

Abstract
Reactive oxygen species (ROS), in particular H2O2, regulate intracellular signaling through reversible oxidation of reactive protein thiols present in a number of kinases and phosphatases. H2O2 has been shown to regulate mitogen-activated protein kinase (MAPK) signaling depending on the cellular context. We report here that in human articular chondrocytes, the MAPK family member c-Jun N-terminal kinase 2 (JNK2) is activated by fibronectin fragments and low physiological levels of H2O2 and inhibited by oxidation due to elevated levels of H2O2. The kinase activity of affinity-purified, phosphorylated JNK2 from cultured chondrocytes was reversibly inhibited by 5–20 μm H2O2. Using dimedone-based chemical probes that react specifically with sulfenylated cysteines (RSOH), we identified Cys-222 in JNK2, a residue not conserved in JNK1 or JNK3, as a redox-reactive site. MS analysis of human recombinant JNK2 also detected further oxidation at Cys-222 and other cysteines to sulfinic (RSO2H) or sulfonic (RSO3H) acid. H2O2 treatment of JNK2 resulted in detectable levels of peptides containing intramolecular disulfides between Cys-222 and either Cys-213 or Cys-177, without evidence of dimer formation. Substitution of Cys-222 to alanine rendered JNK2 insensitive to H2O2 inhibition, unlike C177A and C213A variants. Two other JNK2 variants, C116A and C163A, were also resistant to oxidative inhibition. Cumulatively, these findings indicate differential regulation of JNK2 signaling dependent on H2O2 levels and point to key cysteine residues regulating JNK2 activity. As levels of intracellular H2O2 rise, a switch occurs from activation to inhibition of JNK2 activity, linking JNK2 regulation to the redox status of the cell.
Keywords: mitogen-activated protein kinase (MAPK), redox regulation, redox signaling, chondrocyte, integrin, reactive oxygen species (ROS), hydrogen peroxide, c-Jun N-terminal kinase (JNK), disulfide, cellular redox status
Introduction
Intracellular signaling mediated by the MAPK5 family member c-Jun N-terminal kinase (JNK), also known as stress-activated kinase, plays a central role in controlling the response to cytokines and a variety of stress stimuli. JNK signaling is important in the regulation of diverse processes, including cell proliferation, cell death and survival, oxidative stress, insulin resistance, and inflammation underlying conditions such as diabetes, cancer, arthritis, and aging (1–4). A total of 10 JNK isoforms have been described that result from alternative splicing of three JNK genes, JNK1, JNK2, and JNK3 (MAPK8, MAPK9, and MAPK10, respectively), producing JNK1α1, -α2, -β1, and -β2; JNK2α1, -α2, -β1, and -β2; and JNK3α1 and -α2 (5). The JNK1α1, JNK1β1, JNK2α1, and JNK2β1 isoforms have a shorter C-terminal region and when expressed with an epitope tag in Chinese hamster ovary cells were detected as 46-kDa proteins on immunoblots, whereas the JNK1α2, JNK1β2, JNK2α2, and JNK2β2 isoforms exhibited apparent molecular masses of ∼55 kDa (5). JNK3 has an extended N terminus, resulting in a slighter higher molecular weight for the associated isoforms. JNK1 and JNK2 are expressed by most cell types, whereas JNK3 is primarily found in the brain. All JNK isoforms are activated by dual phosphorylation of conserved Thr and Tyr residues mediated by MAPK kinases MKK4 and MKK7 and can be inactivated by MAPK phosphatases (6). Given the importance of JNK signaling in biological systems, the mechanisms responsible for regulation of JNK activity are important to understand.
Reactive oxygen species (ROS) have been increasingly recognized for their role in the regulation of intracellular signaling pathways (3, 4, 7). Hydrogen peroxide (H2O2) is particularly well suited for redox signaling because of its relatively higher half-life, allowing it to diffuse over longer distances in cells. There is evidence that H2O2 can positively regulate the JNK signaling pathway upstream of the MKKs through activation of Src and Cas (8, 9) as well as Ask1 (10). Oxidative inhibition of MAPK phosphatase 1 (MKP1), resulting in sustained JNK activation, has also been described (11, 12). JNK activation is suppressed by the formation of a complex with GSH S-transferase Pi, which can be further stabilized by the binding of peroxiredoxin 1 (Prx1), but these interactions have not been shown to be directly influenced by JNK redox status (13). JNK signaling may also in turn regulate the redox environment of the cell. JNK signaling in Drosophila was found to increase expression of genes that facilitated an oxidative stress response, resulting in increased lifespan (14), whereas in HeLa cells JNK signaling in the mitochondria increased ROS production (15). These studies point to the need for a better understanding of the interplay between JNK signaling and redox metabolism in cells, including the question of whether there is direct regulation of JNK by oxidants like H2O2.
One of the most commonly observed mechanisms by which H2O2 regulates intracellular signaling is via the reversible oxidation of protein thiols. This occurs when reactive cysteines in the thiolate state (Cys-S−) are oxidized by H2O2 to form cysteine sulfenic acids (Cys-SOH), a process also known as sulfenylation (4, 16, 17). Sulfenic acids are highly labile species, often progressing to disulfide formation upon reaction with thiol groups. Both sulfenic acids and disulfides can be reduced back to their thiol(ate) states by cellular reducing systems. Ask1 and MKP1, two key regulators of JNK phosphorylation, have been shown to be regulated by thiol oxidation, enhancing JNK phosphorylation through oxidative activation of the kinase Ask1 (18) and inhibition of the phosphatase MKP1 (12). Although redox regulation of signaling kinases like Src, p38, Akt, and ERK is becoming an increasingly common theme (9, 19–21), it is still unknown whether or not JNK is directly regulated by H2O2.
We have previously shown that exposure of human articular chondrocytes to fragments of the extracellular matrix protein fibronectin stimulates JNK signaling in chondrocytes in an ROS-dependent fashion (22). Fibronectin fragments (FN-fs), including fragments containing the Arg-Gly-Asp (RGD) cell binding sequence, are found in the articular cartilage and synovial fluid of patients with arthritis, likely the result of increased activity of matrix-degrading enzymes (23, 24). These FN-fs can activate cell surface receptors, including the α5β1 integrin on chondrocytes and synovial fibroblasts, leading to increased production of matrix metalloproteinases (25–27). Relevant to the present study, FN-f stimulation was found to increase sulfenylation of multiple proteins in chondrocytes, including the kinase c-Src (28). In those experiments, pretreatment with dimedone, a reagent that reacts selectively with sulfenylated cysteines (29), blocked FN-f–induced phosphorylation of JNK and its substrate c-Jun, indicating that protein sulfenylation was required for signaling that resulted in JNK and c-Jun phosphorylation.
Based on these studies, FN-f stimulation of human articular chondrocytes was used as a biologically relevant model system to further study the role of protein thiol oxidation in the regulation of JNK activity. FN-f stimulation was compared with treating cells with various concentrations of exogenous H2O2 as a direct oxidant and with the redox cycling agent menadione, which generates higher levels of intracellular H2O2. We found that both FN-f treatment, which generated low levels of H2O2, and treatment with low micromolar levels of H2O2 stimulated phosphorylation of JNK and its substrate c-Jun. Menadione treatment and treatment with high levels of exogenous H2O2 reversibly inhibited JNK2 kinase activity through oxidation of cysteine residues in JNK2. The mechanism of this inhibition was further explored using mass spectrometry (MS) and site-directed mutagenesis to identify the sites of oxidation on JNK2α2.
Results
Increasing H2O2 levels are associated with differential regulation of JNK signaling in chondrocytes
To determine conditions in which JNK signaling is regulated by H2O2 in chondrocytes, treatment of chondrocytes with FN-f was compared with addition of exogenous H2O2 or the redox cycling agent menadione. We had previously found that menadione generates high levels of endogenous H2O2 in chondrocytes, resulting in oxidative stress, at least in part through inactivation of Prxs that are critical to cellular ROS detoxification (30). Using the Orp1-roGFP H2O2 biosensor, menadione was found to rapidly generate increased levels of intracellular H2O2 that were similar to what we had shown previously (30), whereas FN-f generated a more gradual and lower level of H2O2 production (Fig. 1A). To further examine relative levels of intracellular H2O2, we measured Prx hyperoxidation (i.e. formation of sulfinic or sulfonic forms, referred to as PrxSO2/3H, which are inactive) by immunoblotting cell lysates with an antibody specific to PrxSO2/3H. Using this technique, we had previously shown high levels of PrxSO2/3H in chondrocytes treated with menadione (30), which was confirmed here (Fig. 1B). In contrast, the lower levels of H2O2 generated by FN-f induced little, if any, increase in PrxSO2/3H, whereas the addition of exogenous H2O2 led to a dose-dependent increase in PrxSO2/3H (Fig. 1, B and C).
Figure 1.

JNK phosphorylation and activity are differentially regulated by the level of H2O2 in chondrocytes. A, human articular chondrocytes were transduced with an Orp1-roGFP–expressing baculovirus for H2O2-specific redox sensing. Cells were treated for 30 min with either 1 μm FN-f or 25 μm menadione. Data from 10 individual cells from three independent donors were quantified and are presented showing ±S.D. (error bars) of relative mean (Rel. Ave.) 405/488-nm excitation (ex) ratio. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001 relative to untreated control; ††††, p < 0.001 relative to FN-f. B, human articular chondrocytes were treated with menadione or FN-f for a time course from 0 to 60 min and then lysed in NEM-containing lysis buffer for analysis of Prx hyperoxidation. Immunoblots from three independent donors were quantified using ImageJ. *, p < 0.05; **, p < 0.01. C, human articular chondrocytes were treated with FN-f and a dose curve of H2O2 for 30 min, and lysates were analyzed by immunoblotting. Results shown are representative of three independent experiments using cells from independent donors. D, human articular chondrocytes were treated with either menadione or FN-f for 30 min or pretreated with menadione for 30 min before FN-f treatment. Lysates were analyzed by immunoblotting. Immunoblots are representative of three independent experiments using cells from independent donors. p, phospho; t, total.
