger domain modulates the response of the epithelial sodium channel to shear stress. J. Biol. Chem. 287, 15439–15444 10.1074/jbc.M112.346551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jasti J., Furukawa H., Gonzales E. B., and Gouaux E. (2007) Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 449, 316–323 10.1038/nature06163 [DOI] [PubMed] [Google Scholar]
- 32. Krauson A. J., Rued A. C., and Carattino M. D. (2013) Independent contribution of extracellular proton binding sites to ASIC1a activation. J. Biol. Chem. 288, 34375–34383 10.1074/jbc.M113.504324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baconguis I., Bohlen C. J., Goehring A., Julius D., and Gouaux E. (2014) X-ray structure of acid-sensing ion channel 1-snake toxin complex reveals open state of a Na+-selective channel. Cell 156, 717–729 10.1016/j.cell.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baconguis I., and Gouaux E. (2012) Structural plasticity and dynamic selectivity of acid-sensing ion channel-spider toxin complexes. Nature 489, 400–405 10.1038/nature11375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yue G., Malik B., Yue G., and Eaton D. C. (2002) Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J. Biol. Chem. 277, 11965–11969 10.1074/jbc.M108951200 [DOI] [PubMed] [Google Scholar]
- 36. Ma H. P., Saxena S., and Warnock D. G. (2002) Anionic phospholipids regulate native and expressed epithelial sodium channel (ENaC). J. Biol. Chem. 277, 7641–7644 10.1074/jbc.C100737200 [DOI] [PubMed] [Google Scholar]
- 37. Suh B. C., and Hille B. (2008) PIP2 is a necessary cofactor for ion channel function: how and why? Annu. Rev. Biophys. 37, 175–195 10.1146/annurev.biophys.37.032807.125859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahn Y. J., Brooker D. R., Kosari F., Harte B. J., Li J., Mackler S. A., and Kleyman T. R. (1999) Cloning and functional expression of the mouse epithelial sodium channel. Am. J. Physiol. 277, F121–F129 [DOI] [PubMed] [Google Scholar]
- 39. Sheng S., Bruns J. B., and Kleyman T. R. (2004) Extracellular histidine residues crucial for Na+ self-inhibition of epithelial Na+ channels. J. Biol. Chem. 279, 9743–9749 10.1074/jbc.M311952200 [DOI] [PubMed] [Google Scholar]
- 40. Kashlan O. B., and Kleyman T. R. (2011) ENaC structure and function in the wake of a resolved structure of a family member. Am. J. Physiol. Renal Physiol. 301, F684–F696 10.1152/ajprenal.00259.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kota P., García-Caballero A., Dang H., Gentzsch M., Stutts M. J., and Dokholyan N. V. (2012) Energetic and structural basis for activation of the epithelial sodium channel by matriptase. Biochemistry 51, 3460–3469 10.1021/bi2014773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Passero C. J., Mueller G. M., Myerburg M. M., Carattino M. D., Hughey R. P., and Kleyman T. R. (2012) TMPRSS4-dependent activation of the epithelial sodium channel requires cleavage of the γ-subunit distal to the furin cleavage site. Am. J. Physiol. Renal Physiol. 302, F1–F8 10.1152/ajprenal.00330.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Patel A. B., Chao J., and Palmer L. G. (2012) Tissue kallikrein activation of the epithelial Na channel. Am. J. Physiol. Renal Physiol. 303, F540–F550 10.1152/ajprenal.00133.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Passero C. J., Mueller G. M., Rondon-Berrios H., Tofovic S. P., Hughey R. P., and Kleyman T. R. (2008) Plasmin activates epithelial Na+ channels by cleaving the γ subunit. J. Biol. Chem. 283, 36586–36591 10.1074/jbc.M805676200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adebamiro A., Cheng Y., Rao U. S., Danahay H., and Bridges R. J. (2007) A segment of γ ENaC mediates elastase activation of Na+ transport. J. Gen. Physiol. 130, 611–629 10.1085/jgp.200709781 [DOI] [PMC free article] [PubMed] [Google Scholar]