Examination of JNK and c-Jun phosphorylation state revealed that although FN-f stimulated phosphorylation of both, there was a dose-response effect of H2O2 such that only the lower doses of 0.5 and 1 μm stimulated phosphorylation of both JNK and c-Jun (Fig. 1C). Menadione weakly stimulated JNK and c-Jun phosphorylation and, when added to cells prior to addition of FN-f, blocked the large increase in phosphorylation in response to FN-f (Fig. 1D). This suggested that the high levels of H2O2 generated by menadione might be inhibiting the effects of FN-f. This effect on JNK signaling was distinct from the other two MAP kinase family members, ERK and p38, which were strongly phosphorylated in response to either menadione or FN-f, and with no further increase or decrease in phosphorylation upon addition of both (Fig. 1D).
JNK2 is sulfenylated in chondrocytes
Previous studies have shown that JNK phosphorylation is regulated by H2O2 through the oxidative activation of the upstream kinase Ask1 as well as through oxidative inhibition of JNK phosphatases (12, 18). We next investigated whether JNK could be directly regulated by oxidation with H2O2 at cysteine sites, resulting in sulfenic acid formation. This was prompted by a preliminary experiment where we had observed a protein band consistent with phosphorylated JNK2 in chondrocyte lysates from FN-f–treated cells that were enriched in sulfenylated proteins using the biotin-tagged dimedone analog DCP-Bio1 (Fig. S1). The anti-phospho-JNK antibody used in this experiment recognizes all JNK isoforms due to amino acid conservation around the phosphorylation sites, but only a single, relatively high-molecular-mass band at about 54–55 kDa was detected in the DCP-Bio1 pulldown experiments. Based on the apparent molecular weight, this band could contain one or more of the JNK2α2, JNK1α2, JNK1β2, or JNK2β2 isoforms, whereas the other JNK isoforms are of a lower molecular weight.
In subsequent experiments using a JNK2-specific antibody, FN-f and low doses of H2O2 were observed to increase the levels of JNK2 sulfenylation based on affinity capture after DCP-Bio1 labeling (Fig. 2A). In some experiments, a lower molecular weight JNK2 band was also observed that likely represents the smaller and more minor JNK2α1 or JNK2β1 isoform (5). It is important to note that chondrocytes isolated from different tissue donors exhibited variability in the time points and doses of H2O2 that yielded maximal DCP-Bio1 labeling of JNK2. This is to be expected given the transient nature of cysteine sulfenylation, the heterogeneity of the tissue from different donors, and the variable effects of culture conditions on the redox status of the cell. Oxidized JNK2 was also detected using a second (biotin switch) method in which biotinylated iodoacetamide (BIAM) was used to capture reversibly modified thiols. In this method, free thiols are first blocked, and then after incubation with a reductant, labeling with the biotinylated reagent occurs, revealing sites modified by disulfide bond formation (including glutathionylation), sulfenylation, and potentially other reversible forms (Fig. 2B). Unlike DCP-Bio1, the BIAM method also detected oxidized JNK2 when cells were treated with 10 and 50 μm H2O2, indicating the potential presence of intra- or intermolecular disulfides at the higher doses of H2O2.
Figure 2.
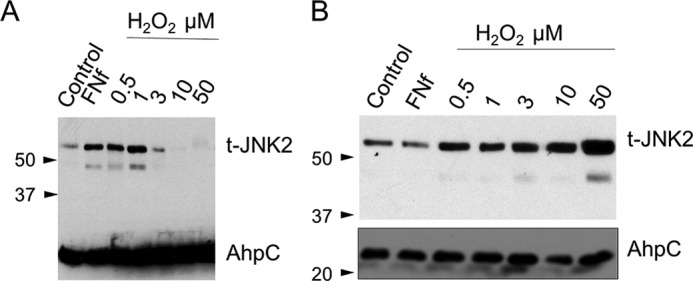
JNK2 is sulfenylated in response to FN-f and H2O2. A, human articular chondrocytes were treated for 60 min with either FN-f or increasing doses of H2O2. DCP-Bio1 (1 mm) was added in the last 3 min of treatment time before lysis. Cells were lysed, and DCP-Bio1–labeled proteins were captured using streptavidin affinity columns with an internal standard of prebiotinylated AhpC added for confirmation of experimental elution efficiency. Enriched samples were analyzed by immunoblotting using antibodies for total JNK2 (t-JNK2) and AhpC. A blot representative of three independent experiments is shown. B, BIAM labeling after thiol blocking and reduction of lysates from human articular chondrocytes treated with FN-f or a dose curve of H2O2 was performed as detailed under “Experimental procedures,” and lysates were analyzed as shown in A. A blot representative of three independent experiments is shown.
JNK2 activity is inhibited by oxidation
Oxidative thiol modifications, including protein sulfenylation, can either activate or inhibit protein kinases, whereas most protein phosphatases and particularly protein-tyrosine phosphatases, which rely on a nucleophilic cysteine for catalysis, are inhibited (31). To evaluate the effect of oxidation on JNK2 kinase activity, primary human chondrocytes were transfected with an expression plasmid for FLAG-tagged JNK2α2. Cells were subsequently stimulated with FN-f for 30 min to activate signaling and phosphorylate JNK. Cell lysates were prepared and treated with the reducing agent dithiothreitol (DTT) so that all proteins would be fully reduced, and then anti-FLAG magnetic beads were used to capture the recombinant JNK2 protein. To assess the effect of H2O2 on JNK2 activity, samples were exposed to various concentrations of H2O2 from 0.5 to 20 μm for 10 min, excess H2O2 was removed by the addition of catalase, and c-Jun and ATP were added to initiate the activity assay (Fig. 3A). Under these conditions, JNK kinase activity was inhibited by H2O2 treatment (Fig. 3, B and C). The amounts of H2O2 needed to inhibit JNK kinase activity using this cell-free method were lower than the amounts needed when H2O2 was added directly to live cells (Fig. 1C). This is partly because a significant proportion of exogenously added H2O2 in cells is undoubtedly removed by efficient cellular antioxidant systems (including peroxiredoxins and catalase).
Figure 3.

JNK2 activity is reversibly inhibited by H2O2. A, JNK2 kinase assay. FLAG-JNK2α2–expressing chondrocytes were treated with FN-f to stimulate JNK phosphorylation. Cellular proteins were reduced prior to affinity capture of JNK2 with anti-FLAG magnetic beads. A single immunoprecipitation (IP) was then split into multiple fractions and treated or not with various amounts of H2O2 for 10 min prior to the addition of catalase to remove excess H2O2. Protein kinase A activity was then initiated by the addition of prereduced c-Jun and ATP, and JNK2 activity was measured as a function of c-Jun phosphorylation. B, WT FLAG-JNK2 was immunoprecipitated from 60 μg of lysate and then split into multiple tubes prior to treatment with 0, 0.5, 1, 3, or 20 μm H2O2 for 10 min, and kinase activity was measured as described in A. C, shown is the mean and S.D. (error bars) for n = 14 replicates for protein expressed from chondrocytes harvested from different donors. The p value for one-way ANOVA comparing different H2O2 concentrations is <0.0001. Dunnett's multiple comparison test was used to identify H2O2 concentrations with significantly different JNK kinase activity from untreated protein; *, p value <0.05; ***, p value <0.005. D, JNK2 inhibition is reversible by reduction. Active, phosphorylated JNK2α2 was purchased from SignalChem and treated or not with H2O2 and then catalase as described above. Each sample was then split in half. DTT was added to one half (+), and an equal amount of water (−) was added to the second prior to the addition of c-Jun and ATP. p, phospho; t, total.
To test whether oxidative JNK2 inhibition is reversible, a separate assay was performed with commercially available, recombinant active JNK2α2. The addition of DTT recovered most or all of the activity of JNK2 after inhibition with H2O2 concentrations up to 100 μm (Fig. 3D). These results indicate reversible inhibition of JNK2 activity over a wide range of H2O2 concentrations and confirmed that this regulation occurred in the absence of other proteins.
JNK2 is sulfenylated at Cys-222 in vitro
To identify the cysteine residue(s) directly targeted by H2O2, we expressed and purified WT JNK2α2 with an N-terminal His6 tag as described under “Experimental procedures.” The resulting proteins were purified in the absence of reducing agent to >95% purity. His-tagged JNK2 was treated with successive additions of 100 μm H2O2 and excess DTT in the presence of dimedone. Like DCP-Bio1, dimedone reacts selectively with sulfenylated cysteine residues and forms irreversible thioether products. Multiple rounds of oxidation and reduction were performed to allow greater opportunity for dimedone to trap and accumulate presumably transient sulfenic acid species that would otherwise resolve into intramolecular disulfides and be unreactive with the dimedone. Free thiols and any unreacted sulfenylated residues were then trapped by iodoacetamide (IAM) (32). Although the Cys-423 peptide was not observed by MS, the other eight cysteine residues present in JNK2 were identified as iodoacetamide products or reduced thiol (Table S1 and Fig. S4). Cys-423 is located at the very C terminus and is part of a 51-residue tryptic peptide that may not be amenable to detection by traditional MS-based proteomics techniques. A dimedone product with Cys-222 was detected in the H2O2-treated sample but not the untreated control (Table S1, Experiment 1, and Fig. S5).
To further assess oxidation-sensitive cysteine residues in the absence of reductant, His-tagged JNK2 was first reduced with DTT and then (after excess DTT was removed) treated with 0 or 100 μm H2O2 in the presence of dimedone. After selectively blocking free thiols with methylsulfonyl benzothiazole (MSBT) (33, 34) and digestion with trypsin, the dimedone-labeled cysteine residues were identified by MS analysis. Again, a dimedone product was only observed at Cys-222 (Fig. 4A and Table S1, Experiment 2). No dimedone product was observed with any other cysteine residue in this experiment. With the exception of Cys-41, Cys-116, and Cys-423, all other cysteine residues were observed to form an MSBT product. Although no cysteine other than Cys-222 was trapped in sulfenylated form by dimedone, we did observe some hyperoxidation of Cys-6, Cys-116, Cys-163, Cys-213, and Cys-222 to sulfinic (SO2H) and/or sulfonic (SO3H) acid in one or both experiments, indicating that these cysteine residues also exhibit a degree of oxidation sensitivity (Table S1 and Fig. S6). It should be noted, however, that the degree of hyperoxidation of any cysteine(s) affecting activity in the intact protein must be minimal based on the results shown in Fig. 3D that demonstrate substantial reversibility of the inhibition.
Figure 4.

Identification of Cys-222 sulfenylation and intramolecular disulfide bonds in JNK2. A, purified, recombinant His-JNK2α2 (30 μm) was prereduced and treated with 100 μm H2O2 for 30 min in the presence of 5 mm dimedone and analyzed by nano-LC-MS/MS. The difference between the mass of b2+ fragmentation ion 299.1060 (dimedone-labeled peptide; top spectrum) and b2+ 294.0365 (MSBT-alkylated Cys-222; bottom spectrum) is consistent with Cys-222 labeling by dimedone. No other dimedone-labeled peptide was observed (Table S1). B, reduced His-JNK2α2 was treated with increasing concentrations of H2O2. The resulting protein was resolved on a nonreducing, denaturing gel. No JNK2 dimer was observed, indicating the absence of any intermolecular disulfide bond. C, the samples from B were analyzed for disulfide bonds using XlinkX 2.0. Disulfide bonds between Cys-222 and either Cys-177 or Cys-213 or with Cys-222 from another subunit were observed with significant scores. Spectra for the disulfide bonds can be found in Fig. S7.
Potential intramolecular disulfide bonds were identified in JNK2
We next explored the possibility of disulfide bond formation given the sulfenylated Cys-222 detected in JNK2. Cysteine sulfenic acids can be stabilized within the protein structure, directly reduced back to the thiol form, or condense with another thiol group to form a disulfide bond (17, 31). Intermolecular disulfide bonds can form with a cysteine in another protein or subunit, or intramolecular disulfides can form within the same subunit. Mixed disulfides may also form with small molecule thiols such as GSH. Any of these modifications could regulate protein function by “locking in” a particular structural conformation or by blocking or altering binding sites (17, 31). To look for the formation of an intermolecular disulfide bond in oxidized JNK2, recombinant, nonphosphorylated, His-tagged JNK2α2 was reduced with DTT, excess reductant was removed using a gel filtration column, and the protein was treated with increasing H2O2 concentrations and analyzed by nonreducing SDS-PAGE. No dimer band was observed at H2O2 concentrations up to 1 mm (Fig. 4B); however, there was a slight shift in the monomer band compared with DTT-treated protein, which may indicate localized, intramolecular disulfide bond formation. These results rule out the formation of intermolecular disulfide bonds between two JNK2 subunits under these conditions and suggest the formation of intramolecular disulfide bonds.
To identify potential disulfide bonds in JNK2, the dimedone- and MSBT-treated, His-tagged JNK2 samples used in the MS experiments described above were analyzed for the presence of intramolecular disulfides using XlinkX 2.0 (35, 36). Using this method, no disulfide bonds were observed in the untreated sample, but peptides containing disulfide bonds corresponding to Cys-222 partnered with either Cys-213 or Cys-177 were observed in the H2O2-treated samples (Fig. 4C, Table S1, Experiment 2, and Fig. S7).
Mutation of Cys-222, Cys-116, or Cys-163 renders JNK2 insensitive to H2O2-mediated inhibition
The combined results of the MS studies suggested that Cys-222 is the cysteine in JNK2 most likely to be directly targeted by H2O2, although there were indications that other cysteine residues (Cys-116, Cys-163, Cys-177, and Cys-213) may also be directly oxidized and/or serve as potential disulfide-bonding partners with Cys-222 (Cys-177 and Cys-213). To confirm the identity of the JNK2 cysteine(s) involved in redox regulation, individual C116A, C163A, C177A, C213A, C222A, and C222S mutants were generated in the FLAG-tagged JNK2α2 construct. The mutant proteins were expressed in human articular chondrocytes obtained from tissue donors, activated through phosphorylation by FN-f stimulation, collected under reducing conditions, immunoprecipitated, and assayed for c-Jun kinase activity. Under these conditions, c-Jun phosphorylation and detectable signal for JNK protein were observed only when the FLAG-tagged constructs were expressed, indicating that the measured activity in our assays arose almost exclusively from the overexpressed mutants and not from endogenously expressed JNK2 (Fig. 5A). For all mutants, the level of reduced JNK protein and kinase activity in the immunoprecipitated material was not significantly different from the WT JNK2–expressing cells derived from the same donor (Fig. 5B). Similar to WT JNK2, the C177A and C213A constructs were inhibited by the addition of H2O2 prior to measuring kinase activity, whereas the C116A, C163A, C222A, and C222S constructs were insensitive to H2O2-mediated inhibition (Fig. 5C).
Figure 5.

Sensitivity of JNK2 cysteine mutants to H2O2-mediated inhibition. A, FLAG-tagged WT JNK2α2, C222S JNK2, or a FLAG-GFP control construct were immunoprecipitated (IP) from FN-f–stimulated chondrocytes, and kinase activity was measured as a function of c-Jun phosphorylation (p) as described in Fig. 3A except that no H2O2 was added and assays included 2 mm DTT. Blots were stripped and reprobed with an antibody to detect the total (t) amount of JNK2 protein added to each assay. All constructs were transfected into the same donor-derived cell line, and all treatments and analysis were performed side by side on the same day. B, c-Jun kinase activity of reduced JNK2 mutants does not differ significantly from WT JNK2. Shown are mean and S.D. (error bars) for the kinase activity for each mutant compared with the activity of untreated WT protein from the same donor-derived cells. Mutant and WT activity assays were performed on the same day and analyzed on the same gel. All values are normalized to the signal for total JNK2 protein. Shown are the mean and S.D. One-way ANOVA indicated no significant difference between the samples (p value = 0.42). C, the immunoprecipitated JNK2 cysteine mutants were assayed for JNK2 kinase activity after treatment with increasing concentrations of H2O2 as described in Fig. 2A. Shown are the mean and S.D. (error bars) for a minimum of four biological replicates for each mutant. Two-way ANOVA followed by Dunnett's post hoc comparison was used to identify mutant constructs that were significantly less inhibited by H2O2 than WT JNK2 with ‡ indicating a p value <0.05. Asterisks represent significant differences: *, p < 0.05; **, p < 0.005 relative to untreated control for the same variant.
Oxidation of JNK2 occurs in regions of the protein important for activity and formation of the closed, active conformation
To better understand the mechanism by which oxidation regulates JNK2 activity, we mapped the location of all cysteine residues in JNK2α2 to the two solved crystal structures of JNK2 (37, 38). The region of the protein containing Cys-6 is not visible in either JNK2 structure, and the C-terminal cysteine (Cys-423 in JNK2) is not visible in any of the JNK1, JNK2, or JNK3 structures. The remaining cysteine residues of JNK2 are scattered throughout the protein with all but Cys-44 found on the surface of the protein, making them potentially accessible to both oxidation by H2O2 and formation of a disulfide with a nearby cysteine residue or small-molecule thiols in cells (Fig. 6A). In both structures, JNK2 is unphosphorylated and complexed with different inhibitors so that the activating phosphorylation sites (Thr-183/Tyr-185) are inaccessible for phosphorylation. The activation loop is found in a more open conformation in Protein Data Bank (PDB) code 3E7O and in a more closed conformation in PDB code 3NPC (Fig. 6B) (38). Cys-116 is located near the ATP-binding site and is the target of a series of JNK-specific inhibitors that covalently bind to JNK1, JNK2, and JNK3 isoforms and inhibit c-Jun phosphorylation in HeLa and A375 cells (39). Cys-222 is located 20 Å away from the ATP-binding site on the surface of the protein (Fig. 6A). This region of the protein has been implicated in modulating dimerization and autophosphorylation of JNK2α2 (40) as well as being responsible for the lower Km for c-Jun observed for JNK2 compared with JNK1 (41). Although there is no JNK structure to show the conformation of the activation loop in the phosphorylated, active protein, it is clear from the closed conformation of the activation loop in 3NPC that oxidation of either Cys-116 or Cys-222 could alter the conformation of the activation loop and thus modulate the activity of JNK2 (Fig. 6B). In contrast, Cys-163 and Cys-137 are located a substantial distance away (>20 Å) from the active site in the middle of the MKK-binding pocket (Fig. 6C). Oxidation of these residues would be expected to either block or change the specificity of substrate binding. Interestingly, we found that the C116A, C222A, and C163A mutations all eliminated the sensitivity of JNK2 to oxidation (Fig. 5C), suggesting that multiple mechanisms may be used to regulate the JNK activity simultaneously in response to an increase in H2O2.
Figure 6.

Location of redox-sensitive cysteine residues in JNK2. A, location of cysteine residues on the structure of JNK2 (PDB code 3NPC, chain A) (38). Cys-222 is found in a region of JNK2 that modulates its affinity for c-Jun substrate (41) (residues 208–230, in blue and cyan). Cys-222 is also near the α-region (residues 218–226, in cyan) shown to mediate dimerization and autophosphorylation (40). Cys-177 is found in the JNK2 activation loop (residues 170–190, in magenta) near the Thr-183 and Tyr-185 phosphorylation sites in red van der Waals surface. The MAP kinase insert (residues 247–278, shown in tan) is only observed in MAP kinases and CDK2 and has been shown to interact with the activation loop in unphosphorylated ERK2 (57). This insert is longer in JNK, and the additional residues (residues 279–290) are indicated in brown. The other cysteines in the structure are shown in yellow. An atrial natriuretic peptide (ANP) molecule (in light green with phosphate groups in orange) is modeled in the ATP-binding site near Cys-116 and the MKK7 peptide (dark green, modeled from PDB code 3UKH) is next to Cys-163. N-terminal residues 1–6, which include Cys-6, and C-terminal residues 364–424, including Cys-423, are not shown in the structure. B, oxidation of JNK2 occurs in regions involved in the conformational rearrangements of the activation loop. The surface representation of the open form of JNK2 containing the C222S and C177S mutations (PDB code 3E7O, chain A) (37) is shown on the right with the activation loop in pink; the closed conformation of the enzyme is shown on the left (PDB code 3NPC, chain A). The surface-accessible sulfur residues are shown in yellow. The putative locations of a bound atrial natriuretic peptide (light green) ball and a bound MKK peptide (dark green) are modeled in ball and stick representation. C, Cys-163 is found in the D-site substrate-binding pocket. The surface model of 3NPC with the MKK peptide from a JNK1 structure (3UKH) is overlaid onto the JNK2 structure and shown in magenta. The surface-accessible sulfurs of Cys-163 and Cys-137 are shown in yellow.
Despite our MS results, it is unclear from the crystal structure how disulfide bond formation in JNK2 would occur without some local rearrangement of the protein structure. Cys-213 and Cys-177 are 11 and >30 Å, respectively, from Cys-222 in the two existing JNK2 structures. Although Cys-213 is closer, Cys-177 is located within the flexible activation loop, and the locations of this residue in 3E7O and 3NPC differ by ∼25 Å. It is not known how posttranslational modifications would change the conformation of this flexible region of the protein because there is no phosphorylated structure for any JNK isoform. The temporal order of disulfide formation and the conformational change triggered by initial disulfide bond formation is also currently unknown. These events may bring the Cys-222 and Cys-177 in closer proximity to allow for formation of a disulfide bond between these residues.
Redox-sensitive cysteine residues show different degrees of conservation across JNK isoforms
Alignment of all JNK2 isoforms indicated that all cysteine residues were conserved between all four JNK2 isoforms with the exception of Cys-222 and Cys-423 (Fig. 7 and Fig. S2). Cys-222 is present in JNK2α1 and JNK2α2 but not JNK2β1 or JNK2β2. In contrast, Cys-423 is found in the JNK2α2 and JNK2β2 isoforms but not the JNK2α1 or JNK2β1 isoforms.
Figure 7.

Cysteine conservation across human MAP kinases. MAP kinase sequences (GenBankTM accession numbers NP_002743.3, NP_620707.1, NP_620708.1, NP_620709.1, NP_620634.1, NP_001310231.1, NP_001265476.1, NP_620637.1, NP_620448.1, NP_002744.1, NP_620407.1, NP_002737.2, NP_002960.2, NP_620581.1, NP_002745.1, and NP_002742.3) were aligned using Clustal Omega (58), and the relative location of cysteine residues in each protein are represented in boxes. Cysteine residues in white are conserved across all MAP kinases. Cysteines in purple are found only in p38 proteins, cysteines in blue are conserved only in ERK1 and ERK2, and cysteines in yellow are conserved across all JNK isoforms. Cysteines in magenta are only found in JNK3, cysteines in orange are found in JNK1 and JNK3, cysteines in brown are only in JNK1, and cysteines in red are only found in JNK2.
Alignment of all JNK1, JNK2, and JNK3 isoforms demonstrated even more variability between the redox-sensitive cysteines (Fig. 7 and Fig. S3). Cys-41, Cys-79, Cys-116, Cys-137, Cys-163, and Cys-213 are conserved in all JNK isoforms, whereas Cys-6 and Cys-177 are only observed in the four JNK2 isoforms. In addition, all JNK1 and JNK3 isoforms contain an additional Cys-245 residue (JNK1 numbering). The α2 isoform in JNK3 and the α2 and β2 isoforms in JNK1 contain two adjacent cysteine residues at the C terminus of the protein (Cys-425 and Cys-426 in JNK1) with Cys-426 in JNK1α2 being equivalent to C423 in JNK2α2. Interestingly, the redox-sensitive C222 residue is not found in any JNK1 or JNK3 isoform (nor in β isoforms of JNK2 as mentioned above), although the two α isoforms in JNK1 do contain a nearby Cys-220 residue. Taken together, this indicates that JNK2α1 and JNK2α2 are the only JNK isoforms that could be regulated by oxidation at Cys-222, although any changes in activity caused by oxidation at Cys-116 or Cys-163 would be expected to apply to all JNK proteins. The molecular weight of the band detected by the antibody against JNK2 in the biotin pulldown of oxidized proteins from FN-f– and H2O2-treated chondrocytes suggests that JNK2α2, the larger of the two α isoforms, is more likely to be the predominant oxidized species in our samples than JNK2α1.
Discussion
In these studies, we identify cysteine oxidation by H2O2 as a novel mechanism to regulate JNK2 activity. We show in human articular chondrocytes and with purified protein that JNK2 can be directly oxidized by H2O2 and that this oxidation inhibits the kinase activity of the protein. These experiments indicate that there is a dual layer of JNK2 regulation; the protein appears to be most active when it is both phosphorylated and reduced. Low levels of H2O2, such as those generated by FN-f and other physiologic stimuli, lead to JNK phosphorylation through the activation of kinases upstream of JNK as well as through inhibition of phosphatases (8–12). As shown here, treatment with menadione or high levels of exogenous H2O2 that result in oxidative stress (detected by Prx hyperoxidation) stunt JNK signaling through both inhibition of the upstream mediators responsible for JNK phosphorylation and direct oxidative inhibition of JNK activity. Oxidation is observed on both unphosphorylated and phosphorylated JNK2, and activity of phosphorylated JNK2 can be restored in the presence of DTT, indicating that the oxidation is reversible, even at high H2O2 concentrations. This suggests that the phosphorylation state of the protein can be maintained during oxidation. This “primed” phospho-JNK2 inhibited by oxidation can then be switched “on” as a kinase immediately upon reduction.
JNK2 is susceptible to oxidation by the addition of <5 μm extracellular H2O2, which can be estimated to correspond to an intracellular concentration between 5 and 500 nm (42). These values are physiologically relevant as H2O2 concentrations are estimated to be 10–100 nm under steady-state conditions and are expected to be even higher under oxidative stress or near the “hot spots” of H2O2 generation (42). Although we do not yet have a full understanding of how JNK2 and other signaling proteins are able to compete for reaction with H2O2 in the presence of abundant and efficient peroxidase proteins such as peroxiredoxins, our data, as well as those of many other investigators, do provide clear indications that such direct oxidation can and does occur during cell signaling events (20, 21, 43, 44).
Activity assays with JNK2α2 cysteine mutants and MS identification of the dimedone product both indicate that Cys-222 is directly sulfenylated by H2O2 to inhibit the kinase activity of the protein. C177A and C213A mutants are still sensitive to H2O2-mediated inhibition, indicating that these residues, although able to form a disulfide with Cys-222, are not directly oxidized by H2O2. It is possible that one or both of these residues may function as a resolving cysteine, but if so, formation of the disulfide bond does not appear to be required for the inhibition observed here.
Interestingly, mutation of either Cys-116 or Cys-163 also inhibits the oxidation sensitivity of JNK2α2 despite the fact that no dimedone product was observed for either of these residues. A highly selective JNK inhibitor, JNK-IN-8, forms a covalent bond with Cys-116, a modification that is required for the inhibition of JNK2 activity by this compound; nonetheless, mutation of Cys-116 to a serine did not affect JNK kinase activity (39) as we also noted here in the C116A mutant.
One hypothesis that may explain these findings is that these cysteines are involved in forming or stabilizing the structural rearrangements required for inhibition of the oxidized JNK2α2. Alternatively, JNK2α2 may be sensitive to multiple oxidation events that could work together to modulate the activity of this protein. The locations of these residues in the peptide-binding cleft (Cys-163), near the ATP-binding site (Cys-116), and packed against the activation loop in the closed form of the protein (Cys-222) support the second hypothesis. Because we identified several redox-active cysteines in JNK2, it is also possible that oxidation at each of these sites might occur at different H2O2 concentrations or in the presence of different binding partners, allowing for further fine-tuning of JNK2 activity in different cellular contexts.
Although this study focused on the JNK2α2 isoform, it is possible that other JNK2 isoforms would also be sensitive to redox regulation, particularly through Cys-116 and Cys-163. It is, however, interesting to note that Cys-222 is not present in the JNK2β1 or JNK2β2 isoforms or in JNK1 or JNK3, indicating that cysteine oxidation modulates JNK activity in an isoform-specific manner. In particular, regulation through Cys-222 will be specific to the JNK2α isoforms. This may have important implications for binding of JNK to its substrates c-Jun and ATF2. JNK2α1 and JNK2α2 have been shown to bind preferentially to c-Jun peptide compared with ATF2 peptide, whereas the opposite was seen with JNK2β1 and JNK2β2 (5). Also, a loop region containing Cys-222 (amino acids 218–226) in JNK2α2 was found to be responsible for JNK2α2 autophosphorylation (45), and a similar JNK2 Cys-222–containing region (amino acids 208–230) was noted to be responsible for the higher binding affinity of JNK2 than JNK1 for c-Jun (by 25-fold) (41).
Redox regulation has also been reported for other MAP kinases. Treatment of HeLa cells with prostaglandin J2 (associated with inflammation) or 1 mm H2O2 led to the reversible oxidation of p38α (19). This oxidation was associated with decreased phosphorylation of the p38α substrate ATF2 in a cell culture model, both in vivo and after pulldown, despite similar levels of phosphorylated p38α. Immunoprecipitation of p38 followed by a kinase activity assay confirmed that p38α kinase activity was inhibited by oxidation and that this inhibition was reversed by the addition of DTT. In another study, ERK2 was shown to be oxidized in response to a variety of growth factors using DCP-Bio1 to capture sulfenic acid–containing proteins (21). Oxidation of immunoprecipitated phospho-ERK2 by H2O2 inhibited the ability of ERK2 to phosphorylate ELK1 polypeptide in an in vitro assay, and this activity could also be recovered with the addition of DTT.
In these MAP kinases, oxidation appears to be regulated independently of phosphorylation, and kinase activity is highest when it is both phosphorylated and reduced. This suggests that the amount of active JNK, ERK, and p38 may be greatly overestimated by studies that simply look at the presence of phosphorylated protein. Even in the case where MAP kinase activity is directly measured, the effect of oxidation is typically masked by the ubiquitous presence of reductants in the assay. A search for commercially available products indicated that DTT or β-mercaptoethanol was present in all of the commercial JNK activity assay buffers, c-Jun peptide substrates, and recombinant, active JNK protein preparations identified.
Although redox regulation appears to be conserved across the MAP kinases, the location and number of cysteine residues varies among the different families of MAP kinases (Fig. 7 and Fig. S3). JNK1, JNK2, and JNK3 have more cysteine residues (10–11) than ERK1 and ERK2 (6 and 7, respectively) or p38α, -β, -γ, and -δ (3–4) (Fig. 7). Cys-222, which is the only residue we observed forming sulfenic acid in our studies, is only found in JNK2. We also observed that the C116A mutant of JNK2 is insensitive to inhibition by H2O2; this cysteine is only found in JNK proteins and not in ERK or p38. Of the three cysteines (Cys-46, Cys-163, and Cys-213; JNK2 numbering) found in all three MAP kinase subfamilies, only Cys-163 was shown to be associated with the oxidation sensitivity of p38α (Cys-162) and JNK2 (Fig. 5C and Ref. 19). In p38α, a second cysteine (Cys-119) located in close proximity to Cys-162 is proposed to form a disulfide bond; however, only p38α and p38β contain this residue, indicating that oxidation of Cys-163 in the peptide-binding pocket of different MAP kinases would exhibit some structural differences. Together, these data suggest that oxidation of the different MAP kinase proteins can result in distinctly different structural perturbations. Although it is still early in our understanding of the mechanism by which oxidation regulates the MAP kinases, it is intriguing to hypothesize that the difference in the number and location of cysteine residues may fine-tune the redox sensitivity of the different MAP kinases.
The cell culture experiments in the present study were performed in primary cultures of human articular chondrocytes with the anticipation that results are transferable to other cell types where JNK signaling plays an important physiologic or pathologic role. Chondrocytes were chosen because of previous studies suggesting that JNK is activated in cartilage from patients with osteoarthritis (46), and activation of MAP kinases, including JNK, promotes matrix metalloproteinase expression in response to matrix fragments such as FN-f (47, 48) that require ROS as secondary messengers (22, 28). In addition, studies with JNK2 knockout mice indicate a role for JNK2 in cartilage destruction in osteoarthritis (49) and collagen-induced arthritis (50), a model of rheumatoid arthritis in humans. The present findings demonstrate that as levels of H2O2 increase in the cell, JNK2 switches from an active state to a less active or inactive state due to both inhibition of factors upstream of JNK2 that mediate JNK2 phosphorylation and direct JNK2 thiol oxidation, suggesting a mechanism of inhibition of JNK under oxidative stress conditions. This indicates that JNK may play different roles in regulating cell functions depending on the redox environment of the cell.
Experimental procedures
Antibodies and reagents
Antibodies purchased from Cell Signaling Technology were to phospho-JNK (Thr-183/Tyr-185), total JNK2, phospho-c-Jun (Ser-73), phospho-ERK (Thr-202/Tyr-204), total ERK, phospho-p38 (Thr-180/Tyr-182), total p38, and β-actin. Antibodies to PrxSO2/3H were from Abcam, and anti-AhpC antibody was purified from rabbit serum (29). Salmonella enterica serovar Typhimurium AhpC C165S protein was expressed and purified as described previously (51), and reduced C165S AhpC was labeled with biotin-maleimide as described previously (29). The PremoTM roGFP-based cellular H2O2 sensor (Orp1-roGFP) was purchased from Molecular Probes, Life Technologies. DCP-Bio1 and MSBT were synthesized as described previously (34, 52). Menadione, dimedone, N-ethylmaleimide (NEM), catalase, and IAM were purchased from Sigma-Aldrich. Endotoxin-free recombinant FN-f was expressed and purified as described previously (28). DTT was purchased from Life Technologies/Thermo Fisher Scientific.
Chondrocyte cell culture and stimulation of JNK signaling
Normal adult human articular cartilage from the talus of tissue donors was obtained within 72 h of death through the Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL) via Rush Medical College (Chicago, IL) or from the National Disease Research Interchange (Philadelphia, PA). Chondrocytes were isolated by enzymatic digestion and cultured as described previously (53). At confluence, cultures were changed to serum-free medium and cultured overnight before use in experiments. Cells were treated for 30 min with either FN-f (1 μm), menadione (25 μm), or a dose curve of H2O2 concentrations from 0.5 to 50.0 μm. In some experiments, cells were pretreated with menadione for 30 min before the FN-f treatment. After treatment, cell lysates were prepared with standard cell signaling lysis buffer (1×) (Cell Signaling Technology) containing phenylmethanesulfonyl fluoride (PMSF; Sigma-Aldrich), Phosphatase Inhibitor Cocktail 2 (PIC; Sigma-Aldrich), and either NEM or IAM. Cell lysates were analyzed by immunoblotting with antibodies to the phosphorylated forms of proteins or antibodies to the hyperoxidized forms of the peroxiredoxins as described (30). Blots were then stripped and reprobed with antibodies to the total protein for a loading control for blots with anti-phosphoantibodies or to β-actin.
Quantification of H2O2 production
Intracellular H2O2 levels were quantified using the Premo cellular H2O2 sensor Orp1-roGFP (Life Technologies) as described previously (30).
DCP-Bio1 and BIAM labeling and affinity capture
Proteins were affinity-tagged with DCP-Bio1 to specifically label sulfenylated proteins using a modification of a previously published procedure (28). Rather than adding the DCP-Bio1 to cells at the time of cell lysis, 1 mm DCP-Bio1 was added to the medium for the last 3 min of cell treatment prior to lysis to reduce the chance of capturing proteins oxidized only during cell lysis. Cell lysates were then prepared in cell signaling lysis buffer (1×) containing catalase (200 units/ml), DTT (8 mm), IAM (20 mm), PMSF, and PIC. The samples were rotated for 30 min at 4 °C and centrifuged (10 min at 13,200 rpm) to remove insoluble proteins. BCA analysis was performed on the supernatants to measure the amount of total protein in the sample.
For BIAM labeling, the lysis buffer was 1× cell signaling buffer containing PMSF, PIC, and 10 mm NEM to alkylate free thiols. The samples were rotated for 30 min at 4 °C, and then the samples were desalted using Zeba columns (Thermo Scientific) to remove excess NEM. The desalted samples were transferred to Eppendorf tubes and reduced with DTT (5 mm) for an hour at 4 °C. The samples were then incubated with BIAM (0.1 mm) overnight while rotating at 4 °C to label nascent thiols generated from DTT-reversible thiol oxidative modifications such as sulfenic acids, disulfides, and glutathionylated and nitrosylated thiols (32). After BIAM labeling, the samples were centrifuged for 10 min at a maximum speed of 13,200 rpm at 4 °C to remove insoluble proteins, and BCA analysis was performed on the supernatant.
High-capacity streptavidin-agarose beads (Thermo Fisher) were added to Pierce spin columns (120 μl each) and centrifuged at 400 × g for 30 s. The beads were washed twice with 600 μl of Dulbecco's phosphate-buffered saline (PBS) and twice with 0.1% (w/v) SDS (600 μl) with a centrifugation step (400 × g for 30 s) after each wash. Lysate protein from either DCP-Bio1– or BIAM-labeled samples (150–200 μg) was then loaded in the Pierce spin columns containing the streptavidin beads. Twenty microliters (0.5 ng) of prebiotinylated AhpC (in PBS with 2% (w/v) SDS) was added as an internal standard to control for the efficiency of the affinity capture, elution, and gel loading steps. The samples were rotated overnight at 4 °C. The next day, the columns were centrifuged at 400 × g for 30 s. The eluent was considered the unbound fraction. Columns were then put through a series of washes, each lasting 5 min. The DCP-Bio1 sample columns were washed three times with Tris-buffered saline containing 1% (v/v) Tween 20, once with 2 m urea, once with 1 m NaCl, once with 0.1% (w/v) SDS plus 10 mm DTT, and twice with PBS. BIAM sample columns were washed three times with PBS. Bound proteins were eluted by boiling the beads for 10 min in 100 μl of 2× Laemmli sample buffer containing 5% (v/v) β-mercaptoethanol. The samples were then cooled on ice for 1 min and centrifuged at 1000 × g for 4 min. The supernatants were then subjected to SDS-PAGE and transferred to nitrocellulose membranes for immunoblotting with antibodies to phospho-JNK or total JNK2.
Generation of JNK2 mutants
Cysteine to alanine or serine change was generated in a WT JNK2α2 cDNA construct (pcDNA3-FLAG-JNK2 plasmid from Dr. Roger Davis (University of Massachusetts) using the QuikChange XL site-directed mutagenesis kit (Stratagene). Primers were designed according to the protocol provided. A QIAprep Spin Miniprep kit (Qiagen) was used to isolate and purify the mutant DNA constructs.
Chondrocyte transfection
The NucleofectionTM method (Lonza) was used as described previously (54) to transfect 2,000,000 primary human chondrocytes with 5 μg of WT or mutant FLAG-JNK2 plasmid. After recovery for 48 h, cells were switched to serum-free medium overnight and stimulated with 1 μm Fn-f for 30 min. Cells were washed twice with PBS; lysed for 30 min at 4 °C in standard lysis buffer with PMSF, PIC, 200 units/ml catalase, and 5 mm DTT; and stored at −80 °C.
Immunoprecipitation and JNK kinase assay
WT JNK2 and the JNK2 mutant constructs were transfected into primary human chondrocytes by nucleofection and then stimulated with recombinant FN-f as noted above for 30 min to activate JNK. Cell lysates were prepared in 1× radioimmune precipitation assay buffer and treated with DTT so that all constructs would be fully reduced. Protein content of human and mouse lysates was quantified using the Pierce Micro BCA kit (Thermo Scientific), and then DTT was removed by passing JNK lysate (60 μg) through a Bio-Gel P-6 spin column (Bio-Rad) equilibrated in PBS supplemented with protease and phosphatase inhibitors. Anti-FLAG magnetic beads were incubated with the resulting lysate overnight at 4 °C to capture the JNK2 constructs. Each immunoprecipitated sample was washed twice with Tris-buffered saline and then twice with kinase buffer (25 mm Tris, pH 7.5, 10 mm MgCl2, 0.1 mm sodium vanadate, and 5 mm β-glycerophosphate) and split into five Eppendorf tubes. Next, immunoprecipitated JNK2 was treated with increasing concentrations of H2O2 from 0.5 to 20 μm for 10 min at 24 °C, and excess H2O2 was removed by the addition of 0.5 unit of catalase for 5 min. JNK activity was then measured in the absence of reductant by the addition of c-Jun substrate and 0.2 mm ATP. The reaction mixture was incubated for 30 min at 30 °C and quenched by the addition of SDS loading buffer. Phosphorylation of c-Jun (Ser-73) was quantified by Western blotting using an antibody against phospho-c-Jun. Blots were then stripped and reprobed with antibodies to the total JNK2.
c-Jun fusion protein was purchased from Cell Signaling Technology and provided in 2 mm DTT. The c-Jun fusion construct (residues 1–89) did not include the Cys-269 residue shown to be regulated by glutathionylation in the intact protein (55). Prior to performing the assays, c-Jun was passed through a Bio-Gel P-6 spin column equilibrated in kinase buffer to remove reductant, aliquoted, and stored at −80 °C until use.
To test whether JNK2 inhibition is reversible, a separate assay was performed with commercially available, recombinant, active JNK2α2. Ten nanograms of active, phosphorylated JNK2 (SignalChem) was mixed with 0, 2, 10, 20, or 100 μm H2O2 in a final volume of 24 μl. The purchased phospho-JNK2 contained both DTT and GSH, with final concentrations (after mixing) of 1 and 50 μm, respectively. Following incubation for 10 min at 24 °C, each sample was incubated with 1 unit of catalase for 5 min prior to initiating the kinase reaction by the addition of ATP and c-Jun. Immediately after initiation, each sample was split with 12 μl of each sample added to a tube containing 2 μl of either water or 10 mm DTT. The reaction was then allowed to proceed for 30 min at 30 °C and was analyzed as described above.
Expression and purification of recombinant His-JNK2
β-Mercaptoethanol and DTT present in commercial preparations of JNK2 can become oxidized and react with protein cysteine residues. Although theoretically these modifications could be reversed, the low amounts of protein present in the commercial preparations made it technically difficult to recover JNK2 after reduction and buffer exchange using a desalting column. To circumvent this problem, we expressed and purified JNK2 using a bacterial expression system. A bacterial expression construct, pGSTag-FLAG-JNK2α2, was provided by Roger Davis (Addgene plasmid 15744) (5). An alternative bacterial expression construct was also created that replaced the GST tag with a His tag to prevent the formation of nonphysiological disulfide bonds between the GST protein and JNK2. The pTHCm1Δp-JNK2 plasmid was created by removing the JNK2 coding sequence from pGSTag-FLAG-JNK2α2 by digestion with HindIII and BamHI and ligating the resulting insert into the pTHCm1Δp vector, derived from pTrcHisA from Invitrogen in which the β-lactamase gene was replaced by a chloramphenicol resistance gene (56).
His-JNK2 was expressed at 37 °C in 4.8 liters of B834 Escherichia coli cells. Cells were washed, resuspended in His purification buffer (50 mm sodium phosphate, pH 8.0, and 300 mm NaCl), and lysed by passage through an Avestin EmulsiFlex-C5. Cell debris was removed by centrifugation at 40,000 × g for 40 min at 4 °C, and nucleic acids were removed from the cleared lysate by the addition of 2% (w/v) streptomycin sulfate and centrifugation at 18,000 × g for 20 min at 4 °C. Imidazole was added to the cleared lysate to a final concentration of 10 mm and stirred with 10–20 ml nickel-nitrilotriacetic acid resin (Thermo Scientific) for 15 min at 4 °C. The slurry was then loaded onto a gravity flow column and washed with 600 ml of 10 mm imidazole in His purification buffer, and His-JNK2 was eluted with His purification buffer supplemented with 0.5 m imidazole. Purified His-JNK2 was dialyzed against 25 mm Tris, pH 7.5, 100 mm NaCl, and 10% (v/v) glycerol; concentrated to 9.9 mg/ml using an Amicon ultracentrifugal filter; and stored at −80 °C.
Mass spectrometry analysis of oxidized cysteines in JNK2 and multiple rounds of oxidation and reduction with iodoacetamide
His-JNK2 (192 μm in 50 mm Tris, pH 7.5) was incubated with 5 mm dimedone and 5 mm DTT and then cycled by the addition of five subsequent aliquots of 95–100 μm H2O2 for a final concentration of ∼500 μm H2O2. Samples were incubated at ambient temperature for a minimum of 15 min between each H2O2 addition. Accessible free thiols were then trapped by the addition of an equal volume of 250 mm IAM in water (in the dark). After incubation for a further 20 min, excess small molecules were removed using a Bio-Gel P-6 spin column equilibrated in 20 mm ammonium acetate. JNK protein was digested with 1.25 ng of tosylphenylalanyl chloromethyl ketone–treated trypsin (Worthington Biochemical) in 20 mm ammonium acetate, 10% (v/v) acetonitrile, and 1 mm CaCl2 overnight at 37 °C. The tryptic peptides were dried using a SpeedVac and resuspended in Optima water (Thermo Fisher) containing 0.1% (v/v) formic acid.
Peptides were analyzed by LC-MS/MS using an Accela Open UPLC coupled to a Thermo Scientific Orbitrap LTQ XL high-resolution mass spectrometer. Separations were achieved using a Thermo Hypersil GOLD C18 column (50 × 2.1 mm, 1.9 μm) at ambient temperature with a gradient of buffer A (0.1% (v/v) formic acid in water) and buffer B (100% (v/v) acetonitrile and 0.1% (v/v) formic acid) at a flow rate of 100 μl/min. Peptides were separated using a gradient elution of 5–100% (v/v) B over 15 min, holding at 100% B for 2 min, followed by a 2-min wash at 100% B. The gradient was then decreased to 5% B over 5 min and held at 5% B for 3 min for a total run time of 25 min. Eluant was introduced to the mass spectrometer via positive electrospray ionization with the following settings: sheath gas of 65, source voltage of 4.0 kV, capillary temperature of 325 °C, capillary voltage of 38.5 V, and tube lens held at 68.0 V. The mass spectrometer was operated in data-dependent acquisition mode using Xcalibur v1.3 (Thermo Scientific). After a full scan (20–2000 m/z range) at high resolution (30,000), the top five most intense precursor ions were isolated and fragmented using collision-induced dissociation with the normalized collision energy set at 35%, activation Q at 0.25, and activation time at 30 ms. Dynamic exclusion was enabled with a repeat duration and exclusion duration of 30 s.
MS spectra were searched using the SEQUEST search algorithm with Proteome Discoverer v1.3 (Thermo Scientific). Search parameters were as follows: FT-trap instrument, parent mass error tolerance of 10 ppm, and fragment mass error tolerance of 0.8 Da (monoisotopic) with variable modifications that included methionine oxidation, cysteine iodoacetamide products, cysteine dimedone products, cysteine dioxidation, cysteine trioxidation, and cysteine conversion to dehydroalanine.
Mass spectrometry analysis of oxidized cysteines in JNK2 and single oxidation experiment with MSBT as thiol blocker
His-JNK2 was first reduced with 10 mm DTT, and excess reducing agent was removed using a Bio-Gel P-6 spin column equilibrated in 20 mm ammonium acetate, pH 7, containing 100 μm DTT to maintain the protein in the reduced state. Reduced JNK2 was diluted to 30 μm, incubated with 5 mm dimedone, and subsequently treated with 0 or 100 μm H2O2. After incubation for 35 min at 24 °C, accessible free thiols were trapped by the addition of 5 mm MSBT and incubation for a further 30 min. To allow for alkylation of buried cysteine residues, samples were denatured by treatment with a final concentration of 6 m guanidinium chloride and heated for 5 min at 95 °C. Samples were diluted to 1 m guanidinium chloride, and protein was precipitated by the addition of acetone. The protein pellet was resuspended in 0.1% (v/v) SDS and digested with Thermo sequencing grade modified trypsin (1:100) overnight at 37 °C. The tryptic peptides were dried using a SpeedVac and resuspended in 5% (v/v) acetonitrile and 0.1% (v/v) formic acid.
Peptides were analyzed by LC-MS/MS using a Q Exactive HF hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific) coupled to a Dionex Ultimate-3000 nano-UPLC system (Thermo Scientific) using a Nanospray Flex Ion Source (Thermo Scientific). Peptides were separated using an Acclaim PepMap 100 (C18, 5 μm, 100 Å, 100 μm × 2 cm) trap column and an Acclaim PepMap RSLC (C18, 2 μm, 100 Å, 75 μm × 15 cm) analytical column. Good chromatographic separation was observed with a 90-min linear gradient consisting of mobile phases A (5% (v/v) acetonitrile with 0.1% (v/v) formic acid) and B (80% acetonitrile with 0.1% formic acid). MS data were acquired in data-dependent acquisition mode. The resolution of full MS scans and MS/MS scans were 60,000 and 15,000, respectively. Full MS scans were acquired over the 300–1500 m/z range. The 20 most abundant peaks were selected for MS/MS with normalized collision energy of 28% with a dynamic exclusion duration of 10.0 s. MS spectra were searched using the SEQUEST search algorithm with Proteome Discoverer v2.1 (Thermo Scientific). Search parameters were as follows: FT-trap instrument, parent mass error tolerance of 10 ppm, and fragment mass error tolerance of 0.02 Da (monoisotopic) with variable modifications that included methionine oxidation, cysteine MSBT products, cysteine dimedone products, cysteine dioxidation, and cysteine trioxidation. MS spectra were also searched for the presence of disulfide bonds using XlinkX 2.0 software.
Statistical analysis
Data were analyzed by Student's t test (for two-sample comparisons) or a one-way or two-way ANOVA (for comparison of more than two samples) with a Dunnett's or Tukey honestly significant difference post hoc correction, as appropriate, using GraphPad Prism version 6 (GraphPad Software, Inc.). Results are presented as mean values ±S.D. from a minimum of three independent biological replicates. The exact number of independent samples for each experiment is provided in the figure legends. A level of p < 0.05 was considered to be significant.
Study approval
Use of human tissue was in accordance with the Institutional Review Board at the Rush University Medical Center and the University of North Carolina at Chapel Hill.
Author contributions
K. J. N., E. A. B., L. B. P., C. M. F., and R. F. L. conceptualization; K. J. N., J. A. B., H. W., J. A. C., E. A. B., J. A. R., C. K., S. T. W., R. R. Y., L. B. P., C. M. F., and R. F. L. data curation; K. J. N., J. A. B., H. W., J. A. C., E. A. B., J. A. R., C. K., S. T. W., R. R. Y., L. B. P., C. M. F., and R. F. L. formal analysis; K. J. N. and R. F. L. writing-original draft; K. J. N., J. A. B., H. W., J. A. C., E. A. B., J. A. R., C. K., S. T. W., R. R. Y., L. B. P., C. M. F., and R. F. L. writing-review and editing; J. A. B., J. A. R., L. B. P., C. M. F., and R. F. L. methodology; R. F. L. supervision; R. F. L. funding acquisition; R. F. L. investigation; R. F. L. project administration.
Supplementary Material
Acknowledgments
We thank S. Bruce King for supplying the DCP-Bio1, Harold Erickson for providing the plasmid construct to produce the FN7–10 fibronectin fragment, and Roger Davis for contributing the pcDNA3-FLAG-JNK2 plasmid. We also thank Dr. Susan Chubinskaya, the Gift of Hope Organ and Tissue Donor Network, and the National Disease Research Interchange for providing donor tissue as well as Robert Currin and the University of North Carolina Hooker Imaging core for assistance with confocal microscopy. We acknowledge the support of the Wake Forest Baptist Comprehensive Cancer Center Proteomics and Metabolomics Shared Resource, supported by the NCI, National Institutes of Health, Cancer Center Support Grant P30 CA012197, and the Mass Spectrometry Facility in the Chemistry Department at Wake Forest University, supported in part by the National Science Foundation.
This work was supported by National Institutes of Health Grants R37 AR049003 and RO1 AG044034 (to R. F. L.), F31 AG032796 (to E. A. E.), R33 CA126659 (to L. B. P.), and R33 CA177461 (to C. M. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7 and Table S1.
- MAPK
- mitogen-activated protein kinase
- ROS
- reactive oxygen species
- JNK
- c-Jun N-terminal kinase
- H2O2
- hydrogen peroxide
- Cys-SOH
- cysteine sulfenic acid
- Cys-S−
- cysteine thiolate
- FN-f
- fibronectin fragment
- DCP-Bio1
- biotin-linked analog of dimedone
- ERK
- extracellular signal-regulated kinase
- MKP1
- MAPK phosphatase 1
- Prx
- peroxiredoxin
- BIAM
- biotinylated iodoacetamide
- MSBT
- methylsulfonyl benzothiazole
- IAM
- iodoacetamide
- NEM
- N-ethylmaleimide
- PIC
- Phosphatase Inhibitor Cocktail 2
- PMSF
- phenylmethanesulfonyl fluoride
- SO2H
- cysteine sulfinic acid
- SO3H
- cysteine sulfonic acid
- MKK
- MAPK kinase
- MAP
- mitogen-activated kinase
- roGFP
- reduction-oxidation–sensitive green fluorescent protein
- ANOVA
- analysis of variance.
References
- 1. Weston C. R., and Davis R. J. (2007) The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149 10.1016/j.ceb.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 2. Thalhamer T., McGrath M. A., and Harnett M. M. (2008) MAPKs and their relevance to arthritis and inflammation. Rheumatology 47, 409–414 10.1093/rheumatology/kem297 [DOI] [PubMed] [Google Scholar]
- 3. Holmström K. M., and Finkel T. (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421 10.1038/nrm3801 [DOI] [PubMed] [Google Scholar]
- 4. Labunskyy V. M., and Gladyshev V. N. (2013) Role of reactive oxygen species-mediated signaling in aging. Antioxid. Redox Signal. 19, 1362–1372 10.1089/ars.2012.4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gupta S., Barrett T., Whitmarsh A. J., Cavanagh J., Sluss H. K., Dérijard B., and Davis R. J. (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15, 2760–2770 10.1002/j.1460-2075.1996.tb00636.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weston C. R., and Davis R. J. (2002) The JNK signal transduction pathway. Curr. Opin. Genet. Dev. 12, 14–21 10.1016/S0959-437X(01)00258-1 [DOI] [PubMed] [Google Scholar]
- 7. Finkel T. (2011) Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15 10.1083/jcb.201102095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoshizumi M., Abe J., Haendeler J., Huang Q., and Berk B. C. (2000) Src and Cas mediate JNK activation but not ERK1/2 and p38 kinases by reactive oxygen species. J. Biol. Chem. 275, 11706–11712 10.1074/jbc.275.16.11706 [DOI] [PubMed] [Google Scholar]
- 9. Giannoni E., and Chiarugi P. (2014) Redox circuitries driving Src regulation. Antioxid. Redox Signal. 20, 2011–2025 10.1089/ars.2013.5525 [DOI] [PubMed] [Google Scholar]
- 10. Nadeau P. J., Charette S. J., Toledano M. B., and Landry J. (2007) Disulfide bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H2O2-induced c-Jun NH2-terminal kinase activation and apoptosis. Mol. Biol. Cell 18, 3903–3913 10.1091/mbc.e07-05-0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kamata H., Honda S., Maeda S., Chang L., Hirata H., and Karin M. (2005) Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120, 649–661 10.1016/j.cell.2004.12.041 [DOI] [PubMed] [Google Scholar]
- 12. Liu R. M., Choi J., Wu J. H., Gaston Pravia K. A., Lewis K. M., Brand J. D., Mochel N. S., Krzywanski D. M., Lambeth J. D., Hagood J. S., Forman H. J., Thannickal V. J., and Postlethwait E. M. (2010) Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor β1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J. Biol. Chem. 285, 16239–16247 10.1074/jbc.M110.111732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim Y. J., Lee W. S., Ip C., Chae H. Z., Park E. M., and Park Y. M. (2006) Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 66, 7136–7142 10.1158/0008-5472.CAN-05-4446 [DOI] [PubMed] [Google Scholar]
- 14. Wang M. C., Bohmann D., and Jasper H. (2003) JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev. Cell 5, 811–816 10.1016/S1534-5807(03)00323-X [DOI] [PubMed] [Google Scholar]
- 15. Chambers J. W., and LoGrasso P. V. (2011) Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 286, 16052–16062 10.1074/jbc.M111.223602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paulsen C. E., and Carroll K. S. (2010) Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 5, 47–62 10.1021/cb900258z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Devarie-Baez N. O., Silva Lopez E. I., and Furdui C. M. (2016) Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic. Res. 50, 172–194 10.3109/10715762.2015.1090571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nadeau P. J., Charette S. J., and Landry J. (2009) REDOX reaction at ASK1-Cys250 is essential for activation of JNK and induction of apoptosis. Mol. Biol. Cell 20, 3628–3637 10.1091/mbc.e09-03-0211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Templeton D. J., Aye M. S., Rady J., Xu F., and Cross J. V. (2010) Purification of reversibly oxidized proteins (PROP) reveals a redox switch controlling p38 MAP kinase activity. PLoS One 5, e15012 10.1371/journal.pone.0015012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wani R., Qian J., Yin L., Bechtold E., King S. B., Poole L. B., Paek E., Tsang A. W., and Furdui C. M. (2011) Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 108, 10550–10555 10.1073/pnas.1011665108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keyes J. D., Parsonage D., Yammani R. D., Rogers L. C., Kesty C., Furdui C. M., Nelson K. J., and Poole L. B. (2017) Endogenous, regulatory cysteine sulfenylation of ERK kinases in response to proliferative signals. Free Radic. Biol. Med. 112, 534–543 10.1016/j.freeradbiomed.2017.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Del Carlo M., Schwartz D., Erickson E. A., and Loeser R. F. (2007) Endogenous production of reactive oxygen species is required for stimulation of human articular chondrocyte matrix metalloproteinase production by fibronectin fragments. Free Radic. Biol. Med. 42, 1350–1358 10.1016/j.freeradbiomed.2007.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Homandberg G. A., Wen C., and Hui F. (1998) Cartilage damaging activities of fibronectin fragments derived from cartilage and synovial fluid. Osteoarthritis Cartilage 6, 231–244 10.1053/joca.1998.0116 [DOI] [PubMed] [Google Scholar]
- 24. Zack M. D., Arner E. C., Anglin C. P., Alston J. T., Malfait A. M., and Tortorella M. D. (2006) Identification of fibronectin neoepitopes present in human osteoarthritic cartilage. Arthritis Rheum. 54, 2912–2922 10.1002/art.22045 [DOI] [PubMed] [Google Scholar]
- 25. Homandberg G. A., Costa V., and Wen C. (2002) Fibronectin fragments active in chondrocytic chondrolysis can be chemically cross-linked to the alpha5 integrin receptor subunit. Osteoarthritis Cartilage 10, 938–949 10.1053/joca.2002.0854 [DOI] [PubMed] [Google Scholar]
- 26. Kheradmand F., Werner E., Tremble P., Symons M., and Werb Z. (1998) Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science 280, 898–902 10.1126/science.280.5365.898 [DOI] [PubMed] [Google Scholar]
- 27. Huhtala P., Humphries M. J., McCarthy J. B., Tremble P. M., Werb Z., and Damsky C. H. (1995) Cooperative signaling by α5β1 and α4β1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J. Cell Biol. 129, 867–879 10.1083/jcb.129.3.867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wood S. T., Long D. L., Reisz J. A., Yammani R. R., Burke E. A., Klomsiri C., Poole L. B., Furdui C. M., and Loeser R. F. (2016) Cysteine-mediated redox regulation of cell signaling in chondrocytes stimulated with fibronectin fragments. Arthritis Rheumatol. 68, 117–126 10.1002/art.39326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nelson K. J., Klomsiri C., Codreanu S. G., Soito L., Liebler D. C., Rogers L. C., Daniel L. W., and Poole L. B. (2010) Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 473, 95–115 10.1016/S0076-6879(10)73004-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Collins J. A., Wood S. T., Nelson K. J., Rowe M. A., Carlson C. S., Chubinskaya S., Poole L. B., Furdui C. M., and Loeser R. F. (2016) Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signalling in aging chondrocytes. J. Biol. Chem. 291, 6641–6654 10.1074/jbc.M115.693523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klomsiri C., Karplus P. A., and Poole L. B. (2011) Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 14, 1065–1077 10.1089/ars.2010.3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reisz J. A., Bechtold E., King S. B., Poole L. B., and Furdui C. M. (2013) Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 280, 6150–6161 10.1111/febs.12535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang D., Devarie-Baez N. O., Li Q., Lancaster J. R. Jr, and Xian M. (2012) Methylsulfonyl benzothiazole (MSBT): a selective protein thiol blocking reagent. Org. Lett. 14, 3396–3399 10.1021/ol301370s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen X., Wu H., Park C. M., Poole T. H., Keceli G., Devarie-Baez N. O., Tsang A. W., Lowther W. T., Poole L. B., King S. B., Xian M., and Furdui C. M. (2017) Discovery of heteroaromatic sulfones as a new class of biologically compatible thiol-selective reagents. ACS Chem. Biol. 12, 2201–2208 10.1021/acschembio.7b00444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu F., van Breukelen B., and Heck A. J. (2014) Facilitating protein disulfide mapping by a combination of pepsin digestion, electron transfer higher energy dissociation (EThcD), and a dedicated search algorithm SlinkS. Mol. Cell. Proteomics 13, 2776–2786 10.1074/mcp.O114.039057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu F., Rijkers D. T., Post H., and Heck A. J. (2015) Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 12, 1179–1184 10.1038/nmeth.3603 [DOI] [PubMed] [Google Scholar]
- 37. Shaw D., Wang S. M., Villaseñor A. G., Tsing S., Walter D., Browner M. F., Barnett J., and Kuglstatter A. (2008) The crystal structure of JNK2 reveals conformational flexibility in the MAP kinase insert and indicates its involvement in the regulation of catalytic activity. J. Mol. Biol. 383, 885–893 10.1016/j.jmb.2008.08.086 [DOI] [PubMed] [Google Scholar]
- 38. Kuglstatter A., Ghate M., Tsing S., Villaseñor A. G., Shaw D., Barnett J. W., and Browner M. F. (2010) X-ray crystal structure of JNK2 complexed with the p38α inhibitor BIRB796: insights into the rational design of DFG-out binding MAP kinase inhibitors. Bioorg. Med. Chem. Lett. 20, 5217–5220 10.1016/j.bmcl.2010.06.157 [DOI] [PubMed] [Google Scholar]
- 39. Zhang T., Inesta-Vaquera F., Niepel M., Zhang J., Ficarro S. B., Machleidt T., Xie T., Marto J. A., Kim N., Sim T., Laughlin J. D., Park H., LoGrasso P. V., Patricelli M., Nomanbhoy T. K., et al. (2012) Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 19, 140–154 10.1016/j.chembiol.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nitta R. T., Chu A. H., and Wong A. J. (2008) Constitutive activity of JNK2α2 is dependent on a unique mechanism of MAPK activation. J. Biol. Chem. 283, 34935–34945 10.1074/jbc.M804970200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kallunki T., Su B., Tsigelny I., Sluss H. K., Dérijard B., Moore G., Davis R., and Karin M. (1994) JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 8, 2996–3007 10.1101/gad.8.24.2996 [DOI] [PubMed] [Google Scholar]
- 42. Antunes F., and Brito P. M. (2017) Quantitative biology of hydrogen peroxide signaling. Redox Biol. 13, 1–7 10.1016/j.redox.2017.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haque A., Andersen J. N., Salmeen A., Barford D., and Tonks N. K. (2011) Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell 147, 185–198 10.1016/j.cell.2011.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heppner D. E., Hristova M., Dustin C. M., Danyal K., Habibovic A., and van der Vliet A. (2016) The NADPH oxidases DUOX1 and NOX2 play distinct roles in redox regulation of epidermal growth factor receptor signaling. J. Biol. Chem. 291, 23282–23293 10.1074/jbc.M116.749028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cui J., Holgado-Madruga M., Su W., Tsuiki H., Wedegaertner P., and Wong A. J. (2005) Identification of a specific domain responsible for JNK2α2 autophosphorylation. J. Biol. Chem. 280, 9913–9920 10.1074/jbc.M412165200 [DOI] [PubMed] [Google Scholar]
- 46. Clancy R., Rediske J., Koehne C., Stoyanovsky D., Amin A., Attur M., Iyama K., and Abramson S. B. (2001) Activation of stress-activated protein kinase in osteoarthritic cartilage: evidence for nitric oxide dependence. Osteoarthritis Cartilage 9, 294–299 10.1053/joca.2000.0388 [DOI] [PubMed] [Google Scholar]
- 47. Forsyth C. B., Pulai J., and Loeser R. F. (2002) Fibronectin fragments and blocking antibodies to α2β1 and α5β1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 46, 2368–2376 10.1002/art.10502 [DOI] [PubMed] [Google Scholar]
- 48. Loeser R. F., Forsyth C. B., Samarel A. M., and Im H. J. (2003) Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J. Biol. Chem. 278, 24577–24585 10.1074/jbc.M304530200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ismail H. M., Miotla-Zarebska J., Troeberg L., Tang X., Stott B., Yamamoto K., Nagase H., Fosang A. J., Vincent T. L., and Saklatvala J. (2016) Brief report: JNK-2 controls aggrecan degradation in murine articular cartilage and the development of experimental osteoarthritis. Arthritis Rheumatol. 68, 1165–1171 10.1002/art.39547 [DOI] [PubMed] [Google Scholar]
- 50. Han Z., Chang L., Yamanishi Y., Karin M., and Firestein G. S. (2002) Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. 46, 818–823 10.1002/art.10104 [DOI] [PubMed] [Google Scholar]
- 51. Poole L. B., and Ellis H. R. (1996) Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry 35, 56–64 10.1021/bi951887s [DOI] [PubMed] [Google Scholar]
- 52. Poole L. B., Klomsiri C., Knaggs S. A., Furdui C. M., Nelson K. J., Thomas M. J., Fetrow J. S., Daniel L. W., and King S. B. (2007) Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug. Chem. 18, 2004–2017 10.1021/bc700257a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Loeser R. F., Pacione C. A., and Chubinskaya S. (2003) The combination of insulin-like growth factor 1 and osteogenic protein 1 promotes increased survival of and matrix synthesis by normal and osteoarthritic human articular chondrocytes. Arthritis Rheum. 48, 2188–2196 10.1002/art.11209 [DOI] [PubMed] [Google Scholar]
- 54. Pulai J. I., Chen H., Im H. J., Kumar S., Hanning C., Hegde P. S., and Loeser R. F. (2005) NF-κB mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J. Immunol. 174, 5781–5788 10.4049/jimmunol.174.9.5781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Klatt P., Molina E. P., and Lamas S. (1999) Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J. Biol. Chem. 274, 15857–15864 10.1074/jbc.274.22.15857 [DOI] [PubMed] [Google Scholar]
- 56. Nelson K. J., Parsonage D., Hall A., Karplus P. A., and Poole L. B. (2008) Cysteine pKa values for the bacterial peroxiredoxin AhpC. Biochemistry 47, 12860–12868 10.1021/bi801718d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang F., Strand A., Robbins D., Cobb M. H., and Goldsmith E. J. (1994) Atomic structure of the MAP kinase ERK2 at 2.3 Å resolution. Nature 367, 704–711 10.1038/367704a0 [DOI] [PubMed] [Google Scholar]
- 58. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.